

Abstract
Background
The genetics underlying body mass and growth are key to understanding a wide range of topics in biology, both evolutionary and developmental. Body mass and growth traits are affected by many genetic variants of small effect. This complicates genetic mapping of growth and body mass. Experimental intercrosses between individuals from divergent populations allows us to map naturally occurring genetic variants for selected traits, such as body mass by linkage mapping. By simultaneously measuring traits and intermediary molecular phenotypes, such as gene expression, one can use integrative genomics to search for potential causative genes.
Results
In this study, we use linkage mapping approach to map growth traits (N = 471) and liver gene expression (N = 130) in an advanced intercross of wild Red Junglefowl and domestic White Leghorn layer chickens. We find 16 loci for growth traits, and 1463 loci for liver gene expression, as measured by microarrays. Of these, the genes TRAK1, OSBPL8, YEATS4, CEP55, and PIP4K2B are identified as strong candidates for growth loci in the chicken. We also show a high degree of sex-specific gene-regulation, with almost every gene expression locus exhibiting sex-interactions. Finally, several trans-regulatory hotspots were found, one of which coincides with a major growth locus.
Conclusions
These findings not only serve to identify several strong candidates affecting growth, but also show how sex-specificity and local gene-regulation affect growth regulation in the chicken.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4441-3) contains supplementary material, which is available to authorized users.
Keywords: Growth, Liver, QTL, Gene expression, eQTL, Genetical genomics
Background
The molecular basis of variation in quantitative traits, such as body mass, is still very much an open question. Knowledge of how variation in body mass is affected by underlying genes and polymorphisms has been an area of intense study, and has ramifications for many diverse areas of biology, ranging from evolutionary theory, to human health, in the form of obesity risks, and to domestic animal production. For example, the genetic regulation of bodyweight in humans has proven to be complex but vital area of study [1], with over 13% of the worldwide population currently obese, whilst this prevalence has doubled since 1980, as estimated by the WHO. Various model species have been used for body weight analysis, ranging from C.elegans to Drosophila [2]. Domestication can be seen as a case-study in artificial selection for growth, and as such is an excellent tool in understanding the genetic basis of body mass regulation and control. Body mass has increased due to breeding in all major livestock species (reviewed by [3]). During domestication, and particularly the later phase of breed improvement, chickens have increased in body mass from between twofold to over fivefold as compared to their wild progenitor, the Red Junglefowl [4]. Similar to other species, chicken body mass is clearly polygenic, affected by variants at many loci. Body mass is a target of direct selection in chickens, particularly meat-type broilers [5]. It is also likely affected by correlated selection for egg production in layers, since there is genetic evidence of pleiotropy between traits [6], with body mass a strong determinant of egg size [7]. Finally, because body mass is likely related to fitness in the wild, wild and domestic chickens are likely to experience different selection pressure on body mass.
As is the case with all complex traits, there are very few known causal sequence variants that affect bodymass, yet some examples do exist. These indicate that diverse molecular processes can contribute to heritable body mass variation, some being of major-effect, far more being of small effect. There are Mendelian loss-of-function mutations that make animals small [8, 9]. For example, the sex-linked dwarfing locus in the chicken is caused by a growth hormone receptor mutation [10]. In mice, the dwarf locus is caused by a mutated Pit-1 transcription factor, and loss of pituitary function, while the little locus is caused by mutation in the growth hormone-releasing hormone receptor [9]. There are also major quantitative trait loci (QTL) that have been resolved. In pigs, a regulatory variation affecting the insulin like growth factor 2 affects muscle growth and mass [11]. In mice, a major quantitative trait locus for body mass is caused by gene-regulatory variation at Glypican 3 [12], and variation in the lipo-oxygenase gene Alox5 affects gene expression and multiple metabolic traits [13]. Both of these loci appear to act through changes to liver gene expression. Genome-wide association studies of obesity in humans have isolated the FTO/IRX3 locus, where it appears that a variant in an intron of the FTO gene affects body mass index by a regulatory effect on the neighbouring gene IRX3 [14, 15].
To understand the mechanisms of chicken domestication, we need to know which genes have been affected by gene-regulatory variation in domestication. The genetic variants that affect traits must do so by means of affecting intermediary molecular phenotypes. A good way to get at the intermediary molecular traits is to genetically map gene expression levels, as measured by transcript abundance. This approach is called expression quantitative trait locus mapping or genetical genomics [16]. So far, it has revealed a plethora of gene-regulatory variation, with the strongest effects being local, putatively cis-regulatory, effects of genetic variants in or close to the gene itself [17, 18]. There is also a tendency for associations to cluster in the genome, into expression quantitative trait locus hotspots. This pattern is consistent with trans-regulatory changes affecting networks or pathways of genes [19] and with phenotypic buffering, where the phenotype is robust to genetic variation yet disruptions to key buffering loci can give rise to hotspots of QTL [20].
The genetical genomics approach allows us to integrate gene expression and trait mapping to find candidate quantitative trait genes that explain loci. Take the case where a regulatory genetic variant changes the expression of a gene, affecting a pathway that influences a phenotypic trait. This means that both the trait value and the gene expression level of the causative gene will be associated with the same genomic region. In this way, the overlap of QTL and expression quantitative trait loci (eQTL) can be used as a first step to filter potentially causative genes. For each overlap, gene expression is then correlated with the phenotype of the relevant growth QTL. In this way, putatively causal genes can be identified, with this pruning the number of candidates, and providing far stronger evidence of causation above basic overlap [7, 21, 22].
The liver is a key metabolic organ, involved in the metabolism of carbohydrates, proteins, and fats. It also both produces and breaks down hormones. Consequently, several studies have successfully used liver gene expression to investigate the genomics of metabolic traits [12, 13, 23, 24]. However, genetic variation in body mass could also act through other mechanisms, such as appetite regulation in the nervous system [25], or muscle growth in broiler chickens [26].
In this paper, we map QTL for body mass by linkage mapping in an advanced intercross of wild Red Junglefowl and domestic White Leghorn layer chickens. The use of an advanced intercross gives us improved mapping resolution for loci affecting body mass in chicken domestication. By allowing individuals to interbreed over multiple generations, the intercross accumulates recombinations, thus improving mapping resolution [27]. In the advanced intercross used for this study, we see approximately a four-fold increase in mapping resolution [28]. Given the key role of the liver as an effector of metabolism, we map eQTL for liver gene expression from a subset of 130 of the phenotyped animals. In this way, we find genes that have been affected by gene-regulatory variation under domestication. We integrate the genetic and gene expression evidence using association overlaps and linear models, with this then generating a final list of putatively causal genes affecting growth.
Methods
Mapping population
The intercross was started in the 1990s by crossing a Red Junglefowl rooster of Thai origin to three White Leghorn L13 layer hens [29]. The Leghorn line stemmed from a Scandinavian breeding experiment. The cross was previously expanded for mapping in the F2 generation. Since then, it has been maintained at a population size of approximately 100 individuals per generation. For the F8 generation (used in this study), chickens were raised in six batches. We weighed the chickens at hatch and days 8, 42, 112 and 212. We have body mass phenotypes for 566 birds at day 8 (270 males and 296 females), out of which 471 remained at day 212 (231 males and 240 females). Since a chick’s body mass at hatch is largely determined by the mass of the egg from which it hatched, which in turn is dependent on maternal factors [30, 31], maternal genetic effects may confound QTL for early growth and body mass. Therefore, we mapped early growth traits only as the difference between mass at day 42 and day 8. The full list of phenotypes is therefore weight at days 42, 112, and 212, and growth between days 8 and 42, 42 and 112, and 112 and 212. Chickens were culled by cervical neck dislocation followed by decapitation (as per the ethical permit). Animal handling was as per the ethical permit for the project.
Quantitative trait locus mapping
All chickens (n = 566) were genotyped at 652 single nucleotide polymorphism markers by the Uppsala SNP&SEQ Technology Platform, using the Illumina Golden Gate assay. 551 markers were fully informative of parental origin, with the remainder being partially informative. The total map length was ~9268 cM, whilst the average marker spacing was ~16 cM. Additional file 1: Table S1 shows the genetic map with physical locations on the reference genome (version Galgal4). QTL for body mass at day 212 have been previously published in connection with brain proportion mapping [32].
We performed quantitative trait locus mapping using Haley-Knott regression [33] in the R/qtl package [34, 35]. Models included batch and sex as covariates, and genotype by sex interactions, where significant, were also included. Both additive and dominance effects were measured. Family effect was controlled for by including covariates of principal components based on global genotypes, with the first ten PCs checked against the phenotype to be tested, and all significant ones included in the model. We used both one-dimensional scans, detecting for single QTL, and two-dimensional scans for epistatic pairs of loci. In this way, we detect both marginal-effect and two-way epistatic loci, and then combined them in multiple-QTL models [36]. In this way, the multiple QTL model included covariates, main effects, sex interactions, and epistasis, starting with the most significant loci and working down for each trait. The power of the study (as calculated using the r/qtlDesign package [36]) was sufficient to give an 80% chance to detect a QTL of 4% effect size.
Permutation tests were carried out to find significance thresholds, using R/qtl, based on the classical and well established method as first outlined in [37, 38]. A 5% genome-wide LOD threshold was considered significant (LOD score ~4.3), whilst a 20% genome-wide threshold was considered as suggestive (LOD score ~3.7) [39]. Genomic confidence intervals were based on a 1.8 logarithm of odds (LOD) drop [40]. We compared the QTL with previously published growth loci from the F2 generation of the intercross, using the estimated physical locations in Animal QTLdb [41]. Epistatic interactions were also assessed using permutation thresholds generated using R/qtl, with a 20% suggestive and 5% significant genome-wide threshold again used. In the case of epistatic loci, the approximate average significance lod threshold for pairs of loci were as follows (as per the guidelines given in [36]): full model ~11, full versus one ~9, interactive ~7, additive ~7, additive versus one ~4. These are described in further depth in [36], but in brief these refer to the following: ‘Full’ refers to the lod threshold for the full two-locus model with additive and dominance effects included, ‘full vs 1’ is the threshold when comparing a two locus model as compared to a model with only one of the QTL pair fitted. ‘Lod.int’ gives the lod score threshold specifically for the interaction between the two QTL, ‘add’ gives the threshold for a two-locus model, but only considering additive effects, whilst ‘add versus 1’ gives the threshold for threshold for the additive two-locus model as compared to a single additive only QTL model.
Liver RNA samples
A randomly chosen subset (130) of the F8 chickens that were used to map growth were also used for expression analysis. The subset used for gene expression were therefore not selected based on phenotype. Chickens were culled at 212 days of age. Liver samples were frozen in liquid nitrogen and stored at −80 °C. Frozen samples were homogenized with TRIzol (Qiagen) in a FastPrep MP-24 homogenizer (MP Biomedical), and total RNA was isolated according to the manufacturer’s protocol. We assayed RNA integrity on a Bioanalyzer (Agilent). The RNA integrity numbers ranged from 7.3 to 9.8 (mean 8.9).
Gene expression microarrays
The eQTL sample consisted of 130 individuals from the intercross (67 males and 63 females). We made labelled cDNA with the Agilent Low Input Quick Amp Two Color Labeling kit. Agilent 8x60K arrays were used (with each array having 60 k probes and with 8 arrays per slide). Two different fluorophores were used (Cy3 and Cy5), enabling a total of 16 samples to be run per slide (2 samples per array and 8 arrays per slide). Arrays were hybridized according to the manufacturer’s protocol and scanned on a NimbleGen MS200 (Roche NimbleGen) scanner. We preprocessed array images with the Agilent Feature Extraction Software (version 12.0). These probe-level background-corrected values were then quantile-normalized, and summarized to probeset-level data by median polish using the preprocessCore R package (version 1.32.0 [42]). We used the ComBat method, implemented in the sva R package (version 3.18.0; [43]) to adjust for batch effects introduced by running multiple samples on the same slide.
Microarray probesets are based on Ensembl transcripts or RefSeq mRNA sequences, with 2–3 probes being used for each probeset, and summarized to one value per probeset. They were designed based on annotation from an older version of the chicken reference genome (WASHUC2.1/galGal3). To update the probesets, we first queried the Ensembl (version 85; [44]) database, through BioMart [45], for the current location of the genes underlying the probeset. In cases where the accession number had been retired, we used Blat [46] to align the probe sequences to Ensembl transcripts. When probe sequences aligned exclusively to transcripts from one Ensembl gene model, we annotated the probeset as belonging to that gene. This left us a total of 20,771 probesets, 16,360 of which were annotated to a genomic location.
Expression quantitative trait locus mapping
We performed expression quantitative trait locus mapping with Haley Knott regression in R/qtl. We analysed local and distal eQTL separately, scanning only the 100 cM region around the gene (i.e. 50 cM upstream and 50 cM downstream), based on the genetic distance in the F8 map, for local eQTL. We estimated genetic effects coefficients for each eQTL using R/qtl’s fitqtl function. Each model was fitted with batch, sex and fluorophore (two different fluorophore colours were used in each microarray, Cy3 or Cy5) as covariates. We also fitted each eQTL model with and without a sex interaction, and compared the logarithm of the odds (LOD). In the cases where the sex interaction QTL has a LOD score greater than 1 this indicates that the individual sex * QTL interaction term in the QTL model has a ~p < 0.05 (the exact number can vary slightly from model to model), and the interaction was included in the final model. Permutation was once again used to generate significance thresholds for the local and trans eQTL. In the case of the local eQTL only the markers surrounding the gene in question were tested, with this reducing the multiple testing correction required. To account for the full number of eQTL probeset phenotypes (20771) and in the case of the trans eQTL the full genetic map, permutations were based on 100 randomly sub-sampled probesets (rather than single phenotypes). A similar method was used for the local eQTL, though in this case only the limited 100 cM region around each gene was used for extracting LOD scores (100 probesets were still considered simultaneously however). This generated a local threshold of LOD ~4.0 and a trans LOD threshold of ~8.0. The power of the study (as calculated using the r/qtlDesign package [36]) was sufficient to give an 80% chance to detect a QTL of 16% effect size.
For each phenotypic quantitative trait locus, we found the overlapping local eQTL, and tested each of them for an association between the growth trait in question and gene expression. We used linear models with the growth trait value as response variable, and the gene expression value as predictor, with fluorophore (Cy3 or Cy5), and batch as covariates.
Gene Ontology annotation was downloaded from Ensembl (version 85) through BioMart.
Expression quantitative trait locus hotspots
We calculated the number of expression quantitative trait locus confidence intervals covering every part of the genome with the GenomicRanges R package [47], and compared it to the coverage of intervals in simulations where the intervals were placed at random. To avoid the risk of spurious hotspots driven by extreme individuals and skewed genotypes, we filtered out potential trans-eQTL where there were ten or fewer individuals of any genotype class. To generate a null distribution of eQTL, we simulated placing the distal eQTL confidence intervals randomly on a 1 Gb interval (the size of the sequenced autosomal chicken genome), and found the highest coverage in each simulation.
We also tested to see whether any trans-eQTL hotspots were potentially controlled by any of the local eQTL that overlapped the trans hotspot location. This was performed using a conditional eQTL model. First, we identified any local eQTL that overlapped the hotspot, and then performed a test of correlation between the expression levels of the respective cis and trans eQTL phenotypes. We used the residual gene expression value from a model that included sex, batch and fluorophore. Finally, we repeated the trans eQTL detection model (i.e. how genotype modifies gene expression), but now also included the expression of the local eQTL gene as a covariate in the trans-eQTL model. If the cis eQTL is a causal to the trans eQTL hotspot the inclusion of the cis eQTL as a covariate should reduce the variance explained by the genotypic effect (i.e they both explain the same variance component), and hence the strength of the trans eQTL should drop. We considered the local eQTL as a potential mediator of the trans-association if 1) there was a significant association between the expression values, and 2) the logarithm of the odds of the trans-eQTL was reduced by more than half when the local gene was included as a covariate in the trans-eQTL model. We used the circlize R package [48] for the circular plot of hotspots, and the igraph R package [49] for network plots. It must also be noted that such loci are only putative – rather than the trans eQTL being downstream targets of the local gene, it is impossible to rule out that the causal element has both local and trans regulatory properties.
Selective sweep overlap
We compared the QTL with selective sweep signals detected by [50] by searching for overlaps between layer and all domestic sweep regions and quantitative trait locus support intervals. We used WASHUC2.1/galGal3 genome coordinates, because the sweep dataset was based on that version of the reference genome. We generated an empirical null distribution by uniformly random placement of sweeps and counting the overlaps.
Results
QTL for body mass
We found a total of four loci for body mass at 42 days, five for body mass at 112 days and six for body mass at 212 days. We also found seven loci for growth between day 42 and 112, and three loci for growth between day 112 and 212. In total, this amounts to 16 genomic regions associated with body mass or growth (Table 1, Fig. 1a). There was some epistasis evident in the cross, in the form of six two-locus interactions (Fig. 1b).
Table 1.
QTL with estimates and position
| Chr | cM | Mb | LOD | Trait | a | SE | d | SE | a_by_sex | SE | d_by_sex | SE | Interactions | Interval start (Mb) | Interval end (Mb) | Variance explained (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 509 | 34 | 27.0 | w42 | 19.9 | 2.1 | 9.6 | 2.7 | 28.9 | 34.7 | 4.0 | |||||
| 1 | 510 | 34 | 5.5 | g112.212 | 31.4 | 6.4 | −4.8 | 8.0 | 28.9 | 34.7 | 2.0 | |||||
| 1 | 510 | 34 | 42.4 | w112 | 83.3 | 13.3 | 64.3 | 16.6 | −34.9 | 13.0 | 0.6 | 17.6 | 1@510.0:27@68.2 | 28.9 | 34.7 | 9.0 |
| 1 | 510 | 34 | 43.6 | w212 | 159.8 | 13.1 | 15.6 | 17.1 | −75.4 | 16.8 | 35.9 | 23.2 | 28.9 | 34.7 | 8.4 | |
| 1 | 512 | 35 | 35.3 | g42.112 | 85.3 | 7.9 | 14.2 | 10.2 | −47.4 | 10.0 | 20.1 | 14.0 | 28.9 | 34.7 | 7.0 | |
| 1 | 1138 | 81 | 4.3 | w112 | −25.6 | 6.0 | −7.0 | 8.0 | 77.6 | 85.0 | 0.8 | |||||
| 1 | 1137 | 81 | 5.1 | g42.112 | −23.1 | 5.2 | −9.0 | 6.9 | 78.6 | 85.0 | 0.9 | |||||
| 2 | 158 | 18 | 9.4 | g112.212 | 1.2 | 7.1 | −6.5 | 9.4 | 11@4.0:2@158.0 | 16.7 | 19.3 | 3.4 | ||||
| 2 | 255 | 34 | 5.2 | w42 | 12.6 | 3.8 | −27.7 | 6.8 | −8.8 | 5.3 | 29.1 | 9.3 | 29.0 | 39.1 | 0.7 | |
| 3 | 631 | 93 | 7.5 | w42 | −19.8 | 4.5 | −7.2 | 6.8 | 8.7 | 5.7 | −6.7 | 8.9 | 92.6 | 95.1 | 1.0 | |
| 4 | 265 | 31 | 8.6 | w212 | 7.8 | 9.8 | 19.0 | 12.3 | 4@265.0:24@14.1 | 30.2 | 32.9 | 1.4 | ||||
| 4 | 493 | 76 | 11.7 | g42.112 | −9.2 | 7.6 | −7.3 | 9.7 | 30.7 | 9.3 | −28.2 | 13.4 | 4@493.0:17@169.0 | 73.8 | 79.3 | 2.1 |
| 6 | 207 | 21 | 5.0 | w212 | 26.2 | 8.2 | 29.3 | 11.1 | 16.0 | 21.6 | 0.8 | |||||
| 6 | 259 | 27 | 13.8 | g42.112 | 15.5 | 5.5 | 53.4 | 7.6 | 6@259.0:12@66.0 | 25.5 | 29.9 | 2.5 | ||||
| 6 | 258.7 | 27 | 9.0 | w112 | 26.6 | 13.1 | 37.3 | 14.5 | 27@68.2:6@258.7 | 25.5 | 29.9 | 1.6 | ||||
| 10 | 177 | 12 | 4.4 | w42 | 8.8 | 2.4 | 8.2 | 2.9 | 11.1 | 15.1 | 0.6 | |||||
| 11 | 4 | 1 | 9.2 | g112.212 | −5.1 | 6.9 | −16.6 | 9.1 | 11@4.0:2@158.0 | 1.3 | 3.8 | 3.4 | ||||
| 12 | 66 | 5 | 8.6 | g42.112 | 12.2 | 7.2 | −35.5 | 10.0 | 6@259.0:12@66.0 | 2.7 | 17.4 | 1.5 | ||||
| 12 | 64 | 5 | 4.4 | w212 | 34.7 | 10.2 | −19.8 | 14.4 | 2.7 | 5.9 | 0.7 | |||||
| 17 | 169 | 9 | 8.3 | g42.112 | 20.6 | 6.0 | −25.5 | 8.7 | 4@493.0:17@169.0 | 7.2 | 9.5 | 1.5 | ||||
| 23 | 147 | 5 | 4.8 | w112 | −31.0 | 7.0 | 7.4 | 11.1 | 3.5 | 4.7 | 0.9 | |||||
| 23 | 154.9 | 5 | 3.8 | g42.112 | −20.4 | 5.1 | 0.8 | 6.8 | 3.5 | 4.7 | 0.7 | |||||
| 24 | 14.1 | 1 | 9.5 | w212 | 1.1 | 9.0 | −38.1 | 10.9 | 4@265.0:24@14.1 | 1.1 | 1.5 | 1.5 | ||||
| 27 | 68.1 | 3 | 4.2 | w212 | 37.8 | 11.1 | −1.7 | 13.3 | 2.4 | 3.4 | 0.7 | |||||
| 27 | 68.2 | 3 | 9.6 | w112 | 43.6 | 15.0 | 19.2 | 16.1 | 1@510.0:27@68.2 27@68.2:6@258.7 | 2.4 | 4.7 | 1.8 |
Columns indicate the trait, the location of the QTL (in cM and Mb), the LOD score for the QTL, the additive and dominance estimates with standard errors, the coefficients for additive and dominance by sex interactions with standard errors, epistatic pairs that the QTL contribute to, the limits of the QTL confidence interval (in Mb), and the variance explained
Fig. 1.
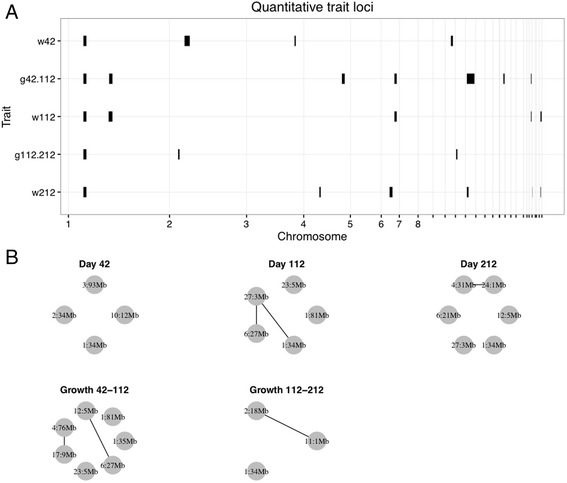
a Genomic confidence intervals of the QTL. The horizontal axis shows physical locations along the chicken genome (chromosomes 1 to 28). The red lines indicate QTL overlap between at least two traits. b Epistatic networks for body mass and growth loci. Nodes represent loci, labelled by their chromosome and physical location, and edges pairwise epistatic interactions between them
The loci for body mass at earlier and later time points suggest a different architecture for early growth and adult body mass. This cross has a major body mass locus on chromosome 1, previously named growth1, which was the only locus that is detected at all ages. Except for growth1, there was no overlap between body mass loci at day 42 and day 112, and only one more overlapping locus (on chromosome 27) between day 112 and day 212. In comparison with previously published QTL from the F2 generation of the intercross [4], we replicated four QTL regions on chromosomes 1, 2, 12 and 27 (Additional file 2: Figure S1). There was also another overlapping locus on chromosome 6 with a previously published reanalysis of the F2 data [51]. We also overlapped the detected QTL with the selective sweeps due to domestication and improvement detected by [50] (overlaps given in Additional file 1: Table S1). Multiple overlaps were observed between selective sweeps and QTL, although there was no significant enrichment of sweeps observed in QTL regions (i.e the numbers were not significantly different from those that would be observed by chance). The most sweeps appeared to be more apparent in the growth phenotypes. For example, g112–212 (growth between 112 and 212 days) had a total of 11 sweeps overlapping 3 QTL (5 sweeps seen in all domestic birds and 6 sweeps observed in layer birds), whilst g42–112 (growth between 42 and 112 days) had 11 sweeps overlapping 7 QTL regions (5 ‘all domestic’ sweeps and 6 layer-specific sweeps). In comparison, weight at 212 days only had 5 sweeps overlapping 6 QTL regions. This could indicate that selection is acting principally on growth (particularly early to intermediate growth stages) rather than final adult weight.
Gene expression quantitative trait loci in liver
We found 1463 eQTL, 1201 local and 262 distal trans-acting (Fig. 2a, Additional file 3: Table S2). Evidence of a sex x QTL interaction was seen in 1077 local (90%) and 260 (99%) distal (trans) loci.
Fig. 2.
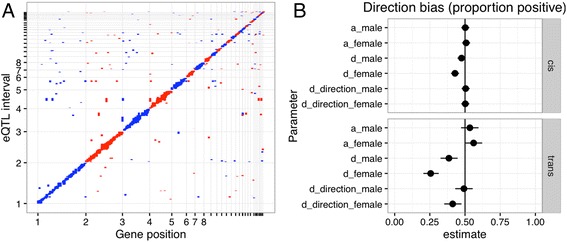
a eQTL local-distal plot. Plot of the genomic position of the expression quantitative trait locus confidence intervals (vertical axis) and their respective gene (horizontal axis). The diagonal represents local eQTL, mapping to the location of the gene itself. b Proportions of positive and negative genetic effects
The additive effects of the eQTL appear to have no bias in the direction of effect. Approximately as many loci were associated with a higher expression in the Junglefowl allele as the White Leghorn allele, in both males and females for both local and distal loci (Fig. 2b). Given that we have no reason to expect biased genotypic effects, this result is as expected. However, when it comes to the dominance effects, local and distal loci seem to differ. The local eQTL in males again appeared unbiased. However, the majority of female local eQTL and trans eQTL in both sexes had a negative dominance effect, meaning that the heterozygote expression was lower than expected by the mid-parental value. We also investigated the direction of dominance in terms of bias of the heterozygote value towards the Red Junglefowl or White Leghorn homozygote, and found no evidence of a directional bias in any sex. The strongest local eQTL include genes that likely effect liver function. We found eQTL affecting mitochondrial NADH dehydrogenases NDUFA8 (LOD = 40) and NDUFS6 (LOD = 16), transforming growth factor beta 2 (TGFB2, LOD =18), and redox enzymes thioredoxin (LOD = 15) and thioredoxin reductase 3 (LOD = 23). We also found a female-specific locus for beta-carotene oxygenase 2 (BCO2, LOD = 21), which causes the yellow-skin phenotype in domestic chickens (Additional file 4: Figure S2). The most common Gene Ontology Biological Process terms among genes with eQTL were related to transcription, signal transduction, transport, and protein phosphorylation (Additional file 5: Table S3).
Genetical genomics search for candidate quantitative trait genes
By overlapping phenotypic and gene eQTL, we searched for candidates for body mass at different ages (Table 2; Fig. 3; Additional file 6: Figure S4; Additional file 7: Figure S5 and Additional file 8: Figure S6). We considered a gene a candidate quantitative trait gene if: 1) It had a local expression quantitative locus overlapping the confidence interval of the phenotypic locus, and 2) the expression level of the gene was also associated with the trait, allowing for sex-interaction and adjusting for technical covariates. In total five candidate genes were identified using this approach. Trafficking protein, kinesin binding 1 is a candidate for body mass at 42 days on chromosome 2 (TRAK1; ENSGALG00000011938, LOD = 5, correlation P = 0.003). YEATS domain-containing protein 4 (YEATS4; ENSGALG00000029135, LOD = 5.3, correlation P = 0.006) and oxysterol binding protein like 8 (OSBPL8; ENSGALG00000010246, LOD = 4.4, correlation P = 0.0035) are candidates for body mass at 42 days on chromosome 1, at growth1. Given its apparent female-specific effect, YEATS4 seems a less likely candidate than OSBPL8 for growth1, which has a clear effect on both sexes. However, growth1 could also be made up of multiple linked loci. There was a second LOD score peak for body mass at day 42 and 112 (Additional file 9: Figure S7). If so, the location of OSBPL8 is suggestive of a role in this second locus. Centrosomal protein 55 (CEP55; ENSGALG00000006639) is a candidate for body mass at 112 days on chromosome 6. On chromosome 27, Phosphatidylinositol-5-phosphate 4-kinase type-2 beta (PIP4K2B; ENSGALG00000001610) is a candidate for the adult body mass locus. The localization of the expression quantitative trait locus, however, is imprecise for PIP4K2B, with a peak relatively far from the peak of the phenotypic locus.
Table 2.
Candidate genes from expression quantitative trait locus mapping
| Gene name | Chr | Start | Trait | LOD | p-value | Interval start | Interval end | Ensembl gene ID |
|---|---|---|---|---|---|---|---|---|
| YEATS4 | 1 | 35.4 | w42 | 5.3 | 0.0060 | 33.6 | 44.1 | ENSGALG00000029135 |
| OSBPL8 | 1 | 38.0 | w42 | 4.4 | 0.0035 | 33.6 | 45.7 | ENSGALG00000010246 |
| TRAK1 | 2 | 44.3 | w42 | 5.0 | 0.0026 | 29.0 | 39.1 | ENSGALG00000011938 |
| CEP55 | 6 | 20.0 | w112 | 4.7 | 0.0042 | 21.6 | 25.5 | ENSGALG00000006639 |
| PIP4K2B | 27 | 4.0 | w212 | 5.7 | 0.0017 | 1.2 | 4.7 | ENSGALG00000001610 |
Columns indicate the name of the gene, its chromosome and location (in Mb), the LOD score for the eQTL, p-value for the regression between gene expression level and trait value, the locations of the eQTL confidence interval (in Mb), and the Ensembl gene ID
Fig. 3.
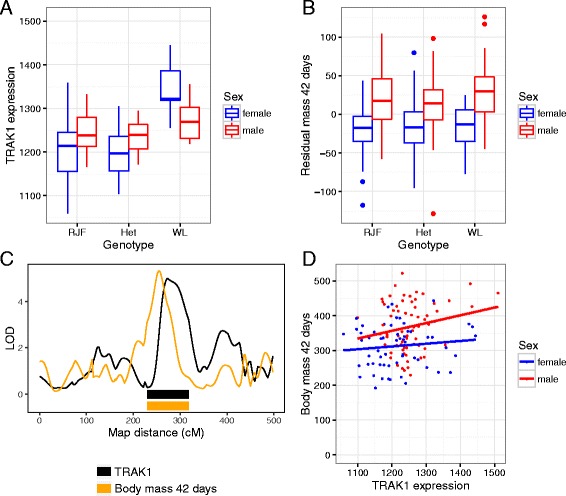
Plots of individual phenotypes versus their genotype (a, b), Logarithm of the odds curves (c), and scatterplots of body mass versus gene expression for TRAK1 for body mass at 42 days (d)
Trans-regulatory hotspots
We found nine putative trans-regulatory hotspots, where more expression quantitative trait locus mapped than would be expected by chance (Fig. 4a, Additional file 10: Table S5). Two of them, on chromosomes 1 and 12, overlapped phenotypic QTL: the chromosome 1 hotspot overlapped growth1, and the chromosome 12 hotspot overlapped a locus for growth from day 42 to 112. We used local eQTL overlapping trans-acting loci to search for genes that may regulate multiple genes (Fig. 4b). The resulting network only included 32 of the distal, putatively-trans acting loci. In one case, the hotspot on chromosome 5, we found a regulatory candidate that may explain the associations (Fig. 4c). At the chromosome 5 hotspot, the analysis suggested that a gene-regulatory variant affecting carbohydrate sulfotransferase 12 (CHST12; ENSGALG00000004276) had downstream effects on collagen 10A1 (COAA1; ENSGALG00000014965), retinoic acid receptor responder protein 1 (RARRES1; ENSGALG00000009594, represented by two probesets), lysyl oxidase homolog 2 (LOXL2; ENSGALG00000000402), RAS p21 protein activator (GTPase activating protein) 1 (RASA1; ENSGALG00000017706), and an uncharacterized gene (ENSGALG00000001885). This hotspot appeared to be female-specific (Additional file 4: Figure S2; Additional file 11: Figure S3). We did not try to infer the connections between the trans eQTL as there may be more complicated direct and indirect regulatory relationships between them.
Fig. 4.
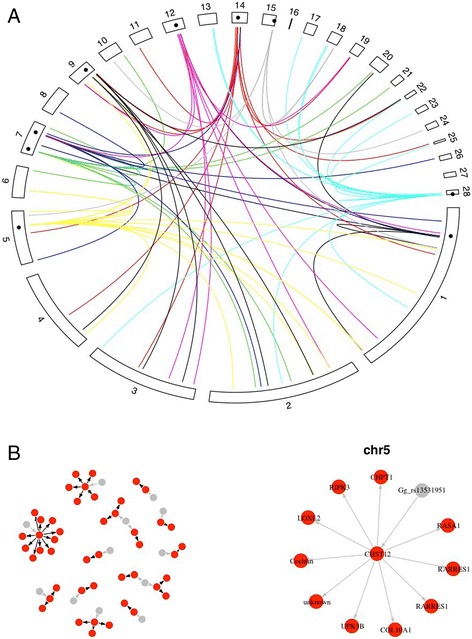
a Circular genome plot of putative trans-eQTL hotspots. The points show the location of hotpots on the genome, and the arcs show trans-eQTL associated with the hotspot. Each hotspot has its own colour. b eQTL network plots with the chromosome 5 hotspot zoomed in. Grey nodes represent markers associated with genes. Red nodes represent probesets. The edges represent potential regulatory relationships from a local eQTL to distal trans-eQTL
Correlations between gene expression and growth traits
In addition to testing the overlapping QTL/eQTL genes, a search for genes whose expression is associated with growth and body mass regardless of the presence of an eQTL was also conducted. To this end, we used the same linear model as for the positional candidate genes above, but applied it across the whole array using a permutation test. In this way, we found 40 probesets associated with traits (Additional file 12: Table S4). The list includes regulatory genes such as SOX5 (ENSGALG00000013204, P = 7 × 10−5), and BDNF (ENSGALG00000012163, P = 2 × 10−5). The list does not overlap with the candidate genes found for the phenotypic loci. However, two of these candidates had local eQTL. One of them, LOC776181 similar to receptor for egg jelly-like protein (ENSGALG00000014240, P = 1 × 10−4) also overlapped a phenotypic quantitative trait locus for the same trait, namely body mass at 112 days, and in this way can also be considered to be a putative candidate gene.
Discussion
In this paper, we perform quantitative trait locus mapping of growth traits, and expression quantitative trait locus of liver gene expression in an advanced intercross of wild Red Junglefowl and domestic White Leghorn layer chickens. We find that the genetic architecture of body mass is specific to different ages, with little overlap between QTL for early growth and adult body mass. The only locus detectable at every age is the major growth locus, growth1, which explains around 9% of the variance in weight. One caveat with this is that smaller, undetected loci may well be shared between early and adult growth traits but have not been detected in this study. By measuring liver gene expression in a subset of the cross (130 individuals), we detected 1463 eQTL.
Using an integrative genomics approach we identified candidate genes for some of the loci for body mass at 42, 112 and 212 days. Two of the candidate genes have known connections to metabolism or cell proliferation. TRAK1 is involved in mitochondrial transport and attachment to microtubules [52, 53]. In broiler chickens, differences in mitochondrial function are associated with feed efficiency [54, 55]. This suggests that genetic effects on the expression of mitochondrial proteins could affect growth in chickens. OSBPL8 binds oxysterols, and changing its expression in transgenic mice, altering lipid levels in liver and blood [56]. The other candidate for this locus, YEATS4 is involved in gene regulation and cell proliferation [57]. It is also located close to a genome-wide association signal for human height that occurs in the neighbouring gene FRS2 [58, 59]. CEP55 encodes a microtubule-associated protein involved in cell division [60] and is required for embryonic development in zebrafish [61]. PIP4K2B is an enzyme that phosphorylates phosphatidylinositol 5-phosphate. The product is involved in regulation of cell proliferation, and overexpression of PIP4K2B can be contribute to cancer [62].
The quantitative trait locus analysis of body mass replicates five out of the 13 loci from the F2 generation of the intercross [4, 63]. As opposed to the marginal effect loci, the patterns of epistasis found in the F2 generation do not replicate. For example, we detect again loci on chromosome 1, 6, 12 and 27 for adult body mass from the F2 reanalysis [51]. In the previous study, these loci were all involved in at least one epistatic interaction, while we find no epistatic interactions between any of them. There are several reasons why epistatic effects in an F2 cross may not replicate in an advanced intercross. Firstly, the previous analyses used different software, and slightly different phenotypes (in that phenotypes were recorded at slightly different ages, though always within a few days of each other between the two studies), though these are both relatively minor differences. Secondly, the longer linkage blocks in an F2 intercross cause lower resolution, and what one detects may be synthetic associations made up of several linked loci. As recombination reduces linkage over generations, the apparent epistatic patterns may change. Another reason for the lack of detected epistasis may be the unbalanced allele frequencies of the advanced intercross. Allele frequencies have changed by drift over the generations, which make genotype frequencies deviate from the near perfect 1:2:1 Mendelian ratios of an F2. This makes some multilocus genotypes rare, reducing the power to detect epistasis. Regardless, our results do not support a major effect for epistasis in chicken growth under domestication. We find some epistatic pairs, but most loci lack significant epistatic effects, and only one locus is involved in more than one interaction. Finally, there was no enrichment of selective sweeps, previously detected in chicken domestication [50], in the growth and body mass QTL regions. However, despite there being no significant enrichment, multiple selective sweep regions were nevertheless detected within the various QTL. These may very well reflect genuine selection signals on growth or weight phenotypes. The clearest way to prove or disprove recent selection is the cause of these QTL is to identify the underlying causal variant(s) for each QTL and determine their position relative to these sweeps, though this is by no means a trivial task. Intriguingly, if these sweeps are causal to the observed QTL, this would indicate that selection has been principally acting on growth rather than final weight.
Most of the eQTL are local. This agrees with the general results of expression quantitative trait studies, from various organisms, that genetic variation in gene expression is abundant, and often local. However, there is also the general issue of the higher significance threshold for trans eQTL making them harder to detect, and potentially downwardly biasing their detection. When it comes to distal, putatively trans-acting eQTL, we find nine potential trans hotspots. Further investigation would be needed to know whether these are genuine trans-regulatory hotspots and what traits they may participate in. Interestingly, most of the eQTL exhibit sex-interactions. This indicates that males and females potentially have distinct architectures for liver-specific gene expression, or at the very least the size of the effect of many of the eQTL varies between the sexes. Given the liver’s role in energy balance, and the extreme size difference between male and female chickens, this may not be unusual. For example, extensive sexual dimorphism in liver gene expression has been observed in the mouse [64]. This study found that ~72% of the genes expressed in mouse liver were sexually dimorphic in expression (in comparison ~13% of genes were sexually dimorphic in the brain). Given such sex-differences seem to be present in the liver transcriptome, sex x genotype interactions (when the eQTL allele has a greater effect in one sex) are less surprising. This consistent relationship between the liver transcriptome and sex bias seen in both chickens and mice could potentially reflect the degree of sexual dimorphism present in body size, with the chickens increased size sexual dimorphism (as compared to the mouse) being reflected in the slightly greater liver transcriptome sex bias, though this remains to be tested further. Sex interaction may also be exaggerated because of the lower power to detect sex-specific eQTL. In particular, because of the signal to noise ratio of the microarrays it may be harder to detect an expression quantitative trait locus in the sex with lower expression (in these instances, usually females). This sex interaction does raise a caveat regarding the candidate genes that were identified, as the eQTL candidates interacted between the sexes, whereas the bodyweight QTL for the most part did not. This could indicate that different genes underlie the same QTL in males and females, or that the lower power in the eQTL scan makes identifying the weaker sex-effect more problematic.
Conclusions
In conclusion, we detected 16 loci affecting growth traits in chicken domestication and 1463 eQTL affecting genes that may change liver function. We also highlight six potential candidates genes for affecting body mass in the chicken.
Additional files
Number of overlaps between quantitative trait locus regions and selective sweep regions from [50]. (CSV 426 bytes)
QTL from this study and previous F2 analysis [4] and reanalysis [51] based on physical locations in Animal QTLdb. Since the entries from the reanalysis lack confidence intervals, points indicate the loci. (PDF 5 kb)
All eQTL. (CSV 124 kb)
Figure S2 Female-specific expression quantitative trait locus for BCO2, showing gene expression level as a function of genotype. (PDF 6 kb)
Most common Gene Ontology Biological process terms among eQTL. (XLSX 8 kb)
YEATS4 and OSBPL8 candidate plots. (PDF 19 kb)
CEP55 candidate plots. (PDF 11 kb)
PIP42KB candidate plots. (PDF 10 kb)
Logarithm of odds curves for the region of chromosome 1 containing the two significant QTL, and a potential second peak, after growth1, which may be a second, imperfectly resolved QTL. The dashed lines indicate genome-wide significance thresholds for the respective trait. (PDF 7 kb)
Trans-expression quantitative trait locus hotspots. (XLSX 35 kb)
Plots of gene expression versus genotype for eQTL of the chromosome 5 hotspot. (PDF 22 kb)
Genes with expression levels associated with growth traits. (CSV 2 kb)
Genotype and phenotype data underlying quantitative trait locus mapping analysis in R/qtl format. (CSV 1191 kb)
Genotype and phenotype data underlying expression quantitative trait locus mapping analysis in R/qtl format. (CSV 39860 kb)
Acknowledgements
The research was carried out within the framework of the Swedish Centre of Excellence in Animal Welfare Science, and the Linköping University Neuro-network. SNP genotyping was performed by the Uppsala Sequencing Center.
Funding
The project was supported by grants from the Carl Tryggers Stiftelse, Swedish Research Council (VR), the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS), the Linköping University Neuro-network and European Research Council (advanced research grant GENEWELL 322206).
Availability of data and materials
The datasets supporting the conclusions of this article are available included in the additional files and in ArrayExpress repository. Phenotype, genotype and preprocessed gene expression values are available in Additional files 13 and 14. The raw gene expression data have been uploaded to ArrayExpress (http://www.ebi.ac.uk/arrayexpress) with accession E-MTAB-5572.
Authors’ contributions
Conceived and designed the experiments: DW, RH, AH, JP, MJ, PJ. Performed the experiments: MJ, RH, AH, JP, DW. Analyzed the data: MJ, DW. Wrote the paper: MJ, RH, AH, JP, PJ, DW. All authors read and approved the final manuscript.
Ethics approval
The animal studies were approved by Jordbruksverket (Swedish Animal Ethics board), with ethical permit Dnr 122–10.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4441-3) contains supplementary material, which is available to authorized users.
Contributor Information
Martin Johnsson, Email: martin.johnsson@slu.se.
Rie Henriksen, Email: rie.henriksen@liu.se.
Andrey Höglund, Email: andrey.hoglund@liu.se.
Jesper Fogelholm, Email: jesper.fogelholm@liu.se.
Per Jensen, Email: perje@ifm.liu.se.
Dominic Wright, Email: domwright@gmail.com.
References
- 1.Loos RJ, Yeo GS. The bigger picture of FTO [mdash] the first GWAS-identified obesity gene. Nat Rev Endocrinol. 2014;10(1):51–61. doi: 10.1038/nrendo.2013.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morton G, Cummings D, Baskin D, Barsh G, Schwartz M. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 3.Thornton PK. Livestock production: recent trends, future prospects. Philos Trans R Soc B Biol Sci. 2010;365(1554):2853–2867. doi: 10.1098/rstb.2010.0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kerje S, Carlborg O, Jacobsson L, Schutz K, Hartmann C, Jensen P, Andersson L. The twofold difference in adult size between the red junglefowl and white leghorn chickens is largely explained by a limited number of QTLs. Anim Genet. 2003;34(4):264–274. doi: 10.1046/j.1365-2052.2003.01000.x. [DOI] [PubMed] [Google Scholar]
- 5.Zuidhof M, Schneider B, Carney V, Korver D, Robinson F. Growth, efficiency, and yield of commercial broilers from 1957, 1978, and 2005. Poult Sci. 2014;93(12):PS4291. [DOI] [PMC free article] [PubMed]
- 6.Festing MF, Nordskog A. Response to selection for body weight and egg weight in chickens. Genetics. 1967;55(2):219. doi: 10.1093/genetics/55.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnsson M, Jonsson KB, Andersson L, Jensen P, Wright D. Quantitative trait locus and genetical genomics analysis identifies putatively causal genes for fecundity and brooding in the chicken. G3 Genes Genomes Genet. 2016;6(2):311–319. doi: 10.1534/g3.115.024299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li S, Crenshaw E, 3rd, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature. 1990;347(6293):528–533. doi: 10.1038/347528a0. [DOI] [PubMed] [Google Scholar]
- 9.Godfrey P, Rahal JO, Beamer WG, Copeland NG, Jenkins NA, Mayo KE. GHRH receptor of little mice contains a missense mutation in the extracellular domain that disrupts receptor function. Nat Genet. 1993;4(3):227–232. doi: 10.1038/ng0793-227. [DOI] [PubMed] [Google Scholar]
- 10.Burnside J, Liou SS, Zhong C, Cogburn LA. Abnormal growth hormone receptor gene expression in the sex-linked dwarf chicken. Gen Comp Endocrinol. 1992;88(1):20–28. doi: 10.1016/0016-6480(92)90190-U. [DOI] [PubMed] [Google Scholar]
- 11.Van Laere AS, Nguyen M, Braunschweig M, Nezer C, Collette C, Moreau L, Archibald A, Haley CS, Buys N, Tally M, et al. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in pigs. Nature. 2003;425:832–836. doi: 10.1038/nature02064. [DOI] [PubMed] [Google Scholar]
- 12.Oliver F, Christians JK, Liu X, Rhind S, Verma V, Davison C, Brown SD, Denny P, Keightley PD. Regulatory variation at glypican-3 underlies a major growth QTL in mice. PLoS Biol. 2005;3(5):e135. doi: 10.1371/journal.pbio.0030135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehrabian M, Allayee H, Stockton J, Lum P, Drake T, Castellani L, Suh M, Armour C, Edwards S, Lamb J, et al. Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat Genet. 2005;37(11):1224–1233. doi: 10.1038/ng1619. [DOI] [PubMed] [Google Scholar]
- 14.Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507(7492):371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ragvin A, Moro E, Fredman D, Navratilova P, Drivenes Ø, Engström PG, Alonso ME, de la Calle ME, Skarmeta JLG, Tavares MJ. Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3. Proc Natl Acad Sci. 2010;107(2):775–780. doi: 10.1073/pnas.0911591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jansen R, Nap J. Genetical genomics: the added value from segregation. Trends Genet. 2001;17(7):388–391. doi: 10.1016/S0168-9525(01)02310-1. [DOI] [PubMed] [Google Scholar]
- 17.Aylor DL, Valdar W, Foulds-Mathes W, Buus RJ, Verdugo RA, Baric RS, Ferris MT, Frelinger JA, Heise M, Frieman MB. Genetic analysis of complex traits in the emerging collaborative cross. Genome Res. 2011;21(8):1213–1222. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schadt E, Monks S, Drake T, Lusis A, Che N, Colinayo V, Ruff T, Milligan S, Lamb J, Cavet G, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422(6929):297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- 19.Breitling R, Li Y, Tesson BM, Fu J, Wu C, Wiltshire T, Gerrits A, Bystrykh LV, de Haan G, Su AI, et al. Genetical genomics: spotlight on QTL hotspots. PLoS Genet. 2008;4(10):e1000232. doi: 10.1371/journal.pgen.1000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu J, Keurentjes JJ, Bouwmeester H, America T, Verstappen FW, Ward JL, Beale MH, De Vos RC, Dijkstra M, Scheltema RA. System-wide molecular evidence for phenotypic buffering in Arabidopsis. Nat Genet. 2009;41(2):166–167. doi: 10.1038/ng.308. [DOI] [PubMed] [Google Scholar]
- 21.Johnsson M, Jonsson KB, Andersson L, Jensen P, Wright D. Genetic regulation of bone metabolism in the chicken: similarities and differences to mammalian systems. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnsson M, Williams MJ, Jensen P, Wright D. Genetical genomics of behavior: a novel chicken genomic model for anxiety behavior. Genetics. 2016;202(1):327–340. doi: 10.1534/genetics.115.179010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, GuhaThakurta D, Sieberts SK, Monks S, Reitman M, Zhang C, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37(7):710–717. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome. 2007;18(6–7):463–472. doi: 10.1007/s00335-007-9043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt C, Persia M, Feierstein E, Kingham B, Saylor W. Comparison of a modern broiler line and a heritage line unselected since the 1950s. Poult Sci. 2009;88(12):2610–2619. doi: 10.3382/ps.2009-00055. [DOI] [PubMed] [Google Scholar]
- 27.Darvasi A, Soller M. Advanced intercross lines, an experimental population for fine genetic-mapping. Genetics. 1995;141(3):1199–1207. doi: 10.1093/genetics/141.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnsson M, Gustafson I, Rubin C-J, Sahlqvist A-S, Jonsson KB, Kerje S, Ekwall O, Kämpe O, Andersson L, Jensen P, et al. A sexual ornament in chickens is affected by pleiotropic alleles at HAO1 and BMP2, selected during domestication. PLoS Genet. 2012;8(8):e1002914. doi: 10.1371/journal.pgen.1002914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schutz K, Kerje S, Carlborg O, Jacobsson L, Andersson L, Jensen P. QTL analysis of a red junglefowl x white leghorn intercross reveals trade-off in resource allocation between behavior and production traits. Behav Genet. 2002;32(6):423–433. doi: 10.1023/A:1020880211144. [DOI] [PubMed] [Google Scholar]
- 30.Tullett S, Burton FG. Factors affecting the weight and water status of the chick at hatch. Br Poult Sci. 1982;23(4):361–369. doi: 10.1080/00071688208447969. [DOI] [Google Scholar]
- 31.Wilson H. Interrelationships of egg size, chick size, posthatching growth and hatchability. Worlds Poult Sci J. 1991;47(01):5–20. doi: 10.1079/WPS19910002. [DOI] [Google Scholar]
- 32.Henriksen R, Johnsson M, Andersson L, Jensen P, Wright D. The domesticated brain: genetics of brain mass and brain structure in an avian species. Sci Rep. 2016;6:34031. doi: 10.1038/srep34031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haley CS, Knott SA. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity. 1992;69:315–324. doi: 10.1038/hdy.1992.131. [DOI] [PubMed] [Google Scholar]
- 34.Broman KW, Wu H, Sen S, Churchill GA. R/QTL: QTL maping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- 35.Team RC . R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2008. [Google Scholar]
- 36.Broman KW, Sen S. A guide to QTL mapping with r/qtl. New York: Springer; 2009. [Google Scholar]
- 37.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doerge RW, Churchill GA. Permutation tests for multiple loci affecting a quantitative character. Genetics. 1996;142:285–294. doi: 10.1093/genetics/142.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lander ES, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 40.Manichaikul A, Dupuis J, Sen S, Broman KW. Poor performance of bootstrap confidence intervals for the location of a quantitative trait locus. Genetics. 2006;174(1):481–489. doi: 10.1534/genetics.106.061549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Z-L, Fritz ER, Reecy JM. AnimalQTLdb: a livestock QTL database tool set for positional QTL information mining and beyond. Nucleic Acids Res. 2007;35(suppl 1):D604–D609. doi: 10.1093/nar/gkl946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 43.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 44.Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L. Ensembl 2016. Nucleic Acids Res. 2016;44(D1):D710–D716. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21(16):3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 46.Kent W. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9(8):e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gu Z, Gu L, Eils R, Schlesner M, Brors B. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19):btu393. [DOI] [PubMed]
- 49.Csardi G, Nepusz T. The igraph software package for complex network research. InterJ Complex Syst. 2006;1695(5):1–9. [Google Scholar]
- 50.Rubin C-J, Zody MC, Eriksson J, Meadows JRS, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–591. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- 51.Le Rouzic A, Álvarez-Castro JM, Carlborg Ö. Dissection of the genetic architecture of body weight in chicken reveals the impact of epistasis on domestication traits. Genetics. 2008;179(3):1591–1599. doi: 10.1534/genetics.108.089300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogawa F, Malavasi EL, Crummie DK, Eykelenboom JE, Soares DC, Mackie S, Porteous DJ, Millar JK. DISC1 complexes with TRAK1 and Miro1 to modulate anterograde axonal mitochondrial trafficking. Hum Mol Genet. 2013;23(4):ddt485. [DOI] [PMC free article] [PubMed]
- 53.Koutsopoulos OS, Laine D, Osellame L, Chudakov DM, Parton RG, Frazier AE, Ryan MT. Human Miltons associate with mitochondria and induce microtubule-dependent remodeling of mitochondrial networks. Biochim Biophys Acta-Mol Cell Res. 2010;1803(5):564–574. doi: 10.1016/j.bbamcr.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 54.Bottje W, Iqbal M, Pumford N, Ojano-Dirain C, Lassiter K. Role of mitochondria in the phenotypic expression of feed efficiency. J Appl Poult Res. 2004;13(1):94–105. doi: 10.1093/japr/13.1.94. [DOI] [Google Scholar]
- 55.Iqbal M, Pumford N, Tang Z, Lassiter K, Ojano-Dirain C, Wing T, Cooper M, Bottje W. Compromised liver mitochondrial function and complex activity in low feed efficient broilers are associated with higher oxidative stress and differential protein expression. Poult Sci. 2005;84(6):933–941. doi: 10.1093/ps/84.6.933. [DOI] [PubMed] [Google Scholar]
- 56.Zhou T, Li S, Zhong W, Vihervaara T, Beaslas O, Perttilä J, Luo W, Jiang Y, Lehto M, Olkkonen VM. OSBP-related protein 8 (ORP8) regulates plasma and liver tissue lipid levels and interacts with the nucleoporin Nup62. PLoS One. 2011;6(6):e21078. doi: 10.1371/journal.pone.0021078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park JH, Roeder RG. GAS41 is required for repression of the p53 tumor suppressor pathway during normal cellular proliferation. Mol Cell Biol. 2006;26(11):4006–4016. doi: 10.1128/MCB.02185-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Luan JA, Kutalik Z. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46(11):1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berndt SI, Gustafsson S, Mägi R, Ganna A, Wheeler E, Feitosa MF, Justice AE, Monda KL, Croteau-Chonka DC, Day FR. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet. 2013;45(5):501–512. doi: 10.1038/ng.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.W-m Z, Seki A, Fang G. Cep55, a microtubule-bundling protein, associates with centralspindlin to control the midbody integrity and cell abscission during cytokinesis. Mol Biol Cell. 2006;17(9):3881–3896. doi: 10.1091/mbc.E06-01-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeffery J, Neyt C, Moore W, Paterson S, Bower NI, Chenevix-Trench G, Verkade H, Hogan BM, Khanna KK. Cep55 regulates embryonic growth and development by promoting Akt stability in zebrafish. FASEB J. 2015;29(5):1999–2009. doi: 10.1096/fj.14-265090. [DOI] [PubMed] [Google Scholar]
- 62.Luoh S-W, Venkatesan N, Tripathi R. Overexpression of the amplified Pip4k2β gene from 17q11–12 in breast cancer cells confers proliferation advantage. Oncogene. 2004;23(7):1354–1363. doi: 10.1038/sj.onc.1207251. [DOI] [PubMed] [Google Scholar]
- 63.Carlborg O, Kerje S, Schutz K, Jacobsson L, Jensen P, Andersson L. A global search reveals epistatic interaction between QTL for early growth in the chicken. Genome Res. 2003;13(3):413–421. doi: 10.1101/gr.528003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, Drake TA, Lusis AJ. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006;16(8):995–1004. doi: 10.1101/gr.5217506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Number of overlaps between quantitative trait locus regions and selective sweep regions from [50]. (CSV 426 bytes)
QTL from this study and previous F2 analysis [4] and reanalysis [51] based on physical locations in Animal QTLdb. Since the entries from the reanalysis lack confidence intervals, points indicate the loci. (PDF 5 kb)
All eQTL. (CSV 124 kb)
Figure S2 Female-specific expression quantitative trait locus for BCO2, showing gene expression level as a function of genotype. (PDF 6 kb)
Most common Gene Ontology Biological process terms among eQTL. (XLSX 8 kb)
YEATS4 and OSBPL8 candidate plots. (PDF 19 kb)
CEP55 candidate plots. (PDF 11 kb)
PIP42KB candidate plots. (PDF 10 kb)
Logarithm of odds curves for the region of chromosome 1 containing the two significant QTL, and a potential second peak, after growth1, which may be a second, imperfectly resolved QTL. The dashed lines indicate genome-wide significance thresholds for the respective trait. (PDF 7 kb)
Trans-expression quantitative trait locus hotspots. (XLSX 35 kb)
Plots of gene expression versus genotype for eQTL of the chromosome 5 hotspot. (PDF 22 kb)
Genes with expression levels associated with growth traits. (CSV 2 kb)
Genotype and phenotype data underlying quantitative trait locus mapping analysis in R/qtl format. (CSV 1191 kb)
Genotype and phenotype data underlying expression quantitative trait locus mapping analysis in R/qtl format. (CSV 39860 kb)
Data Availability Statement
The datasets supporting the conclusions of this article are available included in the additional files and in ArrayExpress repository. Phenotype, genotype and preprocessed gene expression values are available in Additional files 13 and 14. The raw gene expression data have been uploaded to ArrayExpress (http://www.ebi.ac.uk/arrayexpress) with accession E-MTAB-5572.