

 Silver(i) and copper(i) complexes of a
macrocyclic NHC-based pincer ligand, bearing a central lutidine donor and a
dodecamethylene spacer, have been prepared and evaluated under equivalent conditions in
transmetallation reactions.
Silver(i) and copper(i) complexes of a
macrocyclic NHC-based pincer ligand, bearing a central lutidine donor and a
dodecamethylene spacer, have been prepared and evaluated under equivalent conditions in
transmetallation reactions.
Abstract
With a view to use as carbene transfer agents, well-defined silver(i) and copper(i) complexes of a macrocyclic NHC-based pincer ligand, bearing a central lutidine donor and a dodecamethylene spacer [CNC–(CH2)12, 1], have been prepared. Although the silver adduct is characterised by X-ray diffraction as a dinuclear species anti-[Ag(μ-1)]2 2+, variable temperature measurements indicate dynamic structural interchange in solution involving fragmentation into mononuclear [Ag(1)]+ on the NMR time scale. In contrast, a mononuclear structure is evident in both solution and the solid-state for the analogous copper adduct partnered with the weakly coordinating [BArF 4]– counter anion. A related copper derivative, bearing instead the more coordinating cuprous bromide dianion [Cu2Br4]2–, is notable for the adoption of an interesting tetranuclear assembly in the solid-state, featuring two cuprophilic interactions and two bridging NHC donors, but is not retained on dissolution. Coinage metal precursors [M(1)]n[BArF 4]n (M = Ag, n = 2; M = Cu, n = 1) both act as carbene transfer agents to afford palladium, rhodium and nickel complexes of 1 and the effectiveness of these precursors has been evaluated under equivalent reaction conditions.
Introduction
The chemistry of N-heterocyclic carbene (NHC) complexes of the transition elements is rich, varied and at the forefront of contemporary organometallic chemistry and catalysis.1 While the formation of these adducts via direct coordination of the singlet carbene, isolated or generated in situ through deprotonation of the corresponding azolium salt, is conceptually the simplest method, the high chemical reactivity of these free carbene intermediates or incompatibility of the reaction conditions with acidic functionalities have necessitated the development of alternative approaches involving isolable ‘protected’ carbenes and transmetallation procedures. In this context, Ag(i)–NHC complexes have proved to be effective carbene transfer agents for a wide variety of late-transition elements (Scheme 1).2–4 These reagents are conveniently formed through reaction of the respective pro-ligand NHC·HX with Ag2O, resulting in mono- or bis-ligated complexes [Ag(NHC)X] or [Ag(NHC)2]X depending on the nature of the anion (X).2,3 Although comparatively less common, Cu(i)–NHC complexes have also been employed as carbene transfer agents (Scheme 1).2,5 Such copper reagents can be prepared from reactions between Cu2O and NHC·HX, but are generally prepared via free carbene intermediates.2 Interestingly, reflecting the relative M(i)–NHC bond strengths of the coinage metals (Au > Cu > Ag),6 transmetallation of both silver and copper complexes can be used to prepare gold derivatives as a result of the more robust nature of Au(i)–NHC bonds.2–5
Scheme 1. Selected synthesis and reactions of Cu(i) and Ag(i)–NHC complexes.4,5 .

The application of carbene transfer methodology is not limited to monodentate examples and the use of silver-based transmetallation protocols is also prevalent in the coordination chemistry of mer-tridentate “pincer” ligands bearing terminal NHC donors.7,8 While the corresponding silver transfer agents are often generated in situ, well defined and characteristically bimetallic Ag(i)–CEC (E = C, N) complexes such as A–C have been isolated and crystallographically characterised (Scheme 2).9–15 Copper adducts of CEC (E = C, N) ligands have also been prepared (e.g. D and E),10,16–18 however, their application as transfer agents has yet to be realised to our knowledge.
Scheme 2. Examples of isolated Ag(i) and Cu(i) complexes of CNC pincer ligands.12–17 .

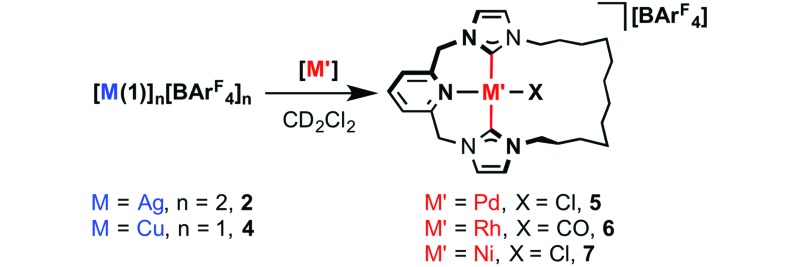
As part of our work involving the organometallic chemistry of macrocyclic CNC pincers,19 we now report the synthesis and characterisation of well-defined Ag(i) and Cu(i) adducts of a lutidine-based pincer ligand bearing a dodecamethylene spacer [CNC–(CH2)12, 1]. The use of these coinage metal species as transfer agents is then detailed for the synthesis of rhodium, palladium, and nickel complexes of 1.
Results and discussion
Our preceding work exploring the coordination chemistry of 1 employed in situ generation of silver transfer agents from the reaction between 1·2HBr and Ag2O in CH2Cl2 solution.19 As a convenient means of incorporating a weakly coordinating [BArF 4]– anion20 and conferring solution solubility, these reactions were carried out in the presence of Na[BArF 4] (ArF = 3,5-C6H3(CF3)2) as halogen ion abstractor. Using a slightly adapted protocol to facilitate isolation, well-defined silver derivative 2 was prepared via reaction in diethyl ether (Scheme 3). Filtration to remove insoluble silver and sodium bromide salts, and subsequent recrystallization from CHCl3/pentane, afforded analytically pure 2 as a white powder in reproducibly high isolated yields of ca. 80%.
Scheme 3. Preparation of 2 and related literature precedents.11 .

Single crystals grown from diethyl ether/pentane and analysed by X-ray crystallography enabled structural elucidation of 2 in the solid-state as a dinuclear complex anti-[Ag(μ-1)]2[BArF 4]2 (Fig. 1) – as for closely related precedents B, F and G.11,13 Interestingly, while the principle geometric metrics about silver in 2 are directly comparable to the aforementioned precedents (e.g. Ag–NHC = 2.088(4), 2.093(4) Å, B; 2.077(3), 2.080(3) Å, 2; NHC–Ag–NHC = 176.78(9)°, B; 178.99(13)°, 2), the ligand topology is significantly altered. At the heart of the structural difference is the adoption of near orthogonal NHC–Ag–NHC geometries in 2 [N25–C24–C18*–N19* = 100.2(4)°], in contrast to coplanar NHC–Ag–NHC arrangements observed in B, F and G. This change in geometry is presumably necessary to accommodate the long aliphatic chain and as a consequence results in a very large Ag···Ag* separation [7.2732(6) cf. 3.7171(5), B; 3.538(2), F; 4.6636(7) Å, G], precluding the adoption of any argentophilic interaction as seen in A and C.12,14,15.
Fig. 1. Solid-state structure of 2. Thermal ellipsoids drawn at the 50% probability level; hydrogen atoms, minor disordered components and anions omitted for clarity. The starred atoms are generated by the symmetry operation 2 – x, 1 – y, 1 – z. Selected bond lengths (Å) and angles (°): Ag1–C18, 2.077(3); Ag1–C24*, 2.080(3); Ag1···Ag1*, 7.2732(6); C24–Ag1–C18*, 178.99(13); N25–C24–C18*–N19*, 100.2(4).

1H and 13C NMR data recorded in CD2Cl2 confirm the expected 1 : 1 ligand to anion ratio and reveal time averaged C 2v symmetry for 2 at 298 K (500 MHz). Such high symmetry is inconsistent with retention of the solid-state structure and instead implies highly fluxional behaviour in solution, involving fragmentation into [Ag(1)]+ (cf. structure of 4 vide infra). Such an assertion is supported by ESI-MS, where only a singly charged species (i.e. integer mass spacing) was evident in the mass spectrum ([Ag(1)]+, 512.1927; calc. 512.1938 m/z). Not surprisingly the carbene signals of 2 were not observed by 13C{1H} NMR spectroscopy at 298 K, although a chemical shift of ca. δ 181 ppm can be inferred from 2D heteronuclear correlation experiments in line with expected values for Ag(i)–NHC complexes.2,3b,21 Progressive cooling to 250 K lead to decoalescence of the (broadened) 1H signals of 2 observed in CD2Cl2 at 298 K and establishment of an equilibrium mixture comprised of three major compounds, one of C 2v symmetry and two of apparent C s symmetry (1 : 0.5 : 1). These species are tentatively assigned as [Ag(1)]+, syn-[Ag(μ-1)]2 2+ and anti-[Ag(μ-1)]2 2+, respectively, on the basis of related behaviour observed for F and G involving rapid equilibration between syn- and anti-isomers in solution (Scheme 3); dynamics that necessitate Ag–NHC bond cleavage and invoke coordination of the central lutidine donor through comparison to the less fluxional m-xylylene bridged analogue H.11 In the case of 2, the NMR data suggests that incorporation of the long dodecamethylene spacer destabilises the dinuclear structures relative to entropically favored fragmentation into [Ag(1)]+ at ambient temperature.22 Further cooling to 200 K resulted in a shift in the equilibrium toward the species assigned to anti-[Ag(μ-1)]2 2+ (ca. 60%) consistent with the pseudo C i symmetric structure observed in the solid-state being enthalpically favoured in solution. In the context of 2 being used as a carbene transfer agent, these NMR data ultimately demonstrate facile Ag–NHC bond cleavage under conditions relevant to synthesis of other transition metal adducts of 1 via transmetallation.
The synthesis of copper adducts of 1 was targeted by low temperature deprotonation of 1·2HBr in THF in the presence of excess copper bromide. In this manner [Cu(1)]2[Cu2Br4] 3 was formed and subsequently isolated in 76% yield (Scheme 4). Further treatment of 3 with Na[BArF 4] in toluene resulted in incorporation of the weakly coordinating [BArF 4]– anion in place of [Cu2Br4]2– to afford 4 in 59% isolated yield.20,23 In CD2Cl2 solution, the 1H and 13C NMR characteristics of both 3 and 4 point towards simple mononuclear complexes of 1, with sharp resonances and apparent C 2v symmetry in the respective spectra at 298 K (400 MHz). Moreover, only minor differences in chemical shift are found for the equivalent 1H (<0.15 ppm) and 13C (<2 ppm) signals of 3/4, and presumably attributed to greater ion pairing in 3. Of most relevance to the coordination of 1, the carbenic centres were readily identified from 13C{1H} NMR spectra by their characteristically high frequency chemical shifts (δ 178.7, 3; δ 180.3, 4).2 Strong parent cation signals are observed by ESI-MS with correct isotope patterns and integer mass spacing (468.2185, 3; 468.2186, 4; calc. 468.2183 m/z), further supporting the presence of discrete [Cu(1)]+ in solution, irrespective of the counter anion.
Scheme 4. Preparation of 3, 4 and a related literature precedent.25 .

Despite similar solution characteristics, in the solid-state the nature of the counter anion impacts significantly on the coordination geometries of 3 and 4 (Fig. 2 and 3). Two independent, but well-separated cation/anion pairs are observed in the solid-state structure of 4. The cationic fragments are structurally similar, displaying near ideal T-shaped coordination geometries with the flexible dodecamethylene spacer notably skewed to one side. Focusing on the metrics associated with the independent cation shown in Fig. 2,24 the complex displays an approximately linear NHC–Cu–NHC angle (176.37(13)°), equivalent Cu–NHC bond lengths within error (1.905(3)/1.906(3) Å), and a Cu–N bond length of 2.233(3) Å. These parameters are in good agreement with those reported for E, although the anionic nature of the component CNC pincer ligand leads to a shorter Cu–N bond length than that in 4 (2.017(2) Å). In the case of 3, an anion bridged dimer/tetranuclear formulation is instead observed, viz [Cu(1)]2{μ-[Cu2Br4]}, featuring two formally bridging NHC donors (Cu1–C24, 1.980(3); Cu2–C24, 2.070(3) Å) and two cuprophilic interactions (Cu–Cu, 2.5209(5) Å). The bonding interaction with the anion results in a significant distortion of the coordination geometry observed in 4, with the NHC–Cu–NHC angle deviating from linearity (167.92(11)°) alongside elongation of the non-bridged Cu–NHC (1.925(3) cf. 1.905(3)/1.906(3) Å) and Cu–N (Cu–N = 2.246(2) cf. 2.233(3)) bonds. The bridging μ2-NHC coordination mode is an unusual feature of 3, but has striking precedence in trinuclear copper clusters, such as I, that contain three ligands in this coordination mode (Cu–NHC ca. 2.026(5)–2.044(5) Å for I).25,26 In these trinuclear clusters, the bridging coordination mode is retained in solution and characterised by δ 13C 167–169 for imidazolyline based variants; values to significantly lower frequency than found for 3 (δ 13C 178.7).25,26
Fig. 2. Solid-state structure of 4. Thermal ellipsoids drawn at the 50% probability level; only one of the unique cations shown (Z′ = 2) and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Cu1–N10, 2.233(3); Cu1–C18, 1.905(3); Cu1–C24, 1.906(3); C18–Cu1–C24, 176.37(13).

Fig. 3. Solid-state structure of 3. Thermal ellipsoids drawn at the 50% probability level; hydrogen atoms omitted for clarity. The starred atoms are generated by the symmetry operation 1 – x, 1 – y, 1 – z. Selected bond lengths (Å) and angles (°): Cu1–Cu2, 2.5209(5); Cu1–N10, 2.246(2); Cu1–C18, 1.925(3); Cu1–C24, 1.980(3); Cu2–Br3, 2.4583(4); Cu2–Br3*, 2.7728(5); Cu2–Br4, 2.4149(4); Cu2–C24, 2.070(3); Cu2–Br3–Cu2*, 83.794(15); Br3–Cu2–Br3*, 96.206(15); Cu1–Cu2–C24, 49.92(7); C18–Cu1–C24, 167.92(11).

With well-defined coinage metal complexes 2 and 4 in hand, we turned to evaluation of their capacity to act as transfer agents of macrocyclic 1 in CH2Cl2. As convenient benchmarks we targeted preparation of known and soluble palladium and rhodium adducts of 1, [Pd(1)Cl][BArF 4] 5 and [Rh(1)(CO)][BArF 4] 6, through reactions with [Pd(NCMe)2Cl2] and [Rh(CO)2Cl]2, respectively (Table 1).19b,c Following these reactions in situ by 1H NMR spectroscopy in CD2Cl2, using the [BArF 4]– resonances as a convenient internal standard, revealed rapid and high yielding transmetallation reactions of 2 in both cases (>70% yield). Consistent with these high yields, 5 and 6 have previously been isolated in 58% and 52% yield, respectively, through in situ generation of the silver transfer agent.19b,c In the case of copper, although complete consumption of 4 was apparent within 30 min in both cases, a large difference in selectivity is apparent: rhodium-based 6 was formed with an excellent yield of 98% (and subsequently isolated in 82% yield), while palladium-based 5 was formed with a significantly inferior yield of 23%.
Table 1. Transmetallation reactions of 2 and 4 a .
| |||||
| [M′] | M | t b /h | T/°C | Product | Yield c |
| [Pd(NCMe)2Cl2] | Ag | 0.5 | 20 | 5 | 73% |
| [Pd(NCMe)2Cl2] | Cu | 0.5 | 20 | 5 | 23% |
| [Rh(CO)2Cl]2 | Ag | 0.5 | 20 | 6 | 72% |
| [Rh(CO)2Cl]2 | Cu | 0.5 | 20 | 6 | 98% |
| [NiCl2(glyme)] | Ag | 20 d | 20 | 7 | 22% |
| [NiCl2(glyme)] | Ag | 20 | 40 | 7 | 76% |
| [NiCl2(glyme)] | Cu | 20 d | 20 | 7 | 86% |
| [NiCl2(glyme)] | Cu | 5 | 40 | 7 | 90% |
aReactions carried out in J. Young's NMR tubes, which were periodically placed in a ultrasound bath during the course of the reaction.
bComplete conversion unless otherwise noted.
cDetermined by integration of 1H NMR data.
dIncomplete reaction.
Seeking to expand the scope of this transmetallation methodology, we targeted the preparation of nickel derivative 7 – the lighter and Earth abundant group 10 congener of 5. Using the aforementioned methodology in combination with [NiCl2(glyme)], resulted in slow dissolution of the largely insoluble nickel(ii) precursor, and gratifying (albeit gradual) formation of 7 at room temperature over 20 hours. The reaction with copper-based 4 notably proceeded ca. 4 times faster, suggesting the explanation for this behaviour is more complex than low solubility of [NiCl2(glyme)] alone. Repeating under more forcing conditions (40 °C) resulted in complete consumption of 2 (20 h) and 4 (5 h) and formation of 7 in 76% and 90% yield, respectively. The new air and moisture stable complex 7 was subsequently isolated from these reactions in ca. 30% yield following purification on alumina and fully characterised. Alternatively, 7 can also be prepared in similar yield using in situ generation of 2 from 1·2HBr and Ag2O (29% isolated yield).
In the solid-state, 7 shows the expected contraction of metal–ligand bond lengths in comparison to 5 (Ni–Cl, 2.145(2); Ni–N, 1.928(5); Ni–C, 1.898(6), 1.916(6) Å; Pd–Cl, 2.287(4); Pd–N, 2.077(10); Pd–C, 2.036(12), 2.056(13) Å), but is otherwise isostructural with the palladium-based analogue (Fig. 4).19c Most notably, the chloride ancillary ligand is easily accommodated within the macrocyclic ring, which is orientated to maintain pseudo C 2 symmetry (cf. twisting observed in 4), with an essentially linear N–Ni–Cl bond angle (179.06(15) cf. 176.2(3)° for the N–Pd–Cl angle in 5). In solution the solid-state structure is fully retained as indicated by the observation of diastereotopic methylene bridge (pyCH2) and N-methylene (N-CH2CH2) resonances at δ 5.14/6.30 (2 J HH = 15.0 Hz) and δ 3.73/4.73, respectively. A single carbenic carbon signal is observed at δ 162.0 (cf. δ 164.5, 5).
Fig. 4. Solid-state structure of 7. Thermal ellipsoids drawn at the 50% probability level; hydrogen atoms and anion omitted for clarity. Selected bond lengths (Å) and angles (°): Ni1–Cl2, 2.145(2); Ni1–N10, 1.928(5); Ni1–C18, 1.898(6); Ni1–C24, 1.916(6); N10–Ni1–Cl2, 179.06(15); C18–Ni1–C24, 176.6(3).

Summary
Well-defined silver(i) and copper(i) complexes of a macrocyclic NHC-based pincer ligand, bearing a central lutidine donor and a dodecamethylene spacer [CNC–(CH2)12, 1], have been prepared. In the solid-state silver adduct 2 is characterised by X-ray diffraction as a dinuclear species anti-[Ag(μ-1)]2 2+, with the two metal centres held distant from each other (Ag···Ag, >7 Å) as a consequence of the conformation of the bridging macrocyclic ligand. However, variable temperature 1H NMR spectroscopy indicates dynamic structural interchange in solution involving fragmentation into mononuclear [Ag(1)]+. In contrast, a mononuclear structure is evident in both solution and the solid-state for the analogous copper adduct 4 partnered with the weakly coordinating [BArF 4]– counter anion. A related copper derivative, bearing instead the more coordinating cuprous bromide dianion [Cu2Br4]2–, is notable for the adoption of an interesting tetranuclear assembly in the solid-state, featuring two cuprophilic interactions (Cu–Cu, 2.5209(5) Å) and two bridging NHC donors, but is not retained in solution. Coinage metal complexes [M(1)]n[BArF 4]n (M = Ag, n = 2, 2; M = Cu, n = 1, 4) both act as carbene transfer agents to afford palladium (5), rhodium (6) and nickel complexes (7) of 1. Although not extensive, these reactions suggest that while silver-based transfer agents are more reliable, copper-based alternatives can result in significantly faster and higher yielding transmetallation reactions.
Experimental
General experimental methods
Manipulations were performed under an inert atmosphere, using Schlenk and glove box techniques unless otherwise stated. Glassware was oven dried and flamed under vacuum prior to use. Anhydrous solvents (<0.005% H2O) were purchased from ACROS or Aldrich and used as supplied: Et2O, CHCl3, pentane, CH2Cl2, THF and toluene. CD2Cl2 was dried over molecular sieves (4 Å) and stored under an atmosphere of argon. Na[BArF 4],27 [Rh(CO)2Cl]2,28 and 1·2HBr19c were synthesised using literature procedures. All other reagents are commercial products and were used as received. NMR spectra were recorded on Bruker HD-300, DPX-400, AV-400, DRX-500 and AVIII-500 HD spectrometers at 298 K unless otherwise stated. Chemical shirts are quoted in ppm and coupling constants in Hz. ESI-MS were recorded on a Bruker MaXis mass spectrometer. Microanalyses were performed at the London Metropolitan University by Stephen Boyer.
Synthesis of 2
A mixture of 1·2HBr (100 mg, 0.176 mmol), Ag2O (42 mg, 0.181 mmol) and Na[BArF 4] (170 mg, 0.192 mmol) was suspended in Et2O (5 mL) and stirred under argon in the absence of light for 48 h. The resulting grey suspension was allowed to settle and the solution filtered through a celite plug (pipette, 3 cm) under a flow of nitrogen. The colourless filtrate was concentrated to afford the crude product as white foam, which was subsequently dissolved in hot chloroform and filtered through a second celite plug (pipette, 3 cm). The analytically pure product was obtained on addition of excess pentane, washed with pentane and dried. Yield: 200 mg (82%, white powder). Crystals suitable for X-ray diffraction were grown from ether/pentane at 20 °C.
1H NMR (400 MHz, CD2Cl2) δ 7.82 (br, 1H, py), 7.70–7.75 (m, 8 H, ArF), 7.56 (br, 4H, ArF), 7.42 (br, 2H, py), 7.17 (br, 2H, imid), 7.05 (s, 2H, imid), 5.25 (br, 4H, pyCH2), 4.06 (app. t, J = 7, 4H, NCH2), 1.79 (app. pent., J = 7, 4H, CH2) 1.10–1.40 (m, 16 H, CH2). 13 C{ 1 H} NMR (101 MHz, CD2Cl2) δ 162.3 (q, 1 J CB = 50, ArF), 155.1 (s, py), 140.5 (br, py), 135.4 (s, ArF), 129.4 (qq, 2 J FC = 32, 3 J CB = 3, ArF), 125.3 (q, 1 J FC = 273, ArF), 125.0 (br, py), 124.1 (br, imid), 120.2 (br, imid), 118.0 (pent., 3 J FC = 4, ArF), 57.1 (s, pyCH2), 54 (obscured, NCH2), 31.4 (s, CH2), 27.5 (br, CH2), 25.8 (s, CH2). The carbene resonance was not unambiguously identified in the 13C{1H} NMR spectrum, but can be located at ca. δ 181 from an HMBC experiment. ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 512.1927 m/z, [M]+ (calc. 512.1938). Anal. Calcd for C57H47AgBF24N5·CHCl3 (1496.05 g mol–1): C, 46.28; H, 3.22; N, 4.64. Found: C, 46.08; H, 3.23; N, 4.58.
Synthesis of 3
A suspension of 1·2HBr (200 mg, 0.353 mmol) and CuBr (152 mg, 1.057 mmol) in THF (7 mL) under argon was cooled to –78 °C before addition of a solution of KOtBu (100 mg, 0.881 mmol) in THF (2 mL) via cannula. The resulting orange suspension was warmed to room temperature and sonicated to produce a yellow suspension, which was stirred for a further 30 h. The reaction mixture was filtered and the product precipitated by addition of excess pentane, isolated by filtration, washed with pentane and dried. Yield: 147 mg (76%, yellow powder). Crystals suitable for X-ray diffraction were grown from CD2Cl2/pentane at 20 °C.
1 H NMR (400 MHz, CD2Cl2) δ 7.78 (t, 3 J HH = 7.7, 1H, py), 7.39 (d, 3 J HH = 7.7, 2H, py), 7.22 (d, 3 J HH = 1.7, 2H, imid), 6.98 (d, 3 J HH = 1.7, 2H, imid), 5.37 (s, 4H, pyCH2), 4.17 (t, 3 J HH = 6.8, 4H, NCH2), 1.83 (app. pent., J = 7, 4H, CH2), 1.14–1.34 (m, 16H, CH2). 13 C{ 1 H} NMR (101 MHz, CD2Cl2) δ 178.7 (s, NCN), 155.5 (s, py), 139.4 (s, py), 123.2 (s, py), 122.5 (s, imid), 120.8 (s, imid), 56.3 (s, pyCH2), 52.1 (s, NCH2), 31.2 (s, CH2), 28.1 (s, CH2), 27.8 (s, CH2), 25.8 (s, CH2). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 468.2185 m/z, [M]+ (Calc. 468.2183). Anal. Calcd for C50H70Br4Cu4N10 (1384.96 g mol–1): C, 43.36; H, 5.09; N, 10.11. Found: C, 43.28; H, 4.97; N, 9.99.
Synthesis of 4
A suspension of 3 (53 mg, 0.0383 mmol) and Na[BArF 4] (90 mg, 0.1016 mmol) was sonicated in toluene (3 mL) and then stirred at room temperature for 2 days before filtration. The filtrate was concentrated to dryness and the crude product crystallised from THF/pentane. Yield: 62 mg (61%, pale yellow powder) as 4·0.5THF. Crystals suitable for X-ray diffraction were grown from ether/pentane at 20 °C.
1 H NMR (400 MHz, CD2Cl2) δ 7.83 (t, 3 J HH = 7.8, 1H, py), 7.73 (bs, 8H, ArF), 7.56 (br, 4H, ArF), 7.42 (d, 3 J HH = 7.8, 2H, py), 7.10 (d, 3 J HH = 1.2, 2H, imid), 7.02 (d, 3 J HH = 1.2, 2H, imid), 5.19 (s, 4H, pyCH2), 4.20 (t, 3 J HH = 7.6, 4H, NCH2), 1.87 (app. pent., J = 7, 4H, CH2), 1.25–1.48 (m, 16 H, CH2).13 C{ 1 H} NMR (101 MHz, CD2Cl2) δ 180.3 (s, NCN), 162.3 (q, 1 J CB = 50, ArF), 153.8 (s, py), 140.2 (s, py), 135.4 (s, ArF), 129.5 (qq, 2 J FC = 32, 3 J CB = 3, ArF), 125.2 (q, 1 J FC = 271, ArF), 124.1 (s, py), 123.2 (s, imid), 119.6 (s, imid), 118.1 (pent., 3 J FC = 4, ArF), 54.9 (s, pyCH2), 52.9 (s, NCH2), 31.4 (s, CH2), 27.2 (s, CH2), 27.1 (s, CH2), 25.8 (s, CH2), 25.6 (s, CH2). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 468.2185 m/z, [M]+ (Calc. 468.2183). Anal. Calcd for C57H47BCuF24N5 (1332.33 g mol–1): C, 51.38; H, 3.56; N, 5.26. Found: C, 51.48; H, 3.47; N, 5.34.
In situ formation of 5 and 6
A J. Young's NMR tube was charged with 0.004/0.008 mmol of 2/4 and 1.1 equivalent (metal per metal) of [Pd(NCMe)2Cl2]/[Rh(CO)2Cl]2. CD2Cl2 (0.5 mL) was added and the reaction monitored by 1H NMR until complete consumption of 2/4. The samples were periodically placed in an ultrasound bath during the course of the reaction. In the case of the reaction between 4 and [Rh(CO)2Cl]2, the crude reaction mixture was subsequently passed through a short silica plug (pipette, 3 cm) with additional CH2Cl2 to afford 6 in 82% yield following removal of the solvent in vacuo.
Synthesis of 7
Following in situ reaction monitoring
A J. Young's NMR tube was charged with 0.004/0.008 mmol of 2/4 and 1.4 equivalent (metal per metal) of [NiCl2(gylme)]. CD2Cl2 (0.5 mL) was added and the reaction monitored by 1H NMR at either 20 °C or 40 °C. The samples were periodically placed in an ultrasound bath during the course of the reaction. On complete consumption of 2/4 at 40 °C, the crude reaction mixture was passed through a short alumina plug (pipette, 3 cm) with additional CH2Cl2 to afford 7 in 27%/26% yield following removal of the solvent in vacuo.
Using in situ silver-based transmetallation reagent
A mixture of 1·2HBr (100 mg, 0.176 mmol), Ag2O (45 mg, 0.194 mmol) and Na[BArF 4] (172 mg, 0.194 mmol) was suspended in CH2Cl2 (3 mL) and stirred under nitrogen in the absence of light for 20 hours. The resulting grey suspension was allowed to settle and the solution filtered into a flask charged with solid [NiCl2(glyme)] (40 mg, 0.182 mmol). After stirring the resulting suspension for 6 h, the solution was passed through a short alumina plug (pipette, 3 cm, washed with CH2Cl2) and the product obtained on removal of the volatiles in vacuo. Yield = 70 mg (29%, yellow powder). Crystals suitable for X-ray diffraction were grown from a mixture of toluene, diethylether, cyclohexane and pentane at 20 °C.
1 H NMR (500 MHz, CD2Cl2): δ 7.82 (t, 3 J HH = 7.7, 1H, py), 7.68–7.76 (m, 8H, ArF), 7.55 (br, 4H, ArF), 7.45 (d, 3 J HH = 7.7, 2H, py), 7.11 (d, 3 J HH = 1.7, 2H, imid), 6.87 (d, 3 J HH = 1.7, 2H, imid), 6.30 (d, 2 J HH = 15.0, 2H, pyCH2), 5.14 (d, 2 J HH = 15.0, 2H, pyCH2), 4.73 (app. t, J = 12, 2H, NCH2), 3.68–3.78 (m, 2H, NCH2), 1.94 (br, 2H, CH2), 1.66 (br, 2H, CH2), 1.18–1.50 (m, 14H, CH2), 1.09 (br, 2H, CH2). 13 C{ 1 H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1 J CB = 50, ArF), 162.0 (s, NCN), 156.5 (s, py), 140.9 (s, py), 135.3 (s, ArF), 129.4 (qq, 2 J FC = 32, 3 J CB = 3, ArF), 125.2 (q, 1 J FC = 271, ArF), 125.1 (s, py), 123.0 (s, imid), 121.4 (s, imid), 118.0 (pent., 3 J FC = 4, ArF), 55.0 (s, pyCH2), 51.3 (s, NCH2), 30.8 (s, CH2), 28.7 (s, CH2), 27.5 (s, CH2), 23.7 (s, CH2). ESI-MS (CH3CN, 180 °C, 4 kV) positive ion: 498.1929 m/z, [M]+ (calc. 498.1929). Anal. Calcd for C57H47BClF24N5Ni (1362.95 g mol–1): C, 50.23; H, 3.48; N, 5.14. Found: C, 50.62; H, 3.74; N, 5.05.
Crystallography
Full details about the collection, solution and refinement are documented in the CIF, which have been deposited with the Cambridge Crystallographic Data Centre under CCDC 1470494–1470497.
Acknowledgments
We thank the University of Warwick (R. E. A.), European Research Council (ERC, grant agreement 637313; C. M. S, A. B. C) and Royal Society (A. B. C.) for financial support. Crystallographic and high-resolution mass-spectrometry data were collected using instruments purchased through support from Advantage West Midlands and the European Regional Development Fund. Crystallographic data for 7 were collected using an instrument that received funding from the ERC under the European Union's Horizon 2020 research and innovation programme (grant agreement No 637313).
Footnotes
References
- (a) Hopkinson M. N., Richter C., Schedler M., Glorius F. Nature. 2014;510:485–496. doi: 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]; (b) Soleilhavoup M., Bertrand G. Acc. Chem. Res. 2015;48:256–266. doi: 10.1021/ar5003494. [DOI] [PubMed] [Google Scholar]; (c) Díez-González S., Marion N., Nolan S. P. Chem. Rev. 2009;109:3612–3676. doi: 10.1021/cr900074m. [DOI] [PubMed] [Google Scholar]; (d) de Frémont P., Marion N., Nolan S. P. Coord. Chem. Rev. 2009;253:862–892. [Google Scholar]; (e) Hahn F. E., Jahnke M. C. Angew. Chem., Int. Ed. 2008;47:3122–3172. doi: 10.1002/anie.200703883. [DOI] [PubMed] [Google Scholar]
- Lin J. C. Y., Huang R. T. W., Lee C. S., Bhattacharyya A., Hwang W. S., Lin I. J. B. Chem. Rev. 2009;109:3561–3598. doi: 10.1021/cr8005153. [DOI] [PubMed] [Google Scholar]
- (a) Lin I. J. B., Vasam C. S. Coord. Chem. Rev. 2007;251:642–670. [Google Scholar]; (b) Garrison J. C., Youngs W. J. Chem. Rev. 2005;105:3978–4008. doi: 10.1021/cr050004s. [DOI] [PubMed] [Google Scholar]
- (a) Yu X.-Y., Patrick B. O., James B. R. Organometallics. 2006;25:2359–2363. [Google Scholar]; (b) de Frémont P., Scott N. M., Stevens E. D., Nolan S. P. Organometallics. 2005;24:2411–2418. [Google Scholar]
- Furst M. R. L., Cazin C. S. J. Chem. Commun. 2010;46:6924–6922. doi: 10.1039/c0cc02308f. [DOI] [PubMed] [Google Scholar]
- Boehme C., Frenking G. Organometallics. 1998;17:5801–5809. [Google Scholar]
- (a) Andrew R. E., González-Sebastián L., Chaplin A. B. Dalton Trans. 2016;45:1299–1305. doi: 10.1039/c5dt04429d. [DOI] [PubMed] [Google Scholar]; (b) Poyatos M., Mata J. A., Peris E. Chem. Rev. 2009;109:3677–3707. doi: 10.1021/cr800501s. [DOI] [PubMed] [Google Scholar]; (c) Pugh D., Danopoulos A. A. Coord. Chem. Rev. 2007;251:610–641. [Google Scholar]; (d) Peris E., Crabtree R. H. Coord. Chem. Rev. 2004;248:2239–2246. [Google Scholar]
- Representative examples of transmetallation reactions involving other polydentate NHC ligands: ; (a) Weiss D. T., Altmann P. J., Haslinger S., Jandl C., Pöthig A., Cokoja M., Kühn F. E. Dalton Trans. 2015;44:18329–18339. doi: 10.1039/c5dt02386f. [DOI] [PubMed] [Google Scholar]; (b) Rit A., Pape T., Hahn F. E. J. Am. Chem. Soc. 2010;132:4572–4573. doi: 10.1021/ja101490d. [DOI] [PubMed] [Google Scholar]; (c) Hahn F. E., Radloff C., Pape T., Hepp A. Chem. – Eur. J. 2008;14:10900–10904. doi: 10.1002/chem.200801877. [DOI] [PubMed] [Google Scholar]; (d) Hu X., Castro-Rodriguez I., Olsen K., Meyer K. Organometallics. 2004;23:755–764. [Google Scholar]; (e) Wanniarachchi Y. A., Khan M. A., Slaughter L. M. Organometallics. 2004;23:5881–5884. [Google Scholar]
- (a) Vaughan J., Carter D. J., Rohl A. L., Ogden M. I., Skelton B. W., Simpson P. V., Brown D. H. Dalton Trans. 2016;45:1484–1495. doi: 10.1039/c5dt04213e. [DOI] [PubMed] [Google Scholar]; (b) Hernández-Juárez M., López-Serrano J., Lara P., Morales-Cerón J. P., Vaquero M., Álvarez E., Salazar V., Suárez A. Chem. – Eur. J. 2015;21:7540–7555. doi: 10.1002/chem.201406040. [DOI] [PubMed] [Google Scholar]; (c) Clark W. D., Tyson G. E., Hollis T. K., Valle H. U., Valente E. J., Oliver A. G., Dukes M. P. Dalton Trans. 2013;42:7338–7344. doi: 10.1039/c3dt32862g. [DOI] [PubMed] [Google Scholar]; (d) Wei S., Li X., Yang Z., Lan J., Gao G., Xue Y., You J. Chem. Sci. 2012;3:359–363. [Google Scholar]; (e) Inamoto K., Kuroda J.-I., Kwon E., Hiroya K., Doi T. J. Organomet. Chem. 2009;694:389–396. [Google Scholar]; (f) Brown D. H., Nealon G. L., Simpson P. V., Skelton B. W., Wang Z. Organometallics. 2009;28:1965–1968. [Google Scholar]; (g) Pugh D., Boyle A., Danopoulos A. A. Dalton Trans. 2008:1087–1094. doi: 10.1039/b715769j. [DOI] [PubMed] [Google Scholar]; (h) Chen W., Wu B., Matsumoto K. J. Organomet. Chem. 2002;654:233–236. [Google Scholar]; (i) Caballero A., Díez-Barra E., Jalón F. A., Merino S., Rodríguez A. M., Tejeda J. J. Organomet. Chem. 2001;627:263–264. [Google Scholar]
- (a).Charra V., de Frémont P., Breuil P.-A. R., Olivier-Bourbigou H., Braunstein P., Liu X., Pattacini R., Deglmann P., Braunstein P. J. Organomet. Chem. Organometallics. 2015;2011;79530:25–33. 3302–3310. [Google Scholar]
- (a) Biffis A., Cipani M., Tubaro C., Basato M., Costante M., Bressan E., Venzo A., Graiff C. New J. Chem. 2013;37:4176–4184. [Google Scholar]; (b) Radloff C., Gong H.-Y., Schulte to Brinke C., Pape T., Lynch V. M., Sessler J. L., Hahn F. E. Chem. – Eur. J. 2010;16:13077–13081. doi: 10.1002/chem.201002276. [DOI] [PubMed] [Google Scholar]
- (Scheme 2, A): Simons R. S., Custer P., Tessier C. A., Youngs W. J., Organometallics, 2003, 22 , 1979 –1982 . [Google Scholar]
- (Scheme 2, B): Nielsen D. J., Cavell K. J., Skelton B. W., White A. H., Inorg. Chim. Acta, 2002, 327 , 116 –125 . [Google Scholar]
- (Scheme 2, C and D): Nakamura T., Ogushi S., Arikawa Y., Umakoshi K., J. Organomet. Chem., 2016, 803 , 67 –72 . [Google Scholar]
- (Scheme 2, C): Herbst A., Bronner C., Dechambenoit P., Wenger O. S., Organometallics, 2013, 32 , 1807 –1814 . [Google Scholar]
- (Scheme 2, D): Lake B. R. M., Willans C. E., Chem. – Eur. J., 2013, 19 , 16780 –16790 Liu B., Ma X., Wu F., Chen W., Dalton Trans., 2015, 44 , 1836 –1844 . [DOI] [PubMed] [Google Scholar]
- (Scheme 2, E): Bezuidenhout D. I., Kleinhans G., Guisado-Barrios G., Liles D. C., Ung G., Bertrand G., Chem. Commun., 2014, 50 , 2431 –2433 . [DOI] [PubMed] [Google Scholar]
- Knishevitsky A. V., Korotkikh N. I., Cowley A. H., Moore J. A., Pekhtereva T. M., Shvaika O. P., Reeske G. J. Organomet. Chem. 2008;693:1405–1411. [Google Scholar]
- (a) Andrew R. E., Ferdani D. W., Ohlin C. A., Chaplin A. B. Organometallics. 2015;34:913–917. [Google Scholar]; (b) Andrew R. E., Chaplin A. B. Inorg. Chem. 2015;54:312–322. doi: 10.1021/ic5024828. [DOI] [PubMed] [Google Scholar]; (c) Andrew R. E., Chaplin A. B. Dalton Trans. 2014;43:1413–1423. doi: 10.1039/c3dt52578c. [DOI] [PubMed] [Google Scholar]
- Krossing I., Raabe I. Angew. Chem., Int. Ed. 2004;43:2066–2090. doi: 10.1002/anie.200300620. [DOI] [PubMed] [Google Scholar]
- Tapu D., Dixon D. A., Roe C. Chem. Rev. 2009;109:3385–3407. doi: 10.1021/cr800521g. [DOI] [PubMed] [Google Scholar]
- Related observations have been made for Ag(i) and Au(i) complexes of di-NHC ligands: Gil-Rubio J., Cámara V., Bautista D., Vicente J., Inorg. Chem., 2013, 52 , 4071 –4083 . [DOI] [PubMed] [Google Scholar]
- Other instances of the [Cu2Br4]2− dianion see: ; (a) Lake B. R. M., Ariafard A., Willans C. E. Chem. – Eur. J. 2014;20:12729–12733. doi: 10.1002/chem.201404298. [DOI] [PubMed] [Google Scholar]; (b) Boess E., Sureshkumar D., Sud A., Wirtz C., Farès C., Klussmann M. J. Am. Chem. Soc. 2011;133:8106–8109. doi: 10.1021/ja201610c. [DOI] [PubMed] [Google Scholar]; (c) Drożdżewski P., Kubiak M. Polyhedron. 2009;28:1518–1524. [Google Scholar]; (d) Canty A. J., Engelhardt L. M., Healy P. C., Kildea J. D., Minchin N. J., White A. H. Aust. J. Chem. 1987;40:1881–1891. [Google Scholar]
- The other independent cation features a significant degree of disorder about the copper centre
- Catalano V. J., Munro L. B., Strasser C. E., Samin A. F. Inorg. Chem. 2011;50:8465–8476. doi: 10.1021/ic201053t. [DOI] [PubMed] [Google Scholar]
- (a) Chen C., Qiu H., Chen W. J. Organomet. Chem. 2012;696:4166–4172. [Google Scholar]; (b) Liu X., Chen W. Organometallics. 2012;31:6614–6622. [Google Scholar]
- Buschmann W. E., Miller J. S., Bowman-James K., Miller C. N. Inorg. Synth. 2002;33:83–91. [Google Scholar]
- McCleverty J. A., Wilkinson G., Lipson L. G., Maddox M. L., Kaesz H. D. Inorg. Synth. 1990;28:84–86. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.