

Abstract
Nerves closely associate with blood vessels and help to pattern the vasculature during development. Recent work suggests that newly formed nerve fibers may regulate the tumor microenvironment, but their exact functions are unclear. Studying mouse models of prostate cancer, we show that endothelial β-adrenergic receptor signaling via adrenergic nerve-derived noradrenaline in the prostate stroma is critical for activation of an angiogenic switch that fuels exponential tumor growth. Mechanistically this occurs through alteration of endothelial cell metabolism. Endothelial cells typically rely on aerobic glycolysis for angiogenesis. We found that the loss of endothelial Adrb2, the gene encoding the β2-adrenergic receptor, leads to inhibition of angiogenesis through enhancement of endothelial oxidative phosphorylation. Co-deletion of Adrb2 and Cox10, a gene encoding a cytochrome IV oxidase assembly factor, prevented the metabolic shift induced by Adrb2 deletion and rescued prostate cancer progression. This cross-talk between nerves and endothelial metabolism could potentially be targeted as an anti-cancer therapy.
Solid tumors depend on angiogenesis to sustain their growth (1). The transition from hyperplasia to highly vascularized growing tumor, referred to as the “angiogenic switch,” is a state in which pro-angiogenic factors—such as vascular endothelial growth factor (VEGF) and other secreted angiocrine factors— predominate over anti-angiogenic signals (2). During development, peripheral nerves associate closely with growing blood vessels, organizing vascular pattern (3, 4), a phenomenon that has also been described in models of wound healing (5, 6). Emerging studies suggest that nerves can also regulate tumorigenesis (7–11). Sympathetic nerve fibers deliver adrenergic signals that act via β-adrenergic receptors (βAdR) expressed in the tumor microenvironment. However, the cellular target(s) and molecular mechanism(s) responsible for neural regulation of cancer are not known and may provide novel therapeutic avenues.
Adrenergic nerves regulate angiogenesis in early tumor growth
To investigate the interplay between adrenergic nerves and early stage tumor growth, we orthotopically implanted prostate cancer xenografts in immunodeficient Balb/c (nu/nu) mice. Xenografts exhibited exponential growth kinetics from ~18 days post-implantation (βAdRWT) (Fig. 1A). By contrast, no tumor growth occurred in nu/nu mice that were also genetically deficient in both β2-adrenergic (Adrb2) and β3-adrenergic (Adrb3) receptors (βAdRKO) (9). Pathological examination of prostate tissues revealed that tumors formed in βAdRKO recipients (Fig. 1A; fig. S1A) but their growth was arrested at day 18 (Fig.1B), suggesting that βAdR signaling plays a key role in the molecular “switch” enabling exponential tumor growth. Before the switch, day-18 tumors from both control and βAdRKO recipients showed no difference in size, vascular permeability or hypoxic area (fig. S1, A-C). We next investigated the vascular density and tumor vasculature patterns by semi-automated analysis using Simple Neurite Tracer (12) in whole cross-sectional montages of day-18 tumors. These analyses revealed reductions in the density of tumor vessels (Fig. 1, C and D) due to significant alterations in the vascular patterns (reduced length and branching) in prostates βAdRKO mice compared with βAdRWT controls (Fig.1D). This observation suggests that the loss of adrenergic signaling in the tumor microenvironment may regulate angiogenesis.
Fig. 1. Loss of P-adrenergic signaling in the prostate microenvironment arrests tumor growth and angiogenesis.
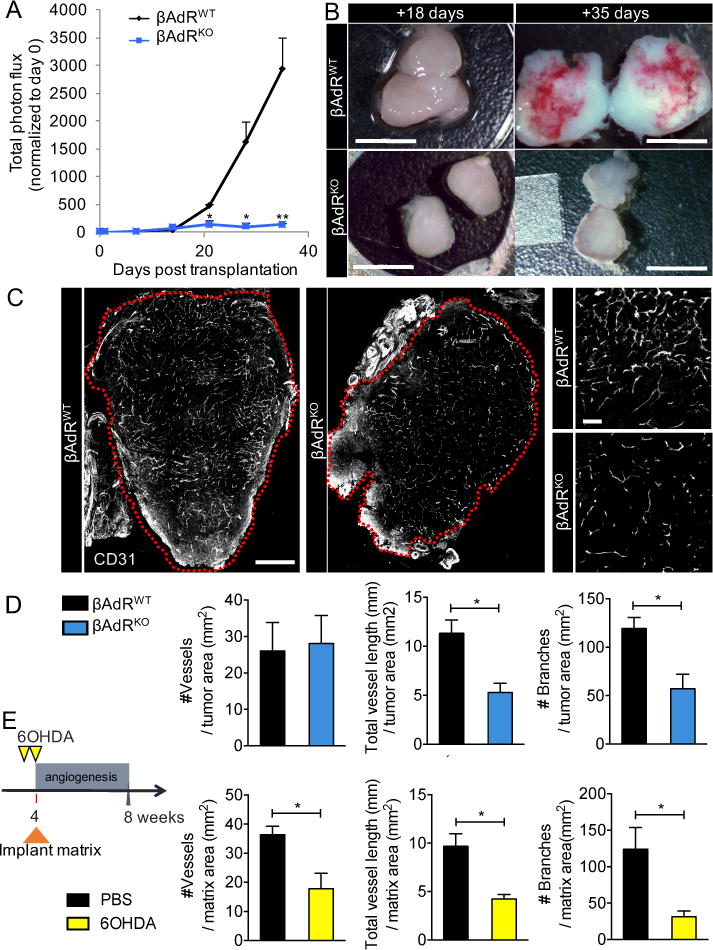
(A to D) PC-3 cells stably expressing luciferase were orthotopically implanted into 8- week-old Balb/c nu/nu prostates. βAdR = Adrb2 + Adrb3. (A) Tumor growth was monitored in vivo weekly by bioluminescence, n = 6 mice per condition. (B) Comparison of orthotopic tumor size pre (+18 days) and post (+35days) angiogenic switch. Scale bars, 5mm. (C) Immunofluorescence analyses of the vasculature in similar-sized tumors (outlined in red) before the angiogenic switch (+18 days). Cross- sectional montage of prostate xenografts from βAdRWT (left) and βAdRKO (middle) mice, and magnified view of βAdRWT vasculature (right, top) and βAdRKO vasculature (right, bottom). CD31 = vasculature. Montage scale bar, 500μm; magnified-view scale bar, 100μm. (D) Quantification of angiogenesis in orthotopic tumors (vessels traced using Simple Neurite Tracer). n = 4 mice per condition. (E) Experimental design and quantification of vessels recruited into orthotopic type I collagen matrix after sympathectomy with 6-hydroxydopamine (60HDA). PBS = phosphate-buffered saline, n = 4 mice per condition. *P<0.05; **P<0.01. Error bars = indicate SEM.
To confirm these results in an independent assay, we orthotopically transplanted a cell-free type I collagen matrix into the prostate capsule of nu/nu mice, between the ventral prostate lobes. Because type I collagen is an angiogenic superpolymer (13) and mimics the changes in composition of the reactive extracellular matrix seen in prostate cancer (14, 15), it promotes angiogenesis without the need to administer exogenous cytokines. We assessed recruitment of host vasculature into the matrix in control immunocompetent C57BL/6 mice and in mice chemically sympathectomized with 6-hydroxydopamine [6OHDA; (9)]. Analyses of vascular patterning revealed significant reductions in vessel migration, density, and branching in the denervated prostate matrices compared with controls (Fig. 1E). These data indicate that the vasculature serves as a stromal target for adrenergic nerves and that adrenergic signals modulate angiogenesis (7).
Endothelial ADRB2 controls the angiogenic switch
The angiogenic switch described above characterizes the progression of prostate cancer from the low-grade pre-neoplastic stage [low-grade prostatic intraepithelial neoplasia(LPIN)] (fig. S2, B and C) to the high-grade malignant stage [high-grade prostatic intraepithelial neoplasia(HPIN)] (Fig. 2A) (16). To explore the role of adrenergic signals in mediating angiogenesis in prostate cancer progression, we used the Hi-Myc (cMyc) spontaneous autochthonous prostate cancer mouse model. In these mice, the human MYC gene, driven by the probasin promoter, is overexpressed in the prostate; this leads to LPIN at 4 weeks, which progresses to HPIN by 8 weeks, followed by adenocarcinoma and finally invasive cancer by 24 weeks (9, 17). This model recapitulates the histological changes seen in human disease (18, 19). Immunofluorescence analyses of adrenergic nerves (tyrosine hydroxylase+, TH+) and vessels (CD31+) showed an increase in both nerve and vessel densities in the HPIN stage (Fig. 2A, and fig. S2A), but not in the pre-angiogenic LPIN stage (fig. S2, B and C). In addition, PIN progression coincided with an increased physical association of nerves and blood vessels (reduced proximity score; Fig. 2A). The levels of the β-adrenergic neurotransmitter noradrenaline were significantly elevated in HPIN prostates, whereas the levels of other catecholamines were unchanged (Fig. 2B). Analyses of Adrb2 expression in prostate stromal populations implicated in tumor angiogenesis (20) revealed that Adrb2 expression levels were highest in endothelial cells (fig. S3A).
Fig. 2. Prostate endothelial cells closely associate with adrenergic nerves and require Adrb2 for cancer progression and angiogenesis.
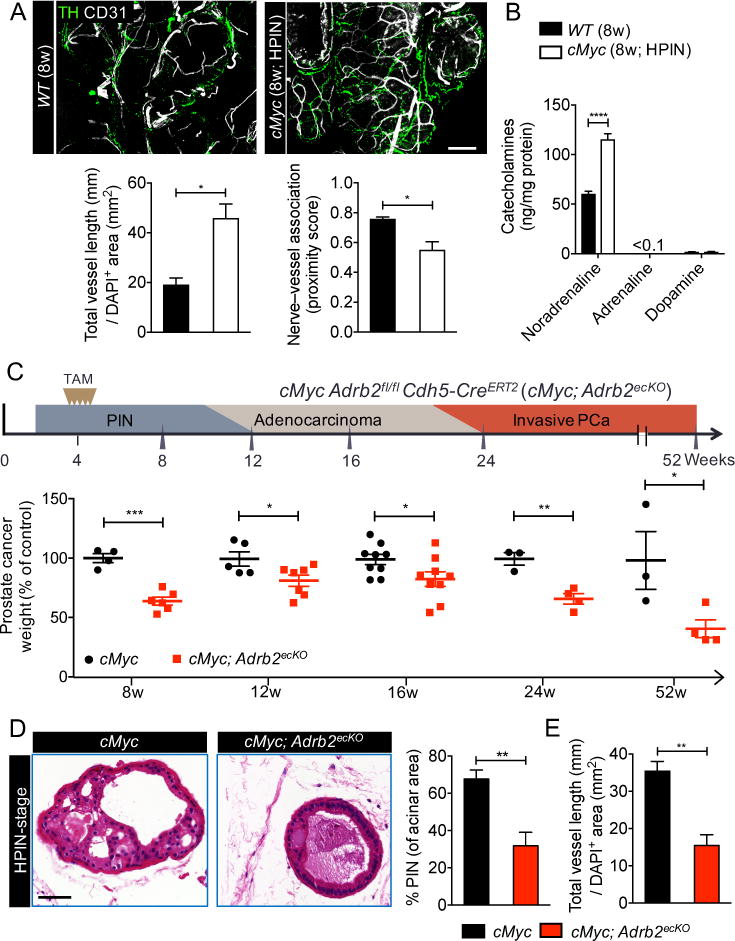
(A) Thick-section images and quantification of the association between adrenergic nerves and the prostate vasculature in the high-grade prostatic intraepithelial neoplasia (HPIN) stage. Representative images of wild-type (WT) prostate (left, top) and HPIN stage prostate (right, top). TH = tyrosine hydroxylase; CD31 = vasculature. Scale bar, 100μm. Quantification of vessel density (left, bottom) and proximity of association between nerves and vessels (right, bottom). n = 4 mice per condition. (B) Catecholamine levels in HPIN-stage prostate quantified by high-performance liquid chromatography. n = 4 mice per condition. (C) Cdh5-CreERT2 deletion of Adrb2 in endothelial cells and its effect at various histopathological stages (schema: top; prostate weight: bottom). TAM = tamoxifen; PCa = prostate cancer. (D and E) Effect of endothelial Adrb2 deletion on HPIN pathology, as shown by (D) representative hematoxylin-and-eosin histology (left). Scale bar, 50μm. Prevalence of PIN (right). n = 4 mice per condition. (E) Quantification of vascular density. n = 4 mice per condition. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. Error bars = indicate SEM.
To assess the specific contributions of Adrb2 in stromal cells to cancer progression in the cMyc model, we conditionally deleted Adrb2 by intercrossing Adrb2fl/fl (21) with Cre-expressing lines to delete the gene in myeloid cells [Csf1riCre (22)], pericytes [NG2CreERTM (23)] or endothelial cells [Cdh5CreERT2 (24)]. Deletion of Adrb2 in the targeted cells occurred at high efficiency (fig. S3, B to D). Deletion of Adrb2 in pericytes during the LPIN stage did not result in a significant difference in prostate cancer weight in the HPIN stage nor at the late adenocarcinoma stage (12 months of age), and no phenotype was also observed when Adrb2 was deleted in myeloid cells (fig. S3, C and D). By contrast, deletion of Adrb2 in endothelial cells during the LPIN stage reduced progression to the HPIN stage (Fig. 2, C and D), and the inhibition of disease progression was sustained for more than 12 months (Fig. 2C). Histological analysis of cMyc Adrb2fl/fl Cdh5-CreERT2 mice (endothelial cell conditional knockout mice, henceforth referred to as cMyc; Adrb2ecKO) revealed a significant reduction in Ki-67+ proliferative prostate epithelial cells (fig. S4A), which we confirmed by fluorescence-activated cell sorting (FACS) analysis (fig. S4B). The proliferation of prostate epithelial cells was reduced to levels seen at the LPIN stage before the angiogenic switch (fig. S2D), but without a significant change in epithelial apoptosis (fig. S4C). Thus, endothelial cells are the major stromal target of adrenergic nerves in the tumor microenvironment.
To evaluate the angiogenic switch during the transition from the LPIN to HPIN stage in prostate cancer progression (16), we induced endothelial Adrb2 deletion in the cMyc model by administering tamoxifen to the mice at 4 weeks of age (LPIN stage). We then assessed angiogenesis and vascular patterning at 8 weeks (HPIN stage). Consistent with our results in the xenograft and denervation models (Fig.1, D and E), we found that cMyc; Adrb2ecKO mice exhibited a significant reduction in vessel density, migration, and branching 4 weeks after Adrb2 gene deletion, as compared with cMyc controls (Fig. 2E, and fig. S4D and S5, A and B). There was no significant change in pericyte coverage, vascular leakage, or tissue hypoxia (fig. S4, E to G). These data collectively suggest that adrenergic signals mediate an early angiogenic progression switch via endothelial Adrb2.
ADRB2 regulates oxidative metabolism in angiogenic prostate endothelial cells
To explore the molecular mechanisms underlying the angiogenic defect in cMyc; Adrb2ecKO mice, we compared the transcriptome of endothelial cells isolated by FACS from cMyc; Adrb2ecKO and cMyc prostates (Fig. 3A, and fig. S6A). Gene set enrichment analysis revealed a highly significant increase in mitochondrial cytochrome c activity (fig. S6B). Among differentially expressed mitochondria-associated genes involved in metabolism, branched chain keto acid dehydrogenase alpha peptide (Bckdha; involved in metabolizing branched-chain amino acids in the tricarboxylic acid cycle) and cytochrome c oxidase assembly factor 6 (Coa6; involved in the electron-transport chain) stood out by hierarchical cluster analysis (fig. S6C). Increased mRNA expression of both genes was validated by quantitative real-time polymerase chain reaction (fig. S6D). Consistent with our observation that Adrb2 deletion in endothelial cells at the LPIN stage sustained inhibition of cancer progression 48 weeks after Cre-mediated recombination (Fig. 2C), we observed that Coa6 expression remained elevated in cMyc; Adrb2ecKO endothelial cells at that timepoint (fig. S6E). In addition, analysis of peri-tumoral and intratumoral endothelial cells isolated from the βAdRKO orthotopic tumor model revealed increased expression of these mitochondrial genes (fig. S7, A and B). The effect of βAdR signals on genes involved in oxidative phosphorylation and vascular branching is reminiscent of recent studies showing that inhibition of glycolysis in endothelial cells alters angiogenesis during the development of retinal and hindbrain vessels (25, 26).
Fig. 3. Adrb2 depletion increases endothelial oxidative metabolism.
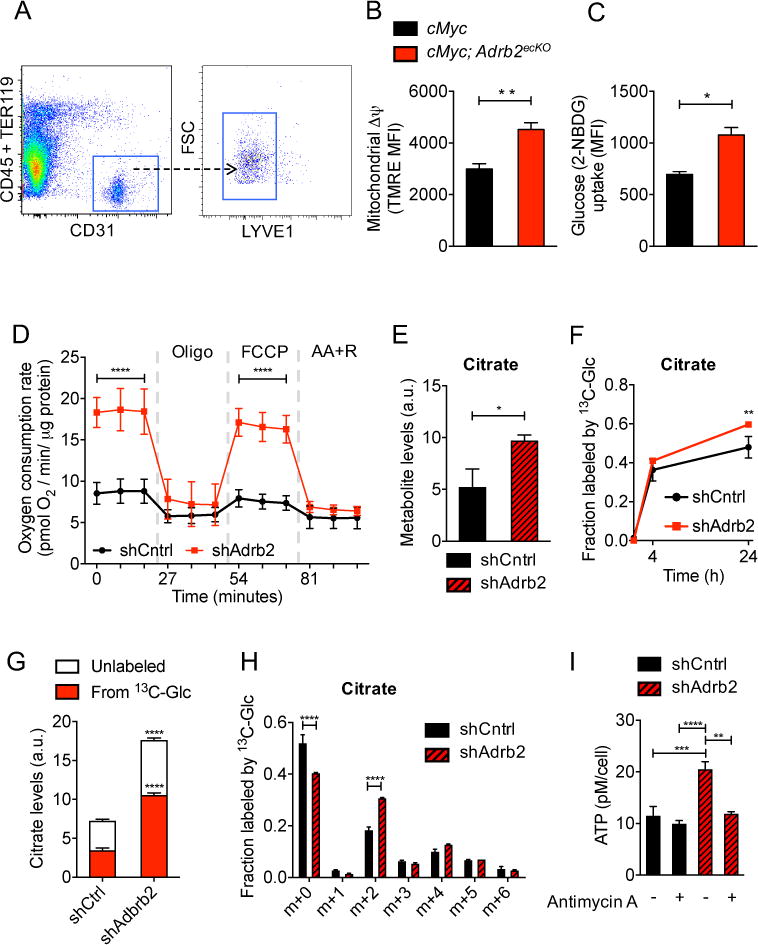
(A to C) FACS analysis of HPIN-stage endothelial cells. (A) Representative endothelial isolation plot from prostates. FSC = forward scatter. (B) Quantification of mitochondrial membrane potential (Δψ) by tetramethylrhodamine ethyl ester (TMRE) staining, n = 5 mice per condition. MFI = mean fluorescent intensity. (C) Quantification of endothelial glucose uptake by 2-NBDG. n = 6 or 7 mice per condition. (D to I) Metabolism and ATP production was assessed in shCntrl and shAdrb2 primary prostate endothelial cells after incubation with noradrenaline. (D) Oxygen consumption rates at baseline and in the presence of oligomycin, FCCP [carbonyl cyanide p-(trifl uoromethoxy) phenylhydrazone], and antimycin A + rotenone (AA+R). n = 4 independent experiments. Error bars indicate = SD. (E to H) Different analyses from the same set of experiments, n = 3 replicates per condition per time point. Metabolite levels were normalized to internal standard and to sample protein content. (E) Intracellular levels of the tricarboxylic acid cycle metabolite citrate, a.u., arbitrary units. (F) Total fraction of citrate containing 13C-label from [U-13C]-glucose. (G) Relative intracellular levels of citrate labeling derived from [U-13C]-glucose at 24 hours. (H) Fraction of each isotopologue of citrate (mass isotopologue distribution) after culture in 5mM [U-13C]-glucose for 24 hours. (I) Measurement of intracellular ATP levels in the presence or absence of antimycin A, an inhibitor of the electron transport chain. n = 3 independent experiments. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. Error bars = indicate SEM.
To investigate the link between nerve-derived adrenergic signals and endothelial metabolism, we evaluated the change in endothelial mitochondrial membrane potential (Δψ), which is associated with the accumulation of positively charged hydrogen ions in the mitochondrial intermembrane space during oxidative phosphorylation. We assessed Δψ in freshly isolated prostate endothelial cells by tetramethylrhodamine ethyl ester (TMRE) and quantified the quotient of basal-to-maximal nictotinamide adenine dinucleotide (reduced form, NADH) levels by autofluorescence, both measures of oxidative phosphorylation (27–29). We found that after progression to the HPIN stage, endothelial cell Δψ was reduced and the NADH quotient was increased without significant change in mitochondrial mass (fig. S8, A to D). Consistent with the notion that prostate endothelial cells operate at maximal respiratory capacity (RC), HPIN and wild-type (WT) prostate-derived endothelial cells did not exhibit spare RC, whereas significant spare RC was apparent in hematopoietic stem and progenitor cells isolated from the same animals (fig. S8E) (29). However, TMRE was significantly increased and NADH quotient reduced in freshly isolated endothelial cells from cMyc; Adrb2ecKO mice (Fig. 3B, and fig. S9A), consistent with enhanced oxidative phosphorylation. This was not due to a change in mitochondrial mass (fig. S9A) or to an effect on endothelial viability (fig. S9, B and C). In addition, we observed a similar increase in endothelial oxidative phosphorylation in orthotopic tumors in βAdRKO recipients, as well as in WT mice subjected to adrenergic denervation (fig. S10, A and B, and S11, A and B). Endothelial glucose uptake was increased in the absence of Adrb2 (Fig. 3C). These results strongly suggest that the loss of βAdR signaling enhances oxidative phosphorylation in endothelial cells.
To evaluate the role of βAdR in endothelial glucose metabolism, the main fuel source of endothelial cells (25, 30), we knocked down Adrb2 expression by lentiviral transduction of short hairpin RNA (shAdrb2) in primary mouse prostate endothelial cells that express the adenoviral E4ORF1 gene; the latter gene allows the cells to survive in culture while maintaining their growth dependence on VEGF and fibroblast growth factor (31). shAdrb2 specifically inhibited Adrb2 expression by 80% compared with control vector (shCntrl), without affecting endothelial cell viability or proliferation (fig. S12, A and B). In the presence of physiological levels of noradrenaline, shAdrb2 endothelial cells exhibited a marked increase in oxidative aerobic metabolism compared with shCntrl endothelial cells as demonstrated by an increase in oxygen consumption (Fig. 3D). Like freshly isolated endothelial cells from cMyc; Adrb2ecKO or βAdRKO mice, endothelial cells treated with shAdrb2 showed an elevation in oxidative phosphorylation activity (increased Δψ and decreased NADH quotient), as well as an increase in glucose uptake, but no change in mitochondrial mass (fig. S13, A to C). Thus, this endothelial culture system recapitulates the metabolic profile of native prostate endothelial cells.
To explore the shift in endothelial metabolism that accompanies the loss of ADRB2 signaling, we assessed glucose uptake and the contributions of glucose-derived carbons to the tricarboxylic acid (TCA) cycle, an integral process that fuels oxidative phosphorylation. We observed increased glucose uptake in cMyc;Adrb2ecKO-isolated endothelial cells compared with controls (Fig. 3C). We then traced the fate of uniformly-labeled [U-13C]-glucose in shCntrl and shAdrb2 endothelial cells cultured in the presence of noradrenaline. These experiments revealed a 1.9-fold increase in citrate levels in shAdrb2 compared with shCntrl endothelial cells (Fig. 3E), whereas lactate levels were unchanged (fig. S13D). Furthermore, shAdrb2 cells incorporated considerably more glucose-derived carbons into citrate than shCntrl cells (Fig. 3, F and G). Examination of isotopologue distribution of glucose-derived citrate revealed an enrichment of m+2 isotopologue in shAdrb2 compared with shCntrl cells, reflective of pyruvate dehydrogenase (PDH)-driven contribution of glucose-derived carbons into the TCA cycle (Fig. 3H). However, shAdrb2 endothelial cells were relatively depleted in the m+3 isotopologue, which may reflect a reduction in anaplerotic flux through pyruvate carboxylase (fig. S14A).
Because glutamine has been reported to be a major anaplerotic substrate in proliferating cells (32), we performed glutamine tracing experiments using uniformly labeled [U-13C]-glutamine. We observed significant elevations in glutamine oxidation as represented by an increase in labeled citrate levels and in the m+4 isotopologue in TCA cycle intermediates (fig. S14, B-D, table S1). Consistent with the notion that accumulation of citrate may provide acetyl–coenzyme A for fatty acid synthesis (33), we also observed higher abundance of fatty acyl carnitines in shAdrb2 compared with control endothelial cells (fig. S14E). Furthermore, we found that adenosine triphosphate (ATP) levels were significantly elevated in shAdrb2 endothelial cells (Fig. 3I).
To dissect the relative contributions of glycolysis versus oxidative phosphorylation to ATP generation, we replaced glucose in the media with galactose (which forces cells to rely on oxidative phosphorylation for ATP production). This led to a substantial reduction in ATP levels in shCntrl cells, whereas no change in ATP levels was observed in shAdrb2 cells (fig. S15A). Inhibition of oxidative phosphorylation with antimycin A also reduced ATP levels to those seen in control cells (Fig. 3I), further indicating that depletion of ADRB2 shifts endothelial metabolism to oxidative phosphorylation. Taken together, the results obtained with multiple model systems and analytical methods show that the loss of βAdR signaling alters endothelial metabolism, enhancing oxidative phosphorylation.
Increased endothelial COA6 activity mediates the shift toward oxidative phosphorylation
To obtain further insight into the mechanism linking adrenergic signaling, altered metabolism, and endothelial cell function, we assessed the migration of Adrb2 sufficient (shCntrl) and Adrb2 deficient (shAdrb2) endothelial cells in an in vitro wound assay. We found that migration of shAdrb2 cells was markedly inhibited compared with that of shCntrl cells (fig. S16, A to C). Staining for α-tubulin revealed altered microtubule directional orientation in the shAdrb2 cells (fig. S16D). Endothelial cells require polarization of their microtubule cytoskeleton for directional migration (34), and microtubule organization regulates ATP-intensive actin cytoskeletal organization (35), which suggests that elevated ATP levels from oxidative metabolism may inhibit endothelial angiogenic function by altering cytoskeletal activity. In line with our transcriptome analysis (fig. S6, C and D), shAdrb2 increased Coa6 mRNA and protein levels (fig. S15, B and C), but did not alter Bckdha expression, indicating that changes in Coa6 expression may drive the observed metabolic alterations. Cyclic adenosine monophosphate, the second messenger generated by adenylyl cyclase (AC) upon βAdR signaling, was undetectable after noradrenaline stimulation in shAdrb2 cells, but was present at high levels in control endothelial cells (fig. S15D). AC inhibition upregulated Coa6 expression in control cells, and conversely, AC stimulation with forskolin repressed Coa6 in shAdrb2 endothelial cells (fig. S15E). These data indicate that ADRB2 signaling in endothelial cells regulates Coa6 expression.
To assess whether these mitochondrial genes were sufficient to increase oxidative phosphorylation and inhibit the angiogenic switch in prostate cancer, we overexpressed Coa6 or Bckdha by lentiviral transduction in culture-expanded prostate endothelial cells. Whereas Bckdha did not induce a significant change in oxidative phosphorylation, Coa6 overexpression (Coa6-GFP) (fig. S17A) substantially augmented endothelial oxidative metabolism (Fig. 4, A and fig. S17B). In addition, Coa6-green fluorescent protein (GFP) significantly inhibited endothelial cell migration and proliferation, but did not affect endothelial cell viability (fig. S17, C and D). To assess the effect of increased oxidative phosphorylation on angiogenesis in vivo, we orthotopically cotransplanted Coa6-GFP endothelial cells with human PC-3 prostate cancer cells into nu/nu males. In the prostate tumors, GFP+ vessel quantification revealed a marked reduction in angiogenesis and tip cell formation in Coa6-GFP xenografts compared with xenografts produced by co-transplantation of PC-3 cells with Cntrl-GFP endothelial cells (Fig. 4, B and C). These results suggest that the loss of adrenergic signaling by Adrb2 deletion increases Coa6 expression, which causes an increase in oxidative phosphorylation, and that this metabolic shift inhibits angiogenesis.
Fig. 4. Increased endothelial oxidative metabolism inhibits angiogenesis, and conditional Cox10 deletion rescues endothelial metabolism, angiogenesis, and cancer progression.
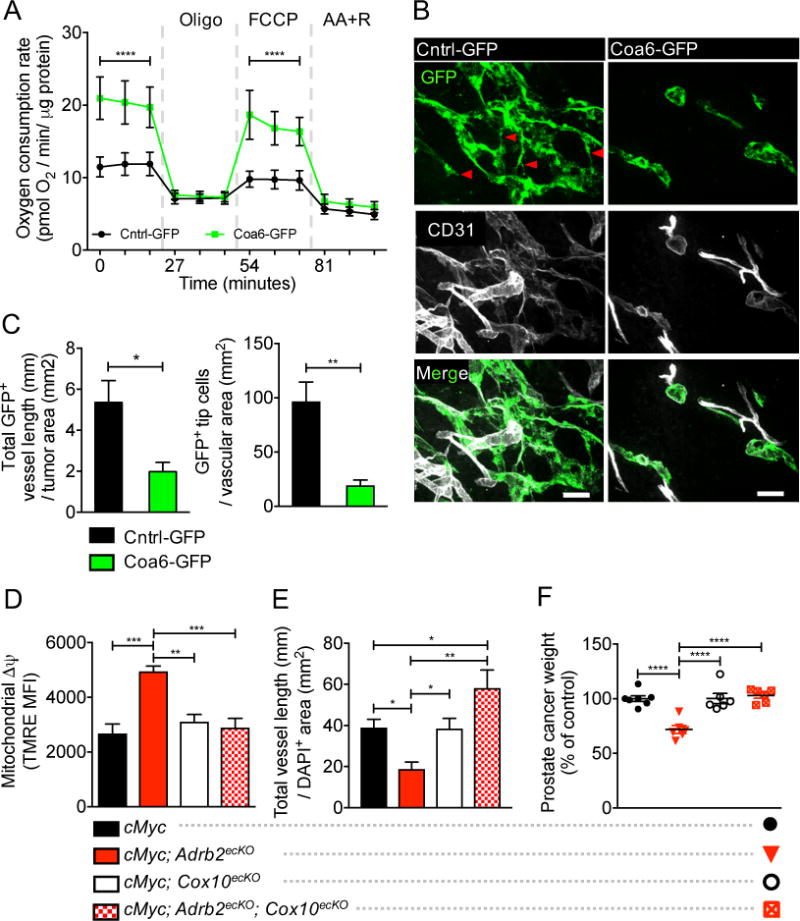
(A) Effect of Coa6 overexpression (Coa6-GFP) on oxygen consumption rates at baseline and in the presence of oligomycin, FCCP, and antimycin A + rotenone (AA+R). n = 4 independent experiments. Error bars indicate = SD. (B and C) Immunofluorescent analysis of orthotopically co-transplanted Cntrl-GFP or Coa6-GFP endothelial cells and PC-3 tumor cells to assess in vivo vessel formation. (B) Cntrl-GFP vessels, left; Coa6-GFP vessels, right. GFP = transplanted GFP-tagged endothelial cells; CD31 = vasculature. GFP+ tip cells are indicated by red arrow heads. Scale bars, 50μm. (C) Quantification of vessel density (left) and tip cell formation (right), n = 3 mice per condition. (D to F) HPIN-stage FACS analysis of Δψ (D), vessel density (E), and prostate cancer weight (F) in cMyc, cMyc; Adrb2ecKO, cMyc; Cox10ecKO, and double cMyc; Adrb2ecKO; Cox10ecKO mice, showing restoration of glycolitic metabolism, angiogenesis, and cancer progression after co-deletion of Adrb2 and Cox10 in endothelial cells, n = 6 or 7 mice per condition. Error bars indicate SEM. *P<0.05. **P<0.01. ***P<0.001. ****P<0.0001.
To assemble cytochrome c oxidase (also known as complex IV), the terminal enzyme in oxidative phosphorylation that transfers electrons to molecular oxygen, COA6 cooperates with several factors, including the protein product of Cox10 [heme A:farnesyltransferase cytochrome c oxidase assembly factor; (36)]. Because conditional Cox10 deletion has been shown to reduce oxidative phosphorylation in vivo in skeletal muscle (37), we interbred the Cox10fl/fl and cMyc; Adrb2fl/fl; Cdh5-CreERT2 mouse strains (referred to as cMyc; Adrb2ecKO; Cox10ecKO). We found that conditional Cox10 deletion in cMyc; Adrb2ecKO; Cox10ecKO prevented the metabolic switch to oxidative phosphorylation in endothelial cells induced by Adrb2 deletion (Fig. 4D and fig. S18, A to C). Additionally, Cox10 deletion rescued vascular perfusion of the prostate (fig. S19A), angiogenesis (Fig. 4E, and fig. S19, B and C), and PIN cancer progression (Fig. 4F, fig. S18, D and E, and fig. S20, A to C), without affecting endothelial proliferation or viability (fig. S20, D and E). Thus, the shift to endothelial oxidative metabolism is sufficient to inhibit the PIN-stage progression switch.
Discussion
Our results elucidate a critical link between neural signals in the tumor microenvironment and angiogenesis, wherein inhibition of adrenergic nerve activity alters endothelial metabolism to prevent the angiogenic switch that supports aggressive prostate cancer. The loss of βAdR signaling increases oxidative phosphorylation in endothelial cells via increased expression of the mitochondrial cytochrome c oxidase assembly factor COA6, inhibiting angiogenesis.
Endothelial cell metabolism represents an emerging targetable pathway for the treatment of vascular diseases (38). For example, activation of oxidative phosphorylation in renal endothelium protects against post-ischemic reperfusion injury in the kidneys (39), and inhibition of endothelial cell glycolysis prevents pathologic angiogenesis in ophthalmologic diseases (40). Our results suggest that endothelial cell metabolism can be locally regulated by nerves.
Nerves and vessels are tightly associated during development, sharing patterning cues (41). As tumors need to re-develop a vascular network to ensure nutrition and communication, neural input may provide a critical set of signals that coordinate cancer progression (9). Our results suggest that adrenergic signals promote the angiogenic switch in prostate cancer and that their inhibition (e.g. using β-blockers) could prevent or delay the dominance of pro-angiogenic factors that allow tumor progression.
Therapies attempting to starve the tumor by inhibiting angiogenesis have had limited long-term therapeutic benefit in various cancers, including that of the prostate (42), most likely due to resistance mechanisms (43). Co-targeting angiogenesis with neural signals and/or endothelial cell metabolism may thus provide a multipronged therapeutic approach with the potential to overcome anti-angiogenic resistance.
Supplementary Material
Acknowledgments
We thank G. Karsenty, R.H. Adams and J.W. Pollard for Adrb2fl/fl, Cdh5-CreERT2, and Csf1r-iCre mice, respectively, and M. Ginsberg (Angiocrine Bioscience) for primary mouse prostate endothelial cells. We also thank C. Prophete, P. Ciero, S. Pierce, J.F. Reidhaar-Olson, P. Guo, X. L. Du, M. Hanoun, L. Tesfa, Y. Qiu, and I. L. Kurland for advice and technical support; A. Beck for assistance with pathology; A. Zahalka for assistance programming in R; and L. Schwartz, S.K. Libutti, H. Pierce, S. Murillo, T. Mizoguchi, and S. Pinho for critical advice. We are grateful for grant support from the NIH (HL097700, DK056638, and HL069438 to P.S.F.) and the New York State Department of Health (C029154 and C030318GG to P.S.F). A.H.Z received the Junior Investigator Neuroscience Research Award from the Albert Einstein College of Medicine and was supported by grant F30CA203446 from the National Cancer Institute and NIH training grants T32 NS007098 and GM007288. M.M. is a New York Stem Cell Foundation-Druckenmiller Fellow and was supported by the EMBO European Commission FP7 (Marie Curie Actions, EMBOCOFUND2012, GA-2012-600394, ALTF 447-2014). F.N. was supported by a Postdoctoral Fellowship for Research Abroad from the Japan Society for the Promotion of Science. This work was also supported by NIH shared equipment grant 1S10OD019961 and NIH Diabetes and Research Training Center grant P60DK020541.
Footnotes
References and notes
- 1.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339:58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 2.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nature reviews Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 3.Mukouyama YS, Shin D, Britsch S, Taniguchi M, Anderson DJ. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell. 2002;109:693–705. doi: 10.1016/s0092-8674(02)00757-2. [DOI] [PubMed] [Google Scholar]
- 4.Li W, et al. Peripheral nerve-derived CXCL12 and VEGF-A regulate the patterning of arterial vessel branching in developing limb skin. Developmental cell. 2013;24:359–371. doi: 10.1016/j.devcel.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ekstrand AJ, et al. Deletion of neuropeptide Y (NPY) 2 receptor in mice results in blockage of NPY-induced angiogenesis and delayed wound healing. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6033–6038. doi: 10.1073/pnas.1135965100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin P. Wound Healing–Aiming for Perfect Skin Regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 7.Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nature reviews Cancer. 2015;15:563–572. doi: 10.1038/nrc3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He D, et al. Biologic effect of neurogenesis in pancreatic cancer. Hum Pathol. 2016;52:182–189. doi: 10.1016/j.humpath.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Magnon C, et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- 10.Saloman JL, et al. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:3078–3083. doi: 10.1073/pnas.1512603113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao CM, et al. Denervation suppresses gastric tumorigenesis. Sci Transl Med. 2014;6 doi: 10.1126/scitranslmed.3009569. 250ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Longair MH, Baker DA, Armstrong JD. Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics (Oxford, England) 2011;27:2453–2454. doi: 10.1093/bioinformatics/btr390. [DOI] [PubMed] [Google Scholar]
- 13.Twardowski T, Fertala A, Orgel J, San Antonio J. Type I Collagen and Collagen Mimetics as Angiogenesis Promoting Superpolymers. Current Pharmaceutical Design. 2007;13:3608–3621. doi: 10.2174/138161207782794176. [DOI] [PubMed] [Google Scholar]
- 14.Tuxhorn JA, et al. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clinical cancer research: an official journal of the American Association for Cancer Research. 2002;8:2912–2923. [PubMed] [Google Scholar]
- 15.Burns-Cox N, Avery NC, Gingell JC, Bailey AJ. Changes in Collagen Metabolism in Prostate Cancer: A Host Response That May Alter Progression. The Journal of urology. 2001;166:1698–1701. doi: 10.1016/s0022-5347(05)65656-x. [DOI] [PubMed] [Google Scholar]
- 16.Huss WJ, Hanrahan CF, Barrios RJ, Simons JW, Greenberg NM. Angiogenesis and prostate cancer: identification of a molecular progression switch. Cancer research. 2001;61:2736–2743. [PubMed] [Google Scholar]
- 17.Ellwood-Yen K, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 18.Ittmann M, et al. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer research. 2013;73:2718–2736. doi: 10.1158/0008-5472.CAN-12-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwata T, et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PloS one. 2010;5:e9427. doi: 10.1371/journal.pone.0009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinoi E, et al. The sympathetic tone mediates leptin’s inhibition of insulin secretion by modulating osteocalcin bioactivity. The Journal of cell biology. 2008;183:1235–1242. doi: 10.1083/jcb.200809113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng L, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am J Pathol. 2010;176:952–967. doi: 10.2353/ajpath.2010.090622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu X, et al. Age-dependent fate and lineage restriction of single NG2 cells. Development. 2011;138:745–753. doi: 10.1242/dev.047951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorensen I, Adams RH, Gossler A. DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood. 2009;113:5680–5688. doi: 10.1182/blood-2008-08-174508. [DOI] [PubMed] [Google Scholar]
- 25.De Bock K, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 26.Wilhelm K, et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529:216–220. doi: 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang YH, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–1323. doi: 10.1016/j.cell.2014.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kocabas F, Zheng J, Zhang C, Sadek HA. Metabolic characterization of hematopoietic stem cells. Methods in molecular biology. 2014;1185:155–164. doi: 10.1007/978-1-4939-1133-2_10. [DOI] [PubMed] [Google Scholar]
- 29.Maryanovich M, et al. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nature communications. 2015;6:7901. doi: 10.1038/ncomms8901. [DOI] [PubMed] [Google Scholar]
- 30.Krutzfeldt A, Spahr R, Mertens S, Siegmund B, Piper HM. Metabolism of exogenous substrates by coronary endothelial cells in culture. J Mol Cell Cardiol. 1990;22:1393–1404. doi: 10.1016/0022-2828(90)90984-a. [DOI] [PubMed] [Google Scholar]
- 31.Seandel M, et al. Generation of a functional and durable vascular niche by the adenoviral E4ORF1 gene. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19288–19293. doi: 10.1073/pnas.0805980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng TL, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munger J, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nature biotechnology. 2008;26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hotchkiss KA, et al. Inhibition of endothelial cell function in vitro and angiogenesis in vivo by docetaxel (Taxotere): association with impaired repositioning of the microtubule organizing center. Molecular cancer therapeutics. 2002;1:1191–1200. [PubMed] [Google Scholar]
- 35.Suzuki R, Hotta K, Oka K. Spatiotemporal quantification of subcellular ATP levels in a single HeLa cell during changes in morphology. Scientific reports. 2015;5:16874. doi: 10.1038/srep16874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghosh A, et al. Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency. Hum Mol Genet. 2014;23:3596–3606. doi: 10.1093/hmg/ddu069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet. 2005;14:2737–2748. doi: 10.1093/hmg/ddi307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Potente M, Carmeliet P. The Link Between Angiogenesis and Endothelial Metabolism. Annu Rev Physiol. 2017;79:43–66. doi: 10.1146/annurev-physiol-021115-105134. [DOI] [PubMed] [Google Scholar]
- 39.Liu S, Soong Y, Seshan SV, Szeto HH. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am J Physiol Renal Physiol. 2014;306:F970–980. doi: 10.1152/ajprenal.00697.2013. [DOI] [PubMed] [Google Scholar]
- 40.Schoors S, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 41.Eichmann A, Brunet I. Arterial innervation in development and disease. Sci Transl Med. 2014;6:252ps259. doi: 10.1126/scitranslmed.3008910. [DOI] [PubMed] [Google Scholar]
- 42.Aragon-Ching JB, Madan RA, Dahut WL. Angiogenesis inhibition in prostate cancer: current uses and future promises. Journal of oncology. 2010;2010:361836. doi: 10.1155/2010/361836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nature reviews Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.