

Short abstract
Background
Diabetic gastropathy is a complex neuromuscular dysfunction of the stomach that commonly occurs in diabetes mellitus. Diabetic patients often present with upper gastrointestinal symptoms, such as epigastric discomfort or pain. The aim of this study was to assess gastric sensation in streptozocin-induced diabetes mellitus (DM) rats and to determine the contribution of C-C motif chemokine receptor 2 (CCR2) signaling to gastric hyperalgesia.
Results
DM rats showed signs of neuropathy (cutaneous mechanical hyperalgesia) from two weeks after streptozocin administration until the end of the experiment. Accelerated solid gastric emptying was observed at two weeks after streptozocin administration compared to the controls. Intense gastric hyperalgesia also developed in DM rats at two weeks after streptozocin administration, which was significantly reduced after intrathecal administration of the CCR2 antagonist INCB3344. Immunochemical analysis indicated that CCR2 expression was substantially upregulated in small and medium-sized dorsal root ganglia neurons of DM rats, although the protein level of monocyte chemoattractant protein-1, the preferred ligand for CCR2, was not significantly different between the control and DM groups.
Conclusions
These data suggest that CCR2 activation in nociceptive dorsal root ganglia neurons plays a role in the pathogenesis of gastric hyperalgesia associated with diabetic gastropathy and that CCR2 antagonist may be a promising treatment for therapeutic intervention.
Keywords: Neuropathic pain, diabetes mellitus, streptozocin, gastric hyperalgesia, CCR2 receptor
Background
Diabetes mellitus (DM) is a major global health problem that affects 415 million people worldwide. Its prevalence is predicted to increase to 642 million people (1 in 10 adults) by 2040.1 Diabetic neuropathy is one of the most common complications of both type 1 and type 2 DM, and comprises sensory neuropathy, focal and multifocal neuropathies, and autonomic neuropathy.2 Approximately 50% of DM patients have some form of neuropathy, with about one third suffering from chronic pain symptoms.3 Diabetic sensory neuropathy frequently coexists with autonomic dysfunction that affects multiple organs in the body, including the gastrointestinal (GI) system.4
Diabetic gastropathy is the term for gastric neuromuscular dysfunctions in DM and is often associated with altered gastric emptying and GI symptoms. In multivariable analysis, significant weight loss and neuropathy are respectively associated with the increased risk of delayed and rapid gastric emptying.5 Upper GI symptoms in DM are also correlated to diabetic complications, particularly peripheral neuropathy.6,7 It is noteworthy that gastropathy in DM not only exacerbates GI symptoms such as nausea, vomiting, bloating, and upper abdominal pain but also impairs glycemic control and has a negative impact on quality of life.8 As gastropathy is a serious health problem in DM, several studies in recent decades have investigated its mechanisms and management. However, most reports focus exclusively on gastric emptying. Gastric sensation remains an important yet under-discussed issue in DM. Furthermore, to our knowledge, the mechanisms of gastric pain in DM have as yet not been investigated.
Chemokines and chemokine receptors, expressed by immune and nerve cells, play a central role in the pathophysiology of acute and chronic pain syndromes.9 They induce neuromodulatory and neurotoxic effects directly through the activation of G-protein-coupled receptors or indirectly through neuron-glial interactions.10 Recent studies have targeted monocyte chemoattractant protein 1 (MCP-1), the so-called C-C motif chemokine ligand 2, and its preferred C-C motif chemokine receptor 2 (CCR2) in the pathogenesis of neuropathic pain. Following peripheral nerve injury, MCP-1 upregulation has been demonstrated in sensory neurons of the dorsal root ganglion (DRG)11–13 and the spinal cord,14–16 as well as in spinal microglia17 and astrocytes.13,15,18 Behavioral studies indicate that MCP-1 neutralizing antibody or CCR2 antagonists can reduce the mechanical allodynia induced by nerve injury in rodent models.12,18–22 In addition, mechanical allodynia after sciatic nerve ligation injury is totally abolished in CCR2 knockout mice,17,23 indicating the essential role of MCP-1/CCR2 signaling in the induction and maintenance of neuropathic pain. Interestingly, Jamali et al.24 and Guan et al.25 further report that serum MCP-1 level is significantly higher in type 1 DM patients with multiple complications, including diabetic neuropathy.
Prompted by the above reports, this study aimed to assess gastric sensation by behavioral assay in the streptozocin (STZ)-induced DM model. The effect of CCR2 antagonist on gastric hyperalgesia in vivo and the localization of MCP-1 and CCR2 proteins in the DRG and spinal cord were also investigated in this model.
Methods
Experimental animals
All experiments were performed on male Sprague-Dawley rats (eight weeks, 250–300 g, obtained from Sankyo Labo Service Corporation, Toyama, Japan). The rats were housed in individual cages with ad libitum access to food and water and maintained under a controlled environment (23 ± 1°C, humidity 60 ± 10%, 12/12 h light/dark cycle). The protocol was approved by Kanazawa University’s Institutional Committee on Animal Research, and all the experiments were performed in accordance with the International Association for the Study of Pain (IASP) guidelines for the care and use of laboratory animals in research.
Induction of diabetes
Diabetes was induced in the experimental rats by a single intraperitoneal (i.p.) injection of STZ (LKT Laboratories, St. Paul, MN) at 45 mg/kg, prepared in 0.1 M citrate buffer (CB; pH 4.5), after overnight fasting. Blood glucose level was determined biweekly from the saphenous vein using a blood glucometer (Life Check, Gunze, Kyoto, Japan). Animals with a level of >250 mg/dl were regarded as hyperglycemic and were placed in the experimental DM group. However, very few (<10%) DM rats were removed from the study for reasons of severe ill health. Exclusion criteria were excessive weight loss (>20%), lack of activity with no responsiveness to behavioral tests, wound sepsis at the site of the abdominal incision (which might interfere with electromyographic (EMG) recording data), and septicemia via the intrathecal catheter. Age-matched control (CT) rats received an equal volume of 0.1 M CB.
Evaluation of pain behavior
All behavioral experiments were performed biweekly for 10 weeks after STZ or CB administration.
Cutaneous mechanical hyperalgesia
Cutaneous mechanical hyperalgesia was assessed by the Randall-Selitto paw pressure test, employing an analgesy meter (Model 37215, Ugo Basile, Varese, Italy). The probe tip of the machine was applied to the dorsal surface of the rat’s left hind paw. The intensity of the pressure on the probe was set to increase linearly from 0 to 250 g. The nociceptive threshold was determined by the force (g) at which the rat showed its withdrawal response. Successive stimuli were applied to the left hind paw at 5-min intervals five times, and the mean value after excluding the lowest and highest intensities was assumed to be the paw withdrawal threshold (PWT) of that animal.
Cutaneous mechanical allodynia
Cutaneous mechanical allodynia was examined using von Frey’s method, employing a dynamic plantar esthesiometer (Model 37450, Ugo Basile). The rats were first acclimated to the testing environment by placement in a clear plastic chamber with a metal mesh floor for about 30 min. Nociceptive threshold was measured by paw withdrawal response to increasing vertical force of a von Frey filament (preset force from 0 to 50 g in 10 s) applied to the plantar surface of the right hind paw. Measurements were repeated five times at approximately 5-min intervals. The PWT was defined as the mean value of the record excluding the lowest and highest forces.
Cutaneous thermal hyperalgesia
The Hargreaves method was used to determine the thermal hyperalgesia using a plantar test apparatus (Model 37370, Ugo Basile). The rats were habituated to the apparatus by placement in a transparent Perspex enclosure with a thin glass floor for about 30 min. A focused beam of infrared heat was applied to the plantar surface of the right hind paw. The intensity of the heat source was maintained at 60 and the cut-off duration was 30 s. Paw withdrawal latency was defined as the time at which an abrupt withdrawal response of the hind paw was observed. The procedure was repeated five times at approximately 5-min intervals. The mean value, excluding the lowest and highest intensities, was taken for each animal.
Gastric hyperalgesia
Gastric sensitivity was assessed by EMG recording from the acromio-trapezius muscle response to gastric balloon distention (GD), as previously described in Ozaki et al.26
Surgical procedure
One week before the recording time, under anesthesia with sodium pentobarbital (i.p., 50 mg/kg), a left lateral oblique epigastric incision was made to permit insertion of a latex balloon (∼ 2.5 cm in diameter) into the rat’s stomach through the fundus. The polyethylene tube connected to the balloon for air inflation was subcutaneously taken out from the nape of the neck. Two wire electrodes (AS632, Cooner Wire, CA) were implanted into the acromio-trapezius muscle for EMG recording.
EMG recording
Each rat was first accommodated to the EMG recording room for at least 30 min. The gastric balloon was inflated to 60 mmHg pressure using the sphygmomanometer. The visceromotor response (VMR) of the acromio-trapezius muscle to GD was recorded twice for 20 s at a 4-minute interval. Each GD trial consisted of two parts: a 10-s pre-distension period (baseline) and a 20-s gastric distension period. Response to GD was defined as an increase in EMG activity above the baseline during GD. The EMG data were amplified, filtered (×10,000, 300–5000 Hz; A-M systems, Everett, WA), digitized, and integrated using a SPIKE2/CED1401 data-acquisition interface. The raw EMG was rectified and quantified by calculating the area under the curve.
Assessment of gastric emptying
The rate of gastric emptying was determined by amberlite pellet assay as previously described in Matsuda et al.27 The rat, which had fasted for 20 to 24 h but had free access to water, was provided 100 amberlite pellets (∼1 mm in diameter) into the stomach by oral tube. The water supply was removed following insertion of the pellets. After 2 h, the rat was euthanized with sodium pentobarbital (i.p., 250 mg/kg). The stomach was then surgically removed and the amberlite pellets remaining inside the stomach were counted. The gastric emptying rate for each rat was calculated according to the following formula.
where A denotes the number of amberlite pellets inserted into the stomach, and B denotes the number of amberlite pellets remaining in the stomach after 2 h.
Intrathecal drug administration
At the time of gastric surgery for EMG recording, a PE10 catheter (Becton, Dickinson and Company, Franklin Lakes, NJ) was implanted into the intrathecal space via a small cut opening on the atlanto-occipital membrane. The tip of the catheter was placed at the T10 level and fixed in the correct position using a few sutures. After abdominal and/or intrathecal surgery, all the rats received ciprofloxacin (i.p., 20 mg/kg in 0.1 N HCl; Sigma-Aldrich, Tokyo, Japan) to prevent the type of infections that usually develop with DM.
To evaluate the effect of the drug, DM rats with ultrasensitive response to GD (10-fold more sensitive than the CT) were selectively placed in the therapeutic (pharmacology) group. Following EMG recording at two weeks after STZ or CB administration, the animals received a single intrathecal injection of CCR2 antagonist (25 µl of INCB3344 in 10% DMSO + saline; MedChem Express, Princeton, NJ) or vehicle (10% DMSO + saline). The VMR to GD was then recorded again at 1, 2, 4, and 24 h after injection.
Immunohistochemistry
Both CT and DM groups were sacrificed at two weeks after STZ or CB administration. The rats were deeply anesthetized with sodium pentobarbital (i.p., 70 mg/kg) and transcardially perfused with heparinized physiological saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer solution. The T10 DRG and spinal cords were removed and post-fixed in the same fixative overnight. The tissues were then cryoprotected in 30% sucrose prepared in 0.1 M phosphate buffer saline at 4°C, then rapidly frozen in optimal cutting temperature compound (Sakura Finetek, Torrance, CA) and stored at –80°C. Spinal cord sections (15 µm) and DRG sections (10 µm) were cut using a cryostat, mounted on glass slides and processed for immunofluorescence as previously described.28
The sections were first allowed heat-mediated antigen retrieval by 0.01 M CB (pH 6.0) and then blocked with 5% normal donkey serum in 0.1 M phosphate buffer saline containing 0.3% Triton X-100 for 30 min at room temperature. The sections were then incubated overnight at 4°C with primary antibodies for MCP-1 (rabbit, 1:500, Torrey Pines Biolabs, Secaucus, NJ), CCR2 (rabbit, 1:200, Novus Biologicals LLC, Littleton, CO), NeuN (mouse, 1:5000, Merck Millipore, Billerica, MA), GFAP (mouse, 1:1000, Cell Signaling Technology, Danvers, MA), and Iba-1 (goat, 1:500, Abcam, Cambridge, UK). The sections were then incubated with donkey anti-rabbit IgG (H + L) Alexa Fluor 594, donkey anti-goat IgG (H + L) Alexa Fluor 488, and goat anti-mouse IgG (H + L) Alexa Fluor 488 conjugated secondary antibodies (1:1000, Thermo Fisher Scientific, Waltham, MA) for 1 h at room temperature. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (Nacalai Tesque, Kyoto, Japan). The slides were cover-slipped with Vectashield mounting media (Vector Laboratories, Burlingame, CA) and examined under a Zeiss LSM 5 Pascal Laser Scanning Confocal Microscope (Carl Zeiss, Advanced Imaging Microscopy, Jena, Germany). The stained sections were analyzed using Image J software.
Western blot
The T10 DRG and spinal cords were taken from the rats as soon as possible after sacrificing by cervical dislocation at two weeks post-injection of STZ or CB. The tissues were homogenized in a lysis buffer (20 mM Tris-HCl pH 7.6, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, and 1 mM 1,4-dithiothreitol) containing protease inhibitor cocktail (Nacalai Tesque). Protein concentrations were determined using a Protein Assay Rapid Kit (Wako Pure Chemicals Industries, Osaka, Japan) with bovine serum albumin as the standard. DRG (5 µg) and spinal cord (10 µg) proteins were separated by SDS-PAGE gel and transferred onto PDVF membrane (Merck Millipore). The blotting membranes were blocked with Blocking One (Nacalai Tesque) for 30 min at room temperature, followed by incubation with primary antibodies for MCP-1 (rabbit, 1:1000) and CCR2 (rabbit, 1:2000) overnight at 4°C. The blots were then incubated with HRP-conjugated goat anti-rabbit IgG (H + L) secondary antibody (1:10,000, Cell Signaling Technology) for 1 hour at room temperature. The GAPDH antibody (mouse, 1:10,000, Acris Antibodies, San Diego, CA) was used as the loading control. The protein bands were visualized by chemiluminescent detection using ImmunoStar LD (Wako Pure Chemicals Industries). The scanned images (ImageQuant LAS 4000, GE Healthcare, Little Chalfont, UK) were analyzed using image J software.28
Data analysis
Statistical analysis was performed using Sigma Plot 12.5 software (Systat Software Inc., San Jose, CA). Graphic data were presented as mean ± SEM. All the behavioral data were analyzed using one-way analysis of variance (ANOVA), two-way ANOVA, or two-way repeated measures ANOVA where appropriate, followed by the Holm-Sidak method for multiple comparisons. Data from immunohistochemistry (IHC) and Western blot were analyzed using Student’s unpaired t-test. A value of p < 0.05 was considered statistically significant.
Results
General changes in STZ-induced DM rats
Following administration of STZ, about 80% of the rats developed DM, suffering from the usual symptoms of diabetes such as polyuria, polydipsia, and polyphagia. Despite a markedly increased food and water intake, the DM rats showed no weight gain: their body weights were constant throughout the experiment, unlike the age-matched CT rats that showed weight gain (Figure 1(a)). The blood glucose level was continuously elevated (>250 mg/dl) in the DM group, starting from three days after STZ administration until the end of the experiment (10 weeks), whereas the CT group maintained their blood glucose level within the normoglycemic range (Figure 1(b)).
Figure 1.

Differences in body weight and blood glucose level between the control and diabetic groups. Body weight (a) and random blood glucose level (b) were measured in the control (CT) and diabetic (DM) rats. Data are shown as mean ± SEM, *p < 0.001 CT versus DM, n = 15–20 rats per group. No weight gain (a) and hyperglycemia (b) were observed in DM rats throughout the experiments.
Assessment of peripheral diabetic neuropathy
The development of neuropathy in STZ-induced DM rats was confirmed by peripheral cutaneous mechanical and thermal hypersensitivity tests. There was no significant difference in cutaneous sensitivity between the experimental groups before the induction of diabetes. STZ induced a sustained and significant reduction of PWT as measured by the Randall-Selitto Paw Pressure Test from two weeks until the end of the experiment (p < 0.001 CT vs. DM, two-way repeated measures ANOVA; p < 0.001, one-way ANOVA for DM group, n = 6), whereas the CT group showed no significant difference between before and after treatment (Figure 2(a)). In von Frey’s filament test, the DM rats showed a lower mechanical nociceptive threshold than the CT, starting from four weeks after STZ administration, but this result was statistically significant only after eight weeks (p = 0.03 CT vs. DM, n = 6) (Figure 2(b)). There was no significant difference in response to thermal stimuli between the CT and DM groups at any time point (p = 0.99, n = 6) (Figure 2(c)). These results indicated that DM rats had cutaneous mechanical hyperalgesia and allodynia, but no change in thermal sensitivity.
Figure 2.

Changes in cutaneous sensitivities after induction of diabetes by STZ. The Randall-Selitto test (a), von Frey’s test (b), and heat sensitivity test (c) were performed in control (CT) and diabetic (DM) rats. Data are shown as mean ± SEM, *p < 0.001, †p < 0.05 CT versus DM, n = 6 rats per group. PWT measured by Randall-Selitto test (a) and von Frey’s test (b) was reduced in the STZ-induced DM group.
Assessment of diabetic gastropathy
Altered gastric emptying and upper abdominal pain are frequent symptoms of diabetic gastropathy.5,29 In this study, the rate of 2-h solid gastric emptying was significantly accelerated in the DM group at two weeks after STZ as compared with the CT group (p < 0.01, n = 6) (Figure 3(a)). During that period, all DM rats discharged almost all their stomach contents into the small intestine within 2 h postprandially, whereas the CT rats emptied about half. The gastric emptying rate after four weeks in the DM group was also faster than the CT, but the data were not statistically significant (p = 0.14, n = 6). From 6 weeks, rapid gastric emptying was no longer observed in DM rats.
Figure 3.

Effect of STZ-induced diabetes on gastric motor and sensory functions. Gastric emptying (a) and gastric sensitivity (b) were assessed in control (CT) and diabetic (DM) rats. Data are shown as mean ± SEM, *p < 0.001, #p < 0.01 CT versus DM. (a) Significantly rapid gastric emptying was observed in DM rats at two weeks after STZ administration (n = 6 rats per group). (b) Distension-induced gastric hyperalgesia developed significantly in DM rats (n = 9 rats per group). (c) Representative EMG recording data from each CT and DM rats.
In addition to gastric motor function, the sensitivity of the stomach was also higher in DM rats than in the CT rats according to the intragastric balloon distension study. VMR to GD was substantially higher in the DM group, especially at two weeks after STZ administration (p < 0.001, n = 9) than in the CT group, with significant levels at 2, 4, 8, and 10 weeks after STZ administration (p < 0.01, n = 9) (Figure 3(b)). At two weeks after STZ administration, approximately 40% of DM rats showed ultrasensitive behavior to GD, i.e., about 10-fold the sensitivity seen in the CT rats (Figure 3(c)). This ultrasensitive response in DM rats was not otherwise observed at any other time point. These findings suggest that rapid gastric emptying and severe gastric hyperalgesia develop in the early phase of DM.
CCR2 and gastric hypersensitivity in DM
Chemokine MCP-1 and its preferred receptor CCR2 are well known to be involved in the development and maintenance of neuropathic pain.9,23 In this study, DM rats that had shown an ultrasensitive response to GD at two weeks after STZ administration were selectively used to continue the therapeutic option for gaining clear evidence on the effects of the drug. Intrathecal application of CCR2 antagonist INCB3344 both time and dose dependently suppressed established gastric hyperalgesia in the STZ-induced DM model (Figure 4). Gastric hyperalgesia in DM rats was significantly attenuated by INCB3344 (1 mM and 0.1 mM) at 1 h (p < 0.01, n = 5–7) and 2 h (p < 0.05) after administration as compared with vehicle-treated rats. Gastric hyperalgesia was also slightly lessened in DM rats at 4 h (p = 0.44 for 1 mM and p = 0.34 for 0.1 mM) and 24 h (p = 0.41 for 1 mM and p = 0.11 for 0.1 mM) after injection, but these data were not significant. A very low dose (0.01 mM) of INCB3344 had no significant effect on gastric hyperalgesia at any time point after injection (p = 0.95 vehicle vs. INCB3344, n = 5–6). There was no change in gastric sensitivity in CT rats before and after treatment. This finding suggests CCR2 signaling to be involved in the development of diabetes-induced gastric hyperalgesia but is not the sole determinant of gastric sensitivity under normal physiological conditions.
Figure 4.
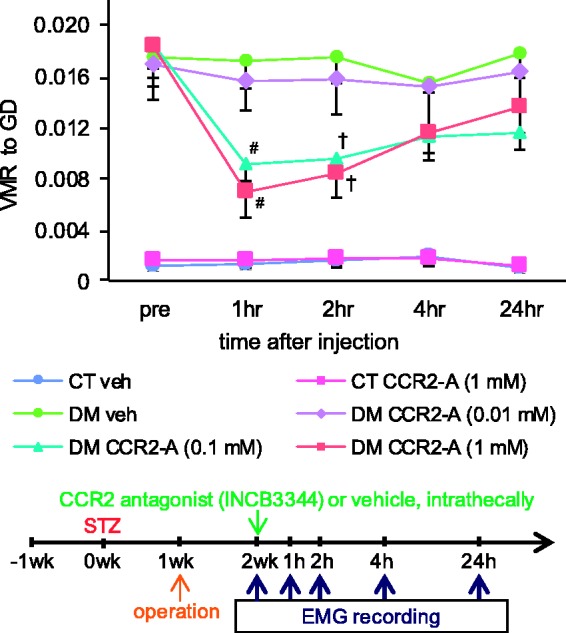
Effect of CCR2 antagonist (INCB3344) on diabetes-induced gastric hyperalgesia. Gastric sensitivity was measured in the control (CT) and diabetic (DM) rats at two weeks after STZ administration. The CCR2 antagonist INCB3344 (CCR2-A) or vehicle (veh) was injected intrathecally. Data are shown as mean ± SEM, #p < 0.01, †p < 0.05 veh versus CCR2-A, n = 5–7 rats per group. INCB3344 (1 mM and 0.1 mM) significantly suppressed distension-induced gastric hyperalgesia in DM rats at 1 and 2 h after administration as compared with vehicle-treated rats.
CCR2 expression in DRG and spinal cord
To determine the mechanism of CCR2 involvement in diabetes-induced gastric hyperalgesia, immunolocalization of CCR2 and its protein level were investigated in T10 DRG and the spinal cord at two weeks after STZ administration. In the DRG sections, CCR2-immunoreactivity (IR) was exclusively observed in neurons rather than in non-neuronal cells in both CT and DM rats (Figure 5(a) and (b)). The mean number of CCR2-expressed neurons per section was significantly greater in the DM group (20% of total, p < 0.001, n = 4) than in the CT (4% of total) (Figure 5(e)), although there was no significant difference in the mean number of total DRG neurons per section between them (p = 0.87, n = 4) (Figure 5(c)). DRG neurons were classified by cell diameter into small (≤ 30 µm), medium (31–44 µm), and large (≥ 45 µm) categories according to Zhang et al.13 There was no significant difference in the size distribution of DRG neurons between the CT and DM rats (p = 0.12, p = 0.11, and p = 0.57 for small-, medium-, and large-sized neurons, respectively, n = 4) (Figure 5(d)). In both groups, a considerable number of CCR2-IR neurons (70%–80%) were small and the remaining (20%–30%) were medium sized, but the numbers were much higher in the DM rats than in the CT (Figure 5(f)). No CCR2-IR was found in the large DRG neurons.
Figure 5.

Cellular localization of CCR2-IR in T10 DRG at two weeks after STZ administration. Representative images of immunostaining with anti-CCR2 antibody in control (CT) (a) and diabetic (DM) (b) rats. Arrows represent CCR2-positive cells. Scale bar: 50 µm. Number of total and CCR2-IR neurons per section (c, e) and their size distribution (d, f) were determined in both the CT and DM groups. Data are shown as mean ± SEM, *p < 0.001 CT versus DM, n = 4 rats per group. CCR2 was exclusively expressed in small- and medium-sized DRG neurons in both the CT and DM rats (a, b, f). CCR2-IR neurons were significantly more numerous in the DM group than in the CT (a, b, e) despite no significant change between them in total DRG neurons per section (c). The number of CCR2-IR cells was relatively greater in small DRG neurons of DM rats according to the size distribution of the DRG neurons (d, f).
In spinal cord sections, the expression of CCR2 protein was restricted exclusively to the superficial layer of the spinal dorsal horn, especially in the lamina I area; only a few were seen in lamina II (Figure 6(a) and (b)). No CCR2 immunolabeling was detected in any other dorsal horn regions, including motor neurons, or in non-neuronal glial cells, as revealed by double-labeling IHC with glial cell markers (data not shown). Western blot analysis indicated that the CCR2 protein level was slightly higher in the DM group than in the CT group, but this was not statistically significant (p = 0.45, n = 3) (Figure 6(c) and (d)). These data show CCR2 signaling to be substantially activated in the DRG neurons of STZ-induced DM rats.
Figure 6.

CCR2 protein expression in T10 spinal cord at two weeks after STZ administration. Representative images of immunostaining with anti-CCR2 antibody in control (CT) (a) and diabetic (DM) (b) rats. Arrows represent CCR2-positive regions. (a2) and (b2) are higher magnification images of white-boxed regions in (a1) and (b1), respectively. Scale bar: 50 µm. CCR2-IR was detected only in the superficial layer of the spinal dorsal horn (n = 4 rats per group). (c) Representative Western blot analyses using anti-CCR2 and GAPDH antibodies. GAPDH was used as a loading control. CT 1–3: CT rats; DM 1–3: DM rats. (d) Semi-quantitative densitometric data for CCR2-IR are shown. Data are shown as mean ± SEM, n = 3 rats per group. The CCR2 protein level in the spinal cord was not significantly different between the CT and DM groups.
Expression of MCP-1 in DRG and the spinal cord
MCP-1 is the preferred ligand for CCR2, and these interactions play an essential role in the pathophysiology of neuroinflammatory diseases.9,10 To determine the possible involvement of MCP-1 in diabetes-induced gastric hyperalgesia, MCP-1 protein expression in T10 DRG and the spinal cord were also identified in both the CT and DM groups. IHC staining indicated MCP-1 immunolabeling to be extensively localized in the DRG neurons of both CT and DM rats, but its upregulation would not be expected in the DM group compared to the CT (Figure 7(a) and (b)). Western blot analysis also supported the IHC finding that the mean expression levels of MCP-1 were closely similar in both CT and DM groups (p = 1.00, n = 3) (Figure 7(c) and (d)).
Figure 7.

Immunolocalization of MCP-1 and its protein level in T10 DRG at two weeks after STZ administration. Representative images of immunostaining with anti-MCP-1 antibody in control (CT) (a) and diabetic (DM) (b) rats. Scale bar: 50 µm. MCP-1 was constitutively expressed in DRG neurons in both CT and DM rats (n = 4 rats per group). (c) Representative Western blot analyses using anti-MCP-1 and GAPDH antibodies are shown. GAPDH was used as a loading control. CT 1–3: CT rats; DM 1–3: DM rats. (d) Semi-quantitative densitometric data for MCP-1-IR is shown. Data are shown as mean ± SEM, n = 3 rats per group. There was no difference in MCP-1 protein levels between the CT and DM groups.
In spinal cord tissues, MCP-1 was widely expressed in the astrocytes of the whole spinal cord area (Figure 8(a) to (e)) and a few were also seen in the motor neurons of the ventral horn regions (data not shown). Western blot analysis indicated MCP-1 protein expression to be higher in DM than in the CT group, but the data were not statistically significant (p = 0.25, n = 3) (Figure 8(f) and (g)). These findings indicate that chemokine MCP-1 is expressed in the neurons of the DRG and in the non-neuronal cells of the spinal cord, but its upregulation was not observed in the DM group.
Figure 8.

Immunolocalization of MCP-1 and its protein level in T10 spinal cord at two weeks after STZ administration. Representative images of immunostaining with anti-MCP-1 antibody in control (CT) (a) and diabetic (DM) (b) rats. (a2) and (b2) are higher magnification images of white-boxed regions in (a1) and (b1), respectively. Scale bar: 50 µm. MCP-1 was expressed in the whole spinal cord (n = 4 rats per group). Double-labeling IHC showed MCP-1 to be co-localized with astrocyte marker (GFAP) (c), but not with microglial (Iba1) (d) and neuronal (NeuN) (e) markers. Scale bar: 50 µm. (f) Representative Western blot analyses using anti-MCP-1 and GAPDH antibodies are shown. GAPDH was used as a loading control. CT 1–3: CT rats; DM 1–3: DM rats. (g) Semi-quantitative densitometric data for MCP-1-IR are shown. Data are shown as mean ± SEM, n = 3 rats per group. The MCP-1 protein level was higher in the DM group than in the CT group, but the data were not significant.
Discussion
Diabetic neuropathy is a chronic deteriorating complication characteristic of both type 1 and type 2 DM, affecting multiple organs and systems throughout the whole body. Diabetic gastropathy is a term that embraces a number of neuromuscular disorders of the stomach, including the gastric emptying dysfunction. The regulation of gastric emptying is undoubtedly multifactorial, consisting of the normal functions of gastric motility, GI hormones, and neuronal reflex.30 Although gastroparesis is a well-known late complication of DM, rapid gastric emptying has also been reported in some clinical and preclinical studies in the early phase of both type 1 and type 2 DM. Solid gastric emptying was accelerated in obese type 2 DM patients without overt autonomic neuropathy when compared with obese non-diabetic patients.31 Schwartz et al.32 also found a rapid gastric emptying of solid pancake meal in type 2 DM patients compared with the controls with similar BMI. Accelerated solid gastric emptying was also observed in the first one to two weeks in NOD/LtJ mice and in STZ-induced DM rats.33,34 In agreement with the previous findings, this study also exhibited unambiguously accelerated solid gastric emptying in DM rats at two weeks after STZ administration.
Diabetic patients often present with GI symptoms that include nausea, vomiting, bloating, and epigastric pain that result from alterations in gastric emptying. In spite of a great deal of research having been conducted on gastric emptying, studies of gastric sensations are rare: only two investigations have reported an increased sensory response to gastric distension in type 1 DM patients compared with healthy controls.35,36 Similar findings were observed in this study: that DM rats had higher sensitivity to GD than did age-matched CT rats (Figure 3(b)), and furthermore, that hypersensitive VMR to GD in DM rats was partially but significantly reversed for at least 2 h by intrathecal injection of the CCR2 antagonist INCB3344 (1 mM and 0.1 mM) (Figure 4). It was consistent with other neuropathic pain studies in which mechanical and thermal hyperalgesia after peripheral nerve injury were inhibited by CCR2 antagonists.18,21,22
Upregulation of CCR2 is convincingly recorded in all studies of neuropathic pain, although its cellular localization remains highly controversial, as it varies according to the species and antibodies used, presenting within DRG neurons,11,13,18,20,37 dorsal horn neurons,15,38 astrocytes,39 and microglia23,40 of the spinal cord. In the present study, CCR2 expression was substantially increased in the DRG neurons of DM rats, whereas it was hardly detectable in CT sections (Figure 5(e)). CCR2-IR cells were notably restricted to small- and medium-sized DRG neurons (Figure 5(f)), as well as in the most superficial laminae of the spinal dorsal horn (Figure 6(a) and (b)). It is well known that almost all nociceptive C and Aδ fibers possess small cell bodies, and their central terminals project to the superficial dorsal horn (lamina I and II) of the spinal cord.41 Both behavioral and IHC findings therefore suggest that increased CCR2 signaling in neurons of the DRG and spinal cord plays a major role in the pathogenesis of distension-induced gastric hyperalgesia in DM.
CCR2 is the preferred receptor, with a higher affinity for MCP-1.42 Several studies have revealed the contribution of MCP-1/CCR2 signaling to the processes of inflammatory and neuropathic pain in animal models. Activation of MCP-1 induces the initiation and maintenance of pain via a dual mechanism that involves the central sensitization of neurons and neuron-glial interactions.9 In primary neuronal culture, pre-incubation of MCP-1 augmented capsaicin-evoked cationic currents in small-diameter DRG neurons.43 Application of MCP-1 increased excitatory synaptic transmission and enhanced NMDA- and AMPA-induced currents in lamina II neurons of the spinal dorsal horn.15 MCP-1 also potentially inhibited GABA-induced currents via GABAA receptors in a primary culture of spinal cord neurons.38
Recent studies have revealed that MCP-1/CCR2 signaling is also mediated by neuron-glial interactions in nociceptive transmission. Ji et al.44 postulate that MCP-1/CCR2-mediated neuron-microglial interaction is involved in the induction of neuropathic pain, whereas MCP-1/CCR2 mediated astrocyte-neuron signaling takes part in the maintenance of neuropathic pain. It has been supported by the following studies: The time course of MCP-1 upregulation in DRG neurons was parallel to that of microglial activation after nerve injury.12,16 Intrathecal injection of MCP-1 neutralizing antibody or CCR2 antagonists inhibited microglial activation in neuropathic pain models.12,17,21 MCP-1-induced microglial activation and nerve injury-induced phosphorylation of p38 mitogen-activated protein kinase (MAPK) in spinal microglia was diminished in CCR2 knockout mice.17,23 MCP-1 expression was primarily upregulated in astrocytes of the spinal cord and trigeminal nucleus, but not in sensory neurons in some neuropathic pain studies.15,19,22 Transgenic mice overexpressing MCP-1 in astrocytes were hypersensitive to pain.45 In addition, Zhang and De Koninck16 noted that early microglial activation induced by MCP-1-expressed damaged neurons contributed to the initiation of neuropathic pain, whereas delayed astrocyte activation was crucial in maintaining long-lasting hypersensitivity.
Based on previous studies of diabetic neuropathy, activation of spinal microglia in the dorsal horn has been demonstrated in the STZ-induced DM model, which was started from the early phase of DM (1–2 weeks) and lasted for six months.46,47,48 Changes in mechanical allodynia in these animals were positively correlated with microglial activation (both morphology and function). Interestingly, spinal astrocytes did not appear to play a part in diabetic neuropathic pain, as they manifested no change47 or even decreased in number.46,48 In this study, neither microglia nor astrocyte changes were significantly apparent in DM rats at two weeks after STZ administration, compared with CT rats (data not shown). It was probably explained by the low concentration and different route of administration of STZ used in this study. Another disagreement with the conclusions of other neuropathic pain studies is that significant upregulation of MCP-1 expression was not likely to occur in neurons and glial cells of DM rats, although its receptor CCR2 expression was extensively increased in DRG neurons at two weeks after STZ administration. However, microglial activation in the previous findings of diabetic neuropathy became more exacerbated and prominent over time,46,47 which does not rule out microglial activation and MCP-1 upregulation in the later phase of DM in this study. Nevertheless, our findings demonstrate that CCR2 activation in nociceptive DRG neurons is somewhat related to early diabetic neuropathy. It can be further concluded from the collected data that MCP-1/CCR2 signaling-mediated gastric hyperalgesia in the early phase of DM is likely to be chiefly the consequence of central sensitization of sensory neurons rather than neuron-glial interactions. DM induces over-sensitization of CCR2 in small- and medium-sized DRG neurons, promoting the transduction and transmission of nociceptive signals to the central nervous system and is involved in the facilitation and modulation of pain via the other chemical mediators produced by the cells along the nociceptive pathways.
In the past decade, a number of preclinical studies have been conducted to clarify the underlying mechanisms of MCP-1/CCR2 signaling in neuropathic pain. As a classical G protein-coupled receptor, CCR2 activation induces the phosphorylation of multiple signal transduction pathways including phospholipase C, protein kinase C, phosphatidylinositol-3 kinase, extracellular signal-regulated kinase (ERK) 1/2, and p38 MAPK.49 Following peripheral nerve injury, CCR2 activation increased the sensitizations of transient receptor potential vanilloid 1 (TRPV1), transient receptor potential cation channel, subfamily A, member 1, and tetrodotoxin-resistant Nav1.8 sodium channels in DRG neurons that were mediated by protein kinase C and phospholipase C activation.11,50 Activated CCR2 receptors on presynaptic terminals lead to increased glutamate release in synaptic junctions that were dependent on TRPV1 receptor activation.51 Moreover, MCP-1/CCR2 signaling potentiated the TRPV1 and Nav1.8 receptor functions, enhanced the excitability of DRG neurons, and promoted nociceptive transmission via the activation of the phosphatidylinositol-3 kinase/Akt (protein kinase B) signaling pathway, but not the ERK activation pathway.43 In contrast, MCP-1 or nerve injury-evoked mechanical allodynia was directly correlated to increased phosphorylation of ERK 1/2 and upregulation of proinflammatory mediators (interleukin-1β, interleukin-6, cyclooxygenase-2, and nitric oxide synthase 2), which were prevented by administration of CCR2 antagonist, ERK inhibitor, or TRPV1 receptor antagonist.21,51 In this study, we suggest that CCR2 activation in small- and medium-sized DRG neurons induces the sensitization of TRPV1 or Nav1.8 channels through the activation of MAPK signaling pathways; however, further investigations are needed.
Conclusion
We have demonstrated for the first time that CCR2 activation in DRG neurons plays an integral role in gastric hyperalgesia related to diabetic gastropathy. The evidence of alleviation of diabetes-induced gastric hyperalgesia by CCR2 antagonist in vivo suggests the potential use of CCR2 antagonist for therapeutic intervention in diabetes gastropathy.
Acknowledgments
The authors are grateful to Ms. Rumi Shima and the participants in this research for their invaluable advice and excellent technical support.
Authors' note
Shinichiro Hiraga is now associated with Department of Functional Anatomy, Graduate School of Medical Science, Kanazawa University, Ishikawa, Japan.
Authors’ contributions
AAM, NO, and HO designed the project and NO, HO, and KH supervised the experiments. AAM, YK, TN, and SH performed the animal models, surgeries, and conducted the behavioral experiments. AAM, TN, and YS carried out the general assessment of diabetes, gastric emptying, and immunochemical studies. AAM, NO, HO, and KH analyzed the data and wrote the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Grants-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, Science and Technology (Grant number 25460718, 2013).
References
- 1.International Diabetes Federation. IDF Diabetes Atlas, 7th ed. 2015, https://www.idf.org/e-library/epidemiology-research/diabetes-atlas.html
- 2.Boulton AJ, Vinik AI, Arezzo JC, et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 2005; 28: 956–962. [DOI] [PubMed] [Google Scholar]
- 3.Feldman EL. Epidemiology and classification of diabetic neuropathy – up to date, https://www.uptodate.com (2015, accessed 5 January 2017).
- 4.Vinik AI, Maser RE, Mitchell BD, et al. Diabetic autonomic neuropathy. Diabetes Care 2003; 26: 1553–1579. [DOI] [PubMed] [Google Scholar]
- 5.Bharucha AE, Camilleri M, Forstrom L, et al. Relationship between clinical features and gastric emptying disturbances in diabetes mellitus. Clin Endocrinol 2009; 70: 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bytzer P, Talley NJ, Hammer J, et al. GI symptoms in diabetes mellitus are associated with both poor glycemic control and diabetic complications. Am J Gastroenterol 2002; 97: 604–611. [DOI] [PubMed] [Google Scholar]
- 7.Enck P, Rathmann W, Spiekermann M, et al. Prevalence of gastrointestinal symptoms in diabetic patients and non-diabetic subjects. Z Gastroenterol 1994; 32: 637–641. [PubMed] [Google Scholar]
- 8.Kong MF andHorowitz M.. Gastric emptying in diabetes mellitus: relationship to blood-glucose control. Clin Geriatr Med 1999; 15: 321–338. [PubMed] [Google Scholar]
- 9.White FA Jung H andMiller RJ.. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci USA 2007; 104: 20151–20158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung H, Toth PT, White FA, et al. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem 2008; 104: 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramesh G MacLean AG andPhilipp MT.. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm 2013; 2013: 480739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thacker MA, Clark AK, Bishop T, et al. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain 2009; 13: 263–272. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, Boyette-Davis JA, Kosturakis AK, et al. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain 2013; 14: 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dansereau MA, Gosselin RD, Pohl M, et al. Spinal CCL2 pronociceptive action is no longer effective in CCR2 receptor antagonist-treated rats. J Neurochem 2008; 106: 757–769. [DOI] [PubMed] [Google Scholar]
- 15.Gao YJ, Zhang L, Samad OA, et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci 2009; 29: 4096–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J andDe Koninck Y.. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem 2006; 97: 772–783. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Shi XQ, Echeverry S, et al. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci 2007; 27: 12396–12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu X, Cao S, Zhu MD, et al. Contribution of chemokine CCL2/CCR2 signaling in the dorsal root ganglion and spinal cord to the maintenance of neuropathic pain in a rat model of lumbar disc herniation. J Pain 2014; 15: 516–526. [DOI] [PubMed] [Google Scholar]
- 19.Dauvergne C, Molet J, Réaux-Le Goazigo A, et al. Implication of the chemokine CCL2 in trigeminal nociception and traumatic neuropathic orofacial pain. EJP 2014; 18: 360–375. [DOI] [PubMed] [Google Scholar]
- 20.Jeon SM Sung JK andCho HJ.. Expression of monocyte chemoattractant protein-1 and its induction by tumor necrosis factor receptor 1 in sensory neurons in the ventral rhizotomy model of neuropathic pain. Neuroscience 2011; 190: 354–366. [DOI] [PubMed] [Google Scholar]
- 21.Van-Steenwinckel J, Réaux-Le Goazigo A, Pommier B, et al. CCL2 released from neuronal synaptic vesicles in the spinal cord is a major mediator of local inflammation and pain after peripheral nerve injury. J Neurosci 2011; 31: 5865–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang ZJ, Dong YL, Lu Y, et al. Chemokine CCL2 and its receptor CCR2 in the medullary dorsal horn are involved in trigeminal neuropathic pain. J Neuroinflamm 2012; 9: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abbadie C, Lindia JA, Cumiskey AM, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. PNAS 2003; 100: 7947–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jamali Z, Nazari M, Khoramdelazad H, et al. Expression of CC chemokines CCL2, CCL5, and CCL11 is associated with duration of disease and complications in Type-1 diabetes: a study on Iranian diabetic patients. Clin Lab 2013; 59: 993–1001. [DOI] [PubMed] [Google Scholar]
- 25.Guan R, Purohit S, Wang H, et al. Chemokine (C-C motif) ligand 2 (CCL2) in sera of patients with type 1 diabetes and diabetic complications. PLoS One 2011; 6: e17822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozaki N, Bielefeldt K, Sengupta JN, et al. Models of gastric hyperalgesia in the rat. Am J Physiol Gastrointest Liver Physiol 2002; 283: G666–G676. [DOI] [PubMed] [Google Scholar]
- 27.Matsuda M, Aono M, Moriga M, et al. Centrally administered neuropeptide Y (NPY) inhibits gastric emptying and intestinal transit in the rat. Digest Dis Sci 1993; 38: 845–850. [DOI] [PubMed] [Google Scholar]
- 28.Okuda H, Tatsumi K, Morita-Takemura S, et al. Hedgehog signaling modulates the release of gliotransmitters from cultured cerebellar astrocytes. Neurochem Res 2016; 41: 278–289. [DOI] [PubMed] [Google Scholar]
- 29.Bytzer P, Talley NJ, Jones MP, et al. Oral hypoglycaemic drugs and gastrointestinal symptoms in diabetes mellitus. Aliment Pharmacol Ther 2001; 15: 137–142. [DOI] [PubMed] [Google Scholar]
- 30.Burks TF, Galligan JJ, Porreca F, et al. Regulation of gastric emptying. Fed Proc 1985; 44: 2897–2901. [PubMed] [Google Scholar]
- 31.Bertin E, Schneider N, Abdelli N, et al. Gastric emptying is accelerated in obese type 2 diabetic patients without autonomic neuropathy. Diabetes Metab 2001; 27: 357–364. [PubMed] [Google Scholar]
- 32.Schwartz JG, Green GM, Guan D, et al. Rapid gastric emptying of a solid pancake meal in type II diabetic patients. Diabetes Care 1996; 19: 468–471. [DOI] [PubMed] [Google Scholar]
- 33.Ariga H, Imai K, Chen C, et al. Does ghrelin explain accelerated gastric emptying in the early stages of diabetes mellitus? Am J Physiol Regul Integr Comp Physiol 2008; 294: R1807–R1812. [DOI] [PubMed] [Google Scholar]
- 34.Choi KM, Zhu J, Stoltz GJ, et al. Determination of gastric emptying in nonobese diabetic mice. Am J Physiol Gastrointest Liver Physiol 2007; 293: G1039–G1045. [DOI] [PubMed] [Google Scholar]
- 35.Rayner CK, Verhagen MA, Hebbard GS, et al. Proximal gastric compliance and perception of distension in type 1 diabetes mellitus: effects of hyperglycemia. Am J Gastroenterol 2000; 95: 1175–1183. [DOI] [PubMed] [Google Scholar]
- 36.Samsom M, Salet GA, Roelofs JM, et al. Compliance of the proximal stomach and dyspeptic symptoms in patients with type I diabetes mellitus. Dig Dis Sci 1995; 40: 2037–2042. [DOI] [PubMed] [Google Scholar]
- 37.Belkouch M, Dansereau MA, Réaux-Le Goazigo A, et al. The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gβγ-dependent mechanism. J Neurosci 2011; 31: 18381–18390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gosselin RD, Varela C, Banisadr G, et al. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J Neurochem 2005; 95: 1023–1034. [DOI] [PubMed] [Google Scholar]
- 39.Knerlich-Lukoschus F, Juraschek M, Blomer U, et al. Force-dependent development of neuropathic central pain and time-related CCL2/CCR2 expression after graded spinal cord contusion injuries of the rat. J Neurotrauma 2008; 25: 427–448. [DOI] [PubMed] [Google Scholar]
- 40.Hu JH, Wu MY, Tao M, et al. Changes in protein expression and distribution of spinal CCR2 in a rat model of bone cancer pain. Brain Res 2013; 1509: 1–7. [DOI] [PubMed] [Google Scholar]
- 41.Bourne S Machado AG andNagel SJ.. Basic anatomy and physiology of pain pathways. Neurosurg Clin N Am 2014; 25: 629–638. [DOI] [PubMed] [Google Scholar]
- 42.White FA Bhangoo SK andMiller RJ.. Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov 2005; 4: 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kao DJ, Li AH, Chen JC, et al. CC-chemokine ligand 2 upregulates the current density and expression of TRPV1 channels and Nav1.8 sodium channels in dorsal root ganglion neurons. J Neuroinflammation 2012; 9: 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ji RR Xu ZZ andGao YJ.. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov 2014; 13: 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Menetski J, Mistry S, Lu M, et al. Mice overexpressing chemokine ligand 2 (CCL2) in astrocytes display enhanced nociceptive responses. Neuroscience 2007; 149: 706–714. [DOI] [PubMed] [Google Scholar]
- 46.Cheng KI, Wang HC, Chuang YT, et al. Persistent mechanical allodynia positively correlates with an increase in activated microglia and increased P-p38 mitogen-activated protein kinase activation in streptozotocin-induced diabetic rats. EJP 2014; 18: 162–173. [DOI] [PubMed] [Google Scholar]
- 47.Tsuda M, Ueno H, Kataoka A, et al. Activation of dorsal horn microglia contributes to diabetes-induced tactile allodynia via extracellular signal-regulated protein kinase signaling. Glia 2008; 56: 378–386. [DOI] [PubMed] [Google Scholar]
- 48.Wodarski R, Clark AK, Grist J, et al. Gabapentin reverses microglial activation in the spinal cord of streptozotocin-induced diabetic rats. Eur J Pain 2009; 13: 807–811. [DOI] [PubMed] [Google Scholar]
- 49.Bajetto A, Bonavia R, Barbero S, et al. Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J Neurochem 2002; 82: 1311–1329. [DOI] [PubMed] [Google Scholar]
- 50.Zhao R, Pei GX, Cong R, et al. PKC-NF-kB are involved in CCL2-induced Nav1.8 expression and channel function in dorsal root ganglion neurons. Biosci Rep 2014; 34: e00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spicarova D, Adamek P, Kalynovska N, et al. TRPV1 receptor inhibition decreases CCL2-induced hyperalgesia. Neuropharmacology 2014; 81: 75–84. [DOI] [PubMed] [Google Scholar]