

Abstract
Chronic pain states are highly prevalent and yet poorly controlled by currently available analgesics, representing an enormous clinical, societal, and economic burden. Existing pain medications have significant limitations and adverse effects including tolerance, dependence, gastrointestinal dysfunction, cognitive impairment, and a narrow therapeutic window, making the search for novel analgesics ever more important. In this article, we review the role of an important endogenous pain control system, the endocannabinoid (EC) system, in the sensory, emotional, and cognitive aspects of pain. Herein, we briefly cover the discovery of the EC system and its role in pain processing pathways, before concentrating on three areas of current major interest in EC pain research; 1. Pharmacological enhancement of endocannabinoid activity (via blockade of EC metabolism or allosteric modulation of CB1 receptors); 2. The EC System and stress-induced modulation of pain; and 3. The EC system & medial prefrontal cortex (mPFC) dysfunction in pain states. Whilst we focus predominantly on the preclinical data, we also include extensive discussion of recent clinical failures of endocannabinoid-related therapies, the future potential of these approaches, and important directions for future research on the EC system and pain.
Keywords: Cannabinoid, endocannabinoid, pain, FAAH, MAGL, stress, stress-induced analgesia, stress-induced hyperalgesia, mGluR5, mPFC, amygdala, cortical control of pain
1. Introduction
1.1 Pain
Pain is complex phenomena comprising an unpleasant sensory and emotional experience associated with actual or potential tissue damage (Loeser and Treede, 2008), serving a vital protective evolutionary function. Whilst acute pain can be considered adaptive, chronic pain in the absence of injury, following its resolution, or following damage to the nervous system (neuropathic pain) is a pathological condition of enormous clinical, societal, and economic significance (Apkarian et al., 2009). Chronic pain is the most commonly presented clinical complaint in the USA, afflicting ~10% of the adult population (Nahin, 2015), and representing the greatest economic burden of any pathological condition with an estimated annual cost of $565–635 billion in this region alone (Gaskin and Richard, 2012). Despite its prevalence, current treatments for pain provide inadequate duration and/or extent of relief (Vardeh et al., 2016), highlighting the urgent need for effective novel analgesic agents. Opioids, nonsteroidal anti-inflammatory drugs, selective COX2 inhibitors (Coxibs), anti-depressants, anticonvulsants and local anaesthetics are all used clinically in the treatment of pain (Guindon et al., 2007b). Opioids, such as morphine derived from the opium poppy, have been utilised in pain relief for millennia (Holden et al., 2005) whereas synthetic opioids (tramadol, fentanyl, remifentanil) are mainstays for neuropathic and post-operative pain (Guindon et al., 2007b). These agents, nonetheless have significant limitations including constipation, tolerance and dependence which have contributed to an epidemic of addiction and drug-related deaths in the US in recent years (Kolodny et al., 2015). In the past few decades, a new target for pain relief, also taking advantage of an ancient pain relieving medication and an endogenous pain control pathway, has emerged. Preparations of the Cannabis sativa plant have been used as analgesics for centuries, but it was only in the 1960s that the major active constituent (Δ9-tetrahydrocannabinol) was identified (Mechoulam and Gaoni, 1967). It was not until the 1990s that the molecular targets mediating its effects were discovered (the cannabinoid receptors CB1 & CB2 - Devane et al., 1988; Munro et al., 1993), and the mechanisms and sites of action elucidated (Gregg et al., 2012; Herkenham et al., 1990; Hohmann et al., 2005; Martin et al., 1999; Walker and Hohmann, 2005; Walker and Huang, 2002). This work revealed the existence of a second ubiquitous endogenous pain control pathway; the endocannabinoid (EC) system. In the past two decades, numerous tools to perturb the EC system have been developed, and a wealth of research has demonstrated the potential efficacy of this approach for pain relief (reviewed in Guindon and Hohmann, 2009; Sagar et al., 2009; Sagar et al., 2012; Woodhams et al., 2015). However, global targeting of the EC system is also associated with undesirable results, including deleterious effects on memory (Hall and Solowij, 1998), cognition (Pattij et al., 2008), and mood (Rubino et al., 2015), and the development of tolerance and dependence (Lichtman and Martin, 2005; Tappe-Theodor et al., 2007). This subject has been the topic of many excellent review articles in the past, and therefore in this Special Issue of Neuropharmacology, we briefly cover the history of EC research before focussing on three areas of current interest in the field of cannabinoid pain research – pharmacological enhancement of EC system activity via enzyme inhibitors (Section 2) or by allosteric modulation of CB1 (Section 3), the role of the EC system in stress-induced modulation of pain (Section 4), and actions on the cognitive aspects of pain via coupling of endocannabinoids and metabotropic glutamate receptors in the medial pre-frontal cortex (mPFC; Section 5).
1.2 Pain & the EC system
The subjective experience of pain involves integration of sensory, emotional, and cognitive aspects. This cannot be reported by the non-human animals on which basic pain research is conducted, and thus it is important to make the distinction between subjective pain and the measurable neuronal events and behavioural outputs underlying it, termed nociception (Loeser and Treede, 2008).
Nociceptive signalling begins with the transduction of a noxious stimulus (thermal, mechanical, or chemical) in the periphery into neuronal activity in specialised classes of sensory afferent neurons. The resultant action potentials travel to cell bodies located in dorsal root ganglia (DRG), then to a synapse in the superficial dorsal horn of the spinal cord. Here, local processing integrates peripheral input with descending supraspinal modulation, before transmitting the output via several ascending pathways to the brainstem, thalamus, and other higher brain regions involved in the sensory and affective components of pain (Millan, 1999). The components of the EC system, described in detail elsewhere in this Special Issue, comprise the G protein-coupled cannabinoid receptors CB1 & CB2, their endogenous ligands anandamide (AEA) and 2-arachidonyl glycerol (2-AG), and their respective major synthetic (N-acylphosphatidylethanolamine phospholipase D [NAPE-PLD] & diacyglyerol lipase α [DAGLα]) and degradative (fatty acid amide hydrolase [FAAH] & monoacyglycerol lipase [MAGL]) enzymes. These components are expressed almost ubiquitously throughout nociceptive pathways, and thus targeting the system via exogenous cannabinoid ligands or enhancement of endogenous signalling can regulate nociceptive signalling at multiple sites; in the periphery (reviewed in Guindon and Beaulieu, 2009), the dorsal horn of the spinal cord (Hohmann, 2002; Nyilas et al., 2009; Richardson et al., 1998; Sagar et al., 2010; Woodhams et al., 2012) and in supraspinal pain-associated regions of the brain, as summarized in Figure 1. Sections 2 & 3 detail effects of systemic or local modulation of EC activity at peripheral and central sites, whilst Sections 4 & 5 focus on the role of the EC system in supraspinal pain-associated regions. In neural circuits, endocannabinoids act as short-term circuit breakers (Katona and Freund, 2008). Endocannabinoids (i.e. 2-AG) are generated on-demand in response to high levels of activity, and produce short-term antinociceptive effects via their actions as retrograde transmitters at presynaptic inhibitory CB1 G protein-coupled receptors (GPCRs), with duration of effect limited by their rapid enzymatic degradation. Endocannabinoids play a key role in the resolution of acute pain states (Alkaitis et al., 2010), and are elevated at various sites in nociceptive pathways in chronic pain (Guindon et al., 2013; Sagar et al., 2009; Sagar et al., 2012), highlighting their role as endogenous analgesics.
Figure 1. Schematic of Nociceptive Pathways & Sites of EC System Expression.
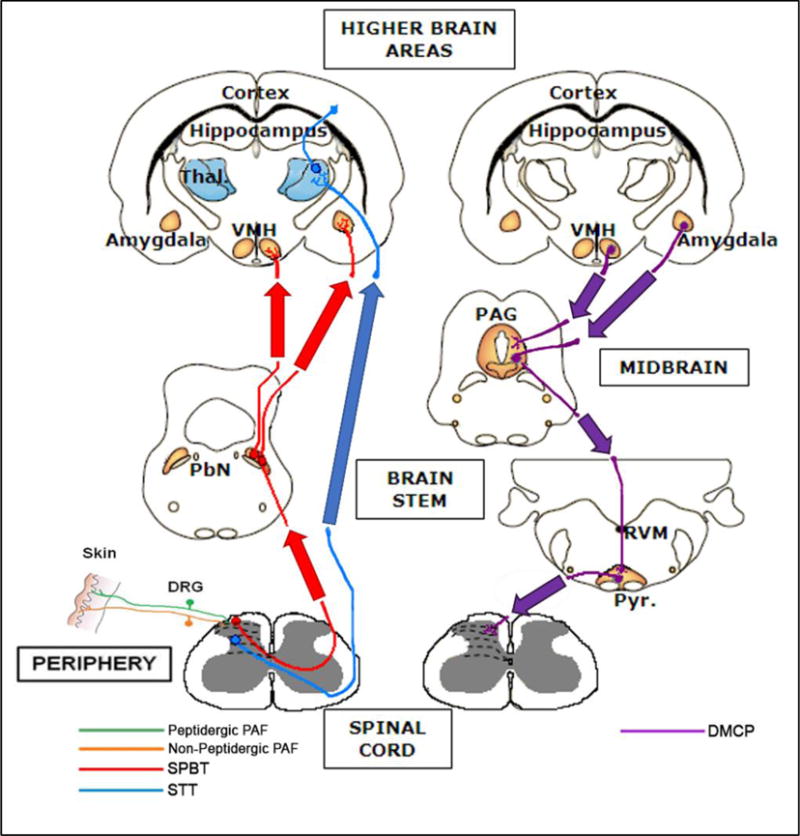
Nociceptive stimuli are conducted from the periphery to the dorsal horn of the spinal cord, and then transmitted to the supraspinal regions via the spinothalamic tract (STT, ) and spinoparabrachial tract (SPBT, ). The major descending modulatory control pathway (DMCP, ) is displayed on the right. This pathway crosses the midline at the level of the medulla. Coloured areas indicate the position of synapses, and therefore sites of additional neuronal processing and endocannabinoid signalling, in each pathway. The positions of laminae I–VI in the dorsal horn are indicated by dotted lines, while the black region in the brain represents the lateral ventricles.
Thal., thalamus; VMH, ventromedial hypothalamus; PbN, parabrachial nucleus; PAF, primary afferent fibre; PAG, periaqueductal grey matter; RVM, rostroventral medulla; Pyr., pyramidal tract; DRG, dorsal root ganglion.
Adapted from (Hunt and Mantyh, 2001).
However, this rather simplistic picture is complicated by the ubiquity of EC system expression – components are localized not just on excitatory neurons, but also inhibitory neurons, peripheral immune cells, and glial cells in the central nervous system (Egertova et al., 2003; Egertova and Elphick, 2000; Gong et al., 2006; Gregg et al., 2012; Hegyi et al., 2009; Horváth et al., 2014).
Furthermore, cannabinoid ligands and their metabolites are promiscuous (Alexander and Kendall, 2007), with some excitatory actions such as AEA agonism at TRPV1 (Ross, 2003), alongside inhibitory actions at CB1 and CB2 and the nuclear family of peroxisome proliferator-activated receptors (PPARs, reviewed in O’Sullivan, 2007; O’Sullivan, 2016). EC activity can therefore induce a complex interplay of actions, the result of which can be antinociceptive (Alkaitis et al., 2010) or pronociceptive (Pernia-Andrade et al., 2009) depending on the site of expression and the underlying physiological state (Zeilhofer, 2010).
2. Blockade of EC Metabolism as an Analgesic Approach: Past Issues and Future Directions
As EC levels are known to be elevated specifically at sites of injury or excessive nociceptive signalling (Sagar et al., 2012), the use of specific enzyme inhibitors to augment their effects has received particular interest in the field of pain research. This approach targets areas of high EC turnover whilst sidestepping the undesirable effects of global cannabinoid receptor activation associated with application of exogenous cannabinoid ligands (reviewed in Ameri, 1999). In the following section we review the historical preclinical successes of this approach, the recent setbacks in translating into the clinic, and the promising future directions.
2.1 FAAH Inhibition for Analgesia
Anadamide (AEA) was the first endocannabinoid to be identified (Devane et al., 1992), and thus formed the central focus of the early years of endocannabinoid pain research, in which its efficacy as an analgesic was established in multiple preclinical models of acute pain (Calignano et al., 1998; Walker et al., 1999). Fatty-acid amide hydrolase (FAAH) was identified as the major enzyme degrading fatty-acid amides which bind to cannabinoid receptors, such as AEA, but also the related compounds oleoylethanolamine (OEA) and palmitoylethanolamine (PEA) which do not (Cravatt et al., 1996). This discovery led to the development of several classes of compounds capable of inhibiting FAAH and thus promoting signalling of AEA (PF3845 - Ahn et al., 2009; PF-04457845 - Ahn et al., 2011; OL135 - Chang et al., 2006; JNJ-1661010 - Karbarz et al., 2009; URB597 - Kathuria et al., 2003) and other fatty-acid amides. These compounds show high specificity for FAAH, significantly elevating levels of AEA, OEA, and PEA in the central nervous system and peripheral tissues. Whilst OEA and PEA are not endocannabinoids as they have no affinity for cannabinoid receptors, PEA has well-established anti-inflammatory properties via peroxisome proliferator-activated receptors (PPARs) (Alhouayek and Muccioli, 2014; Lambert et al., 2002; LoVerme et al., 2006), and PEA and OEA are capable of elevating levels of AEA through substrate competition at FAAH (Di Marzo et al., 1994). Thus, these N-acylethanolamines are of interest to the field of cannabinoid pain research.
FAAH Inhibition in Models of Inflammatory Pain
Due to the dual analgesic and anti-inflammatory properties of FAAH substrates, inhibitors of FAAH have been preferentially evaluated as a therapeutic approach in preclinical models of inflammatory pain. These models involve application of noxious substances to the hindpaw, resulting in inflammation (oedema) and measureable nociceptive behaviour, including allodynia (heightened responses to non-noxious levels of cutaneous stimulation) and hyperalgesia (heightened responses to noxious levels of cutaneous stimulation), although the underlying signalling mechanisms generating inflammation differ. Both brain impermeant (Clapper et al., 2010) and brain permeant inhibitors of FAAH have been shown to suppress inflammatory pain induced by formalin, carrageenan, and Complete Freund’s Adjuvant (CFA) (for review, see Guindon and Hohmann, 2009). Systemic FAAH inhibition is anti-inflammatory via CB2 in the carrageenan model in mice (Holt et al., 2005), and produces CB1-mediated antinociception in the CFA model in rats (Wilson et al., 2005). Both CB1 and CB2 have been implicated in the antinociceptive effects of FAAH inhibition in this model (Ahn et al., 2011; Jayamanne et al., 2006), and a CB2 component is likely of increased importance in models of inflammatory pain due to potential involvement of CB2-expressing peripheral immune cells and glial cells of the CNS. Indeed, several distinct classes of FAAH inhibitors reduce nociceptive behaviour following hindpaw administration of LPS, and a detailed investigation of the mechanism implicates both CB1 and CB2, but not TRPV1, PPARα, or μ-opioid receptors (Booker et al., 2012).
FAAH Inhibition in Models of Neuropathic Pain
The efficacy of FAAH inhibition is not limited to inflammatory pain states, and numerous studies have demonstrated antinociceptive effects of FAAH inhibition in models of neuropathic pain. In the chronic constriction injury (CCI) model in mice, several studies have demonstrated antinociceptive effects of FAAH inhibitors which appear to require both CB1 and CB2 (Kinsey et al., 2010; Kinsey et al., 2009; Russo et al., 2007), as antagonism or genetic deletion of either receptor blocks the anti-allodynic effect (but also see discussion of Carey et al., 2016 below). In contrast, the antinociceptive effects of FAAH inhibition in the partial sciatic nerve ligation (PSNL) model in mice were dependent only on CB1 (Desroches et al., 2013), whilst that seen in the spinal nerve ligation (SNL) model in rats required both CB1 and CB2 (Chang et al., 2006; Jhaveri et al., 2006; Karbarz et al., 2009). Interestingly, another study failed to find any effect of FAAH inhibition in the PSNL model in rats (Jayamanne et al., 2006), suggesting that there may be species- and model-specific mechanisms which complicate the interpretation of these data.
Central and Peripheral Mechanisms of FAAH Inhibition
To investigate the major site of action of FAAH inhibition, several studies have used local administration of compounds into the hindpaw, spinal cord, or brain. Local administration of a FAAH inhibitor into the hindpaw in the rat carrageenan model is antinociceptive, but not anti-inflammatory, blocking both pain behaviour and the receptive field expansion of dorsal horn neurons (Jhaveri et al., 2008; Sagar et al., 2008), a marker of central sensitisation, and thus development of an inflammatory pain state (Latremoliere and Woolf, 2009). Development of the peripherally-restricted FAAH inhibitor URB937 revealed a significant CB1-mediated peripheral component to the antinociceptive effects of FAAH inhibition in models of inflammatory pain, since the effects were completely blocked by administration of a CB1 antagonist (Clapper et al., 2010; Sasso et al., 2015b), in line with the suggestion that exogenous cannabinoids exert much of their pain relieving effects via peripheral CB1 receptors (Agarwal et al., 2007). It should be noted, however, that spinal and supraspinal mechanisms have also been described for FAAH inhibition in models of acute pain. In the surgical incision model of acute resolving pain in rats, spinal levels of AEA are reduced at early time points coinciding with maximal mechanical hypersensitivity, returning to baseline as nociceptive behaviour subsides (Alkaitis et al., 2010), whilst administration of a FAAH inhibitor to the periaqueductal grey matter altered thresholds to thermal stimuli in a biphasic manner (Maione et al., 2006). Global deletion of FAAH in mice results in 15-fold elevations in brain levels of AEA, reduced sensitivity to thermal and inflammatory pain, and augmented responses to exogenous AEA which are mediated by CB1 (Cravatt et al., 2001). However, recent evidence has also demonstrated a pronociceptive phenotype when FAAH KO mice are challenged with the TRPV1 agonist capsaicin (Carey et al., 2016), and show elevations in numerous endovanilloids and other potentially bioactive lipids suggesting that persistent elevation of fatty-acid amides could potentially lead to sensitisation (e.g. of TRPV1 channels) in certain pathological pain states.
FAAH Inhibitors in the Clinic – a bridge too FAAH?
The wealth of preclinical data demonstrating efficacy of FAAH inhibition as an analgesic strategy has led to completion of Phase I trials in humans (PF-04457845 - Li et al., 2012; V158866 - Pawsey et al., 2016). However, despite being well-tolerated and producing significant elevations in peripheral blood levels of AEA, PF-04457845 failed to produce significant analgesia in a Phase II trial in late-stage osteoarthritis (OA) patients (Huggins et al., 2012). Tolerance to persistently elevated AEA levels may occur with chronic FAAH inhibition, as indicated by a lack of effect of chronic FAAH inhibition in the rat carrageenan model of inflammatory pain (Okine et al., 2012), or it could be that AEA levels at key central signalling sites are already maximally elevated in these patients and/or result in sensitisation of TRPV1 receptors. Better stratification of patients and targeting of pain conditions may result in more positive results. In the case of OA, for example, it may be beneficial to assess efficacy in earlier stages of disease when tolerance to chronically-elevated endocannabinoids has not occurred. Furthermore, recent clinical assessments of the efficacy of cannabinoids in pain states (reviewed in Lichtman and Chapman, 2011), have suggested that beneficial effects on pain comorbidities including mood disturbances, loss of appetite, and disturbed sleep, may outweigh those on sensory aspects of pain. Conditions such as fibromyagia, complex regional pain syndrome, MS, or cancer pain in which such factors play a more prominent role may therefore be better clinical targets for these compounds.
Another FAAH inhibitor, BIA-102474, led to severe neurological toxicity in a more recent Phase I trial, resulting in the death of one of the participants and permanent neurological damage in 5 others (Kaur et al., 2016; Moore, 2016). This tragic outcome would have the potential to kill any further clinical interest in FAAH inhibition, were it not for several important considerations. After extensive review of the data arising from this trial, it now seems that these shocking events reflect problems with the design of the trial and the specific molecule utilised, rather than with targeting FAAH in general (Kaur et al., 2016). These issues relate to the supramaximal dose administered, the decision to continue dosing other participants after adverse events first arose, the relatively low selectivity of the compound, potential irreversible interactions with non-FAAH serine hydrolases, its long half-life, and the lack of toxicity studies specifically relating to its known metabolites. Firstly, it should be noted that the dose administered was 20-50 times that required for full FAAH inhibition (Bégaud et al., 2016), and furthermore that this dose was administered repeatedly, allowing for accumulated tissue concentrations at which off-target effects became inevitable. BIA-102474 reportedly showed 50-100 fold selectivity for FAAH over other serine hydrolases, and adverse events arose only on the 5th and 6th days of 50mg administration when concentrations likely exceeded those at which the drug was selective. Single administrations of up to 100mg showed no ill effects, strongly suggesting that the toxicity arose from accumulation of the drug. Although the cause of toxicity has not been fully established, and it remains possible that they may have arisen due to impurities in the batch of compound utilised in the trial, it seems likely that irreversible, off-target effects of BIA-102474 or one of its metabolites at alternative serine hydrolases was responsible. The lack of similar effects in multiple trials of chemically disparate classes of FAAH inhibitors argue strongly against a FAAH-mediated mechanism (Huggins et al., 2012; Li et al., 2012; Pawsey et al., 2016). In light of these trial and compound-specific issues and the wealth of preclinical data supporting efficacy of compounds directed at FAAH, we believe that FAAH inhibition still remains a promising target for the development of novel analgesics.
2.2 MAGL Inhibition for Analgesia
Although discovered a few years after AEA, 2-AG is now considered to be the major endocannabinoid ligand in the CNS (Stella et al., 1997), responsible for most of the well-characterised synaptic properties of CB1 activation (reviewed in Katona and Freund, 2012). Monoacyglycerol lipase (MAGL) was identified as the major enzyme responsible for terminating 2-AG signalling (Dinh et al., 2002; Dinh et al., 2004), and though other enzymes do contribute to 2-AG metabolism in vivo (Blankman et al., 2007; Marrs et al., 2010), ~85% of hydrolytic activity in brain can be attributed to MAGL. Initially, attempts to develop selective MAGL inhibitors were unsuccessful due to off-target interactions with other serine hydrolases, including FAAH (King et al., 2007; Vandevoorde et al., 2007). Local injections of the O-biphenylcarbamate URB602, an MAGL preferring inhibitor, reduced MGL activity, selectively elevated levels of 2-AG without altering levels of AEA, and produced antinociception, but was not potent enough to be used systemically (Hohmann et al., 2005). However, in 2009 a compound with 300-fold selectivity for MAGL over FAAH was developed from activity-based protein profiling of a diverse library of carbamates and lead optimization by the Cravatt group (Long et al., 2009a). JZL184, administered systemically, produces 8-fold elevations in brain 2-AG without markedly elevating AEA levels, and increases acute thermal and mechanical pain thresholds in mice. Similar to FAAH inhibition, substantial antinociception has been demonstrated in rodent models of peripheral inflammatory pain (Ghosh et al., 2013; Guindon et al., 2011; Woodhams et al., 2012), visceral and gastrointestinal pain (Busquets-Garcia et al., 2011; Kinsey et al., 2011), neuropathic pain (Kinsey et al., 2010; Kinsey et al., 2009), chemotherapy-induced neuropathy (Guindon et al., 2013), and bone cancer pain (Khasabova et al., 2011). In models of acute pain, effects of MAGL inhibition appear to be largely mediated by CB1 (Long et al., 2009a) though a more prominent CB2-mediated component has been identified in inflammatory and neuropathic pain models (Guindon et al., 2007a; Guindon et al., 2011; Guindon and Hohmann, 2008), perhaps unsurprisingly since 2-AG is a full agonist at CB2 whilst AEA is only a weak partial agonist (Gonsiorek et al., 2000). However, no sooner had a specific MAGL inhibitor been developed, than findings questioning the long-term efficacy of this approach appeared. Full inhibition of MAGL via JZL184 produces many cannabinoid-like behaviours (Long et al., 2009a), suggesting that this approach may share some of the unwanted side-effects of cannabinoids. Furthermore, several studies have now revealed that sustained global elevation of 2-AG via genetic deletion of MAGL or persistent blockade of MAGL activity with enzyme inhibitors produces functional antagonism of the brain EC system, resulting in profound downregulation and desensitization of CB1 receptors in nociception-associated regions, and a loss of analgesic phenotype (Chanda et al., 2010; Imperatore et al., 2015; Navia-Paldanius et al., 2015; Schlosburg et al., 2010). Chronic MAGL blockade may also result in physical dependence, since a CB1 antagonist precipitated behavioural symptoms of withdrawal following repeated high dose JZL184 treatment (Schlosburg et al., 2009).
However, these problems may not present an insurmountable obstacle. Chronic partial inhibition of MAGL produces sustained analgesia in the absence of cannabinoid side effects in mice (Busquets-Garcia et al., 2011; Kinsey et al., 2011). Furthermore, a new generation of MAGL inhibitors have been developed with more attractive therapeutic profiles (Ignatowska-Jankowska et al., 2014; MJN110 - Niphakis et al., 2013a). These compounds have greater selectivity for MAGL over FAAH than JZL184, producing antinociceptive effects in mouse models of acute and chronic pain with reduced cannabimimetic properties (Ignatowska-Jankowska et al., 2014; Ignatowska-Jankowska et al., 2015b).
2.3 The Future of EC Metabolic Enzyme Inhibitors: Broadening the Appeal
FAAH and MAGL degrade a variety of lipid signalling molecules which do not bind to cannabinoid receptors, and therefore, FAAH and MAGL inhibitors are not selective for the endocannabinoid system. Moreover, endocannabinoids also undergo oxidative metabolism by a variety of different enzymes such as cyclooxygenases (for reviews, see Guindon and Hohmann, 2009; Starowicz and Di Marzo, 2013). These observations raise the possibility that compounds targeting multiple enzymes may produce better analgesic efficacy than targeting a single enzyme in isolation. Complete ablation of EC catabolic enzyme activity may, therefore, not be the most effective therapeutic strategy for pain relief, as evidenced by the recent clinical failure of a FAAH inhibitor in osteoarthritic pain, and the uncovering of functional antagonism following chronic absence of MAGL activity. However, clinical trials fail for many reasons, including choice of target indication and lack of demonstration of target engagement, and there remains significant hope in this area of analgesic drug development due to the high safety profile observed with well-characterized FAAH inhibitors in humans. Many recent preclinical studies have utilized combined inhibition of FAAH and MAGL, or inhibitors of other pain-related enzymes, and/or pain-relieving drugs to produce analgesia in the absence of side-effects. Multifunctional compounds often have improved safety profiles compared to highly specific single target molecules, and this work suggests that a more broad-spectrum approach may finally allow translation of the pre-clinical promise of EC-directed therapies to the clinic. Finally, the allosteric modulatory site (Price et al., 2005) on the cannabinoid CB1 receptor is also a promising target for small molecule analgesic drug development (Ignatowska-Jankowska et al., 2015a).
Combined FAAH/MAGL Inhibition
Alongside JZL184, Long and colleagues developed an analogue compound with dual inhibitory properties at MAGL and FAAH (Long et al., 2009b). Systemic administration of JZL195 produces greater cannabimimetic effects than full inhibition of MAGL or FAAH alone (Long et al., 2009b), but unlike synthetic cannabinoid agonists, the effective dose for antinociception is significantly lower than that producing unwanted side-effects, indicating a potential therapeutic window (Adamson Barnes et al., 2016). Similarly, a recent study employed full FAAH inhibition with partial MAGL inhibition via co-administration of PF-3845 and JZL184 (Ghosh et al., 2015), producing a synergistic augmentation of antinociceptive potency in models of neuropathic and inflammatory pain, in the absence of cannabimimetic side effects, and with an apparent lack of tolerance and dependence following chronic dosing. In accordance with these data, a novel dual FAAH/MAGL inhibitor with markedly greater potency at FAAH than MAGL (SA-57 - Niphakis et al., 2013b), produces antinociception in mouse models of neuropathic and inflammatory pain (Wilkerson et al., 2016a). Interestingly, this compound can be used in combination with morphine to produce synergistic analgesia, and even appears to alleviate tolerance to opioid analgesia. A similar effect has also been noted using low dose MAGL inhibition via MJN110 in combination with morphine in a rodent model of neuropathic pain (Wilkerson et al., 2016b), suggesting this is an exciting avenue for future research.
Combined FAAH Inhibition/TRPV1 Antagonism
FAAH inhibition can produce biphasic effects in pain models, with antinociception via AEA actions at CB1 at low concentrations, and pronociceptive effects via the activation, but not desensitisation, of TRPV1 when AEA levels are highly elevated (Maione et al., 2006; Ross, 2003). Dual FAAH inhibition in combination with TRPV1 antagonism is therefore an attractive therapeutic target (Fowler et al., 2009). Indeed, TRPV1 antagonism in its own right was once a promising analgesic strategy (Ghilardi et al., 2005; Swanson et al., 2004), but clinical development of TRPV1 inhibitors was stalled due to adverse hyperthermic effects (Gavva, 2009; Gavva et al., 2008). In contrast, the dual FAAH inhibitor/TRPV1 antagonist compounds AA-5-HT (Hohmann et al., 2005; Maione et al., 2007) and OMDM-198 (Maione et al., 2013) produce antinociception in the absence of hyperthermia. AA-5-HT has antinociceptive efficacy in the formalin and carrageenan models of inflammation, and the CCI and SNI models of neuropathic pain (Costa et al., 2010; de Novellis et al., 2008; de Novellis et al., 2011; Maione et al., 2007; Malek et al., 2016), whilst OMDM-198 is effective in the formalin and carrageenan models of inflammatory pain (Maione et al., 2013), and a rat model of osteoarthritic pain (Malek et al., 2015). A significant supraspinal component to these effects has been described via microinjection of AA-5-HT into the PAG, resulting in CB1-, and TRPV1-dependent analgesia, and a restoration of the excitation-inhibition balance in prefrontal cortex (de Novellis et al., 2008; de Novellis et al., 2011). Thus, this approach may even be effective at neutralising the affective component of pain (see Section 3) and the cortical dysfunctions associated with chronic pain states (see Section 4), however further research is still ongoing to determine the clinical potential of these preliminary data.
FAAH/COX2 Inhibition
Cyclooxygenase enzymes in CNS neurons play a critical role in inflammatory pains state through their actions in generating prostanoids (Vardeh et al., 2009), and are the central target for the non-steroidal anti-inflammatory drug (NSAID) class of analgesics. However, COX2 can also metabolise AEA when other catabolic pathways are blocked (Fowler, 2007), reducing AEA tone, and potentially contributing to the clinical failure of FAAH (Starowicz and Di Marzo, 2013). Strong evidence demonstrates that NSAIDs can act as FAAH inhibitors (Fowler et al., 1999; Seidel et al., 2003), and co-administration of FAAH inhibitors and NSAIDs produces synergistic antinociceptive effects in preclinical models of pain (Grim et al., 2014; Guindon and Beaulieu, 2006; Guindon et al., 2006), providing a powerful rationale for the design of compounds with dual activity at these two enzymes. One such compound, ARN2508, has recently been developed and showed efficacy in a model of intestinal inflammation (Sasso et al., 2015a). Notably, this drug produced antinociception in the absence of gastric damage, and may thus avoid a major side-effect of chronic NSAID treatment. In addition, substrate-specific inhibitors of COX2 have been developed which selectively block the hydrolysis of AEA, but spare other molecules (Hermanson et al., 2014; Hermanson et al., 2013). These compounds have shown efficacy at treating preclinical models of stress-induced neuropsychiatric disorders (Gamble-George et al., 2016), highlighting the link between the endocannabinoid system and stress, which forms the basis of a subsequent section of this review.
3. Positive Allosteric modulators of cannabinoid CB1 receptor signalling
The identification of allosteric binding site(s) (Price et al., 2005) on the CB1 GPCR has facilitated drug discovery efforts aimed at harnessing the therapeutic potential of endocannabinoid signalling. Positive allosteric modulators enhance the affinity and/or efficacy of the endogenous ligand at the classical (orthosteric) binding site. Because allosteric modulators bind to sites distinct from the orthosteric binding site, they might be expected to show a more limited spectrum of unwanted cannabimimetic effects compared to direct agonists like THC. Positive allosteric modulators that have been evaluated for antinociceptive efficacy in the published literature include lipoxin A4 (Pamplona et al., 2012) and ZCZ011 (Ignatowska-Jankowska et al., 2015a). Of these, the best characterized is ZCZ011, as Lipoxin A4 is an endogenous lipid mediator that could not be administered systemically. Nonetheless, following intraventricular administration Lipoxin A4 produces typical CB1-mediated cannabimimetic effects following intraventricular administration (i.e. reduces locomotor activity in the open field, produces catalepsy in the ring test, hypothermia) and produces antinociception in the hotplate test (Pamplona et al., 2012). ZCZ011 suppressed neuropathic and inflammatory pain behaviour in mice through a CB1 mechanism (Ignatowska-Jankowska et al., 2015a). ZCZ011 also increased the potency of orthosteric cannabinoid agonists in the tetrad (antinociception in tail-flick, hypothermia, catalepsy and locomotor activity) did not exhibit the discriminative stimulus effects of CP55,940 or ananadamide (Ignatowska-Jankowska et al., 2015b). Antinociceptive efficacy was also preserved following 6 days of repeated dosing (Ignatowska-Jankowska et al., 2015b). Moreover, ZCZ011 did not produce conditioned place preference or aversion (Ignatowska-Jankowska et al., 2015b).
GAT211 and GAT229 are other recently described CB1 positive allosteric modulators (Laprairie et al., 2017). The in vivo profile of GAT211 has only recently been reported (Slivicki et al., 2016). GAT211 suppresses both inflammatory and neuropathic pain without producing typical CB1-mediated cannabimimetic effects (Slivicki et al., 2016). Moreover, GAT211 shows synergistic antinociceptive effects with inhibitors of both FAAH and MAGL, suggesting it is not acting in a probe-specific manner to enhance 2-AG over anandamide signaling or vice versa (Laprairie et al., 2017; Slivicki et al., 2016). Unlike orthosteric CB1 agonists and MAGL inhibitors, GAT211 did not produce tolerance over a 20 day interval of once daily dosing (Slivicki et al., 2016). Moreover, GAT211 also failed to produce conditioned place preference or reward (Slivicki et al., 2016). The similar features shared by GAT211 and ZCZ011 support the importance of allosteric modulatory site(s) on the CB1 receptor as a target for analgesic drug development. Although preclinical studies are still at an early stage CB1 positive allosteric modulators show potential for suppressing chronic pain in a manner that is safe, effective and lacks unwanted side effects of orthosteric CB1 agonists. It should also be noted that several exogenous and endogenous substances have also been identified as negative allosteric modulators of CB1, suggesting complex regulation of CB1 signalling. This topic is covered in detail elsewhere in this Special Issue (Khurana et al., 2017).
4. The EC System and Stress-Induced Modulation of Pain
The intensity of perceived pain does not necessarily correlate with the degree of tissue damage, injury or inflammation, and the importance of modulation of pain by context and emotion is now widely recognized. Stress, fear, and anxiety can modulate pain (Asmundson and Katz, 2009; Burke et al., 2015; Butler and Finn, 2009; Fitzgibbon et al., 2015; Ford and Finn, 2008; Jennings et al., 2014; Olango and Finn, 2014; Rhudy and Meagher, 2000; Rhudy and Meagher, 2001; Wiech and Tracey, 2009). Negative emotions with low to moderate arousal tend to exacerbate pain through the phenomenon of stress-induced hyperalgesia (SIH), while negative emotions with high arousal tend to inhibit pain through the phenomenon of stress-induced analgesia (SIA) (de Wied and Verbaten, 2001; Dougher, 1979; Meagher et al., 2001; Rhudy and Meagher, 2000; Rhudy and Meagher, 2001; Rhudy and Meagher, 2003). Cannabinoid receptors are localized in brain regions involved in the modulation of pain, stress, and emotion including the RVM, PAG, amygdala and PFC (Herkenham et al., 1991; Tsou et al., 1998, see figure 1). Stress and fear have been shown to alter levels of endocannabinoids in these brain regions (Carrier et al., 2005; Gregg et al., 2012; Hill et al., 2013; Hill et al., 2005; Hohmann et al., 2005; Morena et al., 2015; Olango et al., 2012; Patel et al., 2005; Rademacher et al., 2008). In this section, we will review the role of the endocannabinoid system in SIA and SIH, with an emphasis on the sites and mechanisms involved.
4.1 Stress-Induced Analgesia (SIA)
SIA is a form of adaptive pain suppression, an evolutionarily conserved response to stress that has survival value (Amit and Galina, 1986; Butler and Finn, 2009; Ford and Finn, 2008). Early studies indicated that SIA was mediated via both opioid and non-opioid mechanisms, and in 2000 it was shown that CB1 receptor knock-out mice do not exhibit opioid-mediated antinociception following a forced swim in water at 34°C (Valverde et al., 2000), suggesting that the endocannabinoid and opioid systems interact to mediate this form of SIA. In 2004, Finn and colleagues demonstrated that systemic administration of the CB1 receptor antagonist/inverse agonist rimonabant completely prevented the suppression of formalin-evoked nociceptive behaviour expressed in rats upon re-exposure to an aversively conditioned context previously paired with footshock (i.e. fear-conditioned analgesia; FCA) (Finn et al., 2004). A series of studies from Hohmann and colleagues demonstrated a key role for the EC system in an opioid-independent form of unconditioned SIA in rats (footshock followed immediately by tail-flick testing) and identified some of the brain regions involved. Systemic administration of CB1 receptor antagonists (Hohmann et al., 2005), or their direct microinjection into the dorsolateral PAG (Hohmann et al., 2005; Suplita et al., 2005), brainstem RVM (Suplita et al., 2005), basolateral amygdala (BLA) (Connell et al., 2006), but not spinal cord (Suplita et al., 2006), attenuated SIA. Footshock stress was shown to increase the formation of AEA and 2-AG in the dorsolateral PAG (Gregg et al., 2012; Hohmann et al., 2005) and either systemic or intra-PAG administration of drugs which inhibit the enzymatic degradation or transport of endocannabinoids potentiated SIA (Hohmann et al., 2005; Suplita et al., 2005). Potentiation of SIA was also observed following direct injection of a FAAH inhibitor into the RVM (Suplita et al., 2005) or intrathecal injection of FAAH and MAGL inhibitors (Suplita et al., 2006). Thus, although endocannabinoids at the spinal level are capable of modulating this form of unconditioned SIA, mediation of this behavioural response was critically dependent on endocannabinoid-CB1 signalling in supra-spinal sites including the PAG and RVM, key components of the descending pain pathway.
The ability of footshock stress to trigger the mobilization of endocannabinoids such as 2-AG led to the use of this model of stress antinociception to probe the biochemical mechanisms underlying on demand formation of 2-AG in vivo (Gregg et al., 2012). The results of behavioural, pharmacological studies, and immunohistochemical studies, along with RNA interference, quantitative PCR and lipidomic analyses of endocannabinoid content also show that activation of type 5 metabotropic glutamate receptors (mGluR5) leads to a Ca2+-dependent increase in the activity of the enzyme diacylglycerol lipase-α (DAGL-α), which in turn initiates both 2-arachidonoylglycerol formation in the dorsolateral PAG and endocannabinoid mediated analgesia (Gregg et al., 2012). Stimulation of mGluR5 receptors in the dlPAG with the group 1 mGluR agonist (S)-3,5-dihydroxyphenylglycine (DHPG) initiated 2-AG formation and enhanced endocannabinoid-mediated stress antinociception through a mechanism that required both presynaptic CB1 receptors and DAGL-α. Pharmacological inhibition of DAGL activity in the dlPAG with tetrathydrolipstin decreased levels of 2-AG in the PAG with no change in levels of anandamide. Moreover, virally mediated RNA silencing of DAGL-α but not the DAGL-β isoform suppressed both 2-AG formation and stress antinociception. These effects of RNA silencing of DAGL-α mRNA were mimicked by multiple pharmacological inhibitors of DAGL injected into the same site. Finally, 2-AG is likely to act as a retrograde messenger in the dlPAG because CB1 was confined to presynaptic terminal whereas DAGL-α, which resided in dendritic spines, colocalized with mGluR5 (Gregg et al., 2012). A similar molecular architecture of 2-AG signalling and CB1-mediated potentiation of stress antinociception was found in the lumbar spinal cord (Nyilas et al., 2009). Thus, stress-induced synthesis of 2-AG within the dorsolateral PAG and lumbar spinal cord is dependent on mGluR5-induced stimulation of postsynaptic DAGLα (Gregg et al., 2012; Nyilas et al., 2009). Although intra-BLA administration of rimonabant suppressed unconditioned SIA, inhibitors of endocannabinoid hydrolysis had no effect on SIA when injected into this brain region (Connell et al., 2006). Unlike SIA, FCA was not prevented by intra-BLA rimonabant (Roche et al., 2010; Roche et al., 2007), but was attenuated by intra-BLA administration of another CB1 receptor antagonist/inverse agonist, AM251 (Rea et al., 2013). The model of FCA used by Finn and co-workers has an opioid-mediated component. Thus, enhancement of FCA in this model, by systemic administration of the FAAH inhibitor URB597, is prevented by co-administration of the opioid receptor antagonist naloxone, as well as by rimonabant and the selective CB2 receptor antagonist SR144528 (Butler et al., 2008). Like SIA, it has been shown that FCA in rats is fully attenuated by intra-dlPAG injection of the CB1 receptor antagonist/inverse agonist rimonabant and is associated with increased tissue levels of AEA in this PAG column (Olango et al., 2012). The ventral hippocampus is another locus of endocannabinoid-mediated FCA in rats (Ford et al., 2011). Additional work in mice has suggested that interactions between the endocannabinoid system and the cholecystokininergic (CCK) system (particularly CCK 2 receptors) are important for expression of an opioid-dependent form of unconditioned SIA (Kurrikoff et al., 2008).
By contrast, endocannabinoid-mediated SIA was attenuated in rats tolerant to the cannabinoid receptor agonists WIN55,212-2 or Δ9-THC (Hohmann et al., 2005; Suplita et al., 2008) but not in rats rendered tolerant to morphine (Hohmann et al., 2005), suggesting that endocannabinoid mediation of this form of SIA occurs independently of μ-opioid receptors. Furthermore, rats acutely exposed to footshock were hypersensitive to the antinociceptive effects of WIN55,212-2 and Δ9-THC and, in turn, acute Δ9-THC and WIN55,212-2 administration potentiated SIA, suggesting a bidirectional sensitization between endocannabinoid-mediated SIA and exogenous cannabinoid-induced antinociception. In summary, it is clear that the endocannabinoid system plays a key role in mediating non-opioid and opioid-dependent forms of endogenous pain suppression in response to either unconditioned or conditioned stressors.
4.2 Stress-Induced Hyperalgesia (SIH)
Stress and anxiety do not invariably suppress pain; they can also enhance nociception and exacerbate pain in a phenomenon referred to as stress-induced hyperalgesia (SIH). Despite the well-established role of the endocannabinoid system in stress, anxiety, and pain (Finn, 2010), few studies have investigated the role of endocannabinoids in SIH. Systemic administration of the CB1 receptor agonist arachidonyl-2-chloro ethylamine (ACEA) significantly reduced the enhanced visceromotor reflex to colorectal distention (i.e. number of abdominal contractions) and also attenuated changes in electromyogram response in rats stressed by partial restraint, whereas, the CB1 receptor antagonist/inverse agonist (rimonabant) had opposing effects on this stress-induced visceral hypersensitivity (Shen et al., 2010). In the same study, a stress-induced up-regulation of CB1 receptors was demonstrated in the colon. In another study, visceral motor response increased significantly in water avoidance stressed rats, indicating hyperalgesia (Hong et al., 2009). Treatment of water avoidance stressed rats with the cannabinoid receptor agonist, WIN 55,212-2, also prevented the development of visceral hyperalgesia. Levels of anandamide in the dorsal root ganglia of stressed rats were increased, while CB1 receptor expression was decreased (Hong et al., 2009). These results suggest that endocannabinoid signalling through CB1 may play an important role in stress-induced visceral hyperalgesia.
Chronic unpredictable stress (CUS), a widely used model for inducing anxiety and depressive-like behaviour in mice, enhances thermal (hot plate test) and mechanical (von Frey) hyperalgesia (Shi et al., 2010). CUS has also been shown to induce long-lasting widespread hyperalgesia in mice following repeated intramuscular injection of nerve growth factor (NGF) which induces spinal sensitization accompanied by hyperalgesia (Lomazzo et al., 2015). The FAAH and MAGL inhibitors URB597 and JZL184 attenuated the CUS-induced anxiety-related behaviour in the light-dark box and thermal hyperalgesia in the hot plate test. URB597 significantly reduced the widespread hyperalgesia induced via combined CUS and NGF in this study, while JZL184 had no significant effect (Lomazzo et al., 2015). These data suggest a role for a FAAH substrate in this form of SIH.
Genetic background plays a key role in determining the effect of stress on pain. The Wistar-Kyoto (WKY) rat displays increased sensitivity to noxious stimuli and exhibits an anxio-depressive phenotype and hyper-sensitivity to stress, compared with other rat strains, including Sprague-Dawley (SD) rats (Burke, et al., 2010; O’Mahony, et al., 2010). Recently, impairment in pain-related mobilization of AEA and 2-AG, along with their synthesising enzymes, NAPE-PLD and DAGL, respectively, has been demonstrated in the RVM of WKY rats compared with SD rats, following intraplantar injection of formalin (Rea et al., 2014). Systemic administration of AM251 potentiated, while systemic administration of the FAAH inhibitor URB597 attenuated, hyperalgesia to formalin injection in WKY rats, but not SD rats, an effect mediated by CB1 receptors in the RVM. These data suggest that impaired endocannabinoid-CB1-dependent signalling in the RVM underlies the hyper-sensitivity to noxious stimuli in WKY rat model of negative affective state (Rea et al., 2014). More recently, further work from Finn and colleagues has identified a role for non-cannabinoid receptor targets of the endocannabinoids in hyperalgesia in WKY rats, specifically PPARγ (Okine et al., 2017), and TRPV1 (Madasu et al., 2016) in the PAG. The role of the EC system in the effects of repeated exposure to forced swim stress on formalin-evoked nociceptive behaviour in stress normo-responsive (SD) and stress hyper-responsive (WKY) rat strains has also been investigated. Formalin-evoked nociceptive behaviour was increased in SD rats following ten days of forced swim stress (Jennings et al., 2016). Anandamide levels were reduced in the contralateral amygdala (relative to formalin injection) of SD rats but not WKY rats. Strain differences in components of the endocannabinoid system within the amygdala were also observed. For example, decreased levels of anandamide and 2-AG were reported in the ipsilateral amygdala of SD, but not WKY, rats. Lower levels of CB1 receptor mRNA were seen in the ipsilateral, but not contralateral, amygdala of WKY rats. These data indicate a role for the endocannabinoid system in the amygdala in SIH, as well as implicating it in the strain differences seen between WKY and SD rats (Jennings et al., 2016). Additional studies are warranted to fully understand the role of the endocannabinoid system in in SIH, particularly in neuropathic pain models and studies in human subjects. Preclinical studies could, for example, investigate the effects of pharmacological or genetic manipulation of the endocannabinoid system on stress-induced enhancement of hyperalgesia in rodent models of neuropathic pain. Clinical studies could assess the potential utility of endocannabinoids (and related lipids) as biomarkers for stress-induced exacerbation of chronic pain, and co-morbidity of chronic pain with affective disorders, in addition to investigating whether mechanisms elucidated in rodents translate to human patients and might be modulated pharmacologically for therapeutic benefit. Taken together, the data to date, reviewed above, suggest that endocannabinoid-CB1 receptor signalling in key components of the descending pain pathway mediates SIA, while a deficit in endocannabinoid-CB1 receptor signalling may underlie SIH.
5. The EC System & Medial Prefrontal Cortex (mPFC) Dysfunction in Pain States: CB1 and mGluR5 in a Model of Arthritic Pain
Alongside key involvement in the sensory and stress-induced aspects of pain processing, the EC system is also involved in the accompanying affective-emotional and cognitive dysfunctions which underlie the negative mood aspects of chronic pain in human patients. In the final section of this review, we introduce mPFC-amygdala interactions as a recently identified major player in chronic pain states, and the potential for alleviating dysfunctions by targeting the functional coupling of cannabinoid and metabotropic glutamate receptors.
5.1 mPFC deactivation in pain
The mPFC is responsible for executive functions and is closely connected to other limbic areas playing key roles in emotion, such as the amygdala. The infralimbic mPFC (area 25) exerts top-down control of amygdala function to inhibit aversive behaviours in a process referred to as “fear extinction” (Herry et al., 2010; Likhtik et al., 2005; Marek et al., 2013; Orsini and Maren, 2012; Pape and Pare, 2010; Sotres-Bayon and Quirk, 2010). Decreased infralimbic activity has been implicated in extinction deficits (Chang and Maren, 2010; Hefner et al., 2008; Kim et al., 2010; Sierra-Mercado et al., 2011). Accumulating evidence suggests that dysfunction in mPFC-amygdala interactions plays an important role in pain (Neugebauer, 2015). Functional and structural abnormalities in the mPFC have been detected in human pain patients (Apkarian et al., 2004b; Mayer et al., 2005) and in acute and chronic models of pain states (Cardoso-Cruz et al., 2013; Ji et al., 2010; Metz et al., 2009). Decreased activity of output neurons (pyramidal cells) in the infralimbic and prelimbic (area 32) regions of the mPFC has been demonstrated in rodent models of arthritis (Ji and Neugebauer, 2011, 2014; Ji et al., 2010) and neuropathic pain (Wang et al., 2015; Zhang et al., 2015). Brain slice physiology studies show that decreased infralimbic and prelimbic mPFC output is the consequence of abnormally enhanced glutamatergic activation of parvalbumin-expressing GABAergic interneurons (Ji and Neugebauer, 2011; Ji et al., 2010; Kiritoshi et al., 2016; Zhang et al., 2015). Mechanisms of this abnormal feedforward inhibition, however, remained to be determined, and form the subject of the following two sections.
5.2 Synaptic mechanisms
A major source of input to the infralimbic and prelimbic mPFC are long-range projections from the basolateral amygdala (BLA) (Cheriyan et al., 2016; Little and Carter, 2013; Senn et al., 2014). The BLA is a major site for the integration of sensory and other inputs in the amygdala, forwarding this highly processed information to the amygdala output region (central nucleus, CeA) as well as to extra-amygdalar brain regions such as the mPFC. In contrast to pain-related deactivation of the mPFC, the amygdala (CeA and BLA) develops synaptic plasticity and hyperactivity in pain states, which critically contribute to the emotional-affective aspects of pain and the facilitation of pain sensitivity (Neugebauer, 2015). Increased activity in the amygdala has also been described in experimental and clinical human pain states (reviewed by Neugebauer, 2015).
Increased BLA output has been causally linked to mPFC deactivation. Pharmacological studies in an arthritis pain model (knee joint monoarthritis induced by intraarticular kaolin/carrageenan, 5-6 hours post-induction; Neugebauer et al., 2007) showed that normalizing activity of BLA neurons with stereotaxic intra-amygdala injection of a corticotropin-releasing factor 1 (CRF1) receptor antagonist partially restored the decreased activity of layer V mPFC pyramidal neurons (Ji et al., 2010). The mechanism by which BLA projection neurons decrease mPFC pyramidal cell activity is glutamate-driven activation of GABAergic mPFC interneurons, resulting in feedforward inhibition of mPFC pyramidal cells (Figure 2). Whole-cell patch-clamp recordings of layer V pyramidal cells in the infralimbic and prelimbic mPFC showed increased inhibitory synaptic transmission (inhibitory postsynaptic currents, IPSCs) in brain slices from rats with a knee joint monoarthritis (5-6 hours post-induction by intraarticular kaolin/carrageenan) compared to controls (Ji et al., 2010; Kiritoshi et al., 2016). IPSCs were evoked by focal electrical stimulation of fiber tracts containing anterogradely labeled BLA axons, and by more selective optogenetic activation of BLA terminals in the mPFC expressing a light sensitive channel (channel rhodopsin 2, ChR2) 4 weeks after injection of viral vector (rAAV5/CaMKIIa-ChR2(H134R)-eYFP) into the BLA (Ji et al., 2010; Kiritoshi et al., 2016). Increased feedforward inhibition of prelimbic layer V pyramidal cells was also detected 10 days after induction of SNI neuropathic pain in the mouse (Zhang et al., 2015).
Figure 2. Schematic of mGLuR5-CB1 coupling in mPFC-amygdala interaction in pain states.
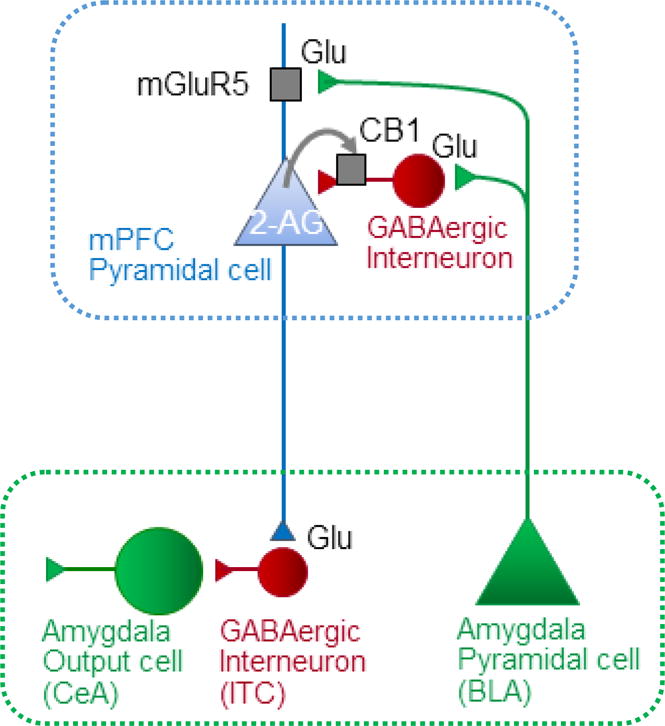
The amygdala sends glutamatergic (Glu) projections from the basolateral nucleus, (BLA) to mPFC pyramidal cells activating mGluR5, and to mPFC GABAergic interneurons inhibiting mPFC pyramidal cells (“feedforward inhibition”). mGluR5 couples to 2-AG synthesis via DAGLα. 2-AG release acts on presynaptic CB1 to inhibit synaptic inhibition hence increasing mPFC output. mPFC pyramidal cells send glutamatergic projections to GABAergic interneurons in the amygdala (intercalated cells, ITC) to control amygdala output from the central nucleus (CeA).
Feedforward inhibition of mPFC pyramidal cells involves activation of GABAergic interneurons mediated by metabotropic glutamate receptor subtype mGluR1, but not mGluR5 (Ji and Neugebauer, 2011; Sun and Neugebauer, 2011). Since blockade of mGluR1 restored mPFC activity only partially (Ji and Neugebauer, 2011), other mechanisms and targets to increase and restore mPFC activity were explored. The EC system has subsequently emerged as a likely candidate, based on the evidence for an important role of CB1 receptors in the infralimbic mPFC in fear extinction (Lin et al., 2009; Marsicano et al., 2002).
5.3 Role of CB1 and mGluR5 Interaction in Control of Cortical Feedforward Inhibition
ECs act as retrograde signalling molecules on presynaptic CB1 receptors to inhibit excitatory or inhibitory synaptic transmission (Di Marzo, 2011; Guindon and Hohmann, 2009; Kano et al., 2009; Lovinger, 2008). In the rodent mPFC, CB1 is predominantly if not exclusively expressed in GABAergic interneurons (Marsicano and Lutz, 1999; Wedzony and Chocyk, 2009) and therefore well positioned to reduce the abnormal synaptic inhibition observed in pain models (see 4.2). Indeed, CB1-mediated depolarization-induced suppression of synaptic inhibition (DSI) has been detected in layer V infralimbic pyramidal cells in brain slices from normal rats (Kiritoshi et al., 2016; Kiritoshi et al., 2013). DSI is a form of short-term synaptic plasticity, involving postsynaptic calcium influx following depolarization, activation of DAGLα, synthesis and release of 2-AG, and retrograde activation of CB1 receptors on the presynaptic terminal to inhibit transmitter release (Di Marzo, 2011; Kano et al., 2009; Lovinger, 2008; Rivera et al., 2014). A selective CB1 agonist (ACEA) decreased frequency, but not amplitude, of miniature IPSCs and increased synaptically-evoked spiking (E–S coupling, a measure of output function), suggesting that presynaptic CB1 controls inhibitory transmission onto mPFC pyramidal cells (Kiritoshi et al., 2013).
In addition to DSI, mGluR5 has been linked to the production of ECs and synaptic depression (Robbe et al., 2002). As previously discussed (see Section 4), mGluR5 can activate the phospholipase C - DAGLα pathway that leads to the formation of 2-AG (Gregg et al., 2012; Nyilas et al., 2009). Complementary to pre-synaptically-expressed CB1, mGluR5 in the mPFC is expressed mostly on post-synaptic elements (Muly et al., 2003), has excitatory effects on layer V pyramidal cells (Fontanez-Nuin et al., 2011; Kiritoshi et al., 2013; Marek and Zhang, 2008), and plays a role in fear extinction (Fontanez-Nuin et al., 2011; Sepulveda-Orengo et al., 2013; Xu et al., 2009). CB1-expressing axon terminals synapse onto mPFC pyramidal cells expressing mGluR5 and DAGLα (Lafourcade et al., 2007), and a selective mGluR5 activator (VU0360172 - Rodriguez et al., 2010) decreased synaptic inhibition of infralimbic pyramidal cells, in a CB1-sensitive manner (Kiritoshi et al., 2013). VU0360172 increased pyramidal output (E-S coupling), which was blocked by intracellular inhibition of DAGLα with tetrahydrolipstatin (THL) or by the CB1 antagonist/inverse agonist AM251 (Kiritoshi et al., 2016; Kiritoshi et al., 2013). These data suggest that mGluR5 increases pyramidal output by controlling synaptic inhibition through 2-AG-CB1 signalling (Figure 2).
Unexpectedly, this mGluR5-CB1-mediated effect is absent in a model of arthritic pain. In brain slices from arthritic animals (kaolin/carrageenan model, 5-6 h post-induction), DSI was impaired, mGluR5 activation with VU0360172 had no effect on the output (E-S coupling) of infralimbic pyramidal cells, and ACEA no longer decreased feedforward inhibition (Kiritoshi et al., 2016; Kiritoshi et al., 2013). This breakdown of mGluR5-CB1 signalling in the mPFC leads to abnormally enhanced feedforward inhibition, and may explain the decreased mPFC output seen in this model. The mechanism underlying the loss of CB1-mediated control of synaptic inhibition appears to be a lack of available 2-AG, rather than of functional CB1 receptors or impaired release of endocannabinoids. This has been demonstrated by the restoration of mGluR5-CB1 facilitation of mPFC output via use of specific enzyme inhibitors. Increasing availability of 2-AG in the postsynaptic cell via inhibition of the postsynaptic 2-AG hydrolyzing enzyme ABHD6 with intracellular WWL70, MAGL via JZL184, or by blocking GABAergic inhibition with intracellular picrotoxin were all effective at restoring mGLuR5-CB1 disinhibition of mPFC output (Kiritoshi et al., 2016).
A rescue strategy has been devised to increase 2-AG availability, thus removing abnormal inhibition and increasing mPFC output. In the arthritic pain state (kaolin/carrageenan model), the combined application of VU0360172 and ACEA into the mPFC restored DSI, decreased IPSCs and increased output of mPFC pyramidal cells measured as synaptically-evoked spiking (E-S coupling) in rat brain slices (Kiritoshi et al., 2016; Kiritoshi et al., 2013). This combination strategy also increased background and evoked activity (action potentials) of infralimbic mPFC neurons recorded extracellularly in anesthetized rats in vivo (Ji and Neugebauer, 2014). Importantly, a TRPV1 receptor antagonist (AMG9810) did not block the restored control of synaptic inhibition by the combination strategy, arguing against the involvement of TRPV1 receptors, which have been implicated in some actions of the endocannabinoid AEA.
5.4 Role of CB1 and mGluR5 Interaction in Control of Amygdala Function
A major limbic target of mPFC output is the amygdala (Gabbott et al., 2005; McDonald, 1998). Cortical control of amygdala function has been identified as a critical mechanism of fear extinction (Herry et al., 2010; Likhtik et al., 2005; Marek et al., 2013; Orsini and Maren, 2012; Pape and Pare, 2010; Sotres-Bayon and Quirk, 2010). In various pain models, amygdala activity and output have been shown to be abnormally increased due to uncontrolled excitatory glutamatergic and peptidergic drive onto neurons of the CeA (Neugebauer, 2015). At the center of this dysfunction is impaired activation of an inhibitory gating mechanism centered on feedforward inhibition of CeA neurons by a cluster of GABAergic neurons in the intercalated (ITC) cell mass, which is in turn driven by glutamatergic projections from BLA and mPFC (Ren et al., 2013) (Figure 2). The infralimbic mPFC is known to target these GABAergic ITC cells (Amir et al., 2011; Berretta et al., 2005; Busti et al., 2011; Pinard et al., 2012).
Co-activation of CB1 and mGluR5 in the infralimbic mPFC with stereotaxic application of VU0360172 and ACEA increased background and evoked activity of infralimbic pyramidal cells in a model of arthritic pain (kaolin/carrageenan monoarthritis, 5-6 h postinduction) and inhibited the pain-related increase of background and evoked activity in CeA neurons, but had no effect under normal conditions (Ji and Neugebauer, 2014). The data suggest that infralimbic mPFC neurons are inversely linked to amygdala output and that restoring mPFC output with a combination strategy of mGluR5-CB1 activation can engage cortical control of abnormally enhanced amygdala output in pain.
5.5 Role of CB1 and mGluR5 Interaction in Cortical Control of Pain Behaviour
Pain is a multidimensional disorder with sensory, emotional-affective, and cognitive aspects. Pain-related neuroplastic changes in the amygdala circuitry generate emotional responses such as vocalizations, and anxiety-like and depression-related behaviours, but also contribute to increased pain sensitivity (Neugebauer, 2015). mPFC dysfunction has been linked to fear extinction deficits (Fontanez-Nuin et al., 2011; Sepulveda-Orengo et al., 2013; Xu et al., 2009) as well as to pain-related cognitive deficits such as in decision-making (Apkarian et al., 2004a,b; Ji et al., 2010; Moriarty et al., 2011; Pais-Vieira et al., 2009 (Apkarian et al., 2004a; Apkarian et al., 2004b; Ji et al., 2010; Moriarty et al., 2011). Restoring mPFC output would therefore be expected to mitigate the cognitive deficits as well as the amygdala-dependent aspects of pain.
Co-activation of CB1 and mGluR5 in the infralimbic mPFC via stereotaxic application of VU0360172 and ACEA increases pyramidal cell output, and effectively inhibits spinal withdrawal reflexes and audible and ultrasonic vocalizations (reflecting sensory and emotional-affective responses, respectively), evoked by mechanical compression of the knee joint in the kaolin-carrageenan model of arthritic pain (5-6 h post-induction). Importantly, stereotaxic injections of VU0360172 and ACEA into the anterior cingulate cortex (area 24b) had no significant effect on hindlimb withdrawal thresholds and vocalizations of arthritic rats, suggesting that this is a region-specific mechanism.
This rescue strategy also mitigated cognitive deficits observed in this arthritic pain model by measuring reward-based decision-making in a rodent gambling task model (Ji et al., 2010; Sun and Neugebauer, 2011). In this task, normal animals switch from preferring the “high-risk” lever that provides 3 chocolate coated food pellets in only 3 of 10 trials to preferring the “low-risk” lever that provides one food reward consistently in 9 of 10 trials. Arthritic rats persist in preferring the high-risk lever, suggesting that they fail to switch strategies. Stereotaxic co-application of VU0360172 and ACEA into the infralimbic mPFC restored the ability of arthritic rats to switch strategies and to prefer the low-risk lever like normal rats (Kiritoshi et al., 2016).
Consistent with these observations, optogenetic silencing of parvalbumin-expressing interneurons in the mPFC decreased mechanical and thermal hypersensitivity of mice in SNI neuropathic pain model, while optogenetic activation of these interneurons exacerbated pain sensitivity (Zhang et al., 2015). In the conditioned place preference test, optogenetic activation or silencing of parvalbumin-expressing interneurons in the mPFC decreased or increased, respectively, preference of neuropathic mice for the conditioned chamber compared to sham controls (Zhang et al., 2015). This optogenetic silencing of inhibitory mPFC interneurons also attenuated escape behaviour in neuropathic mice (Zhang et al., 2015). These studies collectively suggest that pain is associated with the disruption of mGluR5-endocannabinoid signalling in the mPFC, resulting in decreased output due to uncontrolled feedforward inhibition driven by amygdala (BLA) inputs. Restoring mPFC output by co-activation of mGluR5 and CB1 inhibits abnormally enhanced amygdala activity, hypersensitivity and emotional-affective pain responses and also restores cognitive functions in a decision-making task, suggesting that this approach may have utility in treating the non-sensory aspects of chronic pain states.
Conclusions
The EC system is a major endogenous pain control system, running in parallel to the opioid system and playing crucial roles the development and resolution of pain states, and the affective and cognitive aspects of pain. The initial promise of augmenting EC signalling via specific enzyme inhibitors has been diminished by recent clinical failures. However, greater understanding of the role of the EC system in non-opioid and opioid-dependent forms of endogenous pain suppression and exacerbation in response to stress, and the dysfunction of forebrain-limbic circuitry in pain states in humans will aid the development of future analgesic strategies, especially with respect to targeting particular populations of patients. Because FAAH and MAGL inhibitors are not specific for the endocannabinoid system, more work is also necessary to understand the biological roles of other lipid mediators that are generated by these inhibitors that do not bind to cannabinoid receptors. Allosteric modulators of CB1 receptors may therefore be a useful strategy for amplifying effects of endocannabinoids only at sites where they are produced and released on demand.
Nonetheless, multifunctional compounds targeting the EC system allied to inhibition of COX2, antagonism of TRPV1, or in combination with opioids or NSAIDs have great potential to produce a superior therapeutic profile that harnesses the therapeutic potential of the endocannabinoid signalling system while minimizing unwanted cannabimimetic side effects.
Future Directions
Within the field of endocannabinoid research, significant fundamental questions remain unanswered. We still do not completely understand how these hydrophobic lipid signalling molecules are transported across aqueous environments such as the synaptic cleft or cytoplasm to reach their target receptors. Fatty acid binding proteins have been strongly implicated (e.g. Kaczocha et al., 2009; Kaczocha et al., 2015; Kaczocha et al., 2014), but their roles have yet to be full elucidated. Furthermore, not only are ECs promiscuous, but CB1 has been demonstrated to have constitutive activity in the absence of ligand signalling (Howlett et al., 2011; Lee et al., 2015), and can form heterodimers with several other GPCRs (Hudson et al., 2010), further complicating interpretation of the pharmacology of EC-directed therapeutics. Some mystery still surrounds the role of AEA in EC signalling. Anatomical evidence suggests that its purported major synthetic enzyme, NAPE-PLD, is localized pre-synaptically (Nyilas et al., 2008) whilst FAAH is found post-synaptically (Gulyas et al., 2004), in stark contrast to the established complementary post-synaptic position of the 2-AG synthesizing enzyme DAGLα (Gregg et al., 2012; Nyilas et al., 2009) and pre-synaptic position of MAGL (Gulyas et al., 2004; Horváth et al., 2014). Revealing the answers to these questions will facilitate better understanding of the function of EC signalling under physiological and pathological situations, and greatly aid the development of future therapeutic interventions.
Whilst much has been achieved in the past few decades, more work is necessary to characterize both efficacy and safety profiles of existing EC-directed therapeutic strategies, and to answer these fundamental questions about EC function, so that the clinical potential of modulating the endocannabinoid system for analgesia can be realized.
Highlights.
The endocannabinoid (EC) system is a key endogenous pain control system.
Elevating or enhancing EC signalling is antinociceptive in preclinical models.
Recent clinical failures suggest study limitations and gaps in basic knowledge.
Future directions and strategies to improve clinical translation are discussed.
Acknowledgments
DPF is supported by a grant from Science Foundation Ireland (10/IN.1/B2976). AGH is supported by (DA041229).
Footnotes
Chemical Compounds Studied in this Article: AA-5-HT (PubChem CID: 10027372), BIA-102474 (PubChem CID: 46831476), JZL184 (CID: 25021165), JZL195 (PubChem CID: CID 46232606), MJN110 (PubChem CID: 71722059), PF-04457845 (PubChem CID: 24771824), URB597 (PubChem CID: 1383884), URB937 (PubChem CID: 53394762)
References
- Adamson Barnes NS, Mitchell VA, Kazantzis NP, Vaughan CW. Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine neuropathic pain model. Br J Pharmacol. 2016;173:77–87. doi: 10.1111/bph.13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, Rubino T, Michalski CW, Marsicano G, Monory K, Mackie K, Marian C, Batkai S, Parolaro D, Fischer MJ, Reeh P, Kunos G, Kress M, Lutz B, Woolf CJ, Kuner R. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nature Neuroscience. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Discovery and Characterization of a Highly Selective FAAH Inhibitor that Reduces Inflammatory Pain. Chemistry & Biology. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, Young T, Wren P, Zhang Y, Swaney S, Van Becelaere K, Blankman JL, Nomura DK, Bhattachar SN, Stiff C, Nomanbhoy TK, Weerapana E, Johnson DS, Cravatt BF. Mechanistic and Pharmacological Characterization of PF-04457845: A Highly Potent and Selective Fatty Acid Amide Hydrolase Inhibitor That Reduces Inflammatory and Noninflammatory Pain. Journal of Pharmacology and Experimental Therapeutics. 2011;338:114–124. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kendall DA. The complications of promiscuity: endocannabinoid action and metabolism. Br J Pharmacol. 2007;152:602–623. doi: 10.1038/sj.bjp.0707456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhouayek M, Muccioli GG. Harnessing the anti-inflammatory potential of palmitoylethanolamide. Drug Discovery Today. 2014 doi: 10.1016/j.drudis.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Alkaitis MS, Solorzano C, Landry RP, Piomelli D, DeLeo JA, Romero-Sandoval EA. Evidence for a Role of Endocannabinoids, Astrocytes and p38 Phosphorylation in the Resolution of Postoperative Pain. PLoS ONE. 2010;5:e10891. doi: 10.1371/journal.pone.0010891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameri A. The effects of cannabinoids on the brain. Progress in Neurobiology. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- Amir A, Amano T, Pare D. Physiological identification and infralimbic responsiveness of rat intercalated amygdala neurons. J Neurophysiol. 2011;105:3054–3066. doi: 10.1152/jn.00136.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit Z, Galina ZH. Stress-induced analgesia: adaptive pain suppression. Physiological Reviews. 1986;66:1091–1120. doi: 10.1152/physrev.1986.66.4.1091. [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Progress in Neurobiology. 2009;87:81–97. doi: 10.1016/j.pneurobio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, Levy RE, Harden RN, Chialvo DR. Chronic pain patients are impaired on an emotional decision-making task. Pain. 2004a;108:129–136. doi: 10.1016/j.pain.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, Gitelman DR. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. The Journal of Neuroscience. 2004b;24:10410–10415. doi: 10.1523/JNEUROSCI.2541-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmundson GJG, Katz J. Understanding the co-occurrence of anxiety disorders and chronic pain: state-of-the-art. Depression and Anxiety. 2009;26:888–901. doi: 10.1002/da.20600. [DOI] [PubMed] [Google Scholar]
- Bégaud, B., Bousser, M. G., Cohen, P., Diquet, B., Duprat, P., Janssens, W., Mallaret, M., Mazué, G., Micalle, J., Monneret, C., Montastruc, J. L., Venance, L., 2016. Report by the Temporary Specialist Scientific Committee (TSSC), ‘FAAH (Fatty Acid Amide Hydrolase)’, on the causes of the accident during a Phase 1 clinical trial in Rennes in January 2016.
- Berretta S, Pantazopoulos H, Caldera M, Pantazopoulos P, Pare D. Infralimbic cortex activation increases c-Fos expression in intercalated neurons of the amygdala. Neuroscience. 2005;132:943–953. doi: 10.1016/j.neuroscience.2005.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chemistry & Biology. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker L, Kinsey SG, Abdullah RA, Blankman JL, Long JZ, Ezzili C, Boger DL, Cravatt BF, Lichtman AH. The FAAH Inhibitor PF-3845 Acts in the Nervous System to Reverse Lipopolysaccharide-induced Tactile Allodynia in Mice. Br J Pharmacol. 2012 doi: 10.1111/j.1476-5381.2011.01445.x. no-no. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke NN, Finn DP, Roche M. Neuroinflammatory Mechanisms Linking Pain and Depression. Mod Trends Pharmacopsychiatri. 2015;30:36–50. doi: 10.1159/000435931. [DOI] [PubMed] [Google Scholar]
- Busquets-Garcia A, Puighermanal E, Pastor A, de la Torre R, Maldonado R, Ozaita As. Differential Role of Anandamide and 2-Arachidonoylglycerol in Memory and Anxiety-like Responses. Biological Psychiatry. 2011;70:479–486. doi: 10.1016/j.biopsych.2011.04.022. [DOI] [PubMed] [Google Scholar]
- Busti D, Geracitano R, Whittle N, Dalezios Y, Manko M, Kaufmann W, Satzler K, Singewald N, Capogna M, Ferraguti F. Different fear states engage distinct networks within the intercalated cell clusters of the amygdala. The Journal of Neuroscience. 2011;31:5131–5144. doi: 10.1523/JNEUROSCI.6100-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler RK, Finn DP. Stress-induced analgesia. Progress in Neurobiology. 2009;88:184–202. doi: 10.1016/j.pneurobio.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Butler RK, Rea K, Lang Y, Gavin AM, Finn DP. Endocannabinoid-mediated enhancement of fear-conditioned analgesia in rats: Opioid receptor dependency and molecular correlates. Pain. 2008;140:491–500. doi: 10.1016/j.pain.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of Pain Initiation by Endogenous Cannabinoids. Letters to Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Cardoso-Cruz H, Sousa M, Vieira JB, Lima D, Galhardo V. Prefrontal cortex and mediodorsal thalamus reduced connectivity is associated with spatial working memory impairment in rats with inflammatory pain. Pain. 2013;154:2397–2406. doi: 10.1016/j.pain.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Carey LM, Slivicki RA, Leishman E, Cornett B, Mackie K, Bradshaw H, Hohmann AG. A pro-nociceptive phenotype unmasked in mice lacking fatty-acid amide hydrolase. Molecular Pain. 2016;12 doi: 10.1177/1744806916649192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier EJ, Patel S, Hillard CJ. Endocannabinoids in neuroimmunology and stress. Curr Drug Targets CNS Neurol Disord. 2005;4:657–665. doi: 10.2174/156800705774933023. [DOI] [PubMed] [Google Scholar]
- Chanda PK, Gao Y, Mark L, Btesh J, Strassle BW, Lu P, Piesla MJ, Zhang MY, Bingham B, Uveges A, Kowal D, Garbe D, Kouranova EV, Ring RH, Bates B, Pangalos MN, Kennedy JD, Whiteside GT, Samad TA. Monoacylglycerol Lipase Activity Is a Critical Modulator of the Tone and Integrity of the Endocannabinoid System. Molecular Pharmacology. 2010;78:996–1003. doi: 10.1124/mol.110.068304. [DOI] [PubMed] [Google Scholar]
- Chang CH, Maren S. Strain difference in the effect of infralimbic cortex lesions on fear extinction in rats. Behavioural Neuroscience. 2010;124:391–397. doi: 10.1037/a0019479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, Breitenbucher JG, Chaplan SR, Webb M. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheriyan J, Kaushik MK, Ferreira AN, Sheets PL. Specific Targeting of the Basolateral Amygdala to Projectionally Defined Pyramidal Neurons in Prelimbic and Infralimbic Cortex. eNeuro. 2016;3 doi: 10.1523/ENEURO.0002-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nature Neuroscience. 2010;13:6. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell K, Bolton N, Olsen D, Piomelli D, Hohmann AG. Role of the basolateral nucleus of the amygdala in endocannabinoid-mediated stress-induced analgesia. Neuroscience Letters. 2006;397:180–184. doi: 10.1016/j.neulet.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Costa B, Bettoni I, Petrosino S, Comelli F, Giagnoni G, Di Marzo V. The dual fatty acid amide hydrolase/TRPV1 blocker, N-arachidonoyl-serotonin, relieves carrageenan-induced inflammation and hyperalgesia in mice. Pharmacological Research. 2010;61:537–546. doi: 10.1016/j.phrs.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- de Novellis V, Palazzo E, Rossi F, de Petrocellis L, Petrosino S, Guida F, Luongo L, Migliozzi A, Cristino L, Marabese I, Starowicz K, Di Marzo V, Maione S. The analgesic effect of N-arachidonoyl-serotonin, a FAAH inhibitor and TRPV1 receptor antagonist, associated with changes in rostral ventromedial medulla and locus coeruleus cell activity in rats. Neuropharmacology. 2008;55:1105–1113. doi: 10.1016/j.neuropharm.2008.06.023. [DOI] [PubMed] [Google Scholar]
- de Novellis V, Vita D, Gatta L, Luongo L, Bellini G, De Chiaro M, Marabese I, Siniscalco D, Boccella S, Piscitelli F, Di Marzo V, Palazzo E, Rossi F, Maione S. The blockade of the transient receptor potential vanilloid type 1 and fatty acid amide hydrolase decreases symptoms and central sequelae in the medial prefrontal cortex of neuropathic rats. Molecular Pain. 2011;7:1–19. doi: 10.1186/1744-8069-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wied M, Verbaten MN. Affective pictures processing, attention, and pain tolerance. Pain. 2001;90:163–172. doi: 10.1016/s0304-3959(00)00400-0. [DOI] [PubMed] [Google Scholar]
- Desroches J, Charron S, Bouchard JF, Beaulieu P. Endocannabinoids decrease neuropathic pain-related behavior in mice through the activation of one or both peripheral CB1 and CB2 receptors. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Determination and Characterization of a Cannabinoid Receptor in Rat-Brain. Molecular Pharmacology. 1988;34:605–613. [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and Structure of a Brain Constituent That Binds to the Cannabinoid Receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoid signaling in the brain: biosynthetic mechanisms in the limelight. Nature Neuroscience. 2011;14:9–15. doi: 10.1038/nn.2720. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Kathuria S, Piomelli D. RNA Interference Suggests a Primary Role for Monoacylglycerol Lipase in the Degradation of the Endocannabinoid 2-Arachidonoylglycerol. Molecular Pharmacology. 2004;66:1260–1264. doi: 10.1124/mol.104.002071. [DOI] [PubMed] [Google Scholar]
- Dougher MJ. Sensory decision theory analysis of the effects of anxiety and experimental instructions on pain. J Abnorm Psychol. 1979;88:137–144. doi: 10.1037//0021-843x.88.2.137. [DOI] [PubMed] [Google Scholar]
- Finn DP. Endocannabinoid-mediated modulation of stress responses: Physiological and pathophysiological significance. Immunobiology. 2010;215:629–646. doi: 10.1016/j.imbio.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Finn DP, Beckett SRG, Richardson D, Kendall DA, Marsden CA, Chapman V. Evidence for differential modulation of conditioned aversion and fear-conditioned analgesia by CB1 receptors. European Journal of Neuroscience. 2004;20:848–852. doi: 10.1111/j.1460-9568.2004.03509.x. [DOI] [PubMed] [Google Scholar]
- Fitzgibbon M, Finn DP, Roche M. High Times for Painful Blues: The Endocannabinoid System in Pain-Depression Comorbidity. The International Journal of Neuropsychopharmacology. 2015 doi: 10.1093/ijnp/pyv095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanez-Nuin DE, Santini E, Quirk GJ, Porter JT. Memory for fear extinction requires mGluR5-mediated activation of infralimbic neurons. Cerebral Cortex. 2011;21:727–735. doi: 10.1093/cercor/bhq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford GK, Finn DP. Clinical correlates of stress-induced analgesia: Evidence from pharmacological studies. Pain. 2008;140:3–7. doi: 10.1016/j.pain.2008.09.023. [DOI] [PubMed] [Google Scholar]
- Ford GK, Kieran S, Dolan K, Harhen B, Finn DP. A role for the ventral hippocampal endocannabinoid system in fear-conditioned analgesia and fear responding in the presence of nociceptive tone in rats. Pain. 2011;152:2495–2504. doi: 10.1016/j.pain.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Fowler CJ. The contribution of cyclooxygenase-2 to endocannabinoid metabolism and action. Br J Pharmacol. 2007;152:594–601. doi: 10.1038/sj.bjp.0707379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Janson U, Johnson RM, Wahlström G, Stenström A, Norström Å, Tiger G. Inhibition of Anandamide Hydrolysis by the Enantiomers of Ibuprofen, Ketorolac, and Flurbiprofen. Archives of Biochemistry and Biophysics. 1999;362:191–196. doi: 10.1006/abbi.1998.1025. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, Naidu PS, Lichtman A, Onnis V. The case for the development of novel analgesic agents targeting both fatty acid amide hydrolase and either cyclooxygenase or TRPV1. Br J Pharmacol. 2009;156:412–419. doi: 10.1111/j.1476-5381.2008.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Gamble-George JC, Baldi R, Halladay L, Kocharian A, Hartley N, Silva CG, Roberts H, Haymer A, Marnett LJ, Holmes A, Patel S. Cyclooxygenase-2 inhibition reduces stress-induced affective pathology. eLife. 2016;5:e14137. doi: 10.7554/eLife.14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaskin DJ, Richard P. The economic costs of pain in the United States. The Journal of Pain. 2012;13:715–724. doi: 10.1016/j.jpain.2012.03.009. [DOI] [PubMed] [Google Scholar]
- Gavva NR. Setbacks in the Clinical Development of TRPV1 Antagonists: What Next? The Open Drug Discovery Journal. 2009;1:35. [Google Scholar]
- Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A, Alvarez F, Bak A, Darling M, Gore A, Jang GR, Kesslak JP, Ni L, Norman MH, Palluconi G, Rose MJ, Salfi M, Tan E, Romanovsky AA, Banfield C, Davar G. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain. 2008;136:202–210. doi: 10.1016/j.pain.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Ghilardi JR, Rohrich H, Lindsay TH, Sevcik MA, Schwei MJ, Kubota K, Halvorson KG, Poblete J, Chaplan SR, Dubin AE, Carruthers NI, Swanson D, Kuskowski M, Flores CM, Julius D, Mantyh PW. Selective Blockade of the Capsaicin Receptor TRPV1 Attenuates Bone Cancer Pain. The Journal of Neuroscience. 2005;25:3126–3131. doi: 10.1523/JNEUROSCI.3815-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Kinsey SG, Liu Q-s, Hruba L, McMahon LR, Grim TW, Merritt CR, Wise LE, Abdulla RA, Selley DE, Sim-Selley L, Cravatt BF, Lichtman AH. Full FAAH inhibition combined with partial monoacylglycerol lipase inhibition: Augmented and sustained antinociceptive effects with negligible cannabimimetic side effects in mice. Journal of Pharmacology and Experimental Therapeutics. 2015 doi: 10.1124/jpet.115.222851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Wise LE, Chen Y, Gujjar R, Mahadevan A, Cravatt BF, Lichtman AH. The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sciences. 2013;92:498–505. doi: 10.1016/j.lfs.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsiorek W, Lunn C, Fan X, Narula S, Lundell D, Hipkin RW. Endocannabinoid 2-Arachidonyl Glycerol Is a Full Agonist through Human Type 2 Cannabinoid Receptor: Antagonism by Anandamide. Molecular Pharmacology. 2000;57:1045–1050. [PubMed] [Google Scholar]
- Gregg LC, Jung KM, Spradley JM, Nyilas R, Suplita RL, Zimmer A, Watanabe M, Mackie K, Katona I, Piomelli D, Hohmann AG. Activation of Type 5 Metabotropic Glutamate Receptors and Diacylglycerol Lipase-Alpha Initiates 2-Arachidonoylglycerol Formation and Endocannabinoid-Mediated Analgesia. The Journal of Neuroscience. 2012;32:9457–9468. doi: 10.1523/JNEUROSCI.0013-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grim TW, Ghosh S, Hsu KL, Cravatt BF, Kinsey SG, Lichtman AH. Combined inhibition of FAAH and COX produces enhanced anti-allodynic effects in mouse neuropathic and inflammatory pain models. Pharmacology Biochemistry and Behavior. 2014;124:405–411. doi: 10.1016/j.pbb.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon G, Hohmann A. The Endocannabinoid System and Pain. CNS Neurological Disorders Drug Targets. 2009;8:18. doi: 10.2174/187152709789824660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Beaulieu P. Antihyperalgesic effects of local injections of anandamide, ibuprofen, rofecoxib and their combinations in a model of neuropathic pain. Neuropharmacology. 2006;50:814–823. doi: 10.1016/j.neuropharm.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Guindon J, Beaulieu P. The role of the endogenous cannabinoid system in peripheral analgesia. Current Molecular Pharmacology. 2009;2:5. doi: 10.2174/1874467210902010134. [DOI] [PubMed] [Google Scholar]
- Guindon J, Desroches J, Beaulieu P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br J Pharmacol. 2007a;150:693–701. doi: 10.1038/sj.bjp.0706990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Guijarro A, Piomelli D, Hohmann AG. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol. 2011;163:1464–1478. doi: 10.1111/j.1476-5381.2010.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Lai Y, Takacs SM, Bradshaw HB, Hohmann AG. Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: Effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacological Research. 2013;67:94–109. doi: 10.1016/j.phrs.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, LoVerme J, De Lean A, Piomelli D, Beaulieu P. Synergistic antinociceptive effects of anandamide, an endocannabinoid, and nonsteroidal anti-inflammatory drugs in peripheral tissue: A role for endogenous fatty-acid ethanolamides? European Journal of Pharmacology. 2006;550:68–77. doi: 10.1016/j.ejphar.2006.08.045. [DOI] [PubMed] [Google Scholar]
- Guindon J, Walczak JS, Beaulieu P. Recent advances in the pharmacological management of pain. Drugs. 2007b;67:2121–2133. doi: 10.2165/00003495-200767150-00002. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. European Journal of Neuroscience. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- Hall W, Solowij N. Adverse effects of cannabis. Lancet. 1998;352:1611–1616. doi: 10.1016/S0140-6736(98)05021-1. [DOI] [PubMed] [Google Scholar]
- Hefner K, Whittle N, Juhasz J, Norcross M, Karlsson RM, Saksida LM, Bussey TJ, Singewald N, Holmes A. Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. The Journal of Neuroscience. 2008;28:8074–8085. doi: 10.1523/JNEUROSCI.4904-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, Decosta BR, Rice KC. Characterization and Localization of Cannabinoid Receptors in Rat-Brain - a Quantitative Invitro Autoradiographic Study. The Journal of Neuroscience. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, Decosta BR, Rice KC. CANNABINOID RECEPTOR LOCALIZATION IN BRAIN. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson DJ, Gamble-George JC, Marnett LJ, Patel S. Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends in Pharmacological Sciences. 2014 doi: 10.1016/j.tips.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson DJ, Hartley ND, Gamble-George J, Brown N, Shonesy BC, Kingsley PJ, Colbran RJ, Reese J, Marnett LJ, Patel S. Substrate-selective COX-2 inhibition decreases anxiety via endocannabinoid activation. Nature Neuroscience. 2013;16:1291–1298. doi: 10.1038/nn.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Luthi A. Neuronal circuits of fear extinction. European Journal of Neuroscience. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- Hill MN, Kumar SA, Filipski SB, Iverson M, Stuhr KL, Keith JM, Cravatt BF, Hillard CJ, Chattarji S, McEwen BS. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Molecular Psychiatry. 2013;18:1125–1135. doi: 10.1038/mp.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Patel S, Carrier EJ, Rademacher DJ, Ormerod BK, Hillard CJ, Gorzalka BB. Downregulation of Endocannabinoid Signaling in the Hippocampus Following Chronic Unpredictable Stress. Neuropsychopharmacology. 2005;30:508–515. doi: 10.1038/sj.npp.1300601. [DOI] [PubMed] [Google Scholar]
- Hohmann AG. Spinal and peripheral mechanisms of cannabinoid antinociception: behavioral, neurophysiological and neuroanatomical perspectives. Chemistry and Physics of Lipids. 2002;121:173–190. doi: 10.1016/s0009-3084(02)00154-8. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Holden JE, Jeong Y, Forrest JM. The endogenous opioid system and clinical pain management. AACN Clin Issues. 2005;16:291–301. doi: 10.1097/00044067-200507000-00003. [DOI] [PubMed] [Google Scholar]
- Holt S, Comelli F, Costa B, Fowler CJ. Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br J Pharmacol. 2005;146:467–476. doi: 10.1038/sj.bjp.0706348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Fan J, Kemmerer ES, Evans S, Li Y, Wiley JW. Reciprocal changes in vanilloid (TRPV1) and endocannabinoid (CB1) receptors contribute to visceral hyperalgesia in the water avoidance stressed rat. Gut. 2009;58:202–210. doi: 10.1136/gut.2008.157594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth E, Woodhams SG, Nyilas R, Henstridge CM, Kano M, Sakimura K, Watanabe M, Katona I. Heterogeneous presynaptic distribution of monoacylglycerol lipase, a multipotent regulator of nociceptive circuits in the mouse spinal cord. European Journal of Neuroscience. 2014;39:419–434. doi: 10.1111/ejn.12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Reggio PH, Childers SR, Hampson RE, Ulloa NM, Deutsch DG. Endocannabinoid tone versus constitutive activity of cannabinoid receptors. Br J Pharmacol. 2011;163:1329–1343. doi: 10.1111/j.1476-5381.2011.01364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BD, Hébert TE, M Kelly ME. Ligand- and Heterodimer-Directed Signaling of the CB1 Cannabinoid Receptor. Molecular Pharmacology. 2010;77:1–9. doi: 10.1124/mol.109.060251. [DOI] [PubMed] [Google Scholar]
- Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–1846. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nature Reviews Neuroscience. 2001;2:83–91. doi: 10.1038/35053509. [DOI] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA, Damaj IM, Poklis J, Wiley JL, Zanda M, Zanato C, Greig IR, Lichtman AH, Ross RA. A Cannabinoid CB1 Receptor-Positive Allosteric Modulator Reduces Neuropathic Pain in the Mouse with No Psychoactive Effects. Neuropsychopharmacology. 2015a;40:2948–2959. doi: 10.1038/npp.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Ghosh S, Crowe MS, Kinsey SG, Niphakis MJ, Abdullah RA, Tao Q, O’Neal ST, Walentiny DM, Wiley JL, Cravatt BF, Lichtman AH. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: Antinociceptive activity without cannabimimetic side effects. Br J Pharmacol. 2014:1392–1407. doi: 10.1111/bph.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Wilkerson JL, Mustafa M, Abdullah R, Niphakis M, Wiley JL, Cravatt BF, Lichtman AH. Selective monoacylglycerol lipase inhibitors: Antinociceptive vs. cannabimimetic effects in mice. Journal of Pharmacology and Experimental Therapeutics. 2015b doi: 10.1124/jpet.114.222315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperatore R, Morello G, Luongo L, Taschler U, Romano R, De Gregorio D, Belardo C, Maione S, Di Marzo V, Cristino L. Genetic deletion of monoacylglycerol lipase leads to impaired cannabinoid receptor CB1R signaling and anxiety-like behavior. Journal of Neurochemistry. 2015;135:799–813. doi: 10.1111/jnc.13267. [DOI] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings EM, Okine BN, Olango WM, Roche M, Finn DP. Repeated forced swim stress differentially affects formalin-evoked nociceptive behaviour and the endocannabinoid system in stress normo-responsive and stress hyper-responsive rat strains. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2016;64:181–189. doi: 10.1016/j.pnpbp.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Jennings EM, Okine BN, Roche M, Finn DP. Stress-induced hyperalgesia. Progress in Neurobiology. 2014 doi: 10.1016/j.pneurobio.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Kendall DA, Barrett DA, Chapman V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. The Journal of Neuroscience. 2006;26:13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Robinson I, Garle MJ, Patel A, Sun Y, Sagar DR, Bennett AJ, Alexander SPH, Kendall DA, Barrett DA, Chapman V. Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacology. 2008;55:85–93. doi: 10.1016/j.neuropharm.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Pain-related deactivation of medial prefrontal cortical neurons involves mGluR1 and GABA(A) receptors. J Neurophysiol. 2011;106:2642–2652. doi: 10.1152/jn.00461.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. CB1 augments mGluR5 function in medial prefrontal cortical neurons to inhibit amygdala hyperactivity in an arthritis pain model. European Journal of Neuroscience. 2014;39:455–466. doi: 10.1111/ejn.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Sun H, Fu Y, Li Z, Pais-Vieira M, Galhardo V, Neugebauer V. Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. The Journal of Neuroscience. 2010;30:5451–5464. doi: 10.1523/JNEUROSCI.0225-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Glaser ST, Deutsch DG. Identification of intracellular carriers for the endocannabinoid anandamide. Proceedings of the National Academy of Sciences. 2009;106:6375–6380. doi: 10.1073/pnas.0901515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Glaser ST, Maher T, Clavin B, Hamilton J, O’Rourke J, Rebecchi M, Puopolo M, Owada Y, Thanos PK. Fatty acid binding protein deletion suppresses inflammatory pain through endocannabinoid/N-acylethanolamine-dependent mechanisms. Mol Pain. 2015;11:52. doi: 10.1186/s12990-015-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Rebecchi MJ, Ralph BP, Teng YH, Berger WT, Galbavy W, Elmes MW, Glaser ST, Wang L, Rizzo RC, Deutsch DG, Ojima I. Inhibition of Fatty Acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS ONE. 2014;9:e94200. doi: 10.1371/journal.pone.0094200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-Mediated Control of Synaptic Transmission. Physiological Reviews. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Karbarz MJ, Luo L, Chang L, Tham CS, Palmer JA, Wilson SJ, Wennerholm ML, Brown SM, Scott BP, Apodaca RL, Keith JM, Wu JJ, Breitenbucher JG, Chaplan SR, Webb M. Biochemical and Biological Properties of 4-(3-phenyl-[1,2,4]thiadiazol-5-yl)-piperazine-1-carboxylic acid phenylamide, a Mechanism-Based Inhibitor of Fatty Acid Amide Hydrolase. Anesthesia and Analgesia. 2009;108:316–329. doi: 10.1213/ane.0b013e31818c7cbd. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nature Medicine. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund T. Multiple Functions of Endocannabinoid Signaling in the Brain. Annual Review of Neuroscience. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nature Medicine. 2008;14:923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- Kaur R, Sidhu P, Singh S. What failed BIA 10–2474 Phase I clinical trial? Global speculations and recommendations for future Phase I trials. Journal of Pharmacology & Pharmacotherapeutics. 2016;7:120–126. doi: 10.4103/0976-500X.189661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Chandiramani A, Harding-Rose C, Simone DA, Seybold VS. Increasing 2-arachidonoyl glycerol signaling in the periphery attenuates mechanical hyperalgesia in a model of bone cancer pain. Pharmacological Research. 2011;64:60–67. doi: 10.1016/j.phrs.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L, Mackie K, Piomelli D, Kendall DA. Modulation of CB1 cannabinoid receptor by allosteric ligands: Pharmacology and therapeutic opportunities. Neuropharmacology. 2017 doi: 10.1016/j.neuropharm.2017.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Jo YS, Kim IH, Kim H, Choi JS. Lack of medial prefrontal cortex activation underlies the immediate extinction deficit. The Journal of Neuroscience. 2010;30:832–837. doi: 10.1523/JNEUROSCI.4145-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AR, Duranti A, Tontini A, Rivara S, Rosengarth A, Clapper JR, Astarita G, Geaga JA, Luecke H, Mor M, Tarzia G, Piomelli D. URB602 Inhibits Monoacylglycerol Lipase and Selectively Blocks 2-Arachidonoylglycerol Degradation in Intact Brain Slices. Chemistry & Biology. 2007;14:1357–1365. doi: 10.1016/j.chembiol.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, Cravatt BF, Lichtman AH. Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase Inhibitors Produce Anti-Allodynic Effects in Mice Through Distinct Cannabinoid Receptor Mechanisms. The Journal of Pain. 2010;11:1420–1428. doi: 10.1016/j.jpain.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. Blockade of Endocannabinoid-Degrading Enzymes Attenuates Neuropathic Pain. Journal of Pharmacology and Experimental Therapeutics. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Nomura DK, O’Neal ST, Long JZ, Mahadevan A, Cravatt BF, Grider JR, Lichtman AH. Inhibition of Monoacylglycerol Lipase Attenuates Nonsteroidal Anti-Inflammatory Drug-Induced Gastric Hemorrhages in Mice. Journal of Pharmacology and Experimental Therapeutics. 2011;338:795–802. doi: 10.1124/jpet.110.175778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritoshi T, Ji G, Neugebauer V. Rescue of Impaired mGluR5-Driven Endocannabinoid Signaling Restores Prefrontal Cortical Output to Inhibit Pain in Arthritic Rats. The Journal of Neuroscience. 2016;36:837–850. doi: 10.1523/JNEUROSCI.4047-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritoshi T, Sun H, Ren W, Stauffer SR, Lindsley CW, Conn PJ, Neugebauer V. Modulation of pyramidal cell output in the medial prefrontal cortex by mGluR5 interacting with CB1. Neuropharmacology. 2013;66:170–178. doi: 10.1016/j.neuropharm.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodny A, Courtwright DT, Hwang CS, Kreiner P, Eadie JL, Clark TW, Alexander GC. The prescription opioid and heroin crisis: a public health approach to an epidemic of addiction. Annual review of public health. 2015;36:559–574. doi: 10.1146/annurev-publhealth-031914-122957. [DOI] [PubMed] [Google Scholar]
- Kurrikoff k, Inno J, Matsui T, Vasar E. Stress-induced analgesia in mice: evidence for interaction between endocannabinoids and cholecystokinin. European Journal of Neuroscience. 2008;27:8. doi: 10.1111/j.1460-9568.2008.06160.x. [DOI] [PubMed] [Google Scholar]
- Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni OJ. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS ONE. 2007;2:e709. doi: 10.1371/journal.pone.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DM, Vandevoorde S, Jonsson KO, Fowler CJ. The palmitoylethanolamide family: a new class of anti-inflammatory agents? Curr Med Chem. 2002;9:663–674. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Kulkarni PM, Deschamps JR, Kelly ME, Janero DR, Cascio MG, Stevenson LA, Pertwee RG, Kenakin TP, Denovan-Wright EM, Thakur GA. Enantiospecific Allosteric Modulation of Cannabinoid 1 Receptor. ACS Chem Neurosci. 2017 doi: 10.1021/acschemneuro.6b00310. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. The Journal of Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Ledri M, Tóth B, Marchionni I, Henstridge CM, Dudok B, Kenesei K, Barna L, Szabó SI, Renkecz T, Oberoi M, Watanabe M, Limoli CL, Horvai G, Soltesz I, Katona I. Multiple Forms of Endocannabinoid and Endovanilloid Signaling Regulate the Tonic Control of GABA Release. The Journal of Neuroscience. 2015;35:10039–10057. doi: 10.1523/JNEUROSCI.4112-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GL, Winter H, Arends R, Jay GW, Le V, Young T, Huggins JP. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. British Journal of Clinical Pharmacology. 2012;73:706–716. doi: 10.1111/j.1365-2125.2011.04137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Chapman V. A FAAH-fetched approach to treat osteoarthritis pain. Pain. 2011;152:959–960. doi: 10.1016/j.pain.2011.02.034. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Martin BR. Cannabinoid tolerance and dependence. Handb Exp Pharmacol. 2005:691–717. doi: 10.1007/3-540-26573-2_24. [DOI] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Paré D. Prefrontal Control of the Amygdala. The Journal of Neuroscience. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Mao SC, Su CL, Gean PW. The role of prefrontal cortex CB1 receptors in the modulation of fear memory. Cerebral Cortex. 2009;19:165–175. doi: 10.1093/cercor/bhn075. [DOI] [PubMed] [Google Scholar]
- Little JP, Carter AG. Synaptic mechanisms underlying strong reciprocal connectivity between the medial prefrontal cortex and basolateral amygdala. The Journal of Neuroscience. 2013;33:15333–15342. doi: 10.1523/JNEUROSCI.2385-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser JD, Treede RD. The Kyoto protocol of IASP Basic Pain Terminology. Pain. 2008;137:473–477. doi: 10.1016/j.pain.2008.04.025. [DOI] [PubMed] [Google Scholar]
- Lomazzo E, Bindila L, Remmers F, Lerner R, Schwitter C, Hoheisel U, Lutz B. Therapeutic Potential of Inhibitors of Endocannabinoid Degradation for the Treatment of Stress-Related Hyperalgesia in an Animal Model of Chronic Pain. Neuropsychopharmacology. 2015;40:488–501. doi: 10.1038/npp.2014.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li WW, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nature Chemical Biology. 2009a;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2009b;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. Journal of Pharmacology and Experimental Therapeutics. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol. 2008:435–477. doi: 10.1007/978-3-540-74805-2_14. [DOI] [PubMed] [Google Scholar]
- Madasu MK, Okine BN, Olango WM, Rea K, Lenihan R, Roche M, Finn DP. Genotype-dependent responsivity to inflammatory pain: A role for TRPV1 in the periaqueductal grey. Pharmacological Research. 2016;113(Part A):44–54. doi: 10.1016/j.phrs.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Maione S, Bisogno T, de Novellis V, Palazzo E, Cristino L, Valenti M, Petrosino S, Guglielmotti V, Rossi F, Di Marzo V. Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. Journal of Pharmacology and Experimental Therapeutics. 2006;316:969–982. doi: 10.1124/jpet.105.093286. [DOI] [PubMed] [Google Scholar]
- Maione S, Costa B, Piscitelli F, Morera E, De Chiaro M, Comelli F, Boccella S, Guida F, Verde R, Ortar G, Di Marzo V. Piperazinyl carbamate fatty acid amide hydrolase inhibitors and transient receptor potential channel modulators as “dual-target” analgesics. Pharmacological Research. 2013;76:98–105. doi: 10.1016/j.phrs.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Maione S, De Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E, Rossi FS, Woodward DF, Di Marzo V. Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol. 2007;150:766–781. doi: 10.1038/sj.bjp.0707145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek N, Kostrzewa M, Makuch W, Pajak A, Kucharczyk M, Piscitelli F, Przewlocka B, Di Marzo V, Starowicz K. The multiplicity of spinal AA-5-HT anti-nociceptive action in a rat model of neuropathic pain. Pharmacological Research. 2016 doi: 10.1016/j.phrs.2016.06.012. [DOI] [PubMed] [Google Scholar]
- Malek N, Mrugala M, Makuch W, Kolosowska N, Przewlocka B, Binkowski M, Czaja M, Morera E, Di Marzo V, Starowicz K. A multi-target approach for pain treatment - dual inhibition of fatty acid amide hydrolase and TRPV1 in a rat model of osteoarthritis. Pain. 2015 doi: 10.1097/j.pain.0000000000000132. [DOI] [PubMed] [Google Scholar]
- Marek GJ, Zhang C. Activation of metabotropic glutamate 5 (mGlu5) receptors induces spontaneous excitatory synaptic currents in layer V pyramidal cells of the rat prefrontal cortex. Neuroscience Letters. 2008;442:239–243. doi: 10.1016/j.neulet.2008.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek R, Strobel C, Bredy TW, Sah P. The amygdala and medial prefrontal cortex: partners in the fear circuit. J Physiol. 2013;591:2381–2391. doi: 10.1113/jphysiol.2012.248575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SSJ, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Moller T, Mackie K, Manzoni OJ, Cravatt BF, Stella N. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nature Neuroscience. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. European Journal of Neuroscience. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Coffin PO, Attias E, Balinsky M, Tsou K, Walker JM. Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Research. 1999;822:237–242. doi: 10.1016/s0006-8993(98)01368-7. [DOI] [PubMed] [Google Scholar]
- Mayer EA, Berman S, Suyenobu B, Labus J, Mandelkern MA, Naliboff BD, Chang L. Differences in brain responses to visceral pain between patients with irritable bowel syndrome and ulcerative colitis. Pain. 2005;115:398–409. doi: 10.1016/j.pain.2005.03.023. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Cortical pathways to the mammalian amygdala. Progress in Neurobiology. 1998;55:257–332. doi: 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- Meagher MW, Arnau RC, Rhudy JL. Pain and emotion: effects of affective picture modulation. Psychosom Med. 2001;63:79–90. doi: 10.1097/00006842-200101000-00010. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Gaoni Y. The absolute configuration of delta-1-tetrahydrocannabinol, the major active constituent of hashish. Tetrahedron Letters. 1967;12:1109–1111. doi: 10.1016/s0040-4039(00)90646-4. [DOI] [PubMed] [Google Scholar]
- Metz AE, Yau HJ, Centeno MV, Apkarian AV, Martina M. Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2423–2428. doi: 10.1073/pnas.0809897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. The induction of pain: an integrative review. Progress in Neurobiology. 1999;57:1–164. doi: 10.1016/s0301-0082(98)00048-3. [DOI] [PubMed] [Google Scholar]
- Moore N. Lessons from the fatal French study BIA- BMJ. 2016;353:10–2474. doi: 10.1136/bmj.i2727. [DOI] [PubMed] [Google Scholar]
- Morena M, Patel S, Bains JS, Hill MN. Neurobiological Interactions Between Stress and the Endocannabinoid System. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty O, McGuire BE, Finn DP. The effect of pain on cognitive function: A review of clinical and preclinical research. Progress in Neurobiology. 2011;93:385–404. doi: 10.1016/j.pneurobio.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Muly EC, Maddox M, Smith Y. Distribution of mGluR1alpha and mGluR5 immunolabeling in primate prefrontal cortex. J Comp Neurol. 2003;467:521–535. doi: 10.1002/cne.10937. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abushaar M. Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Nahin RL. Estimates of Pain Prevalence and Severity in Adults: United States, 2012. The Journal of Pain. 2015;16:769–780. doi: 10.1016/j.jpain.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navia-Paldanius D, Aaltonen N, Lehtonen M, Savinainen JR, Taschler U, Radner FPW, Zimmermann R, Laitinen JT. Increased tonic cannabinoid CB1R activity and brain region-specific desensitization of CB1R Gi/o signaling axis in mice with global genetic knockout of monoacylglycerol lipase. European Journal of Pharmaceutical Sciences. 2015 doi: 10.1016/j.ejps.2015.06.005. [DOI] [PubMed] [Google Scholar]
- Neugebauer V. Amygdala pain mechanisms. Handb Exp Pharmacol. 2015;227:261–284. doi: 10.1007/978-3-662-46450-2_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Han JS, Adwanikar H, Fu Y, Ji G. Techniques for assessing knee joint pain in arthritis. Molecular Pain. 2007;3:8–8. doi: 10.1186/1744-8069-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis MJ, Cognetta AB, Chang JW, Buczynski MW, Parsons LH, Byrne F, Burston JJ, Chapman V, Cravatt BF. Evaluation of NHS Carbamates as a Potent and Selective Class of Endocannabinoid Hydrolase Inhibitors. ACS Chemical Neuroscience. 2013a doi: 10.1021/cn400116z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis MJ, Johnson DS, Ballard TE, Stiff C, Cravatt BF. O-Hydroxyacetamide Carbamates as a Highly Potent and Selective Class of Endocannabinoid Hydrolase Inhibitors. ACS Chemical Neuroscience. 2013b;3:418–426. doi: 10.1021/cn200089j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyilas R, Dudok B, Urban GM, Mackie K, Watanabe M, Cravatt BF, Freund TF, Katona I. Enzymatic Machinery for Endocannabinoid Biosynthesis Associated with Calcium Stores in Glutamatergic Axon Terminals. The Journal of Neuroscience. 2008;28:1058–1063. doi: 10.1523/JNEUROSCI.5102-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyilas R, Gregg LC, Mackie K, Watanabe M, Zimmer A, Hohmann AG, Katona I. Molecular architecture of endocannabinoid signaling at nociceptive synapses mediating analgesia. European Journal of Neuroscience. 2009;29:1964–1978. doi: 10.1111/j.1460-9568.2009.06751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan SE. An update on PPAR activation by cannabinoids. Br J Pharmacol. 2016;173:1899–1910. doi: 10.1111/bph.13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okine BN, Gaspar JC, Madasu MK, Olango WM, Harhen B, Roche M, Finn DP. Characterisation of peroxisome proliferator-activated receptor signalling in the midbrain periaqueductal grey of rats genetically prone to heightened stress, negative affect and hyperalgesia. Brain Research. 2017;1657:185–192. doi: 10.1016/j.brainres.2016.11.022. [DOI] [PubMed] [Google Scholar]
- Okine BN, Norris LM, Woodhams S, Burston J, Patel A, Alexander SP, Barrett DA, Kendall DA, Bennett AJ, Chapman V. Lack of effect of chronic pre-treatment with the FAAH inhibitor URB597 on inflammatory pain behaviour: evidence for plastic changes in the endocannabinoid system. Br J Pharmacol. 2012;167:627–640. doi: 10.1111/j.1476-5381.2012.02028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olango WM, Finn DP. Neurobiology of stress-induced hyperalgesia. Current Topics in Behavioural Neurosciences. 2014;20:251–280. doi: 10.1007/7854_2014_302. [DOI] [PubMed] [Google Scholar]
- Olango WM, Roche M, Ford GK, Harhen B, Finn DP. The endocannabinoid system in the rat dorsolateral periaqueductal grey mediates fear-conditioned analgesia and controls fear expression in the presence of nocicpetive tone. Br J Pharmacol. 2012 doi: 10.1111/j.1476-5381.2011.01478.x. no-no. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini CA, Maren S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev. 2012;36:1773–1802. doi: 10.1016/j.neubiorev.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona FA, Ferreira J, Menezes de Lima O, Duarte FS, Bento AF, Forner S, Villarinho JG, Bellocchio L, Wotjak CT, Lerner R, Monory K, Lutz B, Canetti C, Matias I, Calixto JB, Marsicano G, Guimarães MZP, Takahashi RN. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proceedings of the National Academy of Sciences. 2012;109:21134–21139. doi: 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiological Reviews. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Cravatt BF, Hillard CJ. Synergistic interactions between cannabinoids and environmental stress in the activation of the central amygdala. Neuropsychopharmacology. 2005;30:497–507. doi: 10.1038/sj.npp.1300535. [DOI] [PubMed] [Google Scholar]
- Pattij T, Wiskerke J, Schoffelmeer ANM. Cannabinoid modulation of executive functions. European Journal of Pharmacology. 2008;585:458–463. doi: 10.1016/j.ejphar.2008.02.099. [DOI] [PubMed] [Google Scholar]
- Pawsey S, Wood M, Browne H, Donaldson K, Christie M, Warrington S. Safety, Tolerability and Pharmacokinetics of FAAH Inhibitor V158866: A Double-Blind, Randomised, Placebo-Controlled Phase I Study in Healthy Volunteers. Drugs in R&D. 2016;16:181–191. doi: 10.1007/s40268-016-0127-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernia-Andrade AJ, Kato A, Witschi R, Nyilas R, Katona I, Freund TF, Watanabe M, Filitz J, Koppert W, Schuttler J, Ji G, Neugebauer V, Marsicano G, Lutz B, Vanegas H, Zeilhofer HU. Spinal Endocannabinoids and CB1 Receptors Mediate C-Fiber-Induced Heterosynaptic Pain Sensitization. Science. 2009;325:760–764. doi: 10.1126/science.1171870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinard CR, Mascagni F, McDonald AJ. Medial prefrontal cortical innervation of the intercalated nuclear region of the amygdala. Neuroscience. 2012;205:112–124. doi: 10.1016/j.neuroscience.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG, Ross RA. Allosteric Modulation of the Cannabinoid CB1 Receptor. Molecular Pharmacology. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Rademacher DJ, Meier SE, Shi L, Vanessa Ho WS, Jarrahian A, Hillard CJ. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology. 2008;54:108–116. doi: 10.1016/j.neuropharm.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Rea K, Olango WM, Harhen B, Kerr DM, Galligan R, Fitzgerald S, Moore M, Roche M, Finn DP. Evidence for a role of GABAergic and glutamatergic signalling in the basolateral amygdala in endocannabinoid-mediated fear-conditioned analgesia in rats. Pain. 2013;154:576–585. doi: 10.1016/j.pain.2012.12.021. [DOI] [PubMed] [Google Scholar]
- Rea K, Olango WM, Okine BN, Madasu MK, McGuire IC, Coyle K, Harhen B, Roche M, Finn DP. Impaired endocannabinoid signalling in the rostral ventromedial medulla underpins genotype-dependent hyper-responsivity to noxious stimuli. Pain. 2014;155:69–79. doi: 10.1016/j.pain.2013.09.012. [DOI] [PubMed] [Google Scholar]
- Ren W, Kiritoshi T, Gregoire S, Ji G, Guerrini R, Calo G, Neugebauer V. Neuropeptide S: a novel regulator of pain-related amygdala plasticity and behaviors. J Neurophysiol. 2013;110:1765–1781. doi: 10.1152/jn.00874.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhudy JL, Meagher MW. Fear and anxiety: divergent effects on human pain thresholds. Pain. 2000;84:65–75. doi: 10.1016/S0304-3959(99)00183-9. [DOI] [PubMed] [Google Scholar]
- Rhudy JL, Meagher MW. Noise Stress and Human Pain Thresholds: Divergent Effects in Men and Women. The Journal of Pain. 2001;2:57–64. doi: 10.1054/jpai.2000.19947. [DOI] [PubMed] [Google Scholar]
- Rhudy JL, Meagher MW. Individual differences in the emotional reaction to shock determine whether hypoalgesia is observed. Pain Medicine. 2003;4:244–256. doi: 10.1046/j.1526-4637.2003.03028.x. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Aanonsen L, Hargreaves KM. Antihyperalgesic effects of spinal cannabinoids. European Journal of Pharmacology. 1998;345:145–153. doi: 10.1016/s0014-2999(97)01621-x. [DOI] [PubMed] [Google Scholar]
- Rivera P, Arrabal S, Cifuentes M, Grondona JM, Perez-Martin M, Rubio L, Vargas A, Serrano A, Pavon FJ, Suarez J, Rodriguez de Fonseca F. Localization of the cannabinoid CB1 receptor and the 2-AG synthesizing (DAGLalpha) and degrading (MAGL, FAAH) enzymes in cells expressing the Ca(2+)-binding proteins calbindin, calretinin, and parvalbumin in the adult rat hippocampus. Front Neuroanat. 2014;8:56. doi: 10.3389/fnana.2014.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche M, Johnston P, Mhuircheartaigh ON, Olango WM, Mackie K, Finn DP. Effects of intra-basolateral amygdala administration of rimonabant on nociceptive behaviour and neuronal activity in the presence or absence of contextual fear. European Journal of Pain. 2010;14:487–495. doi: 10.1016/j.ejpain.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Roche M, O’Connor E, Diskin C, Finn DP. The effect of CB1 receptor antagonism in the right basolateral amygdala on conditioned fear and associated analgesia in rats. European Journal of Neuroscience. 2007;26:2643–2653. doi: 10.1111/j.1460-9568.2007.05861.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Molecular Pharmacology. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003;140:790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino T, Zamberletti E, Parolaro D. Endocannabinoids and Mental Disorders. In: Pertwee RG, editor. Endocannabinoids. Springer International Publishing; Cham: 2015. pp. 261–283. [DOI] [PubMed] [Google Scholar]
- Russo R, LoVerme J, La Rana G, Compton TR, Parrott JA, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. The Fatty Acid Amide Hydrolase Inhibitor URB597 (Cyclohexylcarbamic Acid 3′-Carbamoylbiphenyl-3-yl Ester) Reduces Neuropathic Pain after Oral Administration in Mice. Journal of Pharmacology and Experimental Therapeutics. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- Sagar D, Gaw AG, Okine B, Woodhams S, Wong A, Kendall D, Chapman V. Dynamic regulation of the endocannabinoid system: implications for analgesia. Molecular Pain. 2009;5:59. doi: 10.1186/1744-8069-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar DR, Burston JJ, Woodhams SG, Chapman V. Dynamic changes to the endocannabinoid system in models of chronic pain. Philosophical Transactions of the Royal Society B: Biological Sciences. 2012;367:3300–3311. doi: 10.1098/rstb.2011.0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar DR, Jhaveri M, Richardson D, Gray RA, De Lago E, Fernandez-Ruiz J, Barrett D, Kendall D, Chapman V. Endocannabinoid regulation of spinal nociceptive processing in a model of neuropathic pain. European Journal of Neuroscience. 2010;31:8. doi: 10.1111/j.1460-9568.2010.07162.x. [DOI] [PubMed] [Google Scholar]
- Sagar DR, Kendall DA, Chapman V. Inhibition of fatty acid amide hydrolase produces PPAR-alpha-mediated analgesia in a rat model of inflammatory pain. Br J Pharmacol. 2008 doi: 10.1038/bjp.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasso O, Migliore M, Habrant D, Armirotti A, Albani C, Summa M, Moreno-Sanz G, Scarpelli R, Piomelli D. Multitarget fatty acid amide hydrolase/cyclooxygenase blockade suppresses intestinal inflammation and protects against nonsteroidal anti-inflammatory drug-dependent gastrointestinal damage. The FASEB Journal. 2015a doi: 10.1096/fj.15-270637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasso O, Wagner K, Morisseau C, Inceoglu B, Hammock BD, Piomelli D. Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive. Pharmacological Research. 2015b;97:7–15. doi: 10.1016/j.phrs.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg J, Carlson B, Ramesh D, Abdullah R, Long J, Cravatt B, Lichtman A. Inhibitors of Endocannabinoid-Metabolizing Enzymes Reduce Precipitated Withdrawal Responses in THC-Dependent Mice. The AAPS Journal. 2009;11:342–352. doi: 10.1208/s12248-009-9110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu Q-s, Lichtman AH, Cravatt BF. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nature Neuroscience. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Hamza M, Ates M, Gühring H. Flurbiprofen inhibits capsaicin induced calcitonin gene related peptide release from rat spinal cord via an endocannabinoid dependent mechanism. Neuroscience Letters. 2003;338:99–102. doi: 10.1016/s0304-3940(02)01366-6. [DOI] [PubMed] [Google Scholar]
- Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Grundemann J, Fadok JP, Muller C, Letzkus JJ, Luthi A. Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron. 2014;81:428–437. doi: 10.1016/j.neuron.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Sepulveda-Orengo MT, Lopez AV, Soler-Cedeno O, Porter JT. Fear extinction induces mGluR5-mediated synaptic and intrinsic plasticity in infralimbic neurons. The Journal of Neuroscience. 2013;33:7184–7193. doi: 10.1523/JNEUROSCI.5198-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Yang XJ, Qian W, Hou XH. The role of peripheral cannabinoid receptors type 1 in rats with visceral hypersensitivity induced by chronic restraint stress. J Neurogastroenterol Motil. 2010;16:281–290. doi: 10.5056/jnm.2010.16.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Qi WJ, Gao G, Wang JY, Luo F. Increased thermal and mechanical nociceptive thresholds in rats with depressive-like behaviors. Brain Research. 2010;1353:225–233. doi: 10.1016/j.brainres.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slivicki R, Kulkarni P, Mackie K, Thakur G, A H. Positive allosteric modulation of cannabinoid CB1 receptor signaling suppresses chemotherapy-induced peripheral neuropathy. Society for Neuroscience Annual Meeting; San Diego, CA, USA. 2016. [Google Scholar]
- Sotres-Bayon F, Quirk GJ. Prefrontal control of fear: more than just extinction. Curr Opin Neurobiol. 2010;20:231–235. doi: 10.1016/j.conb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starowicz K, Di Marzo V. Non-psychotropic analgesic drugs from the endocannabinoid system: “Magic bullet” or “multiple-target” strategies? European Journal of Pharmacology. 2013 doi: 10.1016/j.ejphar.2013.01.075. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Sun H, Neugebauer V. mGluR1, but not mGluR5, activates feed-forward inhibition in the medial prefrontal cortex to impair decision making. J Neurophysiol. 2011;106:960–973. doi: 10.1152/jn.00762.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suplita RL, Eisenstein SA, Neely MH, Moise AM, Hohmann AG. Cross-sensitization and cross-tolerance between exogenous cannabinoid antinociception and endocannabinoid-mediated stress-induced analgesia. Neuropharmacology. 2008;54:161–171. doi: 10.1016/j.neuropharm.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suplita RL, Farthing JN, Gutierrez T, Hohmann AG. Inhibition of fatty-acid amide hydrolase enhances cannabinoid stress-induced analgesia: Sites of action in the dorsolateral periaqueductal gray and rostral ventromedial medulla. Neuropharmacology. 2005;49:1201–1209. doi: 10.1016/j.neuropharm.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Suplita RL, Gutierrez T, Fegley D, Piomelli D, Hohmann AG. Endocannabinoids at the spinal level regulate, but do not mediate, nonopioid stress-induced analgesia. Neuropharmacology. 2006;50:372–379. doi: 10.1016/j.neuropharm.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Swanson DM, Dubin AE, Shah C, Nasser N, Chang L, Dax SL, Jetter M, Breitenbucher JG, Liu C, Mazur C, Lord B, Gonzales L, Hoey K, Rizzolio M, Bogenstaetter M, Codd EE, Lee DH, Zhang SP, Chaplan SR, Carruthers NI. Identification and Biological Evaluation of 4-(3-Trifluoromethylpyridin-2-yl)piperazine-1-carboxylic Acid (5-Trifluoromethylpyridin-2-yl)amide, a High Affinity TRPV1 (VR1) Vanilloid Receptor Antagonist. Journal of Medicinal Chemistry. 2004;48:1857–1872. doi: 10.1021/jm0495071. [DOI] [PubMed] [Google Scholar]
- Tappe-Theodor A, Agarwal N, Katona I, Rubino T, Martini L, Swiercz J, Mackie K, Monyer H, Parolaro D, Whistler J, Kuner T, Kuner R. A Molecular Basis of Analgesic Tolerance to Cannabinoids. The Journal of Neuroscience. 2007;27:4165–4177. doi: 10.1523/JNEUROSCI.5648-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Valverde O, Ledent C, Beslot F, Parmentier M, Roques BP. Reduction of stress-induced analgesia but not of exogenous opioid effects in mice lacking CB1 receptors. European Journal of Neuroscience. 2000;12:533–539. doi: 10.1046/j.1460-9568.2000.00929.x. [DOI] [PubMed] [Google Scholar]
- Vandevoorde S, Jonsson KO, Labar G, Persson E, Lambert DM, Fowler CJ. Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br J Pharmacol. 2007;150:186–191. doi: 10.1038/sj.bjp.0706971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardeh D, Mannion RJ, Woolf CJ. Toward a Mechanism-Based Approach to Pain Diagnosis. The Journal of Pain. 2016;17:T50–T69. doi: 10.1016/j.jpain.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardeh D, Wang D, Costigan M, Lazarus M, Saper CB, Woolf CJ, Fitzgerald GA, Samad TA. COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. Journal of Clinical Investigation. 2009;119:287–294. doi: 10.1172/JCI37098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM, Hohmann AG. Cannabinoid mechanisms of pain suppression. Handb Exp Pharmacol. 2005:509–554. doi: 10.1007/3-540-26573-2_17. [DOI] [PubMed] [Google Scholar]
- Walker JM, Huang SM, Strangman NM, Tsou K, Sanudo-Pena MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12198–12203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M, Huang S. Cannabinoid Analgesia. Pharmacology & Therapeutics. 2002;95:127–135. doi: 10.1016/s0163-7258(02)00252-8. [DOI] [PubMed] [Google Scholar]
- Wang GQ, Cen C, Li C, Cao S, Wang N, Zhou Z, Liu XM, Xu Y, Tian NX, Zhang Y, Wang J, Wang LP, Wang Y. Deactivation of excitatory neurons in the prelimbic cortex via Cdk5 promotes pain sensation and anxiety. Nat Commun. 2015;6:7660. doi: 10.1038/ncomms8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedzony K, Chocyk A. Cannabinoid CB1 receptors in rat medial prefrontal cortex are colocalized with calbindin- but not parvalbumin- and calretinin-positive GABA-ergic neurons. Pharmacological Reports. 2009;61:1000–1007. doi: 10.1016/s1734-1140(09)70161-6. [DOI] [PubMed] [Google Scholar]
- Wiech K, Tracey I. The influence of negative emotions on pain: Behavioral effects and neural mechanisms. NeuroImage. 2009;47:987–994. doi: 10.1016/j.neuroimage.2009.05.059. [DOI] [PubMed] [Google Scholar]
- Wilkerson JL, Ghosh S, Mustafa M, Abdullah RA, Niphakis MJ, Cabrera R, Maldonado RL, Cravatt BF, Lichtman AH. The endocannabinoid hydrolysis inhibitor SA-57 Intrinsic antinociceptive effects, augmented morphine-induced antinociception, and attenuated heroin seeking behavior in mice. Neuropharmacology. 2016a doi: 10.1016/j.neuropharm.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JL, Niphakis MJ, Grim TW, Mustafa MA, Abdullah RA, Poklis JL, Dewey WL, Akbarali HI, Banks ML, Wise LE, Cravatt BF, Lichtman AH. The selective monoacylglycerol lipase inhibitor MJN 110 produces opioid sparing effects in a mouse neuropathic pain model. Journal of Pharmacology and Experimental Therapeutics. 2016b doi: 10.1124/jpet.115.229971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AW, Clayton NM, Medhurst SJ, Bountra C, Chessell IP. The FAAH inhibitor URB597 reverses inflammatory pain through a CB1 receptor mediated mechanism. British Pharmacological Society Winter Meeting; Newcastle. 2005. [Google Scholar]
- Woodhams SG, Sagar DR, Burston JJ, Chapman V. The role of the endocannabinoid system in pain. Handb Exp Pharmacol. 2015;227:119–143. doi: 10.1007/978-3-662-46450-2_7. [DOI] [PubMed] [Google Scholar]
- Woodhams SG, Wong A, Barrett DA, Bennett AJ, Chapman V, Alexander SPH. Spinal administration of the monoacylglycerol lipase inhibitor JZL184 produces robust inhibitory effects on nociceptive processing and the development of central sensitization in the rat. Br J Pharmacol. 2012;167:1609–1619. doi: 10.1111/j.1476-5381.2012.02179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhu Y, Contractor A, Heinemann SF. mGluR5 has a critical role in inhibitory learning. The Journal of Neuroscience. 2009;29:3676–3684. doi: 10.1523/JNEUROSCI.5716-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU. Spinal cannabinoids–a double-edged sword? (Commentary on Zhang et al.) European Journal of Neuroscience. 2010;31:223–224. doi: 10.1111/j.1460-9568.2010.07125.x. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gadotti VM, Chen L, Souza IA, Stemkowski PL, Zamponi GW. Role of Prelimbic GABAergic Circuits in Sensory and Emotional Aspects of Neuropathic Pain. Cell Rep. 2015;12:752–759. doi: 10.1016/j.celrep.2015.07.001. [DOI] [PubMed] [Google Scholar]