

Abstract
The fundamental pathology in Alzheimer’s disease (AD) is neuronal dysfunction leading to cognitive impairment. The amyloid-β peptide (Aβ), derived from amyloid precursor protein, is one driver of AD, but how it leads to neuronal dysfunction is not established. In this Review, I discuss the complexity of AD and possible cause-and-effect relationships between Aβ and the vascular and hemostatic systems. AD can be considered a multifactorial syndrome with various contributing pathological mechanisms. Therefore, as is routinely done with cancer, it will be important to classify patients with respect to their disease signature so that specific pathologies, including vascular pathways, can be therapeutically targeted.
Introduction
Alzheimer’s disease (AD) is the most prevalent form of dementia, affecting approximately 5.5 million Americans. With an aging population worldwide, it is predicted to be a public health crisis in the coming decades. In spite of extensive preclinical and clinical efforts, little progress has been made in preventing or reversing this disease. A better understanding of AD requires reconceptualizing the disorder as an amalgam of dysfunctions rather than one pathology. An analogous evolution has occurred in cancer biology; decades ago, the search for a “magic bullet” for cancer treatment suggested that cancer was one disease and that it would be possible to identify a single drug to cure all forms. With today’s advanced knowledge of the many mechanisms that lead to cancer, that original concept is very dated. Yet, researchers today still discuss “a” cure for AD, implying that a single pathological pathway is responsible for all cases. For example, the tag line on a 2016 cover of Time magazine read, “The Alzheimer’s Pill.”
It is becoming obvious that AD, like cancer, is a complex disease with multiple pathogenic mechanisms. One pathway that has been largely overlooked is vascular dysregulation. For example, a relatively recent and highly cited hypothetical temporal ordering of AD pathologies did not include vascular pathology in the possible contributors to the disease (1). Accumulating evidence, detailed in this review, indicates that vascular dysregulation plays a major role in cognitive decline. Undoubtedly, attacking the disease successfully will involve identifying and targeting various mechanisms, as has been done in the revolution in cancer treatment.
Multiple mechanisms can contribute to AD pathology
In cancer, the fundamental pathology is unregulated cell division. However, this aspect by itself is not necessarily lethal. Other mechanisms — for example, immune system evasion and metastasis — can compound too much cell division and exacerbate the morbidity of the disease. Cancer treatments have been developed to counteract these ancillary pathways, and these treatments can improve patient outcome. One can envisage a similar situation for AD. The fundamental pathology is neuronal dysfunction causing cognitive decline, and this pathway can be accelerated by many other abnormalities such as defects in autophagy (2), synaptic toxicity (3), and oxidative stress/mitochondrial dysfunction (4). Of all the many possible contributors, inflammation (5) and vascular abnormalities (6, 7) appear to be especially significant (Figure 1 and see below).
Figure 1. Multiple pathogenic pathways contribute to and provide therapeutic targets for AD.

While neuronal dysfunction is the defining pathology underlying cognitive decline in AD, this disease is likely the product of multiple pathogenic mechanisms and might benefit from combination therapy (128–130). Increasing evidence implicates vascular dysfunction in AD pathogenesis. Aβ plaques and tau tangles drive neuronal dysfunction and neuroinflammation through mechanisms that are not fully established. Aβ is also implicated in vascular pathology, having been shown to interact with fibrin(ogen) and the contact system. The prevalence of vascular dysfunction in patients exhibiting cognitive decline suggests that combining existing treatment strategies targeting neuronal dysfunction (e.g., cholinesterase inhibitors) and vascular dysfunction (e.g., antiinflammatory, antihypertensive, or anticoagulant medications) may advance AD treatment efficacy.
Could vascular mechanisms benefit AD diagnosis or treatment?
If ancillary mechanisms can contribute to AD, one consideration is how to determine the relevance of these pathways in individual patients, especially if the mechanisms are not specific to AD. Again, cancer provides a useful analogy. Estrogen and progesterone receptor expression is not specific to breast cancer, and also occurs in normal tissues and other cancers. Nevertheless, in patients with breast cancer, hormone receptor expression predicts response to antiestrogen therapy such as tamoxifen and such therapy improves patient outcome. Likewise, patients with cognitive impairment could be tested for vascular abnormalities and inflammation. Those that exhibit these ancillary pathologies could then be treated for those conditions. As with cancer, it seems likely that this approach would benefit this subset of patients.
Is there a connection between vascular pathology and AD?
The classical pathological hallmarks of AD are amyloid-β peptide (Aβ) plaques, tau tangles, neuroinflammation, and neuronal loss (8). The Aβ deposition can be in the brain parenchyma or in and around cerebral blood vessels, a condition known as cerebral amyloid angiopathy (CAA), discussed below (9). Less discussed is that AD is very often associated with cerebrovascular abnormalities (6, 10–13). These cerebral pathologies include microinfarcts, hemorrhage, decreased cerebral blood flow, small vessel disease, and white matter abnormalities (14, 15). Three recent studies of vascular disease in AD autopsy samples showed that concurrent vascular disease is very common in AD and strongly correlates with cognitive dysfunction (16–18).
Reinforcing the concept that the vascular system influences AD, multiple studies have reported that exercise, which improves cerebrovascular health, can decrease the risk and/or delay progression of dementia. Benefits of increased physical activity include improved memory performance and reduced hippocampal atrophy (19–21), increased gray matter volume and production of neurotrophic factors (22), lower risk of mortality (23, 24), and reduced risk of AD (25). However, this association is complex (26), as exercise could also have nonvascular benefits, and other studies have found no improvement in risk of dementia with exercise (27). Mechanisms by which exercise could influence AD are discussed below.
Another link between AD and the vascular system is CAA, the deposition of Aβ in and around blood vessels of the brain (9), which affects 80% to 95% of AD patients (15, 16). CAA can cause blood vessel occlusion, microinfarcts, ischemia, microbleeds, and inflammation, conditions that can weaken the blood vessel wall and cause life-threatening hemorrhage (8, 15, 28). All of these conditions could contribute to neuronal death that is associated with AD progression.
AD and vascular dysfunction: independent or causal pathologies?
It is clear that vascular dysfunction can itself lead to cognitive decline. However, the coexistence of AD and cerebrovascular pathology prompts questions of whether these conditions are always independent comorbidities, as they are both more prevalent in the aged population. If so, there could be synergistic effects on brain function. Alternatively, there could be a mechanistic link between AD and vascular pathology (29, 30).
Early vascular pathologies in late-onset AD patients
A recent study used a multifactorial, data-driven analysis to examine various pathologies in 1,171 healthy and late-onset AD (LOAD) subjects and assign a temporal ordering of disease progression (31). The conclusion of this study was that vascular dysregulation, measured by arterial spin labeling of cerebral blood flow (CBF), is the earliest and strongest brain pathology associated with LOAD. A tentative ordering of abnormalities was (a) initial vascular dysregulation, (b) Aβ deposition, (c) metabolic dysfunction, (d) functional impairment, and (e) gray matter atrophy. Given thresholds for detection, the exact order of the pathologies is not certain, but these analyses strongly “suggest that intra-brain vascular dysregulation is an early pathological event during disease development” (31).
Vascular pathologies appear early in early-onset AD patients
An analysis of early-onset AD (EOAD) patients who harbor autosomal-dominant mutations provides strong evidence for a connection between AD and vascular pathology (32). EOAD-associated mutations occur in amyloid precursor protein (APP), from which the Aβ peptide is derived, or in γ-secretase, which helps excise Aβ from APP (33). These mutations are virtually fully penetrant, and by analyzing DNA sequences, one can identify at the time of birth which patients will develop EOAD at a predictable age decades later. A comparison of mutation carriers and noncarriers reveals that the first abnormalities detected in these EOAD patients are alterations in Aβ levels, which occur approximately 30 years before the onset of symptoms (estimated year of onset, EYO). The next pathology to emerge is white matter hyperintensities (WMHs), which are abnormalities observed during brain imaging. In carrier brains, WMH abnormalities were evident in general 6.6 years before EYO and in the occipital lobes as early as 22 years before EYO (32). This study did not measure CBF, so it cannot be compared to the results for LOAD patients discussed above.
WMHs are part of the spectrum of small vessel cerebrovascular disease (34, 35), including ischemic and hemorrhagic stroke, microbleeds, brain atrophy (36), chronic hypoperfusion (37), and an increase in blood-brain barrier (BBB) permeability, which causes fluid to leak into the surrounding brain tissue (38). What is notable is that these lesions predict the clinical outcome of AD as well as or more reliably than established markers such as hippocampal volume and atrophy (39, 40). In the EOAD study (32), mutation carriers and noncarriers were relatively young, virtually identical demographically, and at equal risk for inheriting an autosomal-dominant mutation. Thus, these findings provide strong evidence that vascular dysfunction does not reflect comorbidity or an independent pathophysiology, but rather that vascular dysfunction and Aβ pathology are causally related (32). In recent years, there has been evidence that WMHs are also associated with LOAD (41–43). In particular, WMH volume and amyloid pathology are linked in LOAD (42–44). However, it should be noted that one study found no association between WMHs and AD pathology (45).
Another link between AD and cerebrovascular disease stems from pathological outcomes in patients with autosomal-dominant mutations. Mutations in the Aβ sequence at residues 21–23 (for example, Flemish, Dutch, Iowa, and Italian mutations) are associated with massive CAA, leading to weakening of the vessel wall and frequent infarcts and hemorrhage (46, 47). Mutations in presenilin-1, a protein component of γ-secretase that helps release Aβ from APP, can also show severe cerebrovascular effects (48). All of this evidence indicates a connection between vascular pathology and AD.
Mechanistic links between AD and vascular pathology
Aβ interaction with fibrin.
Fibrin is the major protein component of blood clots and is critical for normal hemostasis (49). It is well established that in addition to its beneficial functions, persistent fibrin can lead to or exacerbate many pathological conditions, including atherosclerosis, rheumatoid arthritis, stroke, spinal cord injury, multiple sclerosis, muscular dystrophy, peripheral nerve regeneration, and even bacterial infection (50–52).
Postmortem AD patient brains have been analyzed with antibodies that do not distinguish between soluble fibrinogen and insoluble fibrin. These studies showed extensive fibrinogen/fibrin deposition in the brain (53–60), which in some cases was attributed to a leaky BBB. Further studies using extraction procedures that remove all soluble fibrinogen and staining with fibrin-specific antibodies showed that insoluble fibrin is greatly increased in AD patient brains compared with nondemented controls, with differences in some regions reaching 100-fold (61). The fibrin deposition often colocalized with parenchymal Aβ deposits and lysosome-associated membrane protein 1 (LAMP-1), which is upregulated in the human AD brain (62) and cerebrospinal fluid (CSF) (63) and enriched in dystrophic areas surrounding amyloid plaques in AD mice (64, 65) and human AD patient brains (62, 66). Fibrin is also found codeposited with Aβ in areas of CAA (53, 67).
High levels of fibrinogen in plasma increase the risk for dementia (68, 69), and fibrinogen in CSF (70–72) and plasma (73, 74) can serve as a useful biomarker to identify AD progression. In addition, fibrinogen has been proposed to be one of the few blood-based biomarkers that is specific for AD and does not apply to other brain disorders (75).
The association of brain fibrin and AD prompts the question of whether this deposition is a result of AD, or whether it contributes to the pathology. AD has been linked to the APP-derived Aβ peptide, a known driver of AD pathology (76–78). Aβ binds to fibrinogen and fibrin (79, 80), leading to blood clots that are structurally abnormal and harder to degrade in vitro and in vivo than normal clots (53, 80). Thus, fibrin clots formed in AD patients and mice might be persistent and cause vessel occlusion and neuroinflammation, which could contribute to neuronal death and other disease pathologies (Figure 2).
Figure 2. Possible influence of Aβ on fibrin deposition and AD pathology.
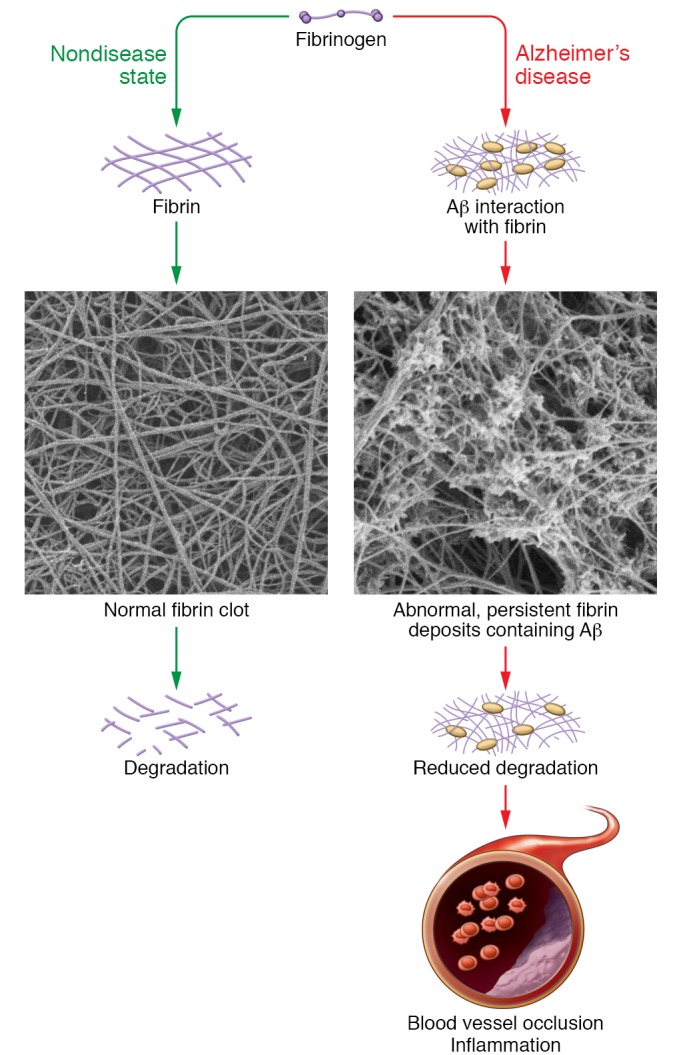
The interaction of Aβ with fibrinogen leads to increased formation of structurally abnormal fibrin clots that are resistant to degradation. This persistent fibrin and resulting predisposition towards blood vessel occlusion and inflammation could contribute to the neurodegeneration observed in AD. Images reproduced with permission from Cortes-Canteli et al. (53).
Data in humans and mice indicate that fibrin deposition in AD brains is not simply a comorbidity due to the aging population at risk, but is instead a driving factor of the disease. Studies in atrial fibrillation patients with and without warfarin treatment showed that anticoagulants can protect against dementia (81). Earlier work with dementia patients reached the same conclusion (82). In AD mice, anticoagulant treatment is protective (83–85) and injection of fibrinogen into the brain induces demyelination (86), reminiscent of the white matter abnormalities observed in AD patients discussed above. More specifically, in AD mice treatment with a small molecule that blocks the Aβ-fibrin(ogen) interaction significantly improves the course of disease (87) and reducing fibrinogen levels results in reduced pathology and better cognitive ability (53, 60, 61). Both of these studies substantiate a role for fibrin.
The mechanism by which fibrin accelerates neuronal degeneration remains unknown. There are two likely possibilities: (a) occlusion — fibrin clots are deposited in the vascular and perivascular space, resulting in reduced blood flow, increased Aβ accumulation due to binding to clots, and neuronal damage due to deprivation of oxygen and nutrients; and (b) inflammation — fibrin deposits drive a chronic inflammatory state that leads to cellular damage (50).
AD is associated with inflammation, which in some cases can be a beneficial response but in other cases can be toxic to cells (88, 89). AD patient brains have increased inflammation, mutations in immune-related molecules lead to an increased risk of AD, and nonsteroidal antiinflammatory drugs show some effects at reducing AD risk (5). Furthermore, AD mice that are incapable of mounting a proinflammatory response show improvement in pathology and cognition (90, 91). Since fibrin is a driver of inflammation, it could therefore contribute to the initiation of the inflammation observed in AD (88).
The contact system, intrinsic blood coagulation, and Aβ.
The contact system is initiated by activation of the serine protease factor XII (FXII). Once converted to its active serine protease form, FXIIa can launch both prothrombotic and proinflammatory pathways (92). Regarding the prothrombotic arm, FXIIa can activate factor XI (FXI), which leads to thrombin generation and fibrin formation via the intrinsic blood coagulation pathway (93). In the proinflammatory arm, FXII can activate plasma prekallikrein (PPK), which leads to the release of bradykinin via cleavage of high molecular weight kininogen (HK). Bradykinin is a potent nonapeptide vasodilator that can activate inflammatory processes (Figure 3 and ref. 94). Thus, the contact system can initiate vascular pathology and inflammation (92, 95), both of which have been implicated in AD, and could contribute to disease pathology.
Figure 3. The contact activation system.
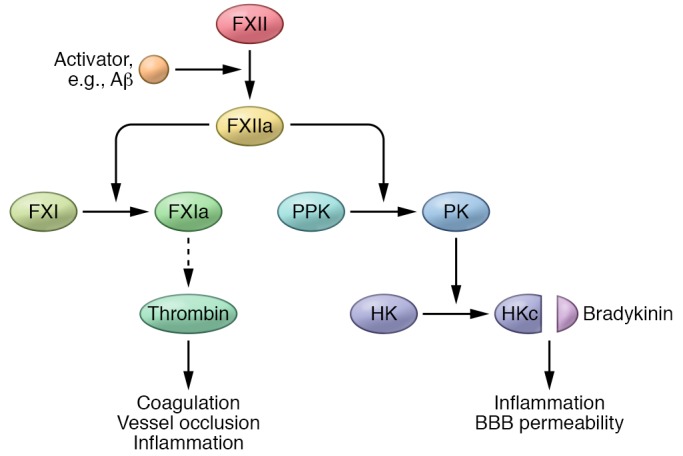
Aβ-mediated dysregulation or overactivation of the contact system could contribute to the coagulopathy and inflammation observed in AD. Aβ can trigger activation of factor XII (FXII) into FXIIa. FXIIa’s activation of FXI initiates the intrinsic coagulation system and fibrin formation, whereas its activation of plasma prekallikrein leads to inflammation. PPK, plasma prekallikrein; PK, plasma kallikrein; HK, high molecular weight kininogen; HKc, cleaved HK. a = activated form.
Many connections between FXII and AD have been reported, suggesting that this system may play a role in disease development. Aβ plaques contain FXII (96), the AD brain parenchyma exhibits higher plasma kallikrein (PK) activity (97), and AD patients have increased HK cleavage in their CSF (98). Consistent with a role for the contact system, AD patients have higher plasma levels of FXIIa and increased HK cleavage compared with nondemented individuals (99). Experiments with a mouse model of AD also show increased contact system activation (99, 100).
The contact system can be triggered by Aβ, which can activate FXII (101–104). This increase in FXIIa leads to elevated thrombin generation, kallikrein activity, and HK cleavage in AD patient plasma (100). Increased contact system activation is also observed in AD mouse model plasma and in plasma from wild-type mice injected intravenously with Aβ42. These results demonstrate that Aβ42-mediated contact system activation can occur in the AD circulation and suggest new pathogenic mechanisms, diagnostic tests, and therapies for AD.
Experiments in mice suggest that the contact system is a direct effector of AD pathology. Depletion of FXII using antisense oligonucleotide treatment ameliorates pathology in AD mice in early-stage disease (100). These results indicate that dysregulated contact system activation contributes to AD pathology.
Contact system activation could be used in conjunction with other diagnostic procedures such as PET and MRI imaging, CSF analysis, and/or cognitive testing to help stratify patients by their pathological profile and help guide therapy. Furthermore, a link between FXII activation and the pathogenesis of AD provides a possible novel approach to treatment since the contact system is an attractive target for therapy (105). Humans deficient in FXII and mice with knockout of the FXII, FXI, or Kng1 (encoding HK) gene all have normal hemostasis (95). However, deficiencies in the contact system protect mice from clotting after arterial injury and experimental cerebral ischemia (92, 106).
Based on these considerations, a promising therapeutic approach to slow disease progression without affecting normal hemostasis would be to block activation of the contact system. This approach would also block bradykinin release from HK and reduce inflammation. Thus, the contact system may reveal new targets to suppress both thrombotic and inflammatory contributions to AD progression. Positive results could be applied to AD patients rapidly; a small molecule inhibitor of PK, ecallantide, is already FDA approved for treatment of hereditary angioedema, a condition that results from excess contact system activation (107). Furthermore, an antibody inhibitor of PK (108) is slated for a phase 3 trial and possible FDA approval by 2018. Some of these reagents might be useful for the treatment of AD in the future.
Could vascular pathology trigger AD?
The above discussion concentrates on mechanisms by which Aβ can drive vascular pathology. The reverse scenario is also a possibility, i.e., that vascular pathology could accelerate AD.
AD ⇄ Vascular Dysfunction
Clearance of Aβ is critical to keep its concentration low in the brain and the cerebrovascular circulation, and decreased clearance may be a major cause of increased Aβ deposition in the AD brain (109, 110). Mechanisms that remove Aβ from the brain include BBB transport and movement from the CSF and parenchyma into the blood (111–114). If the cerebrovascular system were compromised, it could impede removal of Aβ and lead to increased concentration in the brain. Thus, one could envisage a vicious cycle whereby Aβ negatively affects the circulation, which in turn reduces clearance of Aβ and increases its toxic effects.
Another possible contribution of vascular pathology leading to AD could be BBB breakdown. Patients with mild cognitive impairment have been shown to have a compromised BBB (115). This condition could allow plasma proteins, including fibrinogen, to gain access to the brain parenchyma (116) where they could contribute to inflammation and promote neurodegeneration.
Finally, decreases in blood flow can lead to hypoxic tissues and the induction of the hypoxia-inducible factor HIF-1α. This transcription factor can activate γ-secretase, which could lead to increased Aβ production (117, 118). Thus, one can envisage a vicious cycle whereby Aβ causes vascular insufficiency, which in turn leads to increased Aβ.
Diagnostic possibilities stemming from vascular contributions to AD
As mentioned, if vascular dysregulation is a significant factor in some AD cases, it opens a therapeutic window to treat one aspect of the pathology. In addition, vascular involvement offers a possibility of blood-based biomarkers that could help identify contributing pathologies. Analysis of plasma has shown that AD patients and AD mice have increased contact system activation, as evidenced by increased FXII activation and HK cleavage (99, 100). To accurately assess contact activation in blood requires careful attention to many variables, including blood collection, anticoagulation, plasma preparation, sample storage, and analytical procedures (119). Nevertheless, it is possible to standardize procedures to maximize reproducibility. Having a biomarker in blood as an early diagnostic to help guide treatment could lead to a significant benefit for some patients.
Genetics of vascular involvement in AD
This review has concentrated on vascular and inflammatory drivers of AD. In genetic studies, inflammatory genes have emerged as risk factors for AD (120, 121). If vascular dysfunction is also a contributing factor in AD pathogenesis, why haven’t genes associated with clotting and hemostasis turned up in screens for risk factors? One possible reason is that genes that inhibit clotting might prevent or delay AD, so they would not be observed in significant numbers in AD populations, but rather, in individuals at low risk for AD. One would need to look for protective genes; such a study has been done, and it uncovered an APP mutation that confers protection against AD (122). However, mutations that induce changes in clotting may also carry a risk of bleeding, so these patients might be present in low numbers in the aged population. It is clear that several genes identified as risk factors for AD, including Apo E4 (123) and PICALM (124, 125), can affect cerebrovascular function via the BBB (126, 127).
Conclusions
Multiple contributing pathologies affect the risk of developing AD. Just as treating multiple pathways in cancer has improved outcomes, a similar evolution of therapy can be envisaged for AD. The development of assays to classify patients and treat their specific constellation of pathologies will be required to make progress in treatment. Although reversal of cognitive decline would be the ideal treatment, and next to that a complete inhibition of progression, simply slowing the rate of progression of symptoms by 50% would be a very significant advance. Accomplishing this goal is going to require, in general, a wider range of analysis and treatment options than are currently employed, and specifically, more thorough investigations into correlations between AD and vascular dysfunction.
Acknowledgments
I am very grateful to Marta Cortes-Canteli, Erin H. Norris, Carol C. Strickland, Harold Varmus, and Daria Zamolodchikov for comments on the manuscript. I thank the following for generous support of the work from our laboratory: NIH NS 50537; Robertson Therapeutic Discovery Institute; Alzheimer’s Drug Discovery, Rudin Family, Mellam Family, and Mary & James G. Wallach Foundations; Cure Alzheimer’s Fund; Louis Herlands; John A. Herrmann Jr.
Version 1. 02/01/2018
Print issue publication
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(2):556–563.
https://doi.org/10.1172/JCI97509.
References
- 1.Jack CR, Jr, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease — locating the primary defect. Neurobiol Dis. 2011;43(1):38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener. 2014;9:48. doi: 10.1186/1750-1326-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mamelak M. Energy and the Alzheimer brain. Neurosci Biobehav Rev. 2017;75:297–313. doi: 10.1016/j.neubiorev.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2(1):a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada M. Cerebral amyloid angiopathy: emerging concepts. J Stroke. 2015;17(1):17–30. doi: 10.5853/jos.2015.17.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64(6):575–611. doi: 10.1016/S0301-0082(00)00068-X. [DOI] [PubMed] [Google Scholar]
- 11.Kalaria RN. Small vessel disease and Alzheimer’s dementia: pathological considerations. Cerebrovasc Dis. 2002;13(suppl 2):48–52. doi: 10.1159/000049150. [DOI] [PubMed] [Google Scholar]
- 12.Humpel C. Chronic mild cerebrovascular dysfunction as a cause for Alzheimer’s disease? Exp Gerontol. 2011;46(4):225–232. doi: 10.1016/j.exger.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017;18(7):419–434. doi: 10.1038/nrn.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder HM, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement. 2015;11(6):710–717. doi: 10.1016/j.jalz.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinters HV. Emerging concepts in Alzheimer’s disease. Annu Rev Pathol. 2015;10:291–319. doi: 10.1146/annurev-pathol-020712-163927. [DOI] [PubMed] [Google Scholar]
- 16.Serrano-Pozo A, Qian J, Monsell SE, Frosch MP, Betensky RA, Hyman BT. Examination of the clinicopathologic continuum of Alzheimer disease in the autopsy cohort of the National Alzheimer Coordinating Center. J Neuropathol Exp Neurol. 2013;72(12):1182–1192. doi: 10.1097/NEN.0000000000000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toledo JB, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 2013;136(pt 9):2697–2706. doi: 10.1093/brain/awt188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 2016;15(9):934–943. doi: 10.1016/S1474-4422(16)30029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris JK, et al. Aerobic exercise for Alzheimer’s disease: a randomized controlled pilot trial. PLoS One. 2017;12(2):e0170547. doi: 10.1371/journal.pone.0170547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lautenschlager NT, et al. Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA. 2008;300(9):1027–1037. doi: 10.1001/jama.300.9.1027. [DOI] [PubMed] [Google Scholar]
- 21.Erickson KI, et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A. 2011;108(7):3017–3022. doi: 10.1073/pnas.1015950108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruscheweyh R, et al. Physical activity and memory functions: an interventional study. Neurobiol Aging. 2011;32(7):1304–1319. doi: 10.1016/j.neurobiolaging.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Liu R, et al. Cardiorespiratory fitness as a predictor of dementia mortality in men and women. Med Sci Sports Exerc. 2012;44(2):253–259. doi: 10.1249/MSS.0b013e31822cf717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elwood P, et al. Healthy lifestyles reduce the incidence of chronic diseases and dementia: evidence from the Caerphilly cohort study. PLoS One. 2013;8(12):e81877. doi: 10.1371/journal.pone.0081877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buchman AS, Boyle PA, Yu L, Shah RC, Wilson RS, Bennett DA. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology. 2012;78(17):1323–1329. doi: 10.1212/WNL.0b013e3182535d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snowden M, et al. Effect of exercise on cognitive performance in community-dwelling older adults: review of intervention trials and recommendations for public health practice and research. J Am Geriatr Soc. 2011;59(4):704–716. doi: 10.1111/j.1532-5415.2011.03323.x. [DOI] [PubMed] [Google Scholar]
- 27.Sink KM, et al. Effect of a 24-month physical activity intervention vs health education on cognitive outcomes in sedentary older adults: The LIFE Randomized Trial. JAMA. 2015;314(8):781–790. doi: 10.1001/jama.2015.9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reijmer YD, van Veluw SJ, Greenberg SM. Ischemic brain injury in cerebral amyloid angiopathy. J Cereb Blood Flow Metab. 2016;36(1):40–54. doi: 10.1038/jcbfm.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalaria RN, Akinyemi R, Ihara M. Does vascular pathology contribute to Alzheimer changes? J Neurol Sci. 2012;322(1–2):141–147. doi: 10.1016/j.jns.2012.07.032. [DOI] [PubMed] [Google Scholar]
- 30.de la Torre JC. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004;3(3):184–190. doi: 10.1016/S1474-4422(04)00683-0. [DOI] [PubMed] [Google Scholar]
- 31.Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC, Alzheimer’s Disease Neuroimaging Initiative Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun. 2016;7:11934. doi: 10.1038/ncomms11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee S, et al. White matter hyperintensities are a core feature of Alzheimer’s disease: evidence from the dominantly inherited Alzheimer network. Ann Neurol. 2016;79(6):929–939. doi: 10.1002/ana.24647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68(2):270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 34.Shim YS, et al. Pathological correlates of white matter hyperintensities on magnetic resonance imaging. Dement Geriatr Cogn Disord. 2015;39(1–2):92–104. doi: 10.1159/000366411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erten-Lyons D, et al. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology. 2013;81(11):977–983. doi: 10.1212/WNL.0b013e3182a43e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wardlaw JM, Valdés Hernández MC, Muñoz-Maniega S. What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc. 2015;4(6):001140. doi: 10.1161/JAHA.114.001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9(7):689–701. doi: 10.1016/S1474-4422(10)70104-6. [DOI] [PubMed] [Google Scholar]
- 38.Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol. 2013;12(5):483–497. doi: 10.1016/S1474-4422(13)70060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindemer ER, et al. White matter signal abnormality quality differentiates mild cognitive impairment that converts to Alzheimer’s disease from nonconverters. Neurobiol Aging. 2015;36(9):2447–2457. doi: 10.1016/j.neurobiolaging.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brickman AM, et al. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch Neurol. 2012;69(12):1621–1627. doi: 10.1001/archneurol.2012.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brickman AM. Contemplating Alzheimer’s disease and the contribution of white matter hyperintensities. Curr Neurol Neurosci Rep. 2013;13(12):415. doi: 10.1007/s11910-013-0415-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grimmer T, et al. White matter hyperintensities predict amyloid increase in Alzheimer’s disease. Neurobiol Aging. 2012;33(12):2766–2773. doi: 10.1016/j.neurobiolaging.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Y, Yu F, Duong TQ, Alzheimer’s Disease Neuroimaging Initiative White matter lesion load is associated with resting state functional MRI activity and amyloid PET but not FDG in mild cognitive impairment and early Alzheimer’s disease patients. J Magn Reson Imaging. 2015;41(1):102–109. doi: 10.1002/jmri.24550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brickman AM, et al. Cerebral autoregulation, beta amyloid, and white matter hyperintensities are interrelated. Neurosci Lett. 2015;592:54–58. doi: 10.1016/j.neulet.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vemuri P, et al. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain. 2015;138(pt 3):761–771. doi: 10.1093/brain/awu393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Natté R, Maat-Schieman ML, Haan J, Bornebroek M, Roos RA, van Duinen SG. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol. 2001;50(6):765–772. doi: 10.1002/ana.10040. [DOI] [PubMed] [Google Scholar]
- 47.Tomidokoro Y, et al. Iowa variant of familial Alzheimer’s disease: accumulation of posttranslationally modified AbetaD23N in parenchymal and cerebrovascular amyloid deposits. Am J Pathol. 2010;176(4):1841–1854. doi: 10.2353/ajpath.2010.090636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lleó A, Berezovska O, Growdon JH, Hyman BT. Clinical, pathological, and biochemical spectrum of Alzheimer disease associated with PS-1 mutations. Am J Geriatr Psychiatry. 2004;12(2):146–156. doi: 10.1097/00019442-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 49.Kattula S, Byrnes JR, Wolberg AS. Fibrinogen and fibrin in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2017;37(3):e13–e21. doi: 10.1161/ATVBAHA.117.308564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol. 2012;34(1):43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- 51.Litvinov RI, Weisel JW. What is the biological and clinical relevance of fibrin? Semin Thromb Hemost. 2016;42(4):333–343. doi: 10.1055/s-0036-1571342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bardehle S, Rafalski VA, Akassoglou K. Breaking boundaries-coagulation and fibrinolysis at the neurovascular interface. Front Cell Neurosci. 2015;9:354. doi: 10.3389/fncel.2015.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cortes-Canteli M, et al. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron. 2010;66(5):695–709. doi: 10.1016/j.neuron.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fiala M, et al. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002;32(5):360–371. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- 55.Cullen KM, Kócsi Z, Stone J. Microvascular pathology in the aging human brain: evidence that senile plaques are sites of microhaemorrhages. Neurobiol Aging. 2006;27(12):1786–1796. doi: 10.1016/j.neurobiolaging.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 56.Lipinski B, Sajdel-Sulkowska EM. New insight into Alzheimer disease: demonstration of fibrin(ogen)-serum albumin insoluble deposits in brain tissue. Alzheimer Dis Assoc Disord. 2006;20(4):323–326. doi: 10.1097/01.wad.0000213844.21001.a2. [DOI] [PubMed] [Google Scholar]
- 57.Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med. 2009;13(9A):2911–2925. doi: 10.1111/j.1582-4934.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Viggars AP, et al. Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: a study in the MRC-CFAS population neuropathology cohort. Neurosci Lett. 2011;505(1):25–30. doi: 10.1016/j.neulet.2011.09.049. [DOI] [PubMed] [Google Scholar]
- 59.Hultman K, Cortes-Canteli M, Bounoutas A, Richards AT, Strickland S, Norris EH. Plasmin deficiency leads to fibrin accumulation and a compromised inflammatory response in the mouse brain. J Thromb Haemost. 2014;12(5):701–712. doi: 10.1111/jth.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J Exp Med. 2007;204(8):1999–2008. doi: 10.1084/jem.20070304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cortes-Canteli M, Mattei L, Richards AT, Norris EH, Strickland S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol Aging. 2015;36(2):608–617. doi: 10.1016/j.neurobiolaging.2014.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barrachina M, Maes T, Buesa C, Ferrer I. Lysosome-associated membrane protein 1 (LAMP-1) in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2006;32(5):505–516. doi: 10.1111/j.1365-2990.2006.00756.x. [DOI] [PubMed] [Google Scholar]
- 63.Armstrong A, et al. Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromolecular Med. 2014;16(1):150–160. doi: 10.1007/s12017-013-8269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Condello C, Schain A, Grutzendler J. Multicolor time-stamp reveals the dynamics and toxicity of amyloid deposition. Sci Rep. 2011;1:19. doi: 10.1038/srep00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hashimoto T, Ogino K, Shin RW, Kitamoto T, Kikuchi T, Shimizu N. Age-dependent increase in lysosome-associated membrane protein 1 and early-onset behavioral deficits in APPSL transgenic mouse model of Alzheimer’s disease. Neurosci Lett. 2010;469(2):273–277. doi: 10.1016/j.neulet.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 66.Pérez-Gracia E, Torrejón-Escribano B, Ferrer I. Dystrophic neurites of senile plaques in Alzheimer’s disease are deficient in cytochrome c oxidase. Acta Neuropathol. 2008;116(3):261–268. doi: 10.1007/s00401-008-0370-6. [DOI] [PubMed] [Google Scholar]
- 67.Hultman K, Strickland S, Norris EH. The APOE ε4/ε4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J Cereb Blood Flow Metab. 2013;33(8):1251–1258. doi: 10.1038/jcbfm.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Oijen M, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke. 2005;36(12):2637–2641. doi: 10.1161/01.STR.0000189721.31432.26. [DOI] [PubMed] [Google Scholar]
- 69.Xu G, Zhang H, Zhang S, Fan X, Liu X. Plasma fibrinogen is associated with cognitive decline and risk for dementia in patients with mild cognitive impairment. Int J Clin Pract. 2008;62(7):1070–1075. doi: 10.1111/j.1742-1241.2007.01268.x. [DOI] [PubMed] [Google Scholar]
- 70.Craig-Schapiro R, et al. Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer’s disease diagnosis and prognosis. PLoS One. 2011;6(4):e18850. doi: 10.1371/journal.pone.0018850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vafadar-Isfahani B, et al. Identification of SPARC-like 1 protein as part of a biomarker panel for Alzheimer’s disease in cerebrospinal fluid. J Alzheimers Dis. 2012;28(3):625–636. doi: 10.3233/JAD-2011-111505. [DOI] [PubMed] [Google Scholar]
- 72.Lee JW, et al. Fibrinogen gamma-A chain precursor in CSF: a candidate biomarker for Alzheimer’s disease. BMC Neurol. 2007;7:14. doi: 10.1186/1471-2377-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thambisetty M, et al. Plasma biomarkers of brain atrophy in Alzheimer’s disease. PLoS One. 2011;6(12):e28527. doi: 10.1371/journal.pone.0028527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang H, Lyutvinskiy Y, Herukka SK, Soininen H, Rutishauser D, Zubarev RA. Prognostic polypeptide blood plasma biomarkers of Alzheimer’s disease progression. J Alzheimers Dis. 2014;40(3):659–666. doi: 10.3233/JAD-132102. [DOI] [PubMed] [Google Scholar]
- 75.Chiam JT, Dobson RJ, Kiddle SJ, Sattlecker M. Are blood-based protein biomarkers for Alzheimer’s disease also involved in other brain disorders? A systematic review. J Alzheimers Dis. 2015;43(1):303–314. doi: 10.3233/JAD-140816. [DOI] [PubMed] [Google Scholar]
- 76.Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat Neurosci. 2015;18(6):800–806. doi: 10.1038/nn.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(10):a006296. doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahn HJ, Zamolodchikov D, Cortes-Canteli M, Norris EH, Glickman JF, Strickland S. Alzheimer’s disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci U S A. 2010;107(50):21812–21817. doi: 10.1073/pnas.1010373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zamolodchikov D, Strickland S. Aβ delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood. 2012;119(14):3342–3351. doi: 10.1182/blood-2011-11-389668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barber M, Tait RC, Scott J, Rumley A, Lowe GD, Stott DJ. Dementia in subjects with atrial fibrillation: hemostatic function and the role of anticoagulation. J Thromb Haemost. 2004;2(11):1873–1878. doi: 10.1111/j.1538-7836.2004.00993.x. [DOI] [PubMed] [Google Scholar]
- 82.Walsh AC. Anticoagulant therapy for Alzheimer’s disease. J Neuropsychiatry Clin Neurosci. 1996;8(3):361–362. doi: 10.1176/jnp.8.3.361. [DOI] [PubMed] [Google Scholar]
- 83.Bergamaschini L, et al. Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and beta-amyloid accumulation in a mouse model of Alzheimer’s disease. J Neurosci. 2004;24(17):4181–4186. doi: 10.1523/JNEUROSCI.0550-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Timmer NM, van Dijk L, van der Zee CE, Kiliaan A, de Waal RM, Verbeek MM. Enoxaparin treatment administered at both early and late stages of amyloid β deposition improves cognition of APPswe/PS1dE9 mice with differential effects on brain Aβ levels. Neurobiol Dis. 2010;40(1):340–347. doi: 10.1016/j.nbd.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 85.Tripathy D, Sanchez A, Yin X, Luo J, Martinez J, Grammas P. Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front Aging Neurosci. 2013;5:19. doi: 10.3389/fnagi.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ryu JK, et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun. 2015;6:8164. doi: 10.1038/ncomms9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ahn HJ, et al. A novel Aβ-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer’s disease mice. J Exp Med. 2014;211(6):1049–1062. doi: 10.1084/jem.20131751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bagyinszky E, Giau VV, Shim K, Suk K, An SSA, Kim S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J Neurol Sci. 2017;376:242–254. doi: 10.1016/j.jns.2017.03.031. [DOI] [PubMed] [Google Scholar]
- 89.Akiyama H, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Spangenberg EE, et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain. 2016;139(pt 4):1265–1281. doi: 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Asai H, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18(11):1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Renné T. The procoagulant and proinflammatory plasma contact system. Semin Immunopathol. 2012;34(1):31–41. doi: 10.1007/s00281-011-0288-2. [DOI] [PubMed] [Google Scholar]
- 93. White GC II, Marder VJ, Schulman S, Aird WC, Bennett JS. Overview of basic coagulation and fibrinolysis. In: Marder VJ, Aird WC, Bennett JS, Schulman S, White GC, eds. Hemostasis and thrombosis: basic principles and clinical practice. 6th ed. Philadelphia, Pennsylvania, USA: Wolters Kluwer/Lippincott, Williams & Wilkins; 2013:103–109. [Google Scholar]
- 94.Leeb-Lundberg LM, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57(1):27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- 95. Renné T. The Factor XII–driven plasma contact system. In: Marder VJ, Aird WC, Bennett JS, Schulman S, White GC, eds. Hemostasis and thrombosis: basic principles and clinical practice. 6th ed. Philadelphia, Pennsylvania, USA: Wolters Kluwer/Lippincott, Williams & Wilkins; 2013:242–253. [Google Scholar]
- 96.Yasuhara O, Walker DG, McGeer PL. Hageman factor and its binding sites are present in senile plaques of Alzheimer’s disease. Brain Res. 1994;654(2):234–240. doi: 10.1016/0006-8993(94)90484-7. [DOI] [PubMed] [Google Scholar]
- 97.Ashby EL, Love S, Kehoe PG. Assessment of activation of the plasma kallikrein-kinin system in frontal and temporal cortex in Alzheimer’s disease and vascular dementia. Neurobiol Aging. 2012;33(7):1345–1355. doi: 10.1016/j.neurobiolaging.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 98.Bergamaschini L, Donarini C, Foddi C, Gobbo G, Parnetti L, Agostoni A. The region 1-11 of Alzheimer amyloid-beta is critical for activation of contact-kinin system. Neurobiol Aging. 2001;22(1):63–69. doi: 10.1016/S0197-4580(00)00174-3. [DOI] [PubMed] [Google Scholar]
- 99.Zamolodchikov D, Chen ZL, Conti BA, Renné T, Strickland S. Activation of the factor XII-driven contact system in Alzheimer’s disease patient and mouse model plasma. Proc Natl Acad Sci U S A. 2015;112(13):4068–4073. doi: 10.1073/pnas.1423764112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen ZL, Revenko AS, Singh P, MacLeod AR, Norris EH, Strickland S. Depletion of coagulation factor XII ameliorates brain pathology and cognitive impairment in Alzheimer disease mice. Blood. 2017;129(18):2547–2556. doi: 10.1182/blood-2016-11-753202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zamolodchikov D, Renné T, Strickland S. The Alzheimer’s disease peptide β-amyloid promotes thrombin generation through activation of coagulation factor XII. J Thromb Haemost. 2016;14(5):995–1007. doi: 10.1111/jth.13209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang L, Li Y, Bhattacharya A, Zhang Y. A plasma proteolysis pathway comprising blood coagulation proteases. Oncotarget. 2016;7(27):40919–40938. doi: 10.18632/oncotarget.7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shibayama Y, Joseph K, Nakazawa Y, Ghebreihiwet B, Peerschke EI, Kaplan AP. Zinc-dependent activation of the plasma kinin-forming cascade by aggregated β amyloid protein. Clin Immunol. 1999;90(1):89–99. doi: 10.1006/clim.1998.4621. [DOI] [PubMed] [Google Scholar]
- 104.Maas C, et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118(9):3208–3218. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Renne T, Schmaier AH, Nickel KF, Blomback M, Maas C. In vivo roles of factor XII. Blood. 2012;120(22):4296–4303. doi: 10.1182/blood-2012-07-292094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Merkulov S, et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008;111(3):1274–1281. doi: 10.1182/blood-2007-06-092338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sheffer AL, MacGinnitie AJ, Campion M, Stolz LE, Pullman WE. Outcomes after ecallantide treatment of laryngeal hereditary angioedema attacks. Ann Allergy Asthma Immunol. 2013;110(3):184–188.e2. doi: 10.1016/j.anai.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 108.Chyung Y, et al. A phase 1 study investigating DX-2930 in healthy subjects. Ann Allergy Asthma Immunol. 2014;113(4):460–466.e2. doi: 10.1016/j.anai.2014.05.028. [DOI] [PubMed] [Google Scholar]
- 109.Mawuenyega KG, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330(6012):1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Iturria-Medina Y, Sotero RC, Toussaint PJ, Evans AC, Alzheimer’s Disease Neuroimaging Initiative Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders. PLoS Comput Biol. 2014;10(11):e1003956. doi: 10.1371/journal.pcbi.1003956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tarasoff-Conway JM, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457–470. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. 2015;163(5):1064–1078. doi: 10.1016/j.cell.2015.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shibata M, et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106(12):1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Deane R, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron. 2004;43(3):333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 115.Montagne A, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Deane R, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9(7):907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 117.Gertsik N, Chiu D, Li YM. Complex regulation of γ-secretase: from obligatory to modulatory subunits. Front Aging Neurosci. 2014;6:342. doi: 10.3389/fnagi.2014.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Villa JC, et al. Nontranscriptional role of Hif-1α in activation of γ-secretase and notch signaling in breast cancer. Cell Rep. 2014;8(4):1077–1092. doi: 10.1016/j.celrep.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Loeffen R, et al. Preanalytic variables of thrombin generation: towards a standard procedure and validation of the method. J Thromb Haemost. 2012;10(12):2544–2554. doi: 10.1111/jth.12012. [DOI] [PubMed] [Google Scholar]
- 120.Zhang ZG, Li Y, Ng CT, Song YQ. Inflammation in Alzheimer’s disease and molecular genetics: recent update. Arch Immunol Ther Exp (Warsz) 2015;63(5):333–344. doi: 10.1007/s00005-015-0351-0. [DOI] [PubMed] [Google Scholar]
- 121.Sims R, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49(9):1373–1384. doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jonsson T, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 123.Strittmatter WJ, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Harold D, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lambert JC, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 126.Zhao Z, et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat Neurosci. 2015;18(7):978–987. doi: 10.1038/nn.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Atri A, et al. Cumulative, additive benefits of memantine-donepezil combination over component monotherapies in moderate to severe Alzheimer’s dementia: a pooled area under the curve analysis. Alzheimers Res Ther. 2015;7(1):28. doi: 10.1186/s13195-015-0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hendrix JA, et al. Challenges, solutions, and recommendations for Alzheimer’s disease combination therapy. Alzheimers Dement. 2016;12(5):623–630. doi: 10.1016/j.jalz.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 130.Schmitt B, Bernhardt T, Moeller HJ, Heuser I, Frölich L. Combination therapy in Alzheimer’s disease: a review of current evidence. CNS Drugs. 2004;18(13):827–844. doi: 10.2165/00023210-200418130-00001. [DOI] [PubMed] [Google Scholar]