

Abstract
Increased sugar consumption is increasingly considered to be a contributor to the worldwide epidemics of obesity and diabetes and their associated cardiometabolic risks. As a result of its unique metabolic properties, the fructose component of sugar may be particularly harmful. Diets high in fructose can rapidly produce all of the key features of the metabolic syndrome. Here we review the biology of fructose metabolism as well as potential mechanisms by which excessive fructose consumption may contribute to cardiometabolic disease.
Introduction
Glucose is the predominant form of circulating sugar in animals, while sucrose, the disaccharide composed of equal portions of glucose and fructose, is the predominant circulating sugar in plants. As plants form the basis of the food chain, herbivores and omnivores are highly adapted to use sucrose for energetic and biosynthetic needs. Because fructose does not circulate at high levels in animals, ingested fructose may be uniquely positioned to convey signals related to sugar consumption. Therefore, understanding mechanisms by which fructose is sensed may be of consequence for understanding the adaptive physiology of sucrose metabolism as well as potential pathophysiological consequences of excessive sugar consumption.
Sugar in the form of sucrose or high-fructose corn syrup, both of which are composed of nearly equal amounts of glucose and fructose, is added to numerous manufactured food products. Sugar-sweetened beverages (SSBs) are a major source of added sugar in diets worldwide and include sodas, fruit-flavored drinks, and sport drinks. On average, SSBs contribute approximately 7% of daily calories (1) and nearly 50% of added sugars in the diet (2). Although trends in SSB consumption have declined in recent years, almost 66% of US youths still consume at least one SSB per day (3). Other major contributors to added sugar intake include candy and desserts, contributing approximately 4% to 9% of daily energy intake depending on age (2, 4).
Whether increased sugar consumption is a major contributor to the epidemics of obesity, type 2 diabetes, and nonalcoholic fatty liver disease remains controversial (5–7). While the relationships between some measures of dietary sugar exposure and cardiometabolic risk factors are inconsistent, greater SSB consumption consistently associates with indices of higher cardiometabolic risk (5). Several large meta-analyses associate increased SSB consumption with increased body weight, and much, though not all, of this increased weight is likely due to increased total energy consumption (5, 8). SSBs may increase cardiometabolic risk by increasing visceral adiposity, which accounts for much of the weight gain. A recent prospective study showed that daily SSB consumers had a 29% greater increase in visceral adipose tissue volume over 6 years compared with nonconsumers (9). A causal association is supported by evidence that intake of 1 liter of SSB daily for 6 months increased visceral and liver fat, but increases were not observed in those consuming isocaloric semiskim milk, noncaloric diet soda, or water (10). While increased visceral adiposity is a major cardiometabolic risk factor, SSBs may increase risk independently of adiposity. For instance, daily SSB consumption is associated with an unhealthy metabolic profile across BMI strata and with increased risk for type 2 diabetes independently of obesity (11, 12).
Hypertriglyceridemia is a major cardiovascular risk factor and is another mechanism by which SSBs might increase cardiovascular risk. Few large cross-sectional studies have examined the risk of dyslipidemia with SSB intake, and these studies show that dyslipidemia prevalence increases with higher SSB intake (13, 14). One prospective study reported that consuming more than 1 soft drink per day increased the odds of developing hypertriglyceridemia by 25% over 4 years compared with consuming less than 1 soft drink per day (15). Moreover, two recent prospective cohort studies showed that daily SSB consumption was associated with approximately 25% greater risk of developing coronary heart disease in both men and women compared with nonconsumers (13, 16).
SSB consumption also associates with hypertension, another major cardiovascular risk factor. A recent meta-analysis found a modest 12% increase in hypertension risk among the highest SSB consumers compared with the lowest (17). Thus, SSB intake may contribute to hypertension, but it may play a lesser role in this risk factor compared with other cardiometabolic risk factors.
On the basis of short-term overfeeding studies conducted predominantly in animals, the fructose component of SSBs and added sugar appears to be particularly harmful. Feeding animals large amounts of fructose can rapidly produce multiple features of the metabolic syndrome, including obesity, dyslipidemia, fatty liver, hypertension, insulin resistance, and diabetes (18, 19). Some, but not all, short-term dietary intervention studies in humans also demonstrate that overfeeding fructose, but not glucose, can increase visceral adiposity, postprandial hypertriglyceridemia, and insulin resistance, and effects on specific traits may be impacted by gender (20, 21). One concern with such studies is that the amount of fructose consumed often exceeds that commonly found in ad libitum diets. The average consumption of fructose in US populations accounts for approximately 9% of total energy intake, while consumers in the 95th percentile average approximately 15% of total energy from fructose (22). In contrast, many interventional studies are short in duration (less than 4 weeks) and include dietary intakes closer to 25% of total energy intake from fructose (23, 24).
Large randomized controlled dietary intervention studies assessing the effects of added sugars on cardiometabolic risk factors over long periods of time are lacking. Complexity, cost, compliance, and potential ethical issues likely prohibit the conducting of such studies. Nevertheless, some short-term interventional studies, even those within the range of “normal” fructose consumption, show that fructose can rapidly impair intermediate physiological endpoints like circulating lipids and insulin sensitivity in humans (25). Several recent reviews comprehensively discuss the physiological effects of added fructose or sugar on pathophysiological endpoints in human subjects (26, 27).
Understanding the mechanisms by which the isolated monosaccharide fructose might contribute to the development of metabolic disease may provide fundamental insights into pathogenic mechanisms that can be used to develop new diagnostic, preventative, and therapeutic strategies. Here we will review the biochemistry and molecular genetics of fructose metabolism as well as potential mechanisms by which excessive fructose consumption contributes to cardiometabolic disease. We hope that lessons learned from improved understanding of fructose metabolism and fructose-induced cardiometabolic risk may also apply to other forms of diet-induced and genetically induced metabolic disease.
Fructose absorption
Ingested fructose is predominantly absorbed passively from the intestinal lumen via the hexose transporter SLC2A5, also known as GLUT5, which has high affinity for fructose (Km = 6 mM). GLUT5 is highly expressed on enterocytes’ luminal membrane and is also expressed basolaterally (28). Deletion of Glut5 in mice reduces fructose absorption by 75% and causes cecum and colon dilatation as well as gas accumulation (29). These features are suggestive of fructose malabsorption, frequently cited as a cause of gastrointestinal symptoms in humans (30, 31). The intestine’s capacity to absorb fructose is saturable (32), and a healthy adult’s ability to absorb free fructose ranges from less than 5 g to more than 50 g (33). Unabsorbed fructose can impose an osmotic load on the distal small intestine and the colon, which may contribute to gastrointestinal symptoms (32). Moreover, fructose can serve as a substrate for bacterial fermentation, leading to formation of gas and other bacterial metabolites, which can affect intestinal motility and cause various symptoms such as abdominal pain and bloating (34).
Intestinal GLUT5 mRNA levels and fructose transport rates are very low prenatally and rapidly increase with weaning independently of diet, but they can be further induced following weaning to diets containing fructose (35). Recent data showed that high-fructose feeding induces intestinal thioredoxin-interacting protein (TXNIP), which binds and regulates GLUT5-mediated intestinal fructose transport (36). Consistent with this, we recently showed that carbohydrate-responsive element–binding protein (ChREBP), a transcription factor that responds to intracellular carbohydrate nutrients and a known transcriptional regulator of TXNIP (37), also regulates intestinal GLUT5 expression and is required for systemic fructose tolerance (38). In the future, it will be interesting to determine whether variability in the expression or function of GLUT5 or its regulatory factors contributes to the variability in fructose absorption in humans.
Intermediary fructose metabolism
Fructose concentrations in peripheral plasma are typically about 0.04 mM, can acutely increase 10-fold after fructose consumption, and return to fasting levels within 2 hours (39–41). This rapid clearance is mediated in large part by efficient extraction by the liver. Whereas the liver extracts only 15% to 30% of an oral glucose load, it is capable of extracting 70% of an oral fructose load (42, 43). Following fructose ingestion, plasma fructose can achieve low millimolar concentrations in the portal vein accompanied by peripheral circulation levels of approximately 0.2 mM, indicating that peripheral fructose concentrations rarely exceed the high micromolar range (44).
The SLC2A2 glucose transporter, also known as GLUT2, has lower affinity for fructose (Km = 11 mM) than GLUT5 (45). GLUT2 is a minor contributor to intestinal fructose transport (45), whereas it is likely a major contributor to hepatic fructose uptake, since GLUT5 is not robustly expressed in the liver (46, 47). SLC2A8, also known as GLUT8, may also contribute to hepatocellular fructose transport (48). Fructose is a poor substrate for the hepatic hexokinase glucokinase (GCK). Instead, ketohexokinase (KHK, also known as fructokinase) rapidly phosphorylates fructose to generate fructose-1-phosphate (F1P). KHK’s high activity and insensitivity to cellular energy status account for the liver’s ability to efficiently extract fructose. F1P is metabolized to dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (G3P), which enter the glycolytic/gluconeogenic metabolite pools (Figure 1).
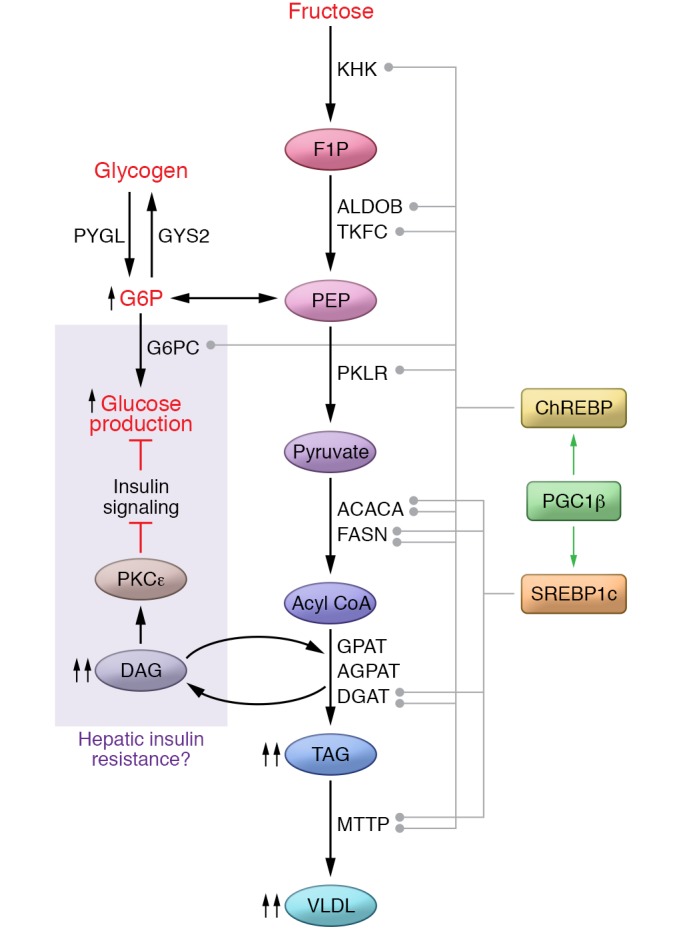
Figure 1. Fructose biochemistry.

Upon entering hepatocytes, fructose is phosphorylated by KHK to F1P. F1P is cleaved to DHAP and glyceraldehyde by ALDOB. Glyceraldehyde is phosphorylated by triose-kinase (TKFC, also known as dihydroxyacetone kinase 2 or DAK) to form the glycolytic intermediate glyceraldehyde 3-phosphate (GA3P). Both fructose-derived DHAP and GA3P enter the glycolytic/gluconeogenic metabolite pool at the triose-phosphate level, and these metabolites have numerous metabolic fates. F1P also allosterically regulates metabolic enzymes (red and green lines) to regulate the disposition of fructose-derived substrate and other metabolic products like uric acid. AMPD3, adenosine deaminase; GA, glyceraldehyde; IMP, inosine monophosphate; MTTP, microsomal triglyceride transfer protein; PYGL, glycogen phosphorylase L; GYS2, glycogen synthase 2; PKLR, pyruvate kinase, liver and red blood cell; PEP, phosphoenolpyruvate; TAG, triacylglycerol.
Cellular metabolic status and energy status tightly regulate the phosphofructokinase (PFK) step in glycolysis, which limits hepatic glycolytic flux (49). In contrast, fructose-derived metabolites enter the triose-phosphate pool distal to PFK and therefore bypass this restriction. As hepatic fructolysis is unrestricted, fructose loads can lead to large, rapid expansions in the hexose- and triose-phosphate pools, potentially providing increased substrate for all central carbon metabolic pathways, including glycolysis, glycogenesis, gluconeogenesis, lipogenesis, and oxidative phosphorylation.
The disposition of fructose-derived carbon among the major metabolic pathways depends on the overall nutritional and endocrine status of the animal as well as the status of key regulatory checkpoints in intermediary metabolism. For instance, in starved animals, low levels of fructose-2,6-biphosphate inhibit PFK activity and glycolysis and activate fructose-1,6-biphosphatase and glucose production (50). Thus, in a starved animal, fructose-derived triose-phosphates are preferentially routed through the gluconeogenic path (51, 52). The fate of ingested fructose may also depend on coingested nutrients. For instance, infusing physiological concentrations of fructose to fed rats and humans increases serum glucose and lactate levels without affecting hepatic glycogen accumulation (53, 54). However, when fructose is infused with glucose, which stimulates insulin secretion, marked glycogen accumulation occurs (55). Chronic fructose consumption can affect metabolic gene expression programs that further affect fructose disposition. These mechanisms will be described in greater detail below.
Although the liver metabolizes the majority of ingested fructose, the intestine itself can metabolize up to 30% of an oral fructose load (56, 57). All of the fructolytic enzymes are highly expressed in the small intestine and notably in the jejunum, where the highest levels of GLUT5 are observed (58). Similarly to GLUT5, intestinal expression of fructolytic and gluconeogenic enzymes including glucose-6-phosphatase (G6PC) increases upon fructose feeding (59) and depends on GLUT5 and KHK activity (60). However, most prandial fructose is not metabolized in the intestine but rather passes via the portal vein to the liver (61, 62).
In addition to providing substrate for metabolic processes, hepatic fructose metabolism generates specific metabolites that also perform signaling functions (Figure 2). Importantly, F1P, the fructose-specific metabolite produced by KHK, exerts strong positive regulatory control on GCK by promoting its release from the inhibitory GCK regulatory protein (GCKR). GCKR sequesters GCK in an inactive state in the nucleus (63–65). “Catalytic” amounts of fructose, in part through activation of GCK, can promote hepatic glucose uptake and phosphorylation, leading to rapid glycogen accumulation (66). F1P may also enhance glycogen synthesis by allosterically inhibiting glycogen phosphorylase (67, 68). Lastly, F1P also allosterically activates pyruvate kinase, the terminal step in glycolysis, contributing to increased circulating lactate levels following fructose ingestion (69). In rodent liver, hepatic F1P levels increase 10-fold to approximately 1 mM within 10 minutes after fructose ingestion and remain elevated for several hours (70). F1P concentrations of only approximately 200 μM are sufficient to alleviate the inhibitory effect of GCKR on GCK (71). Thus, fructose ingestion is likely to have rapid, robust, and sustained effects on hepatic glucose uptake and intermediary metabolism.
Figure 2. Fructose-induced gene expression programs.
Fructose metabolism activates transcription factors including ChREBP and SREBP1c and their coactivator PGC1β to coordinately regulate gene expression of metabolic enzymes that contribute to fructolysis, glycolysis, lipogenesis, and glucose production. These metabolic pathways contribute to steatosis, VLDL packaging and secretion, as well as glucose production and the generation of lipid intermediates that may affect hepatic insulin sensitivity and other biological processes. ACACA, acetyl-CoA carboxylase α; FASN, fatty acid synthase; GPAT, glycerol-3-phosphate acyltransferases; AGPAT, acylglycerol-3-phosphate acyltransferase; DGAT, diacylglycerol acyltransferase; DAG, diacylglycerol.
While the efficiency and rapidity with which the liver can extract and phosphorylate ingested fructose are likely important for its role in integrating nutritional and systemic fuel metabolism, this robust metabolism may also have deleterious consequences. For instance, decreases in intracellular free phosphate due to rapid hepatic fructose phosphorylation can increase uric acid production through activation of AMP deaminase, which leads to catabolism of AMP to uric acid (72, 73). Fructose feeding may also stimulate purine synthesis, contributing to uric acid production (74). Increased circulating uric acid levels increase the risk of gout, a condition characterized by painful inflammation due to deposition of uric acid crystals in joints. Indeed, a growing body of evidence implicates sugar intake as a risk factor for gout (75). Moreover, elevated serum uric acid levels and gout are associated with other cardiometabolic risk factors in diverse populations (76–78). A substantial body of work suggests that increased uric acid levels may independently regulate important aspects of metabolism and contribute to cardiometabolic risk (79–83). However, Mendelian randomization studies do not strongly support a causal role for circulating uric acid in mediating cardiometabolic disease (84). The association between uric acid levels and cardiometabolic risk may be indirect and may reflect activation of distinct fructose-regulated processes that contribute both to uric acid production and cardiometabolic risk.
The liver is at a metabolic crossroads and is crucial for gauging nutrient consumption and integrating peripheral nutrient status to regulate systemic fuel storage versus provisioning. While hormones like insulin and glucagon help inform the liver of systemic fuel status, the liver is also well configured to integrate signals derived directly from fuel substrates. In this sense, the signaling properties of fructose-derived F1P, and particularly its regulation of GCK activity, may function as an evolved mechanism allowing the liver to use fructose metabolism to “sense” sugar (i.e., sucrose or high-fructose corn syrup) consumption. Robust physiological activation of hepatic GCK occurs only when fructose-containing sugars are consumed. This activation enhances net hepatic glucose uptake and storage as glycogen and lipid. Interestingly, at supraphysiological/pathological levels, glucose itself can dissociate GCK from GCKR and may contribute to the increased hepatic GCK activity described in obese diabetics and in genetic models of obesity and diabetes (85, 86). Thus, in the setting of uncontrolled diabetes, the liver may aberrantly sense hyperglycemia as a state of increased sugar consumption. Understanding the metabolic effects of hepatic “sugar sensing” may be of consequence for understanding the pathophysiology of diabetes and hyperglycemia.
Genetic lessons about fructose metabolism
KHK exists as two alternatively spliced isoforms produced by mutual exclusion of the adjacent exons 3C and 3A within the KHK gene (87, 88). The “A” isoform is ubiquitously expressed but has low activity due to relatively low affinity for its substrate (Km = 8 mM) (89). Expression of the “C” isoform is primarily restricted to metabolic tissues including the liver, kidney, and intestine, and this isoform has much higher affinity for fructose (Km = 0.8 mM) (89, 90). Mice deficient in both isoforms were fully protected from fructose-induced metabolic disease even though blood and urinary fructose levels were markedly increased (91). Thus, elevated blood fructose itself is not deleterious; rather, fructose metabolism is essential for fructose-induced metabolic disease. Loss-of-function mutations in KHK cause the benign human disorder essential fructosuria, characterized by impaired hepatic fructose metabolism leading to high blood and urine fructose levels after sucrose or fructose consumption (92). Consistent with observations in mice, there are no documented adverse health effects observed in people with this condition. Altogether, these results suggest that inhibiting KHK may be a safe therapeutic strategy to prevent fructose-induced metabolic disease.
In contrast with global KHK deletion, selective deletion of the A isoform exacerbates the adverse metabolic effects of fructose feeding (91). These results suggest two important hypotheses: (a) fructose metabolism outside of tissues that express the C isoform is non-negligible and contributes to whole-body fructose clearance, and (b) fructose metabolism within the tissues expressing KHK-C is critical for fructose-induced metabolic disease. This is supported by recent data showing that selective knockdown of KHK in mouse liver protects against fructose-induced steatosis (93). Recent data also indicate that altered splicing between KHK-A and KHK-C isoforms may contribute to the development of distinct diseases like hepatocellular carcinoma and heart failure (94, 95).
Hereditary fructose intolerance (HFI) is a rare autosomal recessive disease caused by a deficiency of aldolase B (ALDOB), which is highly expressed in the liver, kidney, and small intestine (96). People with HFI develop abdominal pain, vomiting, diarrhea, symptomatic hypoglycemia, hyperuricemia, and potentially liver failure and death following ingestion of foods containing fructose, sucrose, or sorbitol (97). The precise mechanisms by which ALDOB deficiency causes symptoms are not entirely clear. An Aldob-deficient mouse model mimics the human HFI condition (98). These mice fail to thrive and die when exposed to high-fructose diets. Interestingly, even on a fructose-free diet, Aldob-deficient mice develop steatosis (98), possibly due to impaired metabolism of endogenously synthesized fructose (99).
Endogenous fructose production
While the vast majority of metabolized fructose is derived from dietary sources of sugar, animals including humans are capable of synthesizing fructose endogenously. The sorbitol (polyol) pathway, which is active in a wide range of tissues, is responsible for endogenous fructose formation from glucose (100, 101). In this pathway, glucose is first reduced to sorbitol by aldose reductase (102). Sorbitol is then oxidized to fructose by sorbitol dehydrogenase (103). Physiologically, endogenously synthesized fructose is the primary energy source for sperm and may be important for fertility (104–106). The placenta may also synthesize sorbitol that the developing fetus may use to synthesize fructose, suggesting a broader role for endogenous fructose in reproductive and developmental biology (107).
Sorbitol pathway activity increases during diabetic hyperglycemia (108). Endogenous fructose synthesis and polyol metabolites are considered key players in the development of diabetic microvascular complications (109). Interestingly, semen fructose concentrations are increased in type 1 diabetes and in obesity, in which it is associated with impaired sperm parameters (105, 110). Whether endogenous fructose synthesis might occur at sufficient rates to contribute to other aspects of fructose-induced cardiometabolic risk has only recently been addressed. Glucose dose-dependently induces aldolase reductase in human tissues, and chronic exposure to a high-glucose diet induces polyol pathway activation in mice (99, 111). This may be a mechanism by which severe hyperglycemia may exacerbate cardiometabolic risks. Additionally, Lanaspa et al. report that endogenous fructose production and KHK activation within the kidney contribute to the development of diabetic nephropathy (112). Although sorbitol dehydrogenase is expressed at high levels in human liver (113), whether this pathway is sufficiently active in humans to play an adverse metabolic role will require further investigation.
Fructose effects on lipid homeostasis
As noted above, excessive fructose consumption may have significant effects on lipid metabolism, contributing both to steatosis and to increased circulating triglyceride levels in the form of very low-density lipoprotein (VLDL). Hepatic lipid accumulation results from a combination of increased hepatic de novo lipogenesis (DNL), esterification of preformed fatty acids derived from the diet or adipose stores, decreased VLDL secretion, and decreased hepatic fatty acid oxidation. Activation of the lipogenic program is observed immediately after a single load of fructose and contributes to increased VLDL triglyceride secretion (114, 115). Fructose also acutely suppresses hepatic fatty acid oxidation (116). Thus, fructose contributes to hepatic triglyceride production both by providing substrate for fatty acid and triglyceride synthesis and by activating signaling systems to enhance lipid production (Figure 2).
The liver is the primary site of DNL, the process by which fatty acids are synthesized from dietary precursors, predominantly carbohydrates (117). Due to the differences in hepatic glucose and fructose metabolism, a larger fraction of diet-derived fructose than glucose metabolites are available for conversion to fat in the liver via DNL in animals and humans (20, 118–120). Additionally, fructose metabolites entering the triose-phosphate pool are in equilibrium with glycerol 3-phosphate, which is used to synthesize the glycerol backbone in triglyceride. Moreover, the metabolite malonyl-CoA generated via DNL limits fatty acid oxidation by inhibiting carnitine palmitoyltransferase 1A (CPT1A), the enzyme required for translocation of fatty acids into the mitochondria (121). CPT1A inhibition further increases the availability of fatty acids for triglyceride production. Triglyceride can be incorporated into lipid droplets, leading to steatosis, or can be incorporated into VLDL and secreted from the liver.
In addition to providing substrate for lipogenesis, chronic fructose consumption increases transcriptional regulation of DNL by activating key transcription factors, including sterol regulatory element–binding protein 1c (SREBP1c) and carbohydrate-responsive element–binding protein (ChREBP) (122). SREBP1c promotes lipid synthesis and is regulated at the transcriptional and posttranslational levels by nutrients and hormones. Insulin is a major hormonal activator of hepatic SREBP1c (123, 124). Although acute fructose feeding does not directly stimulate insulin secretion, chronic fructose ingestion can lead to hyperinsulinemia, which may increase hepatic SREBP1c expression and activation (125, 126). Fructose may also activate SREBP1c independently of insulin, since SREBP1c responds to high-fructose feeding in liver-specific insulin receptor–knockout (LIRKO) mice (125). Fructose consumption may also promote ER stress, which may induce proteolytic cleavage of SREBP1c and the lipogenic program (127, 128). Fructose-induced ER stress may also enhance lipogenesis via activation of the transcription factor x box-binding protein 1 independently of other lipogenic transcription factors (129).
ChREBP couples carbohydrate metabolites to lipid synthesis by inducing enzymes required for DNL (130). ChREBP may also suppress fatty acid oxidation by downregulating enzymes like CPT1A, in part by antagonizing peroxisome proliferator–activated receptor α (PPARα), a key transcriptional regulator of the fatty acid oxidation gene program (131, 132). ChREBP is highly expressed in key metabolic tissues, including liver, adipose tissue, small intestine, pancreatic islets, and kidney, where it regulates carbohydrate metabolism in an insulin-independent manner (37, 125, 130). The observation that ChREBP-deficient mice are intolerant to diets containing fructose but not to diets containing dextrose suggests a specific role for ChREBP in regulating fructose metabolism (37, 133). Moreover, ChREBP activity was markedly higher in rats fed high-fructose compared with isocaloric high-glucose diets (126). We recently demonstrated that ingesting fructose, but not glucose, acutely and robustly induces hepatic expression of the potent ChREBP-β isoform along with its lipogenic, fructolytic, and glycolytic targets (133, 134). The mechanism by which sugar metabolites activate ChREBP remains controversial but involves allosteric activation by glucose-6-phosphate as well as modulation by other carbohydrate metabolites and posttranslational modifications (135–137). ChREBP knockdown using antisense oligonucleotides (ASOs) in fructose-fed rats reduced circulating triglyceride levels and confirmed a role for ChREBP in fructose-mediated dyslipidemia, although steatosis was unaffected (138). Consistent with this, GWAS have identified multiple common SNPs within the ChREBP locus associated with increased serum triglyceride and low HDL cholesterol levels (139, 140).
ChREBP knockdown’s selective effect on circulating triglycerides but not steatosis in the experiment described above highlights the fact that fat accretion in lipid droplets and VLDL secretion are distinct processes. ChREBP potently regulates DNL, and fructose-induced DNL strongly correlates with fructose-induced hypertriglyceridemia (141). However, in steatotic human subjects, DNL-derived fatty acids contribute a minor fraction of fatty acids to VLDL (142), and the mechanistic connection between DNL and VLDL secretion remains uncertain. Moreover, ChREBP may have effects to increase circulating triglycerides independently of increasing VLDL secretion. ChREBP may transactivate expression of the apolipoprotein APOC3 as well as angiopoietin-like 8 (ANGPTL8), both of which may inhibit lipoprotein lipase and limit VLDL clearance (refs. 143, 144, and Figure 3). Thus, it is possible that high-fructose feeding may increase circulating VLDL both by enhancing VLDL production and secretion and by reducing VLDL clearance, but the precise mechanisms remain to be determined.
Figure 3. Consequences of fructose overconsumption.

Fructose metabolism in key metabolic tissues including the small intestine, liver, and kidney may contribute to diverse cardiometabolic risk factors including steatosis, increased glucose production, hypertriglyceridemia, increased adiposity, and hypertension. Fructose provides substrate for metabolic processes that contribute to cardiometabolic risk and engages cellular and hormonal signaling systems that regulate these metabolic and pathological processes. LPL, lipoprotein lipase.
PPARγ coactivator 1β (PGC1β) is a transcriptional coactivator that increases the activity of multiple key transcription factors, such as PPARγ, PPARα, estrogen-related receptors (ERRs), and liver X receptor (LXR) (145, 146). PGC1β can also bind SREBP1 and ChREBP and enhance their transcriptional activity (147, 148). ASOs targeting PGC1β prevented SREBP1c expression and lipogenesis, which in turn decreased lipid accumulation in fructose-fed rat livers. PGC1β-targeting ASOs also prevented increases in adiposity, glycemia, and plasma insulin and triglycerides in fructose-fed rats. Thus, PGC1β is uniquely positioned to coordinately regulate both ChREBP and SREBP1c activities in the context of high-fructose feeding.
Increased sugar and fructose consumption is implicated in both simple steatosis and the progression toward more advanced forms of nonalcoholic fatty liver disease (NAFLD), including nonalcoholic steatohepatitis, fibrosis, and hepatocellular carcinoma (149). Important steps in DNL and VLDL synthesis occur at the ER membrane, and fructose-induced lipogenesis may elicit ER stress and the ER stress response (150). Moreover, signaling elements in the ER stress response may contribute to NAFLD pathogenesis and progression (151). Recent work from Zhang et al. suggests that ChREBP may protect the liver against fructose-induced ER stress and hepatic inflammation (152). However, we have recently observed that liver-specific ChREBP-knockout mice do not develop ER stress or hepatic inflammation when challenged with high-fructose diets (38). Mechanisms by which fructose may contribute to progression of NAFLD will require further investigation.
Fructose effects on glucose homeostasis
Fructose does not directly stimulate pancreatic β cell insulin secretion (153, 154). However, high-fructose feeding readily induces hyperinsulinemia in animal models. Moreover, hyperinsulinemia is more pronounced in rodent models with high-fructose compared with high-dextrose feeding despite similar increases in body weight and adiposity (155, 156). Similarly, hypercaloric fructose feeding increases circulating insulin in human subjects (157). Fructose-induced hyperinsulinemia, often considered a proxy for insulin resistance, might be the result of insulin resistance in some combination of liver, muscle, and/or adipose tissue.
The mechanisms by which high-fructose feeding causes hyperinsulinemia and insulin resistance remain uncertain. Fructose-induced steatosis may contribute to hepatic insulin resistance through increased hepatic diacylglycerol accumulation, PKC activation, and impairment of insulin-mediated Akt2 activation (158–160). However, whether steatosis itself can cause hepatic insulin resistance remains controversial (131, 161). In addition to ChREBP’s role in fructose-induced dyslipidemia, Erion et al. demonstrated that ChREBP knockdown enhanced peripheral insulin sensitivity in high-fructose-fed rats (138). Whether the improvement in peripheral insulin sensitivity was directly related to the improvement in circulating lipid levels or adiposity is uncertain. We recently demonstrated that while hepatic ChREBP is essential for fructose-mediated upregulation of fructolytic, glycolytic, and lipogenic enzymes, ChREBP also mediated upregulation of G6PC, the terminal enzyme in glucose production (133). We showed that a fructose-induced, ChREBP-mediated increase in G6PC activity is a major determinant of endogenous glucose production. Moreover, fructose activated ChREBP and induced G6PC in the absence of FOXO1a, indicating that substrate-driven activation of ChREBP and G6PC to enhance glucose production dominates over the suppressive effects of insulin (133). This ChREBP/G6PC signaling axis is also conserved in humans. These results are consistent with dietary intervention studies in humans indicating that either eucaloric substitution or hypercaloric addition of fructose may have more significant effects on hepatic insulin resistance than peripheral insulin resistance (157). However, as hyperinsulinemia itself can induce peripheral insulin resistance (162, 163), we speculate that chronic hyperinsulinemia that compensates for fructose-induced glucose production may subsequently lead to peripheral insulin resistance. This hypothesis remains to be tested experimentally.
Fructose effects on appetite and adiposity
Increased SSB consumption and fructose overconsumption are consistently associated with increased adiposity, which may be attributed to increased caloric intake as well as effects on energy balance and nutrient partitioning that are independent of caloric intake. Fructose is among the sweetest of sugars, and sweetness generally enhances food palatability. This likely contributes to the addition of fructose-containing sugars like sucrose and high-fructose corn syrup to the food supply. Enhanced palatability may increase feeding behavior and thus encourage overeating (164). Moreover, fructose and sucrose can enhance palatability and induce addiction-like behaviors such as binging and dependence in part by stimulating dopaminergic pathways (165–168). Distinct from fructose’s hedonic value, whether fructose impacts additional signaling systems to regulate appetite and feeding behavior has also been studied. For instance, high-fructose feeding may induce leptin resistance, which in turn may lead to increased food intake and obesity (169, 170). Additionally, dietary fructose decreases leptin excursions compared with isocaloric dietary glucose, and fructose is less potent than glucose in suppressing the orexigenic hormone ghrelin (171). In human subjects, fructose versus glucose ingestion has differential effects on hypothalamic blood flow and cerebral cortex reactivity to food cues, suggesting the possibility that fructose and glucose have distinct effects on brain function that may impact feeding behavior (172, 173). The mechanisms by which fructose and glucose differentially regulate appetite and feeding behavior remain to be determined.
Prospective studies in which excess fructose is added on top of habitual diets often document spontaneous reductions in other forms of sugar consumption, suggesting strong feedback mechanisms that specifically regulate sugar consumption (141). Such compensatory mechanisms may contribute to difficulties in accurately assessing dietary sugar consumption in both observational and interventional studies. FGF21 is a liver-derived hormone that regulates energy, glucose, and lipid homeostasis and may also participate in a feedback mechanism regulating macronutrient selection (refs. 174–178 and Figure 3). Increased circulating FGF21 is associated with cardiometabolic risk factors including obesity, NAFLD, type 2 diabetes, and insulin resistance (179–181). FGF21 is a ChREBP transcriptional target (182), and fructose ingestion acutely and robustly induces circulating FGF21, whereas the response to glucose ingestion is less substantial and is delayed (175). Fructose-induced, circulating FGF21 may protect the liver from fructose-induced metabolic disease (183). Interestingly, data from animal models suggest that sugar-induced circulating FGF21 may signal to the brain to suppress additional sugar consumption (184, 185). GWAS also support a role for FGF21 in macronutrient preference, as variants in the FGF21 locus associate with increased dietary carbohydrate consumption relative to dietary fat in human populations (186, 187). However, variants associated with increased carbohydrate consumption also associate with increased circulating FGF21 levels, which is inconsistent with the negative feedback model. More investigation will be required to understand the role of FGF21 in the context of increased sugar and fructose consumption.
Effects of fructose on hypertension
The mechanisms by which fructose contributes to the development of hypertension are less well characterized than its effects on glucose and lipid homeostasis. High-fructose feeding in rodents can increase intestinal salt absorption in part through induction of an intestinal anion exchanger, Slc26a6 (188). Moreover, this induction and associated hypertension are prevented in GLUT5-knockout mice (188). However, these results are confounded by the fact that GLUT5-knockout mice suffer generalized malabsorption and become ill when challenged with fructose. Johnson and colleagues have hypothesized that fructose-induced hyperuricemia may impair kidney function, contributing to hypertension (189). However, as discussed above, genetic data do not strongly support a major role for hyperuricemia in cardiometabolic disease. As fructose is robustly metabolized in the kidney, fructose-mediated changes in renal salt handling may also be important. However, this has yet to be rigorously studied and is an area ripe for further investigation.
Conclusions and future directions
The combination of mechanistic data supporting a role for excessive fructose ingestion and epidemiological data supporting a role for SSBs in the development of cardiometabolic disease supports recent dietary recommendations to limit sugar consumption published by several public health agencies, including the American Heart Association, the World Health Organization, and the Dietary Guidelines Advisory Committee (2, 190, 191). Safe thresholds for sugar consumption and concrete recommendations for targets to reduce cardiometabolic risk remain in dispute. Moreover, implementing effective programs to alter dietary habits remains challenging. However, initial reports indicate that “sugar taxes” may be effective in reducing SSB consumption (192, 193). Time will tell whether such approaches can improve health outcomes. Hopefully, by improving our understanding of the underlying mechanisms by which sugar and fructose can cause disease, we will be able to bring informed, comprehensive approaches to bear on our current metabolic epidemics.
Acknowledgments
This work is supported by American Heart Association 16CSA28590003 (to MAH and NMM), NIH R01DK100425 (to MAH), NIH 5T32HL069772-15 (to DEH), and US Department of Agriculture Agricultural Research Service agreement 58-1950-4-003 (to NMM).
Version 1. 02/01/2018
Print issue publication
Footnotes
Conflict of interest: M.A. Herman has received research support from Eli Lilly and Co.
Reference information: J Clin Invest. 2018;128(2):545–555.
https://doi.org/10.1172/JCI96702.
References
- 1.Kit BK, Fakhouri TH, Park S, Nielsen SJ, Ogden CL. Trends in sugar-sweetened beverage consumption among youth and adults in the United States: 1999–2010. Am J Clin Nutr. 2013;98(1):180–188. doi: 10.3945/ajcn.112.057943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Office of Disease Prevention and Health Promotion. Dietary Guidelines for Americans 2015–2020. ODPHP Website. http://health.gov/dietaryguidelines/2015/guidelines/ Accessed December 12, 2017.
- 3.Rosinger A, Herrick K, Gahche J, Park S. Sugar-sweetened beverage consumption among U.S. youth, 2011–2014. NCHS Data Brief. 2017(271):1–8. [PubMed] [Google Scholar]
- 4.Vos MB, et al. Added sugars and cardiovascular disease risk in children: a scientific statement from the American Heart Association. Circulation. 2017;135(19):e1017–e1034. doi: 10.1161/CIR.0000000000000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan TA, Sievenpiper JL. Controversies about sugars: results from systematic reviews and meta-analyses on obesity, cardiometabolic disease and diabetes. Eur J Nutr. 2016;55(suppl 2):25–43. doi: 10.1007/s00394-016-1345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ter Horst KW, Serlie MJ. Fructose consumption, lipogenesis, and non-alcoholic fatty liver disease. Nutrients. 2017;9(9):E981. doi: 10.3390/nu9090981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanhope KL. Sugar consumption, metabolic disease and obesity: the state of the controversy. Crit Rev Clin Lab Sci. 2016;53(1):52–67. doi: 10.3109/10408363.2015.1084990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malik VS, Pan A, Willett WC, Hu FB. Sugar-sweetened beverages and weight gain in children and adults: a systematic review and meta-analysis. Am J Clin Nutr. 2013;98(4):1084–1102. doi: 10.3945/ajcn.113.058362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma J, McKeown NM, Hwang SJ, Hoffmann U, Jacques PF, Fox CS. Sugar-sweetened beverage consumption is associated with change of visceral adipose tissue over 6 years of follow-up. Circulation. 2016;133(4):370–377. doi: 10.1161/CIRCULATIONAHA.115.018704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maersk M, et al. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: a 6-mo randomized intervention study. Am J Clin Nutr. 2012;95(2):283–289. doi: 10.3945/ajcn.111.022533. [DOI] [PubMed] [Google Scholar]
- 11.Green AK, Jacques PF, Rogers G, Fox CS, Meigs JB, McKeown NM. Sugar-sweetened beverages and prevalence of the metabolically abnormal phenotype in the Framingham Heart Study. Obesity (Silver Spring) 2014;22(5):E157–E163. doi: 10.1002/oby.20724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imamura F, et al. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: systematic review, meta-analysis, and estimation of population attributable fraction. BMJ. 2015;351:h3576. doi: 10.1136/bmj.h3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Koning L, Malik VS, Kellogg MD, Rimm EB, Willett WC, Hu FB. Sweetened beverage consumption, incident coronary heart disease, and biomarkers of risk in men. Circulation. 2012;125(14):1735–1741. doi: 10.1161/CIRCULATIONAHA.111.067017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hert KA, Fisk PS, Rhee YS, Brunt AR. Decreased consumption of sugar-sweetened beverages improved selected biomarkers of chronic disease risk among US adults: 1999 to 2010. Nutr Res. 2014;34(1):58–65. doi: 10.1016/j.nutres.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Dhingra R, et al. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation. 2007;116(5):480–488. doi: 10.1161/CIRCULATIONAHA.107.689935. [DOI] [PubMed] [Google Scholar]
- 16.Fung TT, Malik V, Rexrode KM, Manson JE, Willett WC, Hu FB. Sweetened beverage consumption and risk of coronary heart disease in women. Am J Clin Nutr. 2009;89(4):1037–1042. doi: 10.3945/ajcn.2008.27140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jayalath VH, et al. Sugar-sweetened beverage consumption and incident hypertension: a systematic review and meta-analysis of prospective cohorts. Am J Clin Nutr. 2015;102(4):914–921. doi: 10.3945/ajcn.115.107243. [DOI] [PubMed] [Google Scholar]
- 18.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 19.Reaven GM. Insulin resistance and compensatory hyperinsulinemia: role in hypertension, dyslipidemia, and coronary heart disease. Am Heart J. 1991;121(4 pt 2):1283–1288. doi: 10.1016/0002-8703(91)90434-j. [DOI] [PubMed] [Google Scholar]
- 20.Stanhope KL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119(5):1322–1334. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuzma JN, et al. No differential effect of beverages sweetened with fructose, high-fructose corn syrup, or glucose on systemic or adipose tissue inflammation in normal-weight to obese adults: a randomized controlled trial. Am J Clin Nutr. 2016;104(2):306–314. doi: 10.3945/ajcn.115.129650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marriott BP, Cole N, Lee E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J Nutr. 2009;139(6):1228S–1235S. doi: 10.3945/jn.108.098277. [DOI] [PubMed] [Google Scholar]
- 23.Chung M, Ma J, Patel K, Berger S, Lau J, Lichtenstein AH. Fructose, high-fructose corn syrup, sucrose, and nonalcoholic fatty liver disease or indexes of liver health: a systematic review and meta-analysis. Am J Clin Nutr. 2014;100(3):833–849. doi: 10.3945/ajcn.114.086314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelishadi R, Mansourian M, Heidari-Beni M. Association of fructose consumption and components of metabolic syndrome in human studies: a systematic review and meta-analysis. Nutrition. 2014;30(5):503–510. doi: 10.1016/j.nut.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 25.Stanhope KL, et al. A dose-response study of consuming high-fructose corn syrup-sweetened beverages on lipid/lipoprotein risk factors for cardiovascular disease in young adults. Am J Clin Nutr. 2015;101(6):1144–1154. doi: 10.3945/ajcn.114.100461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campos VC, Tappy L. Physiological handling of dietary fructose-containing sugars: implications for health. Int J Obes (Lond) 2016;40(suppl 1):S6–S11. doi: 10.1038/ijo.2016.8. [DOI] [PubMed] [Google Scholar]
- 27.Stanhope KL, Schwarz JM, Havel PJ. Adverse metabolic effects of dietary fructose: results from the recent epidemiological, clinical, and mechanistic studies. Curr Opin Lipidol. 2013;24(3):198–206. doi: 10.1097/MOL.0b013e3283613bca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel C, Douard V, Yu S, Gao N, Ferraris RP. Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. FASEB J. 2015;29(9):4046–4058. doi: 10.1096/fj.15-272195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barone S, et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem. 2009;284(8):5056–5066. doi: 10.1074/jbc.M808128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson PR, Newnham E, Barrett JS, Shepherd SJ, Muir JG. Review article: Fructose malabsorption and the bigger picture. Aliment Pharmacol Ther. 2007;25(4):349–363. doi: 10.1111/j.1365-2036.2006.03186.x. [DOI] [PubMed] [Google Scholar]
- 31.Heizer WD, Southern S, McGovern S. The role of diet in symptoms of irritable bowel syndrome in adults: a narrative review. J Am Diet Assoc. 2009;109(7):1204–1214. doi: 10.1016/j.jada.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Rumessen JJ. Fructose and related food carbohydrates. Sources, intake, absorption, and clinical implications. Scand J Gastroenterol. 1992;27(10):819–828. doi: 10.3109/00365529209000148. [DOI] [PubMed] [Google Scholar]
- 33.Rumessen JJ, Gudmand-Høyer E. Absorption capacity of fructose in healthy adults. Comparison with sucrose and its constituent monosaccharides. Gut. 1986;27(10):1161–1168. doi: 10.1136/gut.27.10.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skoog SM, Bharucha AE. Dietary fructose and gastrointestinal symptoms: a review. Am J Gastroenterol. 2004;99(10):2046–2050. doi: 10.1111/j.1572-0241.2004.40266.x. [DOI] [PubMed] [Google Scholar]
- 35.Ferraris RP. Dietary and developmental regulation of intestinal sugar transport. Biochem J. 2001;360(pt 2):265–276. doi: 10.1042/0264-6021:3600265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dotimas JR, et al. Diabetes regulates fructose absorption through thioredoxin-interacting protein. Elife. 2016;5:e18313. doi: 10.7554/eLife.18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101(19):7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim M, et al. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight. 2017;2(24):e96703. doi: 10.1172/jci.insight.96703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugimoto K, et al. Lowering of postprandial hyperfructosemia in humans by eucalyptus leaf extract: a randomized, double-blind, placebo-controlled crossover study. Food Sci Technol Res. 2010;16(5):509–512. doi: 10.3136/fstr.16.509. [DOI] [Google Scholar]
- 40.Wahjudi PN, Patterson ME, Lim S, Yee JK, Mao CS, Lee WN. Measurement of glucose and fructose in clinical samples using gas chromatography/mass spectrometry. Clin Biochem. 2010;43(1–2):198–207. doi: 10.1016/j.clinbiochem.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Preston GM, Calle RA. Elevated serum sorbitol and not fructose in type 2 diabetic patients. Biomark Insights. 2010;5:33–38. doi: 10.4137/bmi.s4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam P. Effects of consuming dietary fructose versus glucose on de novo lipogenesis in overweight and obese human subjects. Berkeley Scientific Journal. https://escholarship.org/uc/item/7vv7z7zw [Google Scholar]
- 43.Tappy L, Lê KA. Does fructose consumption contribute to non-alcoholic fatty liver disease? Clin Res Hepatol Gastroenterol. 2012;36(6):554–560. doi: 10.1016/j.clinre.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 44.Sugimoto K, et al. Eucalyptus leaf extract suppresses the postprandial elevation of portal, cardiac and peripheral fructose concentrations after sucrose ingestion in rats. J Clin Biochem Nutr. 2010;46(3):205–211. doi: 10.3164/jcbn.09-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manolescu AR, Witkowska K, Kinnaird A, Cessford T, Cheeseman C. Facilitated hexose transporters: new perspectives on form and function. Physiology (Bethesda) 2007;22:234–240. doi: 10.1152/physiol.00011.2007. [DOI] [PubMed] [Google Scholar]
- 46.Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr. 2003;89(1):3–9. doi: 10.1079/BJN2002763. [DOI] [PubMed] [Google Scholar]
- 47.Karim S, Adams DH, Lalor PF. Hepatic expression and cellular distribution of the glucose transporter family. World J Gastroenterol. 2012;18(46):6771–6781. doi: 10.3748/wjg.v18.i46.6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Debosch BJ, Chen Z, Saben JL, Finck BN, Moley KH. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J Biol Chem. 2014;289(16):10989–10998. doi: 10.1074/jbc.M113.527002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boscá L, Corredor C. Is phosphofructokinase the rate-limiting step of glycolysis? Trends Biochem Sci. 1984;9(9):372–373. doi: 10.1016/0968-0004(84)90214-7. [DOI] [Google Scholar]
- 50.Hers HG, Van Schaftingen E. Fructose 2,6-bisphosphate 2 years after its discovery. Biochem J. 1982;206(1):1–12. doi: 10.1042/bj2060001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sestoft L, Fleron P. Determination of the kinetic constants of fructose transport and phosphorylation in the perfused rat liver. Biochim Biophys Acta. 1974;345(1):27–38. doi: 10.1016/0005-2736(74)90242-9. [DOI] [PubMed] [Google Scholar]
- 52.Björkman O, Felig P. Role of the kidney in the metabolism of fructose in 60-hour fasted humans. Diabetes. 1982;31(6 pt 1):516–520. doi: 10.2337/diab.31.6.516. [DOI] [PubMed] [Google Scholar]
- 53.Sahebjami H, Scalettar R. Effects of fructose infusion on lactate and uric acid metabolism. Lancet. 1971;1(7695):366–369. doi: 10.1016/s0140-6736(71)92208-2. [DOI] [PubMed] [Google Scholar]
- 54.Burch HB, Max P, Ghyu K, Lowry OH. Metabolic intermediates in liver of rats given large amounts of fructose or dihydroxyacetone. Biochem Biophys Res Commun. 1969;34(5):619–626. doi: 10.1016/0006-291X(69)90783-9. [DOI] [PubMed] [Google Scholar]
- 55.Topping DL, Mayes PA. Comparative effects of fructose and glucose on the lipid and carbohydrate metabolism of perfused rat liver. Br J Nutr. 1976;36(1):113–126. doi: 10.1079/BJN19760062. [DOI] [PubMed] [Google Scholar]
- 56.Mavrias DA, Mayer RJ. Metabolism of fructose in the small intestine. 1. The effect of fructose feeding on fructose transport and metabolism in rat small intestine. Biochim Biophys Acta. 1973;291(2):531–537. doi: 10.1016/0005-2736(73)90504-X. [DOI] [PubMed] [Google Scholar]
- 57.Ginsburg V, Hers HG. On the conversion of fructose to glucose by guinea pig intestine. Biochim Biophys Acta. 1960;38:427–434. doi: 10.1016/0006-3002(60)91278-6. [DOI] [PubMed] [Google Scholar]
- 58.Rand EB, Depaoli AM, Davidson NO, Bell GI, Burant CF. Sequence, tissue distribution, and functional characterization of the rat fructose transporter GLUT5. Am J Physiol. 1993;264(6 pt 1):G1169–G1176. doi: 10.1152/ajpgi.1993.264.6.G1169. [DOI] [PubMed] [Google Scholar]
- 59.Cui XL, Soteropoulos P, Tolias P, Ferraris RP. Fructose-responsive genes in the small intestine of neonatal rats. Physiol Genomics. 2004;18(2):206–217. doi: 10.1152/physiolgenomics.00056.2004. [DOI] [PubMed] [Google Scholar]
- 60.Patel C, Douard V, Yu S, Tharabenjasin P, Gao N, Ferraris RP. Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am J Physiol Regul Integr Comp Physiol. 2015;309(5):R499–R509. doi: 10.1152/ajpregu.00128.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bismut H, Hers HG, Van Schaftingen E. Conversion of fructose to glucose in the rabbit small intestine. A reappraisal of the direct pathway. Eur J Biochem. 1993;213(2):721–726. doi: 10.1111/j.1432-1033.1993.tb17812.x. [DOI] [PubMed] [Google Scholar]
- 62. Holdsworth CD, ed. Sugars in Nutrition. New York, New York, USA: Raven Press; 1991. [Google Scholar]
- 63.Brown KS, Kalinowski SS, Megill JR, Durham SK, Mookhtiar KA. Glucokinase regulatory protein may interact with glucokinase in the hepatocyte nucleus. Diabetes. 1997;46(2):179–186. doi: 10.2337/diab.46.2.179. [DOI] [PubMed] [Google Scholar]
- 64.Niculescu L, Veiga-da-Cunha M, Van Schaftingen E. Investigation on the mechanism by which fructose, hexitols and other compounds regulate the translocation of glucokinase in rat hepatocytes. Biochem J. 1997;321(pt 1):239–246. doi: 10.1042/bj3210239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J. 2008;414(1):1–18. doi: 10.1042/BJ20080595. [DOI] [PubMed] [Google Scholar]
- 66.McGuinness OP, Cherrington AD. Effects of fructose on hepatic glucose metabolism. Curr Opin Clin Nutr Metab Care. 2003;6(4):441–448. doi: 10.1097/01.mco.0000078990.96795.cd. [DOI] [PubMed] [Google Scholar]
- 67.Thurston JH, Jones EM, Hauhart RE. Decrease and inhibition of liver glycogen phosphorylase after fructose. An experimental model for the study of hereditary fructose intolerance. Diabetes. 1974;23(7):597–604. doi: 10.2337/diab.23.7.597. [DOI] [PubMed] [Google Scholar]
- 68.Van Den Berghe G, Hue L, Hers HG. Effect of administration of the fructose on the glycogenolytic action of glucagon. An investigation of the pathogeny of hereditary fructose intolerance. Biochem J. 1973;134(2):637–645. doi: 10.1042/bj1340637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eggleston LV, Woods HF. Activation of liver pyruvate kinase by fructose-1-phosphate. FEBS Lett. 1970;6(1):43–45. doi: 10.1016/0014-5793(70)80038-2. [DOI] [PubMed] [Google Scholar]
- 70.Niewoehner CB, Gilboe DP, Nuttall GA, Nuttall FQ. Metabolic effects of oral fructose in the liver of fasted rats. Am J Physiol. 1984;247(4 pt 1):E505–E512. doi: 10.1152/ajpendo.1984.247.4.E505. [DOI] [PubMed] [Google Scholar]
- 71.Van Schaftingen E. A protein from rat liver confers to glucokinase the property of being antagonistically regulated by fructose 6-phosphate and fructose 1-phosphate. Eur J Biochem. 1989;179(1):179–184. doi: 10.1111/j.1432-1033.1989.tb14538.x. [DOI] [PubMed] [Google Scholar]
- 72.Lanaspa MA, Tapia E, Soto V, Sautin Y, Sánchez-Lozada LG. Uric acid and fructose: potential biological mechanisms. Semin Nephrol. 2011;31(5):426–432. doi: 10.1016/j.semnephrol.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 73.van den Berghe G, Bronfman M, Vanneste R, Hers HG. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem J. 1977;162(3):601–609. doi: 10.1042/bj1620601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Raivio KO, Becker MA, Meyer LJ, Greene ML, Nuki G, Seegmiller JE. Stimulation of human purine synthesis de novo by fructose infusion. Metab Clin Exp. 1975;24(7):861–869. doi: 10.1016/0026-0495(75)90133-X. [DOI] [PubMed] [Google Scholar]
- 75.Jamnik J, et al. Fructose intake and risk of gout and hyperuricemia: a systematic review and meta-analysis of prospective cohort studies. BMJ Open. 2016;6(10):e013191. doi: 10.1136/bmjopen-2016-013191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chang CH, et al. Relationship between hyperuricemia (HUC) and metabolic syndrome (MS) in institutionalized elderly men. Arch Gerontol Geriatr. 2009;49(suppl 2):S46–S49. doi: 10.1016/S0167-4943(09)70013-5. [DOI] [PubMed] [Google Scholar]
- 77.Liu M, et al. Association between serum uric acid level and metabolic syndrome and its sex difference in a chinese community elderly population. Int J Endocrinol. 2014;2014:754678. doi: 10.1155/2014/754678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Puig JG, Martínez MA. Hyperuricemia, gout and the metabolic syndrome. Curr Opin Rheumatol. 2008;20(2):187–191. doi: 10.1097/BOR.0b013e3282f4b1ed. [DOI] [PubMed] [Google Scholar]
- 79.Lanaspa MA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem. 2012;287(48):40732–40744. doi: 10.1074/jbc.M112.399899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol. 2007;293(2):C584–C596. doi: 10.1152/ajpcell.00600.2006. [DOI] [PubMed] [Google Scholar]
- 81.Tapia E, et al. Synergistic effect of uricase blockade plus physiological amounts of fructose-glucose on glomerular hypertension and oxidative stress in rats. Am J Physiol Renal Physiol. 2013;304(6):F727–F736. doi: 10.1152/ajprenal.00485.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi YJ, et al. Uric acid induces endothelial dysfunction by vascular insulin resistance associated with the impairment of nitric oxide synthesis. FASEB J. 2014;28(7):3197–3204. doi: 10.1096/fj.13-247148. [DOI] [PubMed] [Google Scholar]
- 83.Lanaspa MA, et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One. 2012;7(10):e47948. doi: 10.1371/journal.pone.0047948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li X, et al. Serum uric acid levels and multiple health outcomes: umbrella review of evidence from observational studies, randomised controlled trials, and Mendelian randomisation studies. BMJ. 2017;357:j2376. doi: 10.1136/bmj.j2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Belfiore F, Romeo F, Iannello S, Salamone C. The glucose-6-phosphatase/glucokinase ratio in the liver of obese-diabetic subjects. Biochem Med Metab Biol. 1989;41(1):77–80. doi: 10.1016/0885-4505(89)90011-X. [DOI] [PubMed] [Google Scholar]
- 86.Huupponen R, Karvonen I, Sotaniemi E. Activity of hepatic glucose phosphorylating and NADPH generating enzymes in Zucker rats. Diabetes Res. 1989;10(3):143–146. [PubMed] [Google Scholar]
- 87.Bonthron DT, Brady N, Donaldson IA, Steinmann B. Molecular basis of essential fructosuria: molecular cloning and mutational analysis of human ketohexokinase (fructokinase) Hum Mol Genet. 1994;3(9):1627–1631. doi: 10.1093/hmg/3.9.1627. [DOI] [PubMed] [Google Scholar]
- 88.Hayward BE, Bonthron DT. Structure and alternative splicing of the ketohexokinase gene. Eur J Biochem. 1998;257(1):85–91. doi: 10.1046/j.1432-1327.1998.2570085.x. [DOI] [PubMed] [Google Scholar]
- 89.Diggle CP, et al. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem. 2009;57(8):763–774. doi: 10.1369/jhc.2009.953190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Asipu A, Hayward BE, O’Reilly J, Bonthron DT. Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenesis of essential fructosuria. Diabetes. 2003;52(9):2426–2432. doi: 10.2337/diabetes.52.9.2426. [DOI] [PubMed] [Google Scholar]
- 91.Ishimoto T, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci U S A. 2012;109(11):4320–4325. doi: 10.1073/pnas.1119908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Steinitz H, Mizrahy O. Essential fructosuria and hereditary fructose intolerance. N Engl J Med. 1969;280(4):222. doi: 10.1056/nejm196901232800421. [DOI] [PubMed] [Google Scholar]
- 93.Softic S, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest. 2017;127(11):4059–4074. doi: 10.1172/JCI94585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mirtschink P, et al. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature. 2015;522(7557):444–449. doi: 10.1038/nature14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li X, et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat Cell Biol. 2016;18(5):561–571. doi: 10.1038/ncb3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hers HG, Joassin G. [Anomaly of hepatic aldolase in intolerance to fructose] Enzymol Biol Clin (Basel) 1961;1:4–14. doi: 10.1159/000458107. [DOI] [PubMed] [Google Scholar]
- 97.Ali M, Rellos P, Cox TM. Hereditary fructose intolerance. J Med Genet. 1998;35(5):353–365. doi: 10.1136/jmg.35.5.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Oppelt SA, Sennott EM, Tolan DR. Aldolase-B knockout in mice phenocopies hereditary fructose intolerance in humans. Mol Genet Metab. 2015;114(3):445–450. doi: 10.1016/j.ymgme.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 99.Lanaspa MA, et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun. 2013;4:2434. doi: 10.1038/ncomms3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Oates PJ. Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol. 2002;50:325–392. doi: 10.1016/s0074-7742(02)50082-9. [DOI] [PubMed] [Google Scholar]
- 101.Hwang JJ, et al. The human brain produces fructose from glucose. JCI Insight. 2017;2(4):e90508. doi: 10.1172/jci.insight.90508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cheng HM, González RG. The effect of high glucose and oxidative stress on lens metabolism, aldose reductase, and senile cataractogenesis. Metab Clin Exp. 1986;35(4 suppl 1):10–14. doi: 10.1016/0026-0495(86)90180-0. [DOI] [PubMed] [Google Scholar]
- 103.Jedziniak JA, Chylack LT, Jr , Cheng HM, Gillis MK, Kalustian AA, and Tung WH. The sorbitol pathway in the human lens: aldose reductase and polyol dehydrogenase. Invest Ophthalmol Vis Sci. 1981;20(3):314–326. [PubMed] [Google Scholar]
- 104.Frenette G, Thabet M, Sullivan R. Polyol pathway in human epididymis and semen. J Androl. 2006;27(2):233–239. doi: 10.2164/jandrol.05108. [DOI] [PubMed] [Google Scholar]
- 105.Martini AC, et al. Overweight and seminal quality: a study of 794 patients. Fertil Steril. 2010;94(5):1739–1743. doi: 10.1016/j.fertnstert.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 106.Jayaraman V, Ghosh S, Sengupta A, Srivastava S, Sonawat HM, Narayan PK. Identification of biochemical differences between different forms of male infertility by nuclear magnetic resonance (NMR) spectroscopy. J Assist Reprod Genet. 2014;31(9):1195–1204. doi: 10.1007/s10815-014-0282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hwang JJ, et al. Fructose levels are markedly elevated in cerebrospinal fluid compared to plasma in pregnant women. PLoS One. 2015;10(6):e0128582. doi: 10.1371/journal.pone.0128582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lorenzi M. The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabetes Res. 2007;2007:61038. doi: 10.1155/2007/61038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mathebula SD. Polyol pathway: a possible mechanism of diabetes complications in the eye. Afr Vision Eye Health. 2015;74(1):Art. #13 [Google Scholar]
- 110.La Vignera S, Condorelli R, Vicari E, D’Agata R, Calogero AE. Diabetes mellitus and sperm parameters. J Androl. 2012;33(2):145–153. doi: 10.2164/jandrol.111.013193. [DOI] [PubMed] [Google Scholar]
- 111.Das B, Srivastava SK. Activation of aldose reductase from human tissues. Diabetes. 1985;34(11):1145–1151. doi: 10.2337/diab.34.11.1145. [DOI] [PubMed] [Google Scholar]
- 112.Lanaspa MA, et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J Am Soc Nephrol. 2014;25(11):2526–2538. doi: 10.1681/ASN.2013080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sobrecases H, et al. Effects of short-term overfeeding with fructose, fat and fructose plus fat on plasma and hepatic lipids in healthy men. Diabetes Metab. 2010;36(3):244–246. doi: 10.1016/j.diabet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 115.Hudgins LC, Parker TS, Levine DM, Hellerstein MK. A dual sugar challenge test for lipogenic sensitivity to dietary fructose. J Clin Endocrinol Metab. 2011;96(3):861–868. doi: 10.1210/jc.2010-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Topping DL, Mayes PA. The immediate effects of insulin and fructose on the metabolism of the perfused liver. Changes in lipoprotein secretion, fatty acid oxidation and esterification, lipogenesis and carbohydrate metabolism. Biochem J. 1972;126(2):295–311. doi: 10.1042/bj1260295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hellerstein MK, Schwarz JM, Neese RA. Regulation of hepatic de novo lipogenesis in humans. Annu Rev Nutr. 1996;16:523–557. doi: 10.1146/annurev.nu.16.070196.002515. [DOI] [PubMed] [Google Scholar]
- 118.Parks EJ, Skokan LE, Timlin MT, Dingfelder CS. Dietary sugars stimulate fatty acid synthesis in adults. J Nutr. 2008;138(6):1039–1046. doi: 10.1093/jn/138.6.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zavaroni I, Chen YD, Reaven GM. Studies of the mechanism of fructose-induced hypertriglyceridemia in the rat. Metab Clin Exp. 1982;31(11):1077–1083. doi: 10.1016/0026-0495(82)90155-X. [DOI] [PubMed] [Google Scholar]
- 120.Crescenzo R, Bianco F, Falcone I, Coppola P, Liverini G, Iossa S. Increased hepatic de novo lipogenesis and mitochondrial efficiency in a model of obesity induced by diets rich in fructose. Eur J Nutr. 2013;52(2):537–545. doi: 10.1007/s00394-012-0356-y. [DOI] [PubMed] [Google Scholar]
- 121.McGarry JD. Banting Lecture 2001: Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51(1):7–18. doi: 10.2337/diabetes.51.1.7. [DOI] [PubMed] [Google Scholar]
- 122.Herman MA, Samuel VT. The sweet path to metabolic demise: fructose and lipid synthesis. Trends Endocrinol Metab. 2016;27(10):719–730. doi: 10.1016/j.tem.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107(8):3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Peterson TR, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Haas JT, et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 2012;15(6):873–884. doi: 10.1016/j.cmet.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Koo HY, Miyashita M, Cho BH, Nakamura MT. Replacing dietary glucose with fructose increases ChREBP activity and SREBP-1 protein in rat liver nucleus. Biochem Biophys Res Commun. 2009;390(2):285–289. doi: 10.1016/j.bbrc.2009.09.109. [DOI] [PubMed] [Google Scholar]
- 127.Kammoun HL, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119(5):1201–1215. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Flamment M, Hajduch E, Ferré P, Foufelle F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab. 2012;23(8):381–390. doi: 10.1016/j.tem.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 129.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320(5882):1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006;4(2):107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 131.Benhamed F, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest. 2012;122(6):2176–2194. doi: 10.1172/JCI41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Boergesen M, Poulsen Ll, Schmidt SF, Frigerio F, Maechler P, Mandrup S. ChREBP mediates glucose repression of peroxisome proliferator-activated receptor alpha expression in pancreatic beta-cells. J Biol Chem. 2011;286(15):13214–13225. doi: 10.1074/jbc.M110.215467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim MS, et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest. 2016;126(11):4372–4386. doi: 10.1172/JCI81993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Herman MA, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484(7394):333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dentin R, et al. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J Hepatol. 2012;56(1):199–209. doi: 10.1016/j.jhep.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 136.Filhoulaud G, Guilmeau S, Dentin R, Girard J, Postic C. Novel insights into ChREBP regulation and function. Trends Endocrinol Metab. 2013;24(5):257–268. doi: 10.1016/j.tem.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 137.Baraille F, Planchais J, Dentin R, Guilmeau S, Postic C. Integration of ChREBP-mediated glucose sensing into whole body metabolism. Physiology (Bethesda) 2015;30(6):428–437. doi: 10.1152/physiol.00016.2015. [DOI] [PubMed] [Google Scholar]
- 138.Erion DM, et al. The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology. 2013;154(1):36–44. doi: 10.1210/en.2012-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kooner JS, et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat Genet. 2008;40(2):149–151. doi: 10.1038/ng.2007.61. [DOI] [PubMed] [Google Scholar]
- 140.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40(2):189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Taskinen MR, et al. Adverse effects of fructose on cardiometabolic risk factors and hepatic lipid metabolism in subjects with abdominal obesity. J Intern Med. 2017;282(2):187–201. doi: 10.1111/joim.12632. [DOI] [PubMed] [Google Scholar]
- 142.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Caron S, et al. Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2011;31(3):513–519. doi: 10.1161/ATVBAHA.110.220723. [DOI] [PubMed] [Google Scholar]
- 144.Fu Z, Berhane F, Fite A, Seyoum B, Abou-Samra AB, Zhang R. Elevated circulating lipasin/betatrophin in human type 2 diabetes and obesity. Sci Rep. 2014;4:5013. doi: 10.1038/srep05013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277(3):1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 146.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1(6):361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 147.Lin J, et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1β coactivation of SREBP. Cell. 2005;120(2):261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 148.Chambers KT, et al. PGC-1β and ChREBP partner to cooperatively regulate hepatic lipogenesis in a glucose concentration-dependent manner. Mol Metab. 2013;2(3):194–204. doi: 10.1016/j.molmet.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7(5):251–264. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 150.Chan SM, et al. Activation of PPARα ameliorates hepatic insulin resistance and steatosis in high fructose-fed mice despite increased endoplasmic reticulum stress. Diabetes. 2013;62(6):2095–2105. doi: 10.2337/db12-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54(4):795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang D, et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J Clin Invest. 2017;127(7):2855–2867. doi: 10.1172/JCI89934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Curry DL. Effects of mannose and fructose on the synthesis and secretion of insulin. Pancreas. 1989;4(1):2–9. doi: 10.1097/00006676-198902000-00002. [DOI] [PubMed] [Google Scholar]
- 154.Adams SH, Stanhope KL, Grant RW, Cummings BP, Havel PJ. Metabolic and endocrine profiles in response to systemic infusion of fructose and glucose in rhesus macaques. Endocrinology. 2008;149(6):3002–3008. doi: 10.1210/en.2007-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Blakely SR, Hallfrisch J, Reiser S, Prather ES. Long-term effects of moderate fructose feeding on glucose tolerance parameters in rats. J Nutr. 1981;111(2):307–314. doi: 10.1093/jn/111.2.307. [DOI] [PubMed] [Google Scholar]
- 156.Beck-Nielsen H, Pedersen O, Lindskov HO. Impaired cellular insulin binding and insulin sensitivity induced by high-fructose feeding in normal subjects. Am J Clin Nutr. 1980;33(2):273–278. doi: 10.1093/ajcn/33.2.273. [DOI] [PubMed] [Google Scholar]
- 157.Ter Horst KW, Schene MR, Holman R, Romijn JA, Serlie MJ. Effect of fructose consumption on insulin sensitivity in nondiabetic subjects: a systematic review and meta-analysis of diet-intervention trials. Am J Clin Nutr. 2016;104(6):1562–1576. doi: 10.3945/ajcn.116.137786. [DOI] [PubMed] [Google Scholar]
- 158.Nagai Y, et al. The role of peroxisome proliferator-activated receptor gamma coactivator 1 beta (PGC-1β) in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009;9(3):252–264. doi: 10.1016/j.cmet.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Samuel VT, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279(31):32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 160.Kumashiro N, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A. 2011;108(39):16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Farese RV, Jr , Zechner R, Newgard CB, Walther TC. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab. 2012;15(5):570–573. doi: 10.1016/j.cmet.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017;23(7):804–814. doi: 10.1038/nm.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Templeman NM, et al. Reduced circulating insulin enhances insulin sensitivity in old mice and extends lifespan. Cell Rep. 2017;20(2):451–463. doi: 10.1016/j.celrep.2017.06.048. [DOI] [PubMed] [Google Scholar]
- 164.Davis JD. The effectiveness of some sugars in stimulating licking behavior in the rat. Physiol Behav. 1973;11(1):39–45. doi: 10.1016/0031-9384(73)90120-0. [DOI] [PubMed] [Google Scholar]
- 165.Avena NM. Examining the addictive-like properties of binge eating using an animal model of sugar dependence. Exp Clin Psychopharmacol. 2007;15(5):481–491. doi: 10.1037/1064-1297.15.5.481. [DOI] [PubMed] [Google Scholar]
- 166.Wideman CH, Nadzam GR, Murphy HM. Implications of an animal model of sugar addiction, withdrawal and relapse for human health. Nutr Neurosci. 2005;8(5–6):269–276. doi: 10.1080/10284150500485221. [DOI] [PubMed] [Google Scholar]
- 167.Spangler R, Wittkowski KM, Goddard NL, Avena NM, Hoebel BG, Leibowitz SF. Opiate-like effects of sugar on gene expression in reward areas of the rat brain. Brain Res Mol Brain Res. 2004;124(2):134–142. doi: 10.1016/j.molbrainres.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 168.Tellez LA, et al. Separate circuitries encode the hedonic and nutritional values of sugar. Nat Neurosci. 2016;19(3):465–470. doi: 10.1038/nn.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shapiro A, Mu W, Roncal C, Cheng KY, Johnson RJ, Scarpace PJ. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am J Physiol Regul Integr Comp Physiol. 2008;295(5):R1370–R1375. doi: 10.1152/ajpregu.00195.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chotiwat C, Sharp C, Teff K, Harris RBS. Feeding a high-fructose diet induces leptin resistance in rats. Appetite. 2007;49(1):284 [Google Scholar]
- 171.Teff KL, et al. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J Clin Endocrinol Metab. 2004;89(6):2963–2972. doi: 10.1210/jc.2003-031855. [DOI] [PubMed] [Google Scholar]
- 172.Luo S, Monterosso JR, Sarpelleh K, Page KA. Differential effects of fructose versus glucose on brain and appetitive responses to food cues and decisions for food rewards. Proc Natl Acad Sci U S A. 2015;112(20):6509–6514. doi: 10.1073/pnas.1503358112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Page KA, et al. Effects of fructose vs glucose on regional cerebral blood flow in brain regions involved with appetite and reward pathways. JAMA. 2013;309(1):63–70. doi: 10.1001/jama.2012.116975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5(6):426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 175.Dushay JR, Toschi E, Mitten EK, Fisher FM, Herman MA, Maratos-Flier E. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol Metab. 2015;4(1):51–57. doi: 10.1016/j.molmet.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Inagaki T, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5(6):415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 177.Sánchez J, Palou A, Picó C. Response to carbohydrate and fat refeeding in the expression of genes involved in nutrient partitioning and metabolism: striking effects on fibroblast growth factor-21 induction. Endocrinology. 2009;150(12):5341–5350. doi: 10.1210/en.2009-0466. [DOI] [PubMed] [Google Scholar]
- 178.Laeger T, et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest. 2014;124(9):3913–3922. doi: 10.1172/JCI74915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dushay J, et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology. 2010;139(2):456–463. doi: 10.1053/j.gastro.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chavez AO, Molina-Carrion M, Abdul-Ghani MA, Folli F, Defronzo RA, Tripathy D. Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care. 2009;32(8):1542–1546. doi: 10.2337/dc09-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fisher FM, et al. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes. 2010;59(11):2781–2789. doi: 10.2337/db10-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Iizuka K, Takeda J, Horikawa Y. Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett. 2009;583(17):2882–2886. doi: 10.1016/j.febslet.2009.07.053. [DOI] [PubMed] [Google Scholar]
- 183.Fisher FM, et al. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol Metab. 2017;6(1):14–21. doi: 10.1016/j.molmet.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.von Holstein-Rathlou S, et al. FGF21 mediates endocrine control of simple sugar intake and sweet taste preference by the liver. Cell Metab. 2016;23(2):335–343. doi: 10.1016/j.cmet.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Talukdar S, et al. FGF21 regulates sweet and alcohol preference. Cell Metab. 2016;23(2):344–349. doi: 10.1016/j.cmet.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tanaka T, et al. Genome-wide meta-analysis of observational studies shows common genetic variants associated with macronutrient intake. Am J Clin Nutr. 2013;97(6):1395–1402. doi: 10.3945/ajcn.112.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chu AY, et al. Novel locus including FGF21 is associated with dietary macronutrient intake. Hum Mol Genet. 2013;22(9):1895–1902. doi: 10.1093/hmg/ddt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Singh AK, et al. Fructose-induced hypertension: essential role of chloride and fructose absorbing transporters PAT1 and Glut5. Kidney Int. 2008;74(4):438–447. doi: 10.1038/ki.2008.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Johnson RJ, et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension. 2003;41(6):1183–1190. doi: 10.1161/01.HYP.0000069700.62727.C5. [DOI] [PubMed] [Google Scholar]
- 190. World Health Organization. Guideline: Sugars Intake For Adults and Children. WHO Website. http://www.who.int/nutrition/publications/guidelines/sugars_intake/en/ Accessed December 12, 2017.
- 191.Johnson RK, et al. Dietary sugars intake and cardiovascular health: a scientific statement from the American Heart Association. Circulation. 2009;120(11):1011–1020. doi: 10.1161/CIRCULATIONAHA.109.192627. [DOI] [PubMed] [Google Scholar]
- 192.Batis C, Rivera JA, Popkin BM, Taillie LS. First-year evaluation of Mexico’s tax on nonessential energy-dense foods: an observational study. PLoS Med. 2016;13(7):e1002057. doi: 10.1371/journal.pmed.1002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Falbe J, Thompson HR, Becker CM, Rojas N, McCulloch CE, Madsen KA. Impact of the Berkeley excise tax on sugar-sweetened beverage consumption. Am J Public Health. 2016;106(10):1865–1871. doi: 10.2105/AJPH.2016.303362. [DOI] [PMC free article] [PubMed] [Google Scholar]