

Abstract
Background
Maternal diabetes induces neural tube defects (NTDs), and oxidative stress is a causal factor for maternal diabetes-induces NTDs. The redox gene Nrf2 (nuclear factor-erythroid 2-related factor 2) is the master regulator of the cellular antioxidant system.
Objective
In the present study, we aimed to determine whether maternal diabetes inhibits Nrf2 expression and Nrf2-controlled antioxidant genes through the redox-sensitive miR-27a.
Study design
We used a well-established type 1 diabetic embryopathy mouse model induced by streptozotocin for our in vivo studies. Embryos at E8.5 were harvested for analysis of Nrf2, Nrf2-controlled antioxidant genes and miR-27a expression. To determine if mitigating oxidative stress inhibits the increase of miR-27a and the decrease of Nrf2 expression, we induced diabetic embryopathy in SOD2 (mitochondrial-associated antioxidant gene)-overexpressing mice. This model exhibits reduced mitochondria reactive oxygen species even in the presence of hyperglycemia. To investigate the causal relationship between miR-27a and Nrf2 in vitro, we examined C17.2 neural stem cells under normal and high glucose conditions.
Results
We observed that the mRNA and protein levels of Nrf2 were significantly decreased in E8.5 embryos from diabetic dams compared to those from nondiabetic dams. High glucose also significantly decreased Nrf2 expression in a dose- and time-dependent manner in cultured neural stem cells. Our data revealed that miR-27a was up-regulated in E8.5 embryos exposed to diabetes, and that high glucose increased miR-27a levels in a dose- and time-dependent manner in cultured neural stem cells. In addition, we found that a miR-27a inhibitor abrogated the inhibitory effect of high glucose on Nrf2 expression, and a miR-27a mimic suppressed Nrf2 expression in cultured neural stem cells. Furthermore, our data indicated that the Nrf2-controlled antioxidant enzymes glutamate-cysteine ligase catalytic subunit (GCLC), glutamate-cyteine ligase modifier subunit (GLCM), and glutathione S-transferase A1 (GSTA1) were downregulated by maternal diabetes in E8.5 embryos and high glucose in cultured neural stem cells. Inhibiting miR-27a restored expression of GCLC, GLCM and GSTA1. Overexpressing SOD2 reversed the maternal diabetes-induced increase of miR-27a and suppression of Nrf2 and Nrf2-controlled antioxidant enzymes.
Conclusions
Our study demonstrates that maternal diabetes-induced oxidative stress increases miR-27a, which, in turn, suppresses Nrf2 and its responsive antioxidant enzymes, resulting in diabetic embryopathy.
Keywords: maternal diabetes, embryopathy, oxidative stress, miR-27a, Nrf2
Introduction
It has been well established that maternal diabetes increases the risk of neural tube defects (NTDs) in offspring1–11. The clinical data show that approximate 8,000 babies born each year in the United States have birth defects in type 1 or 2 diabetic pregnancies11–13. Data from the National Birth Defects Prevention Study shows that the incidence for newborn NTDs are up to 10 times more frequent in women with pregestational diabetes compared to women who never had diabetes or who developed diabetes late in pregnancy (such as gestational diabetes mellitus)14. Maternal diabetes-induced hyperglycemia in the developing embryo has been identified as an adverse factor that impacts embryogenesis and leads to NTD formation1, 2, 5–7. Although evidence from clinical and experimental studies supports the theory that hyperglycemia enhances the generation of reactive oxygen species (ROS) and oxidative stress in developing embryos7, 11, the precise steps by which this occurs are still not fully understood.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a redox sensitive transcription factor and one of the most important cellular defense mechanisms for combating oxidative stress through its ability to regulate phase II detoxifying enzymes and antioxidant proteins15, 16. Many endogenous enzymes that catalyze antioxidant reactions are under the control of Nrf2, including heme oxygenase-1 (HO-1), NAD(P)H dehydrogenase (quinone) 1 (NQO-1), glutathione peroxidase-1 (GPx-1), glutamate-cysteine ligase modifier subunit (GLCM), glutamate-cysteine ligase catalytic subunit (GCLC), and glutathione S-transferase A1 (GSTA1)17. Inhibiting Nrf2 is associated with a reduction of the antioxidant ability of the cell15. Dysregulation of Nrf2 has been involved in many diseases, including alcoholic liver disease, cancer, chronic obstructive pulmonary disease, and neurodegenerative diseases15. It has been shown that suppression of Nrf2 activity leads to oxidative stress-induced insulin resistance in adult cardiomyocytes in the diabetic heart18. Therefore, we wanted to investigate whether inhibition of Nrf2 by maternal diabetes leads to an abundance of ROS and oxidative stress in the developing embryo.
Regulation of Nrf2 can be divided into Kelch-like ECH-associated protein 1 (Keap1)-dependent and Keap1-independent mechanisms19. In normal conditions, the Nrf2 protein level in the cell is maintained at a low level to prevent constitutive activation of the oxidative stress response by its inhibitor Keap1, which sequesters Nrf2 in the cytosol and facilitates its degradation through the proteasome19. Under stressed conditions, the cysteine residues in Keap1 are modified. These modifications lead to a conformational change of Keap1 and result in the release of Nrf2 and disturbed transfer of ubiquitin to Nrf2, ultimately preventing Nrf2 degradation19. Despite the above-mentioned regulation of Nrf2 via Keap1, emerging evidence demonstrates that Nrf2 can be regulated independently of Keap119, 20. The emerging evidence shows that some miRNAs have been shown to be involved in the regulation of Nrf2, including miR-28, -34, -200, and -14420.
In the present study, we observed that miR-27a can regulate Nrf2 expression at the post-transcriptional level and plays a critical role in diabetic embryopathy. Hyperglycemia-increased miR-27a directly affects Nrf2 mRNA stability and results in decreased Nrf2 protein levels, as well as decreased levels of Nrf2-regulated antioxidant genes, including GLCM, GCLC, and GSTA1. Our data reveal a new mechanism by which maternal diabetes induces oxidative stress via the miR-27a-Nrf2 pathway in the developing embryo.
Because oxidative stress is the causal factor for maternal diabetes-induced NTDs7, 11, revealing the role of Nrf2 in diabetic embryopathy will possibly lead to the discovery of new and novel therapeutics for the treatment of this disease. Clinical trials have shown disappointing results in the use of general antioxidants in treating diabetic complications21. Nrf2 is the master regulator of redox homeostasis, and several Nrf2 activators have displayed their potential to activate the Nrf2 pathway and reduce oxidative stress in diabetic complications21, 22. Our study will provide the mechanistic basis for the use of Nrf2 activators as an alternative for the treatment of maternal diabetes-induced NTDs.
Methods and materials
Study design
We used the well-established type 1 diabetic embryopathy mouse model induced by streptozotocin for in vivo studies. Embryos at E8.5 were harvested for analysis of Nrf2, Nrf2-controlled antioxidant genes and miR-27a expression. To determine if mitigating oxidative stress inhibits the increase of miR-27a and the decrease of Nrf2 expression by maternal diabetes, we induced diabetic embryopathy in SOD2 (mitochondrial-associated antioxidant gene)-overexpressing mice. This model exhibits reduced mitochondrial reactive oxygen species (ROS) in the presence of hyperglycemia23. To investigate the relationship between miR-27a and Nrf2 in vitro, we examined C17.2 neural stem cells under normal and high glucose conditions.
Animals
All procedures for animal use were approved by the Institutional Animal Care and Use Committee of University of Maryland School of Medicine. Wild-type (WT) C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). SOD2 transgenic (SOD2-Tg) mice, which overexpress human mitochondrial SOD2 under the β-actin promoter, were obtained from Dr. Robia Pautler at Baylor College of Medicine24.
Model of diabetic embryopathy and morphological assessment of NTDs
We8, 10, 25–29 and others30–32 have used a rodent model of streptozotocin (STZ)-induced diabetes to study diabetic embryopathy. Briefly, ten-week-old WT female mice were intravenously injected daily with 75 mg/kg streptozotocin for two days to induce diabetes. Streptozotocin from Sigma (St. Louis, MO) was dissolved in 0.1 M citrate buffer (pH 4.5). We used a U-100 insulin syringe (Becton Dickinson, Franklin Lakes, NJ) with 281/2-G needles for injections. Approximately 140 μl of STZ solution was injected per mouse. Diabetes was defined as a 12-hour fasting blood glucose level of ≥ 16.7 mM. Male and female mice were paired at 3:00 P.M., and day 0.5 (E0.5) of pregnancy was established at noon on the day that a vaginal plug was present. Embryos were harvested at embryonic day (E) E8.5 (2:00 PM at E8.5) for biochemical and molecular analyses.
Cell culture and treatment
C17.2 mouse neural stem cells (European Collection of Cell Culture, UK) were maintained in DMEM (5 mM glucose) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2. C17.2 cells are newborn mouse cerebellar progenitor cells transformed with retroviral v-myc33. Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was used according to the manufacturer’s protocol for the transfection of miR-27a mimic or inhibitor (Thermo Scientific, Waltham, MA). To investigate the possible effect of miR-27a on Nrf2 and its target gene expression, miR-27a inhibitor or mimic was transfected for 48 hours with or without high glucose (25 mM), and then, cells were harvested for subsequent analysis.
Immunoblotting
Equal amounts of protein (30 or 50 μg) from cultured cells and embryos were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto Immunobilon-P membranes (Millipore, Billerica, MA). Membranes were incubated in 5% nonfat milk for 1 hour, and then incubated for 18 hours at 4°C with Nrf2 primary antibodies (Cell Signaling Technology, Danvers, MA) at dilutions of 1:1000 in 5% nonfat milk. Membranes were then exposed to goat anti-rabbit secondary antibodies. To ensure that equivalent amounts of protein were loaded, membranes were stripped and probed with a mouse antibody against β-actin (1:5000; Abcam, Cambridge, UK). Signals were detected using the SuperSignal West Femto Maximum Sensitivity Substrate kit (Thermo Scientific, Waltham, MA). Quantification of blots was performed using VisionWorksLS software (UVP Company, Upland, CA). All experiments were repeated in triplicate.
RNA extraction and real-time quantitative PCR (RT-qPCR)
Total RNA was isolated from cells using Trizol reagent (Thermo Scientific, Waltham, MA) and reverse transcribed using the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) for mRNA. Reverse transcription of miRNA was performed using a qScript microRNA cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, MD). RT-qPCR for Nrf2, GCLC, GLCM, GSTA1, β-actin, miR-27a and small nuclear RNA U6 was performed using the Maxima SYBR Green/ROX qPCR Master Mix assay (Thermo Scientific, Waltham, MA). The primers for RT-qPCR are listed in Table 1. RT-qPCR and subsequent calculations were performed by a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA).
Table 1.
Primer sequences used in RT-qPCR.
| Primer name | Primer source | Primer sequence |
|---|---|---|
| Nrf2 F | PrimerBank | 5′TAGATGACCATGAGTCGCTTGC3′ |
| Nrf2 R | PrimerBank | 5′GCCAAACTTGCTCCATGTCC3′ |
| GSTA1 F | PrimerBank | 5′AAGCCCGTGCTTCACTACTTC3′ |
| GSTA1 R | PrimerBank | 5′GGGCACTTGGTCAAACATCAAA3′ |
| GCLC F | PrimerBank | 5′GGGGTGACGAGGTGGAGTA3′ |
| GCLC R | PrimerBank | 5′GTTGGGGTTTGTCCTCTCCC3′ |
| GLCM F | PrimerBank | 5′AGGAGCTTCGGGACTGTATCC3′ |
| GLCM R | PrimerBank | 5′GGGACATGGTGCATTCCAAAA3′ |
| mmu - miR - 27a - 3p | Designed by ourselves | 5′TTCACAGTGGCTAAGTTCCGC3′ |
| U6 F | Designed by ourselves | 5′CTCGCTTCGGCAGCACA3′ |
| U6 R | Designed by ourselves | 5′AACGCTTCACGAATTTGCGT3′ |
| β-actin F | Designed by ourselves | 5′GTGACGTTGACATCCGTAAAGA3′ |
| β-actin R | Designed by ourselves | 5′ GCCGGACTCATCGTACTCC3′ |
| Universal primer for miRNA | From Kit | Request from Life technologies |
F: forward; R: reverse; mmu: murine.
Statistical analyses
All experiments were repeated in triplicate. Data are presented as the means ± standard errors (SE). Student’s t-test was used for comparisons between two groups. One-way or two-way ANOVA was performed for more than two group comparisons using the SigmaPlot 12.5 software (SigmaStat). In ANOVA analysis, Tukey’s test was used to estimate significance. Statistical significance was indicated when P < 0.05.
Results
Maternal diabetes in vivo or high glucose in vitro decreases Nrf2 expression
To investigate the possible role of Nrf2 in diabetic embryopathy, we measured Nrf2 expression in the developing embryo. Maternal diabetes significantly decreased Nrf2 mRNA and protein levels in the developing embryo (E8.5) (Fig. 1A and B). To identify the role of fetal hyperglycemia in the suppression of Nrf2, C17.2 neural stem cells were treated with normal (5 mM) or high (16.7, 25, 33.3 mM) glucose, and then, Nrf2 expression was determined. Our experiments confirmed that inhibition of Nrf2 expression resulted from fetal hyperglycemia because high glucose inhibited Nrf2 mRNA and protein levels in a dose- and time-dependent manner (Fig. 1 C–F). Mannitol was used for the osmotic control of glucose. High mannitol concentrations did not affect Nrf2 expression (Fig. 1G and H).
Figure 1. Maternal diabetes in vivo or high glucose in vitro decreases Nrf2 expression.
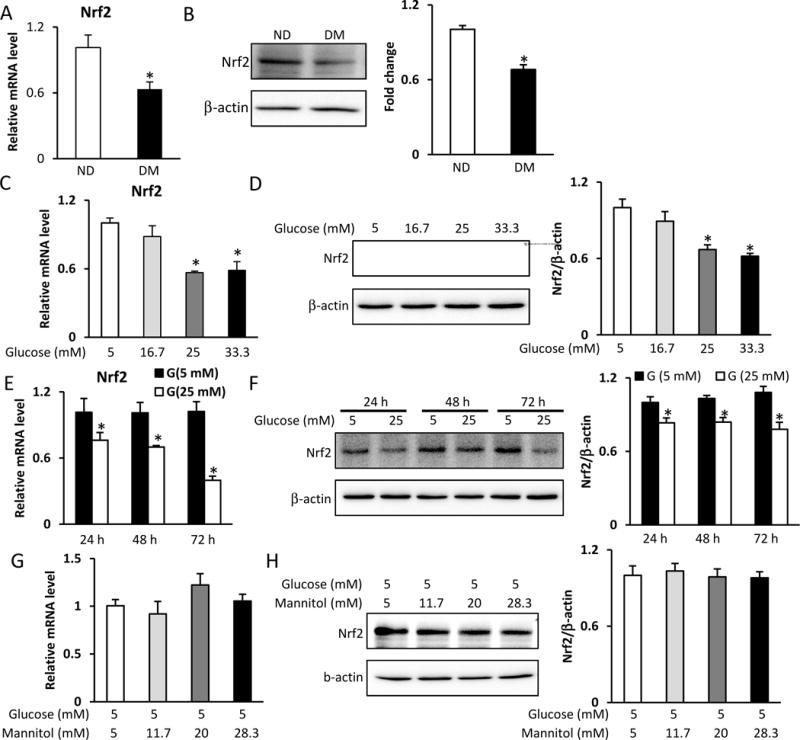
A. Nrf2 mRNA levels in E8.5 embryos assessed by RT-qPCR. B. Nrf2 protein levels in E8.5 embryos assessed by Western blot. C. Nrf2 mRNA levels in C17.2 cells treated with normal (5 mM) or high (16.7, 25, 33.3 mM) glucose for 48 h, assessed by RT-qPCR. D. Nrf2 protein levels in C17.2 cells treated with normal (5 mM) or high (16.7, 25, 33.3 mM) glucose for 48 h, assessed by Western blot. E. Nrf2 mRNA levels in C17.2 cells treated with normal (5 mM) or high (25 mM) glucose for 24 h, 48 h, and 72 h, assessed by RT-qPCR. F. Nrf2 protein levels in C17.2 cells treated with normal (5 mM) or high (25 mM) glucose for 24 h, 48 h, and 72 h, assessed by Western blot. G. Nrf2 mRNA levels in C17.2 cells treated with normal glucose (5 mM) or high mannitol (11.7, 20, 28.3 mM) for 48 h, assessed by RT-qPCR. H. Nrf2 protein levels in C17.2 cells treated with normal glucose (5 mM) or high mannitol (11.7, 20, 28.3 mM), assessed by Western blot. Experiments were repeated using 3 embryos (N =3) from different dams. Experiments were repeated three times. Bar graphs for protein levels show quantitative data from three independent experiments. ND: nondiabetic dams; DM: diabetic dams; G: glucose. * indicates significant difference (P < 0.05) compared with other groups.
Maternal diabetes in vivo or high glucose in vitro increases miR-27a expression
To search for possible miRNAs that may regulate Nrf2 expression, the miRNA target gene prediction tool miRanda (http://www.microrna.org) was used. The prediction showed that miR-27a is a potential regulator of Nrf2 through imperfect complementation with Nrf2 mRNA in the 3′UTR region (Fig. 2A). Next, we examined whether maternal diabetes in vivo or high glucose in vitro affected expression of miR-27a. Maternal diabetes significantly upregulated expression of miR-27a in the developing embryo (E8.5) (Fig. 2B). At the same time, high glucose also significantly increased miR-27a expression in a dose- and time-dependent manner in cultured C17.2 neural stem cells (Fig. 2C and D). High mannitol concentrations did not affect miR-27a expression (Fig. 2E).
Figure 2. Maternal diabetes in vivo or high glucose in vitro decreases miR-27a expression.
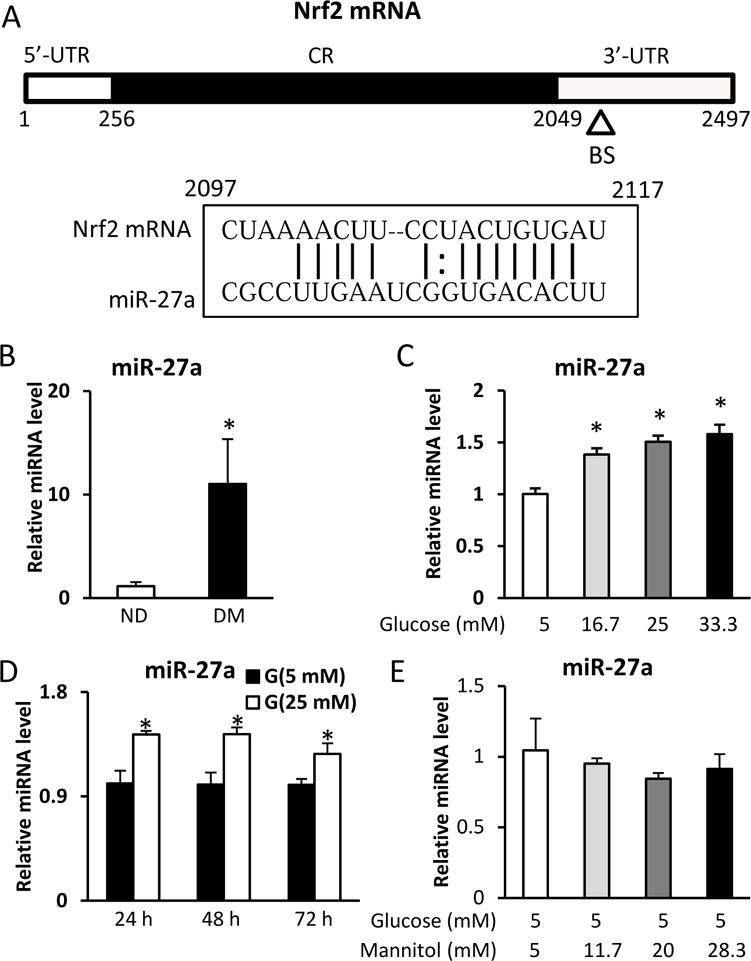
A. Schematic representation of the Nrf2 mRNA depicting the binding site for miR-27a in its 3′-UTR. CR: coding region; BS: binding site; UTR: untranslated region. B. miR-27a levels in E8.5 embryos assessed by RT-qPCR. C. miR-27a levels in C17.2 cells treated with normal (5 mM) or high (16.7, 25, 33.3 mM) glucose for 48 h, assessed by RT-qPCR. D. miR-27a levels in C17.2 cells treated with normal (5 mM) or high (25 mM) glucose for 24 h, 48 h, and 72 h, assessed by RT-qPCR. E. miR-27a levels in C17.2 cells treated with normal glucose (5 mM) or high mannitol (11.7, 20, 28.3 mM) for 48 h, assessed by RT-qPCR. Experiments were repeated using 3 embryos (N =3) from different dams. Experiments were repeated three times. ND: nondiabetic dams; DM: diabetic dams. * indicates significant difference (P < 0.05) compared with other groups.
Nrf2 is the target gene of miR-27a
To determine if miR-27a plays a role in the suppression of Nrf2 by maternal diabetes or high glucose, we used a miR-27a inhibitor or mimic to manipulate the levels of miR-27a in neural stem cells. High glucose (25 mM) increased miR-27a expression and decreased Nrf2 mRNA levels, whereas transfection of miR-27a inhibitor blocked high glucose-increased miR-27a expression and restored high glucose-suppressed Nrf2 mRNA levels in C17.2 neural stem cells (Fig. 3A and B). Moreover, miR-27a inhibitor reversed high glucose-blocked Nrf2 protein levels in C17.2 neural stem cells (Fig. 3C). In addition, transfection of miR-27a mimic increased miR-27a expression and decreased Nrf2 mRNA and protein levels in C17.2 neural stem cells (Fig. 3D–F). Thus, in vitro experiments support the conclusion that high glucose suppressed Nrf2 expression through miR-27a in neural stem cells.
Figure 3. High glucose-increased miR-27a suppresses Nrf2 expression.
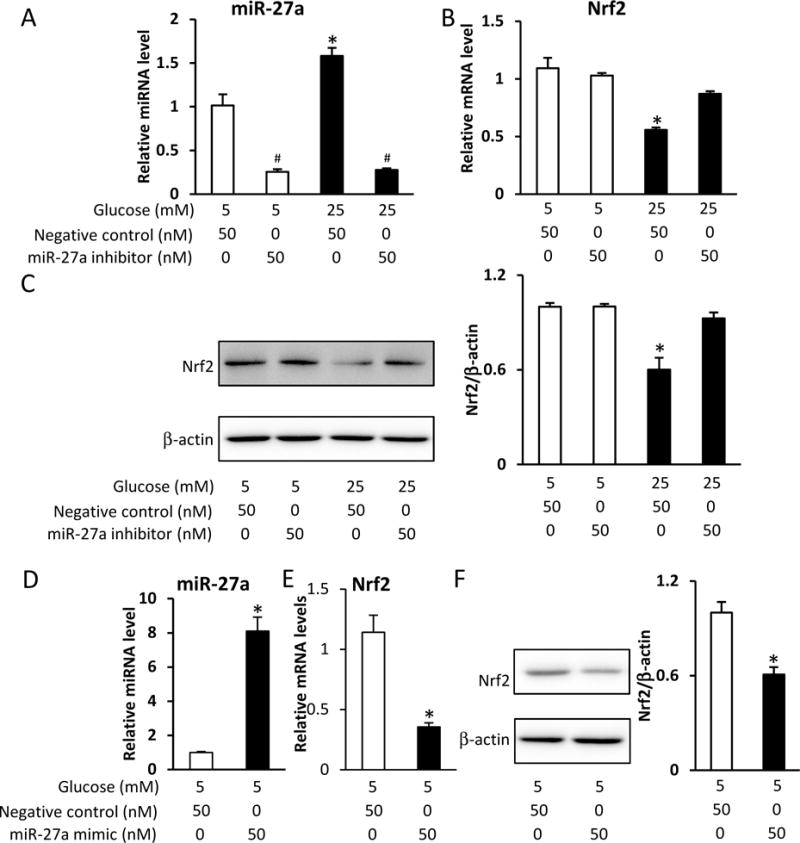
A. miR-27a levels in C17.2 cells treated with normal (5 mM) or high (25 mM) glucose combined with miR-27a inhibitor for 48 h. B. Nrf2 mRNA levels in C17.2 cells treated with normal (5 mM) or high (25 mM) glucose, combined with miR-27a inhibitor for 48 h. C. Nrf2 protein levels in C17.2 cells treated with normal (5 mM) or high (25 mM) glucose, combined with miR-27a inhibitor for 48 h. D. miR-27a levels in C17.2 cells transfected with miR-27a mimic for 48 h. E. Nrf2 mRNA levels in C17.2 cells transfected with miR-27a mimic for 48 h. F. Nrf2 protein levels in C17.2 cells transfected with miR-27a mimic for 48 h. Experiments were repeated three times. Bar graphs for protein levels show quantitative data from three independent experiments. * indicates a significant difference (P < 0.05) compared with other groups. # indicates significant difference (P < 0.05) compared to control group.
The miR-27a-Nrf2 circuit suppresses the expression of Nrf2 responsive genes
Many genes are controlled by Nrf217. In this study, we surveyed these genes and found that three genes (GSTA1, GCLC and GLCM) were inhibited by maternal diabetes in the developing embryo (E8.5) (Fig. 4A). At the same time, high glucose inhibited GSTA1, GCLC and GLCM in a dose-dependent manner in C17.2 neural stem cells (Fig. 4B). Because we demonstrated that miR-27a mediates high glucose-suppressed Nrf2 expression, we tested whether miR-27a mediates high glucose-inhibited Nrf2-controlled gene expression. Indeed, transfection of miR-27a inhibitor significantly restored high glucose-suppressed GSTA1, GCLC and GLCM expression in C17.2 neural stem cells (Fig. 4C). Therefore, our data indicate that miR-27a mediates high glucose-inhibited Nrf2-controlled gene expression in neural stem cells.
Figure 4. High glucose inhibits Nrf2-controlled gene expression through miR-27a.
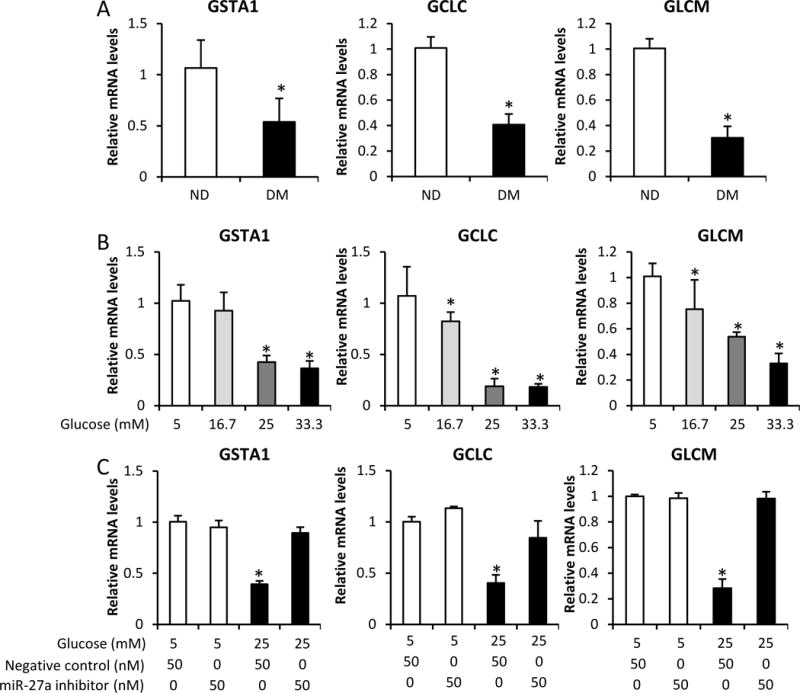
A. mRNA levels of GSTA1, GCLC, and GLCM in E8.5 embryos, assessed by RT-qPCR. B. mRNA levels of GSTA1, GCLC, and GLCM in C17.2 cells treated with normal (5 mM) or high (16.7, 25, 33.3 mM) glucose for 48 h, assessed by RT-qPCR. C. mRNA levels of GSTA1, GCLC, and GLCM in C17.2 cells treated with normal (5 mM) or high (25 mM) glucose, combined with miR-27a inhibitor, for 48 h, assessed by RT-qPCR. Experiments were repeated three times. ND: nondiabetic dams; DM: diabetic dams. * indicates a significant difference (P < 0.05) compared with other groups.
Oxidative stress contributes to the increase of miR-27a and inhibition of Nrf2 expression in diabetic pregnancy
It has been demonstrated that maternal diabetes impairs mitochondrial function by triggering electron leakage and causing ROS production8, 23, 34, 35. Theoretically, Nrf2, as a sensor of ROS, should be increased and promote the expression of antioxidant genes under diabetic conditions. However, Nrf2 does not respond to oxidative stress under hyperglycemic conditions. Therefore, we hypothesized that maternal diabetes causes long-term oxidative stress that may inhibit Nrf2 expression according to an epigenetic mechanism. In the present study, we identified miR-27a as an epigenetic factor that affects Nrf2 expression, as well as its downstream genes. Our previous data demonstrated that overexpression of superoxide dismutase 2 (SOD2), a mitochondrial-located antioxidant gene, blocks maternal diabetes-induced oxidative stress and significantly reduces the incidence of maternal diabetes-induced NTD formation from 25% to 4%23. It is possible that elimination of mitochondrial-generated ROS may reactivate Nrf2 and its downstream genes through the epigenetic factor miR-27a. Our data demonstrates that overexpression of SOD2 blocks the increase of miR-27a by maternal diabetes (Fig. 5A), as well as restores Nrf2 mRNA expression (Fig. 5B) and protein levels (Fig. 5C) in the developing embryo (E8.5), indicating that mitochondrial-generated ROS results in an increase of miR-27a and inhibition of Nrf2. Furthermore, overexpression of SOD2 also restores expression of GSTA1, GCLC and GLCM in the developing embryo (Fig. 5D). Thus, our data indicate that mitochondrial-generated ROS worsens oxidative stress in diabetic embryos through miR-27a-mediated inhibition of Nrf2 expression.
Figure 5. Mitochondrial dysfunction contributes to the increase of miR-27a and the inhibition of Nrf2 expression.
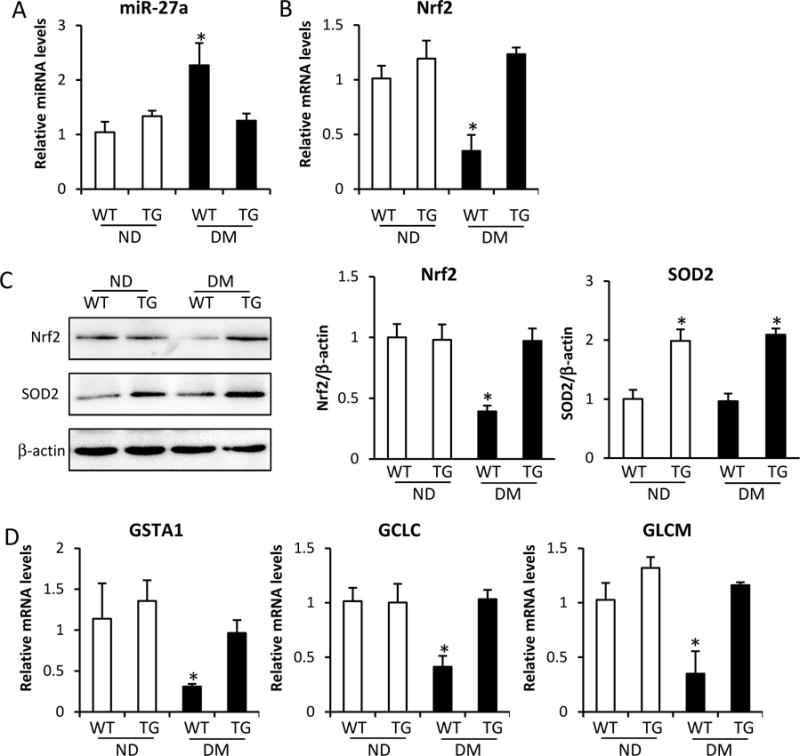
A. miR-27a levels in E8.5 embryos assessed by RT-qPCR. B. Nrf2 mRNA levels in E8.5 embryos assessed by RT-qPCR. C. Nrf2 and SOD2 protein levels in E8.5 embryos assessed by Western blot. D. mRNA levels of GSTA1, GCLC, and GLCM in E8.5 embryos assessed by RT-qPCR. Experiments were repeated using 3 embryos (N =3) from different dams. Experiments were repeated three times. Bar graphs for protein levels show quantitative data from three independent experiments. ND: nondiabetic dams; DM: diabetic dams; WT: wild type; TG: SOD2 transgenic mice. * indicates significant difference (P < 0.05) compared with other groups.
Comment
In the present study, we elucidate a mechanism by which maternal diabetes induces oxidative stress in the developing embryo, thereby causing NTDs. We observed that hyperglycemia in vivo and high glucose in vivo increases expression of the epigenetic factor miR-27a. MiR27a inhibits expression of Nrf2, a major cellular defense transcription factor, and suppresses the overall antioxidant capability of cells. Our results reveal a novel pathway that regulates the cellular redox balance in maternal diabetes-induced embryopathy. We also demonstrated that maternal diabetes inhibits expression of three antioxidant enzymes, GSTA1, GCLC and GLCM, via the miR-27a-Nrf2 pathway to trigger excessive ROS production in the developing embryo.
Nrf2 is subject to complex regulatory mechanisms at both the transcriptional and posttranscriptional levels15–17, 19, 20. The Nrf2 promoter contains two ARE-like sequences that regulate its expression at the transcriptional level36, and Nrf2 protein stability is controlled by Keap1, β-TrCP, and GSK3 at the posttranscriptional level17. In addition, new evidence has demonstrated that Nrf2 mRNA is regulated by various miRNAs20. Eighty-five miRNAs have been predicted to bind to Nrf2 mRNA to downregulate its translation37. Several miRNAs have been shown to directly bind to the 3′UTR of mRNA, including miR-153, -27a, miR-142, and miR-144 in neuronal SH-SY5Y cells38; miR-144 in lymphoblast K562 cells, primary human erythroid progenitor cells, and reticulocytes39; miR-28 in human mammary epithelial cells and breast cancer MCF-7 cells40; and miR-34a in human embryonic kidney HEK293 cells41, have been shown to directly bind to the 3′UTR of mRNA. In addition to the direct downregulation of Nrf2 by miRNAs, miR-200a can target Nrf2 inhibitor Keap1 to affect Nrf2 levels42. Here, we reveal that miR-27a in neuroepithelial cells is increased by maternal diabetes, where it then suppresses Nrf2 expression and contributes to the overproduction of ROS and oxidative stress in the developing embryo.
Mitochondrial damage is thought to be the main source of ROS production observed in various diseases43. Previous studies have demonstrated that developing embryos exposed to hyperglycemia caused by maternal diabetes exhibit morphological damage to mitochondria and long-term excessive ROS production8, 34, 35. We have also shown that repairing mitochondria damaged by high glucose, through overexpression of SOD2, alleviates oxidative stress, indicating that mitochondria are the source of ROS production in diabetic embryopathy23. In this study, we found that maternal diabetes inhibits Nrf2, which should be activated in response to oxidative stress induced by hyperglycemia, by increasing the expression of miR27a. We also found that overexpressing SOD2 in diabetic embryos reduces miR-27a expression and restores Nrf2 expression, compared to the diabetic WT groups. This means that oxidative stress itself results in inhibition of Nrf2 expression through oxidative stress-sensitive miR-27a. We hypothesize that acute oxidative stress may increase Nrf2 expression, thereby eliminating ROS production, but that the long-term oxidative stress caused by maternal diabetes cannot stimulate Nrf2 expression and enhances ROS production in embryos.
Dysregulation of Nrf2 has been shown to be involved in the etiology of diabetes, such as pancreatic islet beta cell dysfunction44 and insulin resistance18, 44, and its complications, including cardiomyopathy44–46, nephropathy44, 45, 47, retinopathy48 and atherosclerosis49. Emerging evidence suggests that the dysregulation of Nrf2 caused by maternal diabetes impairs embryogenesis and placenta development. It has been demonstrated that gestational diabetes impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero50. Insufficient activation of Nrf2 contributes to maternal diabetes-induced renal dysmorphogenesis by increasing renal ROS production in the offspring51. In addition, Nrf2 signaling is involved in maternal diabetes-induced defects in the development of the mouse placenta52. Prepregnancy maternal diabetes combined with obesity impairs placental mitochondrial function, increases oxidative stress of the placenta induced by the Nrf2 pathway and detrimentally alters the metabolism of offspring53. Our data further demonstrate that maternal diabetes-induced oxidative stress results from inhibition of Nrf2 by the epigenetic factor miR-27a in the developing embryo.
Nrf2 regulates the cellular redox balance by controlling expression of antioxidant enzymes17. In the present study, we observed that maternal diabetes suppresses three Nrf2-controlled genes, GSTA1, GCLC and GLCM. GCLC and GLCM are used to synthesize cellular antioxidant glutathione17. Glutathione is a tripeptide antioxidant that directly scavenges ROS within the cell by donating an electron from two molecules of reduced glutathione (GSH), followed by GSH oxidation to oxidized glutathione (GSSG)17. It has been reported that hyperglycemia in vivo or high glucose in vitro lead to decreased concentrations of GSH as well as decreased activity of the rate-limiting GSH-synthesizing enzyme γ-glutamylcysteine synthetase (γ-GCS) in the embryonic tissues of diabetic pregnancy or cultured embryos54, 55. Our data demonstrate that suppression of GCLC and GLCM by maternal diabetes also contributes to the depletion of glutathione in diabetic embryos. GSTA1 is major phase II detoxification enzyme17. In addition to this function, GSTA1 has glutathione peroxidase activity, which uses GSH to reduce H2O217. Previous studies showed that excessive H2O2 accumulation in diabetic embryos may result from the inhibition of GSTA156.
Clinical significance and study limitations
Experiments in animal models have clearly shown that oxidative stress is responsible for maternal diabetes-induced NTDs1, 7, 11, 56–61 and congenital heart defects62, 63, and clinical research has demonstrated that oxidative stress is involved in various adverse pregnancy outcomes64, 65. For example, oxidative stress induced by tobacco smoke is associated with preterm delivery, intrauterine growth restriction, stillbirth, low birth weight, aberrant placental metabolism, syncytial knot formation, and multiple markers of oxidative damage66, 67. A case-control study showed that maternal oxidative stress may be an important contributor to preterm birth, regardless of subtype and timing of exposure during pregnancy68. Another study has shown that oxidative stress markers are found repeatedly in preeclamptic pregnancies, compared with normotensive pregnancies69, 70. Although two studies have reported that Nrf2 dysregulation in deciduas or placentas from preeclamptic patients71, 72, it is still unclear whether impaired Nrf2 function contributes to adverse pregnancy outcomes in various maternal conditions73–77, Therefore, further work is needed to determine the potential involvement of the Nrf2 signaling pathway in the induction of adverse pregnancy outcomes.
Based on the importance of Nrf2 in the antioxidant system of the cell, as well as its association with the etiology of diabetic complications, many Nrf2 activators are being developed as therapeutic agents44–46, 78. For example, the Nrf2 activators sulforaphane and cinnamic aldehyde have been shown to significantly attenuate diabetes-associated metabolic disorders and relieve renal damage in mice79. Another study has revealed that the Nrf2 inducer MG132 can reduce diabetic kidney disease in mice80. A previous study from our laboratory revealed that the Nrf2 activator vinylsulfone reduces high glucose-induced neural tube defects by suppressing cellular stress and apoptosis in cultured mouse embryo81. Taken together, these data indicate that Nrf2 could be a therapeutic target for treating maternal diabetes-associated birth defects.
The limitation of this study is that all data were obtained from animal models. Due to the ethics of research, human fetal samples are difficult to obtain for experiments. Translational studies using nonhuman primates may be carried out in the near future. Future studies may assess the therapeutic effect of Nrf2 activators on maternal diabetes-induced NTDs in rodent and nonhuman primates model. Vinylsulfone, one of the Nrf2 activators that possess the beneficial effect on preventing hyperglycemia-induced NTDs in cultured mouse embryos81, could be a good candidate in our future studies.
Conclusions
Our study reveals that maternal diabetes induces oxidative stress in the developing embryo through increasing miR-27a expression and suppressing Nrf2. miR27a inhibits Nrf2 expression, leading to the repression of Nrf2-controlled antioxidant genes GSTA1, GCLC, and GLCM. These data suggest that Nrf2 activators could be therapeutically effective in treating maternal diabetes-induced structural birth defects.
Supplementary Material
Acknowledgments
This work was supported by the NIH grants NIH R01DK083243, R01DK101972, R01HL131737, R01HL134368, R01HL139060 and R01DK103024. We would like to thank Dr. Julie A. Rosen, at the Dean’s office, University of Maryland School of Medicine, for critical reading and editing.
Source of financial support: This research is supported by NIH R01DK083243, R01DK101972; R01DK103024 and R01HL131737
Glossary of Terms
- Diabetic embryopathy
Embryonic developmental deficiency in prenatal or postnatal fetus caused by maternal diabetes during pregnancy
- Neural tube defects (NTDs)
Congenital abnormalities in the structure where brain and spinal cord form during embryonic development
- Redox
A chemical reaction in which the oxidation states of atoms are changed. Within cell, molecules which the electron is added are defined to be reduced, while molecules which the electron is stripped are defined to be oxidized. Redox is used to describe the status or homeostasis between oxidation and reduction among molecules
- Nuclear factor erythroid 2-related factor 2 (Nrf2)
A redox sensitive transcription factor and one of the most important cellular defense mechanisms for combatting oxidative stress through its ability to regulate phase II detoxifying enzymes and antioxidant proteins
- MiRNA
A small noncoding RNA which can target specific mRNA by complementary binding and lead to the degradation of such mRNA or inhibition of translation
- Glutathione S-transferase A1 (GSTA1)
A major phase II detoxification enzyme and also displays glutathione peroxidase activity
- Glutamate-cysteine ligase catalytic subunit (GCLC)
Catalytic subunit of the enzyme responsible for the rate-limiting step in synthesis of the cellular antioxidant glutathione
- Glutamate-cysteine ligase modifier subunit (GLCM)
Modifier subunit of the enzyme responsible for the rate-limiting step in synthesis of the cellular antioxidant glutathione
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: None of the authors have a conflict of interest.
References
- 1.Dong D, Reece EA, Lin X, Wu Y, AriasVillela N, Yang P. New development of the yolk sac theory in diabetic embryopathy: molecular mechanism and link to structural birth defects. American journal of obstetrics and gynecology. 2016;214:192–202. doi: 10.1016/j.ajog.2015.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabbay-Benziv R, Reece EA, Wang F, Yang P. Birth defects in pregestational diabetes: Defect range, glycemic threshold and pathogenesis. World journal of diabetes. 2015;6:481–8. doi: 10.4239/wjd.v6.i3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Weng H, Xu C, Reece EA, Yang P. Oxidative stress-induced JNK1/2 activation triggers proapoptotic signaling and apoptosis that leads to diabetic embryopathy. Diabetes. 2012;61:2084–92. doi: 10.2337/db11-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li X, Xu C, Yang P. c-Jun NH2-terminal kinase 1/2 and endoplasmic reticulum stress as interdependent and reciprocal causation in diabetic embryopathy. Diabetes. 2013;62:599–608. doi: 10.2337/db12-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loeken MR. Current perspectives on the causes of neural tube defects resulting from diabetic pregnancy. American journal of medical genetics Part C, Seminars in medical genetics. 2005;135C:77–87. doi: 10.1002/ajmg.c.30056. [DOI] [PubMed] [Google Scholar]
- 6.Sukanya S, Bay BH, Tay SS, Dheen ST. Frontiers in research on maternal diabetes-induced neural tube defects: Past, present and future. World journal of diabetes. 2012;3:196–200. doi: 10.4239/wjd.v3.i12.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang F, Reece EA, Yang P. Advances in revealing the molecular targets downstream of oxidative stress-induced proapoptotic kinase signaling in diabetic embryopathy. American journal of obstetrics and gynecology. 2015;213:125–34. doi: 10.1016/j.ajog.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang F, Xu C, Reece EA, et al. Protein kinase C-alpha suppresses autophagy and induces neural tube defects via miR-129-2 in diabetic pregnancy. Nature communications. 2017;8:15182. doi: 10.1038/ncomms15182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilde JJ, Petersen JR, Niswander L. Genetic, epigenetic, and environmental contributions to neural tube closure. Annual review of genetics. 2014;48:583–611. doi: 10.1146/annurev-genet-120213-092208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang P, Li X, Xu C, et al. Maternal hyperglycemia activates an ASK1-FoxO3a-caspase 8 pathway that leads to embryonic neural tube defects. Science signaling. 2013;6:ra74. doi: 10.1126/scisignal.2004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang P, Reece EA, Wang F, Gabbay-Benziv R. Decoding the oxidative stress hypothesis in diabetic embryopathy through proapoptotic kinase signaling. American journal of obstetrics and gynecology. 2015;212:569–79. doi: 10.1016/j.ajog.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feig DS, Palda VA. Type 2 diabetes in pregnancy: a growing concern. Lancet. 2002;359:1690–2. doi: 10.1016/S0140-6736(02)08599-9. [DOI] [PubMed] [Google Scholar]
- 13.Harris MI, Flegal KM, Cowie CC, et al. Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults. The Third National Health and Nutrition Examination Survey, 1988–1994. Diabetes care. 1998;21:518–24. doi: 10.2337/diacare.21.4.518. [DOI] [PubMed] [Google Scholar]
- 14.Correa A, Gilboa SM, Besser LM, et al. Diabetes mellitus and birth defects. American journal of obstetrics and gynecology. 2008;199:237 e1–9. doi: 10.1016/j.ajog.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annual review of pharmacology and toxicology. 2013;53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hybertson BM, Gao B. Role of the Nrf2 signaling system in health and disease. Clinical genetics. 2014;86:447–52. doi: 10.1111/cge.12474. [DOI] [PubMed] [Google Scholar]
- 17.Tebay LE, Robertson H, Durant ST, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free radical biology & medicine. 2015;88:108–46. doi: 10.1016/j.freeradbiomed.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan Y, Ichikawa T, Li J, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60:625–33. doi: 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochemical pharmacology. 2013;85:705–17. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 20.Cheng X, Ku CH, Siow RC. Regulation of the Nrf2 antioxidant pathway by microRNAs: New players in micromanaging redox homeostasis. Free radical biology & medicine. 2013;64:4–11. doi: 10.1016/j.freeradbiomed.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 21.Tan SM, de Haan JB. Combating oxidative stress in diabetic complications with Nrf2 activators: how much is too much? Redox report : communications in free radical research. 2014;19:107–17. doi: 10.1179/1351000214Y.0000000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jimenez-Osorio AS, Gonzalez-Reyes S, Pedraza-Chaverri J. Natural Nrf2 activators in diabetes. Clinica chimica acta; international journal of clinical chemistry. 2015;448:182–92. doi: 10.1016/j.cca.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Zhong J, Xu C, Gabbay-Benziv R, Lin X, Yang P. Superoxide dismutase 2 overexpression alleviates maternal diabetes-induced neural tube defects, restores mitochondrial function and suppresses cellular stress in diabetic embryopathy. Free radical biology & medicine. 2016;96:234–44. doi: 10.1016/j.freeradbiomed.2016.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massaad CA, Washington TM, Pautler RG, Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13576–81. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu H, Yu J, Dong D, et al. High Glucose-Repressed CITED2 Expression Through miR-200b Triggers the Unfolded Protein Response and Endoplasmic Reticulum Stress. Diabetes. 2016;65:149–63. doi: 10.2337/db15-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong D, Fu N, Yang P. MiR-17 Downregulation by High Glucose Stabilizes Thioredoxin-Interacting Protein and Removes Thioredoxin Inhibition on ASK1 Leading to Apoptosis. Toxicological sciences : an official journal of the Society of Toxicology. 2016;150:84–96. doi: 10.1093/toxsci/kfv313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu H, Yu J, Dong D, Zhou Q, Wang JY, Yang P. The miR-322-TRAF3 circuit mediates the pro-apoptotic effect of high glucose on neural stem cells. Toxicological sciences : an official journal of the Society of Toxicology. 2015;144:186–96. doi: 10.1093/toxsci/kfu271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong D, Yu J, Wu Y, Fu N, Villela NA, Yang P. Maternal diabetes triggers DNA damage and DNA damage response in neurulation stage embryos through oxidative stress. Biochemical and biophysical research communications. 2015;467:407–12. doi: 10.1016/j.bbrc.2015.09.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong D, Zhang Y, Reece EA, Wang L, Harman CR, Yang P. microRNA expression profiling and functional annotation analysis of their targets modulated by oxidative stress during embryonic heart development in diabetic mice. Reproductive toxicology. 2016;65:365–74. doi: 10.1016/j.reprotox.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimura Y, Murase T, Oyama K, et al. Prevention of neural tube defects by loss of function of inducible nitric oxide synthase in fetuses of a mouse model of streptozotocin-induced diabetes. Diabetologia. 2009;52:962–71. doi: 10.1007/s00125-009-1312-0. [DOI] [PubMed] [Google Scholar]
- 31.Salbaum JM, Kappen C. Neural tube defect genes and maternal diabetes during pregnancy. Birth defects research Part A, Clinical and molecular teratology. 2010;88:601–11. doi: 10.1002/bdra.20680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamimoto Y, Sugiyama T, Kihira T, et al. Transgenic mice overproducing human thioredoxin-1, an antioxidative and anti-apoptotic protein, prevents diabetic embryopathy. Diabetologia. 2010;53:2046–55. doi: 10.1007/s00125-010-1784-y. [DOI] [PubMed] [Google Scholar]
- 33.Snyder EY, Deitcher DL, Walsh C, Arnold-Aldea S, Hartwieg EA, Cepko CL. Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell. 1992;68:33–51. doi: 10.1016/0092-8674(92)90204-p. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Zhong J, Dong D, Liu G, Yang P. Endoplasmic reticulum stress-induced CHOP inhibits PGC-1alpha and causes mitochondrial dysfunction in diabetic embryopathy. Toxicological sciences : an official journal of the Society of Toxicology. 2017 doi: 10.1093/toxsci/kfx096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu C, Li X, Wang F, Weng H, Yang P. Trehalose prevents neural tube defects by correcting maternal diabetes-suppressed autophagy and neurogenesis. American journal of physiology Endocrinology and metabolism. 2013;305:E667–78. doi: 10.1152/ajpendo.00185.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Molecular and cellular biology. 2002;22:2883–92. doi: 10.1128/MCB.22.9.2883-2892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Papp D, Lenti K, Modos D, et al. The NRF2-related interactome and regulome contain multifunctional proteins and fine-tuned autoregulatory loops. FEBS letters. 2012;586:1795–802. doi: 10.1016/j.febslet.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narasimhan M, Patel D, Vedpathak D, Rathinam M, Henderson G, Mahimainathan L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PloS one. 2012;7:e51111. doi: 10.1371/journal.pone.0051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116:4338–48. doi: 10.1182/blood-2009-04-214817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang M, Yao Y, Eades G, Zhang Y, Zhou Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast cancer research and treatment. 2011;129:983–91. doi: 10.1007/s10549-011-1604-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li N, Muthusamy S, Liang R, Sarojini H, Wang E. Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mechanisms of ageing and development. 2011;132:75–85. doi: 10.1016/j.mad.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 42.Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. The Journal of biological chemistry. 2011;286:40725–33. doi: 10.1074/jbc.M111.275495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Indo HP, Yen HC, Nakanishi I, et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. Journal of clinical biochemistry and nutrition. 2015;56:1–7. doi: 10.3164/jcbn.14-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uruno A, Yagishita Y, Yamamoto M. The Keap1-Nrf2 system and diabetes mellitus. Archives of biochemistry and biophysics. 2015;566:76–84. doi: 10.1016/j.abb.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 45.Li B, Liu S, Miao L, Cai L. Prevention of diabetic complications by activation of Nrf2: diabetic cardiomyopathy and nephropathy. Experimental diabetes research. 2012;2012:216512. doi: 10.1155/2012/216512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J, Zhang Z, Cai L. Diabetic cardiomyopathy and its prevention by nrf2: current status. Diabetes & metabolism journal. 2014;38:337–45. doi: 10.4093/dmj.2014.38.5.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang T, Huang Z, Lin Y, Zhang Z, Fang D, Zhang DD. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:850–60. doi: 10.2337/db09-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhong Q, Mishra M, Kowluru RA. Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Investigative ophthalmology & visual science. 2013;54:3941–8. doi: 10.1167/iovs.13-11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lazaro I, Oguiza A, Recio C, et al. Interplay between HSP90 and Nrf2 pathways in diabetes-associated atherosclerosis. Clinica e investigacion en arteriosclerosis : publicacion oficial de la Sociedad Espanola de Arteriosclerosis. 2017;29:51–59. doi: 10.1016/j.arteri.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 50.Cheng X, Chapple SJ, Patel B, et al. Gestational diabetes mellitus impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero. Diabetes. 2013;62:4088–97. doi: 10.2337/db13-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang SY, Chen YW, Zhao XP, et al. Catalase prevents maternal diabetes-induced perinatal programming via the Nrf2-HO-1 defense system. Diabetes. 2012;61:2565–74. doi: 10.2337/db12-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He MY, Wang G, Han SS, et al. Nrf2 signalling and autophagy are involved in diabetes mellitus-induced defects in the development of mouse placenta. Open biology. 2016;6 doi: 10.1098/rsob.160064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duan Y, Sun F, Que S, Li Y, Yang S, Liu G. Prepregnancy maternal diabetes combined with obesity impairs placental mitochondrial function involving Nrf2/ARE pathway and detrimentally alters metabolism of offspring. Obesity research & clinical practice. 2017 doi: 10.1016/j.orcp.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 54.Trocino RA, Akazawa S, Ishibashi M, et al. Significance of glutathione depletion and oxidative stress in early embryogenesis in glucose-induced rat embryo culture. Diabetes. 1995;44:992–8. doi: 10.2337/diab.44.8.992. [DOI] [PubMed] [Google Scholar]
- 55.Sakamaki H, Akazawa S, Ishibashi M, et al. Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes. 1999;48:1138–44. doi: 10.2337/diabetes.48.5.1138. [DOI] [PubMed] [Google Scholar]
- 56.Wang F, Reece EA, Yang P. Superoxide dismutase 1 overexpression in mice abolishes maternal diabetes-induced endoplasmic reticulum stress in diabetic embryopathy. American journal of obstetrics and gynecology. 2013;209:345 e1–7. doi: 10.1016/j.ajog.2013.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Y, Wang F, Reece EA, Yang P. Curcumin ameliorates high glucose-induced neural tube defects by suppressing cellular stress and apoptosis. American journal of obstetrics and gynecology. 2015;212:802 e1–8. doi: 10.1016/j.ajog.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weng H, Li X, Reece EA, Yang P. SOD1 suppresses maternal hyperglycemia-increased iNOS expression and consequent nitrosative stress in diabetic embryopathy. American journal of obstetrics and gynecology. 2012;206:448 e1–7. doi: 10.1016/j.ajog.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li X, Weng H, Reece EA, Yang P. SOD1 overexpression in vivo blocks hyperglycemia-induced specific PKC isoforms: substrate activation and consequent lipid peroxidation in diabetic embryopathy. American journal of obstetrics and gynecology. 2011;205:84 e1–6. doi: 10.1016/j.ajog.2011.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang P, Li H. Epigallocatechin-3-gallate ameliorates hyperglycemia-induced embryonic vasculopathy and malformation by inhibition of Foxo3a activation. American journal of obstetrics and gynecology. 2010;203:75 e1–6. doi: 10.1016/j.ajog.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang P, Zhao Z, Reece EA. Activation of oxidative stress signaling that is implicated in apoptosis with a mouse model of diabetic embryopathy. American journal of obstetrics and gynecology. 2008;198:130 e1–7. doi: 10.1016/j.ajog.2007.06.070. [DOI] [PubMed] [Google Scholar]
- 62.Wang F, Reece EA, Yang P. Oxidative stress is responsible for maternal diabetes-impaired transforming growth factor beta signaling in the developing mouse heart. American journal of obstetrics and gynecology. 2015;212:650 e1–11. doi: 10.1016/j.ajog.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu Y, Reece EA, Zhong J, et al. Type 2 diabetes mellitus induces congenital heart defects in murine embryos by increasing oxidative stress, endoplasmic reticulum stress, and apoptosis. American journal of obstetrics and gynecology. 2016;215:366 e1–66 e10. doi: 10.1016/j.ajog.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harma M, Harma M, Erel O. Oxidative stress in women with preeclampsia. American journal of obstetrics and gynecology. 2005;192:656–7. doi: 10.1016/j.ajog.2004.07.094. author reply 57. [DOI] [PubMed] [Google Scholar]
- 65.Roberts JM, Hubel CA. Oxidative stress in preeclampsia. American journal of obstetrics and gynecology. 2004;190:1177–8. doi: 10.1016/j.ajog.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 66.Kuper SG, Abramovici AR, Jauk VC, Harper LM, Biggio JR, Tita AT. The effect of omega-3 supplementation on pregnancy outcomes by smoking status. American journal of obstetrics and gynecology. 2017 doi: 10.1016/j.ajog.2017.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sbrana E, Suter MA, Abramovici AR, et al. Maternal tobacco use is associated with increased markers of oxidative stress in the placenta. American journal of obstetrics and gynecology. 2011;205:246 e1–7. doi: 10.1016/j.ajog.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferguson KK, McElrath TF, Chen YH, Loch-Caruso R, Mukherjee B, Meeker JD. Repeated measures of urinary oxidative stress biomarkers during pregnancy and preterm birth. American journal of obstetrics and gynecology. 2015;212:208 e1–8. doi: 10.1016/j.ajog.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blaauw J, Smit AJ, van Pampus MG, et al. Skin autofluorescence, a marker of advanced glycation end products and oxidative stress, is increased in recently preeclamptic women. American journal of obstetrics and gynecology. 2006;195:717–22. doi: 10.1016/j.ajog.2006.06.086. [DOI] [PubMed] [Google Scholar]
- 70.Ferguson KK, Meeker JD, McElrath TF, Mukherjee B, Cantonwine DE. Repeated measures of inflammation and oxidative stress biomarkers in preeclamptic and normotensive pregnancies. American journal of obstetrics and gynecology. 2017;216:527 e1–27 e9. doi: 10.1016/j.ajog.2016.12.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Enquobahrie DA, Meller M, Rice K, Psaty BM, Siscovick DS, Williams MA. Differential placental gene expression in preeclampsia. American journal of obstetrics and gynecology. 2008;199:566 e1–11. doi: 10.1016/j.ajog.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Loset M, Mundal SB, Johnson MP, et al. A transcriptional profile of the decidua in preeclampsia. American journal of obstetrics and gynecology. 2011;204:84 e1–27. doi: 10.1016/j.ajog.2010.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Metz TD, Allshouse AA, Hogue CJ, et al. Maternal marijuana use, adverse pregnancy outcomes, and neonatal morbidity. American journal of obstetrics and gynecology. 2017 doi: 10.1016/j.ajog.2017.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grooten IJ, Den Hollander WJ, Roseboom TJ, et al. Helicobacter pylori infection: a predictor of vomiting severity in pregnancy and adverse birth outcome. American journal of obstetrics and gynecology. 2017;216:512 e1–12 e9. doi: 10.1016/j.ajog.2017.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hacker FM, Whalen PS, Lee VR, Caughey AB. Maternal and fetal outcomes of pancreatitis in pregnancy. American journal of obstetrics and gynecology. 2015;213:568 e1–5. doi: 10.1016/j.ajog.2015.07.031. [DOI] [PubMed] [Google Scholar]
- 76.Faucett AM, Metz TD, DeWitt PE, Gibbs RS. Effect of obesity on neonatal outcomes in pregnancies with preterm premature rupture of membranes. American journal of obstetrics and gynecology. 2016;214:287 e1–87 e5. doi: 10.1016/j.ajog.2015.09.093. [DOI] [PubMed] [Google Scholar]
- 77.Carter EB, Bishop KC, Goetzinger KR, Tuuli MG, Cahill AG. The impact of chorionicity on maternal pregnancy outcomes. American journal of obstetrics and gynecology. 2015;213:390 e1–7. doi: 10.1016/j.ajog.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 78.Bhakkiyalakshmi E, Sireesh D, Rajaguru P, Paulmurugan R, Ramkumar KM. The emerging role of redox-sensitive Nrf2-Keap1 pathway in diabetes. Pharmacological research. 2015;91:104–14. doi: 10.1016/j.phrs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 79.Zheng H, Whitman SA, Wu W, et al. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes. 2011;60:3055–66. doi: 10.2337/db11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cui W, Li B, Bai Y, et al. Potential role for Nrf2 activation in the therapeutic effect of MG132 on diabetic nephropathy in OVE26 diabetic mice. American journal of physiology Endocrinology and metabolism. 2013;304:E87–99. doi: 10.1152/ajpendo.00430.2012. [DOI] [PubMed] [Google Scholar]
- 81.Dong D, Reece EA, Yang P. The Nrf2 Activator Vinylsulfone Reduces High Glucose-Induced Neural Tube Defects by Suppressing Cellular Stress and Apoptosis. Reproductive sciences. 2016;23:993–1000. doi: 10.1177/1933719115625846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.