

ABSTRACT
RNA Binding Proteins (RBPs) are a class of post-transcriptional regulatory molecules which are increasingly documented to be dysfunctional in cancer genomes. However, our current understanding of these alterations is limited. Here, we delineate the mutational landscape of ∼1300 RBPs in ∼6000 cancer genomes. Our analysis revealed that RBPs have an average of ∼3 mutations per Mb across 26 cancer types. We identified 281 RBPs to be enriched for mutations (GEMs) in at least one cancer type. GEM RBPs were found to undergo frequent frameshift and inframe deletions as well as missense, nonsense and silent mutations when compared to those that are not enriched for mutations. Functional analysis of these RBPs revealed the enrichment of pathways associated with apoptosis, splicing and translation. Using the OncodriveFM framework, we also identified more than 200 candidate driver RBPs that were found to accumulate functionally impactful mutations in at least one cancer. Expression levels of 15% of these driver RBPs exhibited significant difference, when transcriptome groups with and without deleterious mutations were compared. Functional interaction network of the driver RBPs revealed the enrichment of spliceosomal machinery, suggesting a plausible mechanism for tumorogenesis while network analysis of the protein interactions between RBPs unambiguously revealed the higher degree, betweenness and closeness centrality for driver RBPs compared to non-drivers. Analysis to reveal cancer-specific Ribonucleoprotein (RNP) mutational hotspots showed extensive rewiring even among common drivers between cancer types. Knockdown experiments on pan-cancer drivers such as SF3B1 and PRPF8 in breast cancer cell lines, revealed cancer subtype specific functions like selective stem cell features, indicating a plausible means for RBPs to mediate cancer-specific phenotypes. Hence, this study would form a foundation to uncover the contribution of the mutational spectrum of RBPs in dysregulating the post-transcriptional regulatory networks in different cancer types.
KEYWORDS: Somatic mutations, cancer genomics, oncogenes, post-transcriptional regulation, gene regulatory networks
Introduction
Post-transcriptional regulation of gene expression is an intricate and essential mechanism that orchestrates the maturation, transport, stability and degradation of all classes of RNAs.1-3 Typically, each of these events are regulated by the formation of diverse ribonucleoprotein (RNA) complexes mediated by RNA binding proteins (RBPs). RBPs bind to the secondary structure or untranslated regions (UTR) or ORF of an RNA in a sequence specific manner to control its fate. Furthermore, most human RBPs are ubiquitously expressed compared to the remaining protein-coding transcriptome with 20% of the expressed transcripts encoding RBPs. Hence, RNA metabolism is not only a conserved cellular process but also has the highest protein copy number demands.1,4-6
Given the importance of regulatory molecules like RBPs in controlling gene expression, it is evident that any deviation from normal function of these proteins can lead to various disorders including cancer.7 Cancer development was often believed to be a result of aberrant transcription and signaling events. Increasing evidence suggests that post transcriptional regulation also controls several important cellular mechanisms including proliferation, differentiation, invasion, metastases, apoptosis and angiogenesis that could lead to a cancer phenotype. RBPs being the central players of post transcriptional control, their dysregulation is a plausible mechanism for mediating cancer initiation and progression.8-10 For instance, KHDRBS1, a KH domain containing splicing factor was shown to be overexpressed in cancers of breast, prostate, kidney and cervix. An increased expression of this protein facilitates the inclusion of exon5 in the pre-mRNA of CD44 – a cell surface protein involved in cancer proliferation.11 Another well studied RBP for its role in tumorogenesis is Musashi-1(MSI1), which is overexpressed in a few central nervous system (CNS) tumors but primarily in glioblastoma. Although the mechanism of action is not well characterized, it is believed that MSI1 acts by regulating the Notch signaling pathway through the translational repression of its mRNA.12,13 Furthermore, RNA binding protein PCBP2, a member of the poly(C) binding protein family was found to be overexpressed in glioma and regulating several targets important for controlling tumor growth.14 Recently, a global analysis on the expression of mRNA levels of genes encoding ∼800 RBPs revealed 30 RBPs to be highly upregulated in several cancers.5 Although there are several studies implicating the changes in the expression of RNA binding proteins in different malignancies, the cause of such a change is not completely established.
However, recent studies signal the contribution of somatic mutations in altering the function of RBPs in several cancers.15 Mutational analysis of all the genes in the human genome across 12 cancer types identified SF3B1, U2AF1 and PCBP1 – RBPs involved in splicing to be significantly mutated in multiple cancers suggesting their role in causing cancer phenotypes.16 Furthermore, APOBEC3B – an important protein in the RNA editing mechanism was found to be upregulated and frequently mutated in cancers of bladder, cervix, lung, head and neck and breast.17 Also notable are the mutations in the gene coding for RBM10 in lung cancer which was found to misregulate the alternative splicing of NUMB protein- a critical regulator of the Notch pathway and hence leading to irregular cell proliferation in lung cancers.18 These studies emphasize the importance of studying the mutational landscape of RBPs in cancer genomes. Hence, to expand the current understanding of mutations in these genes, we performed a systematic analyses of somatic mutations occurring in ∼1300 RBPs in ∼6000 tumor samples across 26 cancer types.
To achieve this, we compiled a list of genes identified to encode RBPs in human cells from several experimental studies (See Materials and Methods). We then analyzed the exome sequencing data of 26 cancer types to identify candidate drivers and integrated their transcriptome profiles to assess alterations in their expression due to mutation. Furthermore, we carried out functional and network analysis of these drivers to identify potentially dysregulated pathways in the cancer genomes and delineate cancer specific interaction networks. Finally, we knocked down two candidate RBP drivers – SF3B1 and PRPF8 in breast cancer cell lines to observe changes in the cellular phenotypes.
Results
General framework
Figure 1 illustrates various steps and methods employed to calculate the mutation frequency, identification of Genes Enriched for Mutations (GEMs) and candidate driver genes. Firstly, the mutation frequency of a gene in a given cancer is calculated by normalizing the number of mutations with the exome length of the gene and the total number of subjects in a cancer type (Fig. 1A). Secondly, we identify Genes Enriched for Mutations (GEMs) in a given cancer using a Fisher's exact test that calculates the probability of observing mutations in a given gene against a genomic background (Fig. 1B). Finally, we identify RBPs that accumulate high functionally-impactful mutations using OncodriveFM approach (Fig. 1C) (See Materials and Methods) that relies on SIFT, PPH2 and Mutation Assessor19-21 to estimate the functional impact of individual mutations. It does so by computing the bias towards the accumulation of high-impact mutations across the cancer in the cohort as a signal of their involvement in tumorogenesis.
Figure 1.
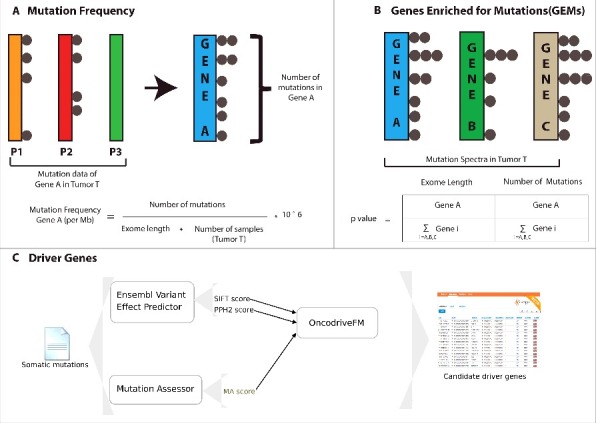
Overview of the different steps for calculating mutation frequency, Genes Enriched in Mutations (GEMs) and candidate drivers from cancer samples. (A) shows the approach adopted to calculate the mutation frequency of a given gene in a given cancer type. (B) shows the approach adopted to identify Genes Enriched for Mutations (GEMs) in a cancer type. (C) illustrates the workflow adopted in OncodriveFM approach, which was used to identify driver genes in cancer cohorts.
RBPs are less frequently mutated than non-RBPs in 70% of the cancers and are mutated at a rate equal to TFs in 50% of the cancers
To identify the mutational frequencies across 26 cancer cohorts for RBPs and Non-RBPs, we first computed the mutational frequencies of all the genes annotated in the human genome as shown in Fig. 1 (see Materials and Methods). We observe RBPs to be mutated at an approximate rate of 3 mutations per Mb across the studied cancer cohorts with the highest and lowest frequencies occurring in cancers of uterine (UCEC) and thyroid (THCA) respectively (Fig. 2A). We then compared the mutational frequencies of RBPs to that of Non-RBPs (any gene that is not included in our RBP repertoire was termed as Non-RBP, See Table S1 for a list of RBPs) to see differences in their mutational spectra. Overall, we find the mutational frequency of Non-RBPs to be significantly higher than that of RBPs in ∼70% of the cancer types studied (Fig. S1A; Exact p-values are listed in the Table S1) and exhibiting an equal rate in LAML, OV, PAAD and UVM. Additionally, comparison of the mutation frequencies of RBPs with ∼3600 Non-RBPs which exhibited similar GC content and exome lengths to RBPs, revealed that in majority of the cancer types (19 out of 26) RBPs exhibited significantly different mutation frequencies than random set of genes with similar properties, at a threshold of p< 0.01 (Wilcoxon test, Fig. S1B). These results suggest that in most cancer types the contribution of GC content and exome length on mutational frequency in RBPs is minimal.
Figure 2.
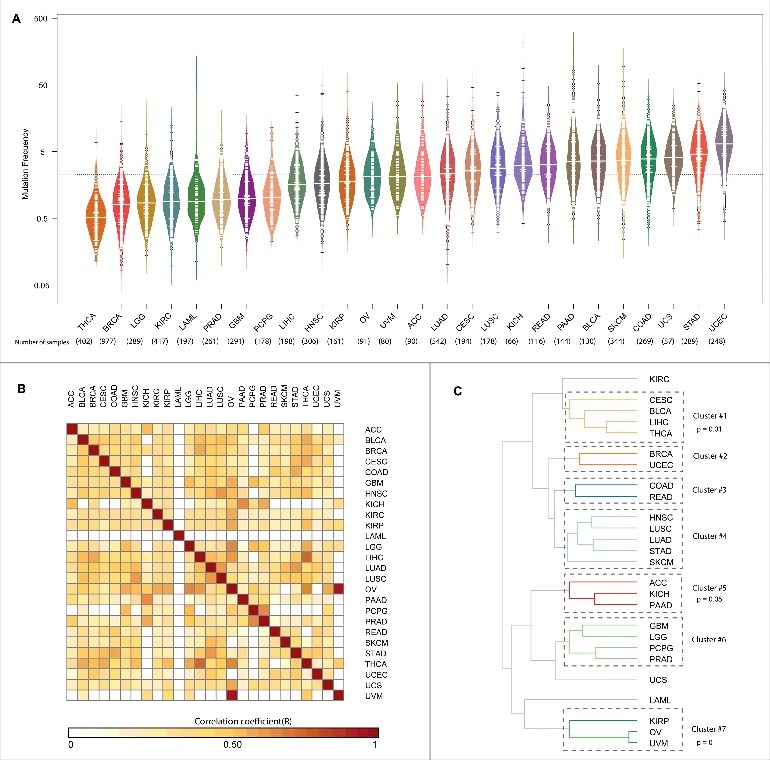
Mutation Frequency of RBPs | (A) The mutation frequency of RBPs across 26 cancers is shown as a bean plot. The thick white line in each bean indicates the median mutation frequency of RBPs in a given cancer and the dashed black line indicates the average mutation frequency of RBPs across all the cancers (B) Heatmap showing the pearson correlation of mutation frequencies of RBPs between 26 cancer types (C) Dendogram obtained by hierarchically clustering the mutation frequencies of RBPs in 26 cancer types. Cancer types shown are abbreviated as follows: Adrenocorticol carcinoma (ACC), Bladder Urothelial carcinoma (BLCA), Breast invasive carcinoma (BRCA), Cervival squamous cell carcinoma an endocervical adenocarcinoma (CESC), Colon adenocarcinoma (COAD), Gliobastoma multiforme (GBM), Head and neck squamous cell carcinoma (HNSC), Kidney Chromophobe (KICH), Kidney renal cell carcinoma (KIRC), Kidney renal papillary cell carcinoma (KIRP), Acute myeloid leukemia (LAML), Lower grade glioma (LGG), Liver hepatocellular carcinoma (LIHC), Lung adenocarcinoma (LUAD), Lung squamous cell carcinoma (LUSC), Ovarian serous cystadenocarcinoma (OV), Pancreatic adenocarcinoma (PAAD), Pheochromocytoma and Paraganglioma (PCPG), Prostrate adenocarcinoma (PRAD), Rectum adenocarcinoma (READ), Skin cutaneous Melanoma (SKCM), Stomach adenocarcinoma (STAD), Thyroid carcinoma (THCA), Uterine corpus endometrial carcinoma (UCEC), Uterine carcinosarcoma (UCS), Uveal Melanoma (UVM).
Although, we have previously shown in multiple studies that RBPs exhibit distinct properties such as high gene expression level and evolutionary conservation compared to other classes of genes/transcripts,4-6 we also found based on Ensembl annotations22 that RBPs exhibit significantly higher exome lengths compared to Non-RBPs albeit comparable in length to Transcription Factors (TFs) (Fig. S2A). In addition, we also noted that RBPs exhibit significantly lower GC content than both Non-RBPs and TFs (Fig. S2B). Hence, warranting the need to study RBPs as a class of regulatory proteins and to consider TFs as an independent biological random set to compare the relative extents of mutations per unit length of a gene, in addition to comparing to the genomic background i.e, Non-RBPs. This involved comparison of the mutational frequencies of two major classes of regulatory molecules – RBPs and TFs (Transcription Factors; obtained from DBD database23) to understand the differences between the mutational landscape of these crucial regulatory genes. While TFs showed higher mutational frequencies than RBPs in 50% of the cancer types (p-values are listed in Table S1), the mutational frequencies of TFs were seen to be equal to RBPs in multiple cancers such as breast (BRCA), bladder (BLCA), cervix (CESC) and brain (GBM). Further, we correlated the mutational frequencies of RBPs using pearson correlation via rcorr function from hmisc package (https://cran.r-project.org/web/packages/Hmisc) across cancers to identify similar mutational loads between cancers. This revealed OV (ovarian cancer) and UVM (uveal melanoma) to be highly correlated (R = 0.94, p = 0) followed by LIHC (Liver hepatocellular) and THCA (Thyroid cancer) (R = 0.75, p = 0) (Fig. 2B). When we performed a similar analysis on the mutational frequencies of Non-RBPs, we observed different correlation patterns; for example – LGG-OV have the highest correlation (R = 0.93, p = 0) followed by OV-UCS (R = 0.88, p = 0, Fig. S1C). Furthermore, to understand if mutational frequency of RBPs segregate cancers into meaningful clusters, we performed hierarchical clustering of the mutational frequencies using pvclust package24 by setting ‘complete’ as the clustering method and ‘correlation’ as the distance metric for 10,000 bootstraps, that revealed 7 different clusters as shown in Fig. 2C. Clusters found to be significant are highlighted with p-values. The mutational frequencies of RBPs cluster cervical, bladder, liver and thyroid cancers viz CESC, BLCA, LIHC and THCA into one cluster (Cluster #1) suggesting a similar mutational spectra of RBPs across these different types of cancers (Fig. 2C). Our results also show clustering of OV and UVM (Cluster #7), suggesting a common origin of these gender-specific cancers. Further, our results although not significant exhibited known relationships between COAD and READ which are commonly studied together as well as the clustering of glandular adenocarcinomas like LUAD and STAD. Interestingly, these patterns were very distinct for Non-RBPs (Fig. S1D), suggesting that these observations could reveal common mechanisms of dysregulation at post-transcriptional level with in members of these cancer type clusters due to mutations in RBPs.
RBPs enriched for mutations are crucial players in translation, splicing and apoptosis mediated pathways
Genes under positive selection, either in individual or multiple cancer types, tend to display higher mutation frequencies above background.16 We employed a statistical approach as shown in Fig. 1B to identify Genes Enriched for Mutations (GEMs) in a given cancer type (see Materials and Methods). This identified 281 genes encoding for RBPs to be significantly enriched for mutations in at least one cancer (see Fig. 3A for RBPs enriched for mutations in at least 4 cancers and Fig. S3 for RBPs enriched for mutations in at least one cancer type; -log[corrected p-values] from the Fisher's exact test are listed in Table S2). Comparison of GEM and non-GEM RBPs with corresponding gene sets for non-RBPs, for differences in the GC content and exome length, revealed that while GC content does not contribute to the extent of mutations in RBPs (p = 0.496, Wilcoxon test), it was found to be significantly different (p = 4.21e-08, Wilcoxon test) between GEM and non-GEM groups for non-RBPs (Fig. S2C). Also, exome length was found to be higher for GEM RBPs compared to non-GEM RBPs (p = 0.0016, Wilcoxon test) which was in sharp contrast to the observation that non-RBPs that are not enriched for mutations have significantly higher exome lengths (Fig. S2D). These observations suggest that while GC content has no significant influence on the extent of mutations in RBPs, exome length might be higher for GEM RBPs, however it does not necessarily determine whether a gene is a GEM since non-RBPs GEMs exhibited significantly lower exome lengths.
Figure 3.
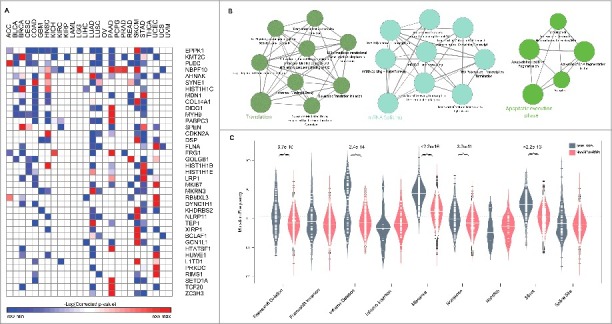
RBPs enriched for mutations | (A) shows the list of RBPs enriched for mutations (Corrected p < 0.01, Fisher's exact test) in at least 4 cancer types. (B) shows the pathways enriched (p < 0.01) in GEM RBPs. (C) illustrates the distribution of mutation frequencies for various variant types between RBPs enriched for mutations (labelled as GEMs-RBPs) and those that are not enriched for mutations (non-GEM-RBPs). All significant differences in the distributions are highlighted with their p-values for p < 0.01 using Wilcox test.
Among the GEM RBPs, we identified KMT2C (MLL3), a histone 3- lysine 4 methyltransferase with tumor-suppressor properties that belongs to a family of chromatin regulator genes, to be enriched for mutations in 40% of the cancer types (Fig. S4). This observation was in accordance with previous studies that showed MLL3 to be significantly mutated in cancers.16,25 Furthermore, PLEC (plectin) – an abundantly expressed versatile protein that links different elements of the cytoskeleton26 was also seen to be enriched for mutations in 11 cancer types including lung, head and neck, bladder and pancreas. Previously, PLEC was identified as a biomarker in cancers of pancreas and was shown to be promoting migration and invasion of cancer cells in head and neck cancers.27,28 These findings together with our observations suggest the importance of PLEC in mediating cancer phenotypes in diverse tissues than currently documented. Also notable is the gene encoding for EPPK1 which was seen to be mutated in ∼40% of the cancers including cancers of cervix, colon, head and neck, pancreas etc. EPPK1 belongs to the plakin family of genes which are known to function in interconnecting cytoskeletal filaments and was identified as a candidate biomarker in the cervical lesions.29,30 Further, functional analysis of RBPs enriched for mutations identified diverse pathways including translation, mRNA splicing and apoptosis to be over-represented (p < 0.01, Fig. 3B, see Materials and Methods), thus uncovering common players and associated mechanisms responsible for cancer phenotypes due to RBPs.
RBPs enriched for mutations undergo frequent frameshift and inframe deletions, missense, nonsense and silent mutations
As different genes are susceptible to undergo different kinds of mutations at varied frequency, we aimed to identify mutation types that RBPs enriched for mutations (GEM-RBPs) frequently undergo when compared to RBPs that are not enriched for mutations (NonGEM-RBPs) (See Materials and Methods). In particular, we quantified the mutation frequencies of nine different classes of mutations namely Frameshift mutations – Deletion and Insertion,31 Inframe Deletion, Inframe Insertion,32 Missense,33 Nonsense,34 Nonstop,35 Silent36 and Splice Site37 for all the RBPs across cancer samples (Materials and Methods, Table S2). Our analysis clearly revealed that RBPs frequently and significantly undergo Frameshift deletion, Inframe deletion, Missense, Nonsense and Silent mutations (Fig. 3C) implying a significant contribution of these mutation types on the function of RBPs in cancer genomes. Abundance of Frameshift deletions in GEM-RBPs clearly indicates that deletion mutations causing change in reading frame thereby resulting in different translation than the original polypeptide, could be a frequent mechanism of dysregulation. Also, a significant difference in the frequency of nonsense mutations – which introduce a premature termination codon (PTC) in the gene; between the two groups indicate the importance of these mutations in triggering the mechanism of nonsense mediate decay (NMD) of RBPs enriched for mutations in several cancers. Earlier, the disengagement between genotype and phenotype in patients with muscular dystrophy was attributed to NMD.38 This study showed that mutations that change the reading frame and introduce a premature termination codon cause a severe form of the disease as the whole transcript is eliminated by NMD, whereas mutations that did not give rise to a PTC resulted in a milder form of muscular dystrophy.38 A similar mechanism could be leading to the dysregulation of RBPs that are enriched for mutations in several cancer phenotypes, due to the higher mutational rate of nonsense mutations in GEM-RBPs (Fig. 3C). Likewise, missense mutations that result in a change in the amino acid composition and inframe deletions which although do not change the frame of transcription but can result in a dysfunctional protein form could contribute to the loss of function phenotypes in RBPs enriched for mutations.
More than 200 RBPs are identified as “candidate” drivers with majority of them specific to cancer types
In addition to identifying Genes Enriched for Mutations (GEMs), which can comprise of both synonymous and nonsynonymous somatic mutations in a cancer genome, we aimed to uncover RBPs that exhibit a bias towards the accumulation of nonsynonymous mutations with high functional impact during tumorigenesis, as a means to uncover likely driver RBPs. Driver genes are known to provide a significant growth advantage to cancer cells. As discussed above, we distinguish these two groups of genes – GEMs and driver genes, based on whether the mutations comprise of any mutation or only nonsynonymous ones respectively. To this end, we used Oncodrive FM39 an approach to detect genes that tend to accumulate functional somatic mutations across a cohort of cancer samples. A significant trend towards the accumulation of such functional mutations is calculated as FM bias – signal of positive selection during cancer development (See Materials and Methods). Hence, we term such genes as “candidate drivers” in the present study. This analysis revealed more than 200 likely driver genes encoding for RBPs in at least one cancer type (See Fig. S5 and Table S3 for extended list of all RBP candidate drivers). However, RBPs were not enriched for drivers when compared to the non-RBPs (p = 1E-10, Fisher exact test, odds ratio = 1.6). Fig. 4 shows the list of RBPs identified as drivers in at least two cancer types. Among these, a notable example is AHNAK, a nucleoprotein initially identified in human neuroblastomas and skin epithelial cells. In addition to being essential for pseudopodia formation and tumoral migration/invasion, AHNAK is known to be a tumor suppressor gene which functions by modulating the TGFβ signaling.40-42 We predict AHNAK to be a candidate driver43 in 12 cancers including head and neck squamous carcinoma, lung adenocarcinoma, lung squamous carcinoma, breast cancers (See Table S3) suggesting the impact of nonsynonymous mutations on protein function and thus misregulating pathways responsible for cellular growth and hence leading to a cancer phenotype. Another striking example is AGO2 which was found to accumulate functional mutations in cancers of breast, skin, lung and stomach. AGO2 is a member of the AGO subfamily that facilitates RNA mediating gene silencing.44 A closer inspection revealed that 5 out of the 11 missense mutations in this gene are seen to be affecting the Piwi domain of the protein that is essential for target cleavage (Fig. S6). These observations suggest the alteration in amino acid composition of the RBPs as a possible mechanism leading to the dysregulation of post transcriptional control in tumorogenesis. Additionally, majority of the RBPs (65%) were predicted to be drivers in only one cancer type with a small fraction of the RBPs identified as drivers in more than five cancer types Fig. S6), suggesting heterogeneous mechanisms and/or pathways might be contributing to the mutational portrait of RBPs in different cancer types.
Figure 4.
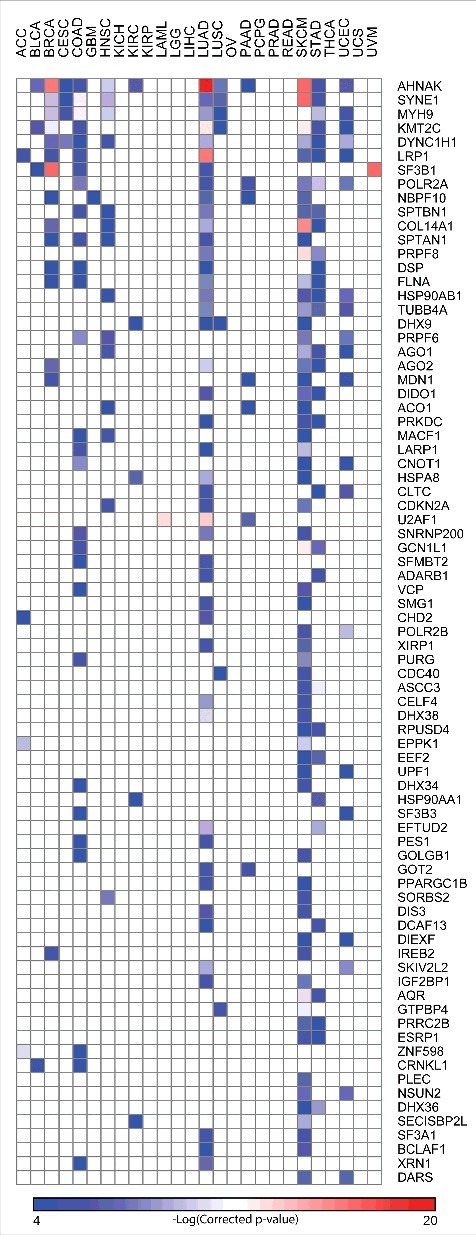
Candidate RBP Drivers | The driver genes in each cancer type were identified using the Oncodrive FM as described in Materials and Methods. Heatmap shows the list of RBPs identified as “candidate drivers” in at least two cancer types.
Further, we tested the overlap between driver genes and those that are GEMs, to identify frequently mutated genes that accumulate functional bias across a given cancer cohort. We found 62 RBPs to be significantly mutated and also possessing deleterious non-synonymous mutations (Enrichment p = 0.0096, Hypergeometric test, complete list shown in Fig. S7). Interestingly, we find the gene encoding SF3B1 – an important splicing factor to be frequently mutated and accumulating functional mutations in uveal melanoma (UVM) which is in accordance with a study that showed the impact of SF3B1 mutations on alternative splicing in uveal melanomas.45 Furthermore, recurrent missense mutation at R625 in patients with uveal melanoma was observed suggesting an oncogenic role of this mutation (Fig. S6C). In addition, another critical protein of the spliceosome machinery, U2AF1 was seen to be frequently mutated and possessing deleterious mutations in acute myeloid leukemia (LAML). Somatic mutations in U2AF1 were previously observed to be contributing to mis-splicing events in myeloid malignancies.46,47 Also, recurrent missense mutations at S34 in the zf-CCCH domain was observed to be highly recurrent in patients with LAML (Fig. S6C).
Pan-cancer expression analysis of candidate RBP drivers shows significant change in RNA levels for 15% of them
To identify if mutations in an RBP gene affects its RNA levels, we performed pan-cancer expression analysis for all the candidate RBP drivers between patient cohorts containing these mutations and cohorts that don't carry such mutations (See Materials and Methods). This identified 30 RBPs that exhibited significant changes in their RNA levels between the pan-cancer cohorts constructed for each candidate RBP as described above (Fig. 5A, Exact p-values are listed in the Table S4). Of these, CDKN2A, a cyclin-dependent kinase inhibitor 2A known to stabilize the tumor suppressor protein p53 was observed to have higher levels of RNA in mutated samples when compared to the non-mutated samples (Fold change = 3.9, p = 1.26E-11, Wilcox test). Furthermore, a nonsense mutation in R80 was seen to be present at a higher frequency (Fig. 5B). Additionally, two distinct RNA helicases – DDX5 and DHX9 were found to have significantly different expression profiles between mutated and non-mutated cohorts. Another interesting example among the genes that showed a significant difference in the expression levels is RBM10 which is a predicted candidate driver in lung adenocarcinoma (LUAD). RBM10 was seen to be 2 fold down regulated in mutated samples when compared to the non-mutated samples. We hypothesize that the lower expression in the mutated samples could be a result of the presence of truncated transcripts due to several non-sense mutations in the gene encoding for RBM10 (Fig. 5B).43
Figure 5.
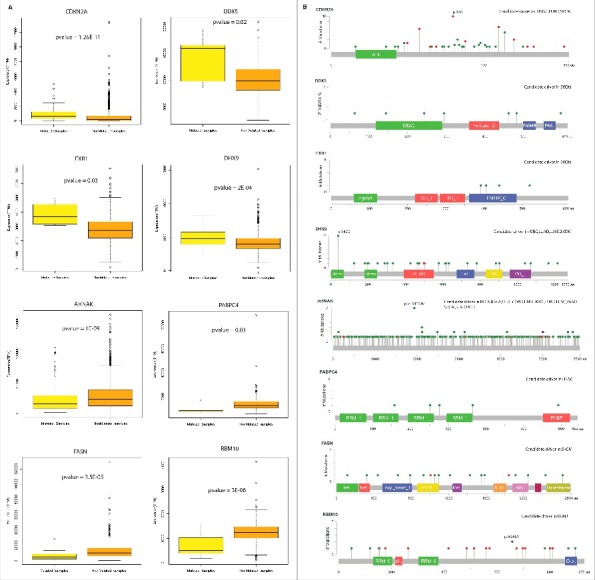
Expression of candidate RBP drivers | (A) Candidate RBP drivers which exhibited significant change in the RNA levels between mutated and non-mutated samples across cancer types. (B) Mutation diagram (Lollipop plots) of RBPs with significant expression change. These were plotted using the mutation mapper tool in the cbioportal.72,73
Candidate RBP drivers form an integral part of the spliceosomal machinery
RNA-binding proteins, often interact with different proteins in the cell to form protein complexes that mediate different events of the post transcriptional regulation. Hence, mutations in the RBP gene might not only lead to abnormal subcellular localization, defective binding to RNA but also lead to altered protein-protein interactions and thus conferring a cancer phenotype.15 Therefore, we analyzed the protein-protein interaction network of those RBPs predicted to be drivers in at least 2 cancers to delineate the common pathways that could be possibly affected by mutations in these genes. We used Reactome Functional Interaction Plugin to analyze such pathways (See Materials and Methods). We constructed a network of interactions among the candidate drivers by allowing the linker proteins. Upon analysis, FLNA – an actin binding protein predicted to be a candidate driver in cancers of breast, colon, stomach, skin and lung was seen to be physically interacting with MAPK14 – one of the important regulator of cancer progression48 (Fig. 6). Interestingly, MAPK14 was not identified as a candidate driver in any of the cancers and hence the interaction of FLNA with MAPK14 might possibly explain the cause of MAPK14 dysregulation in several cancers. Additionally, we performed a functional annotation on the constructed network to identify dense clusters that are functionally important. We observe that proteins important in the spliceosomal machinery to be significantly enriched (Fig. 6, Materials and Methods) suggesting the importance of splicing in causing cancer phenotype. Although post-transcriptional targets of most of these RBPs are unknown especially in the cancerous tissues where they are detected as drivers, we employed Seten,49 a functional analysis tool, which provides a comprehensive summary of the functional role of an RBP based on publicly available CLIP-seq profiles in cell lines, to study whether driver RBPs like PRPF8 and U2AF1 with CLIP-seq data exhibit enrichment to target cancer hallmarks.50,51 This analysis clearly revealed a significant enrichment in controlling several cell cycle progression and apoptosis related processes among the RNA targets of PRPF8 and U2AF1 across two different cancer cell lines K562 (Chronic Myelogenous Leukemia) and HepG2 (Hepatocellular Carcinoma) supporting the notion that pan-cancer driver RBPs might be involved in dysregulating such cancer hallmarks. Hence, we believe this analysis not only helps in unravelling the common drivers across cancers but also improves our understanding of the underlying post-transcriptional mechanisms mediating cancer phenotypes.
Figure 6.
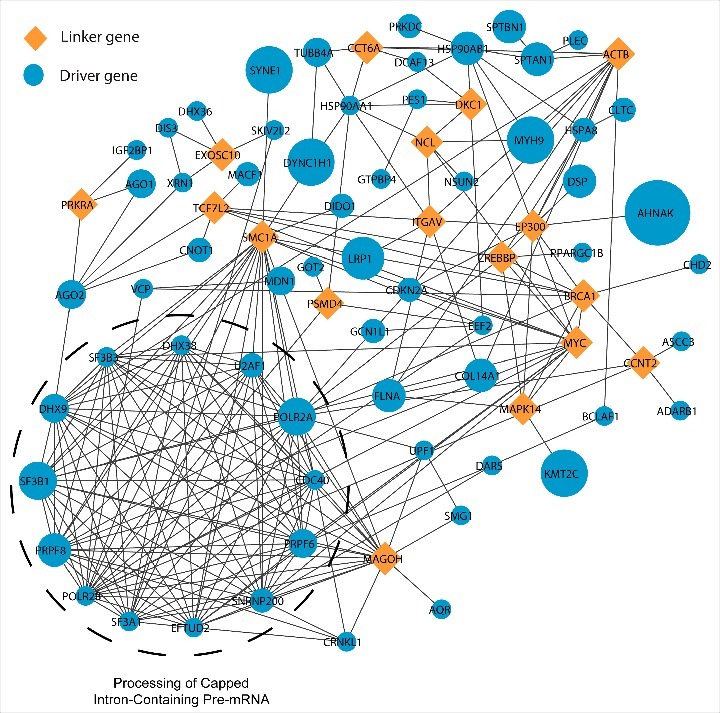
Protein-Protein interaction network among RBPs | The figure illustrates the protein-protein interactions among the RNA Binding proteins with driver mutations in at least two cancer types. The node sizes are proportional to the number of cancers in which a RBP is identified as a candidate driver. The dense cluster annotated as “processing of capped intron-containing pre-mRNA” was identified at p < 1E-03. The analysis was carried out using the ReactomeFI Cytoscape plugin and contains linker genes which connect driver RBPs in this network.
Driver RBPs exhibit significantly higher network centralities and their network analysis reveals several cancer-specific RNP mutational hotspots
We further analyzed the network properties such as degree, betweenness and closeness of RBPs that are predicted to be candidate drivers in at least two cancer types by constructing a protein-protein interaction network among RBPs (see Materials and Methods). Upon comparing the network properties of driver RBPs with that of nondriver RBPs, we found that drivers have significantly higher degree, betweenness and closeness centrality measures compared to the latter (Exact p-values indicated in Fig. 7A, Wilcox test). Higher degree, betweenness and closeness of drivers when compared to the non-drivers indicates that they form an integral part of the protein-protein interaction network of RBPs and thus mutations in them could significantly contribute to causing lethal phenotypes by potentially disrupting the formation of RNP complexes. Indeed, many drivers including RNA helicases like DDX proteins identified in this study were also discovered previously to be highly upregulated in cancer transcriptomes and were found to exhibit different path lengths in the protein interaction network thus suggesting the importance of these drivers in causing cancer phenotype due to disruption in protein complexes.5
Figure 7.
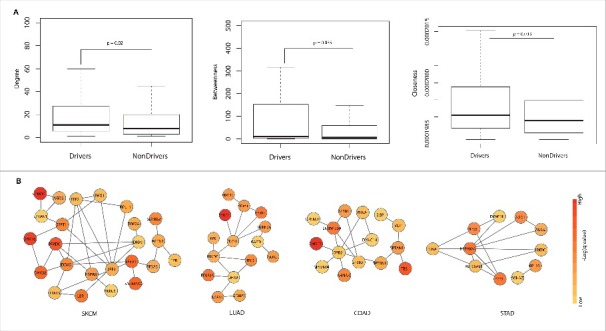
Network analysis and cancer specific subnetworks | (A) Comparison of network properties degree, betweenness and closeness of driver and non-driver RBPs in the protein interaction network among RBPs. All the properties were found to be significantly higher for drivers compared to non-drivers (p< 0.01, Wilcox test). (B) Cancer specific subnetworks in the protein-protein interaction network among candidate drivers in each cancer type – SKCM (A), LUAD (B), COAD (C), STAD (D) are shown. Only the most significant subnetworks are shown for each of these cancer types. The node color in each figure indicates the negative logarithm of the q-value derived from OncodriveFM approach in the respective cancer type.
RBPs often interact with proteins in the cell to form protein complexes that mediate different events of the post transcriptional regulation. Hence, mutations in them may not only affect the expression, defective binding to RNA but can also lead to altered protein-protein interactions and thus conferring a cancer phenotype.15 Also, due to varied mutational frequencies in different cancers, the interactome of an RBP may differ between cancer types. Hence, the topology of the interaction network of RBPs is likely to change among cancers due to the formation of cancer-specific Ribonucleoprotein (RNP) complexes. To address this, we identified cancer-specific subnetworks in the protein-protein interaction network of candidate driver RBPs (see Materials and Methods, Fig. 7B). Fig. 7B shows cancer-specific subnetworks of candidate driver RBPs in SKCM, LUAD, COAD and STAD respectively, obtained using the HotNet2 algorithm (See Materials and Methods, Table S5). PRKDC – a gene that encodes the catalytic subunit of the DNA dependent protein kinase, candidate driver in SKCM, STAD and LUAD was found to show variation in its interacting proteins and topology between these cancer types (Fig. 7B). In SKCM, PRKDC was seen to be interacting with DHX38, DHX9, DDX5 and HSPA8 whereas the interacting partners in LUAD and STAD are (RTCB, TPR, DHX9, CDC5L) and (HSP90AB1, HSP90AA1) respectively. Also notable is SF3B1 which is a candidate driver in LUAD and COAD. This RBP interacts with CDC5L and SF3A1 in LUAD whereas it interacts with SNRNP300 and SF3B3 in COAD suggesting that different combinations of RBP mutated complexes could be contributing to disruption in different post-transcriptional sub-networks leading to varying cancer phenotypes. Therefore, these observations project the prominence of understanding the rewiring of protein-protein interactions of RBPs across different cancers and thus plausibly contributing to the heterogeneity among cancers.
Knockdown of pan-cancer drivers, SF3B1 and PRPF8, in breast cancer cell lines reveals cancer subtype-specific effects
Our analysis at multiple levels indicated that RBPs involved in splicing and splicesomal machinery to be significantly mutated in multiple cancer types. In particular, our functional analysis (Fig. 6) revealed that RBPs such as SF3B1 and PRPF8, which are identified as a driver in at least four different cancer types are an integral part of the splicing machinery. Hence, to understand if the mutations in these proteins are truly deleterious and/or can have a phenotypic impact in breast cancer, we choose SF3B1 and PRPF8 to study their effect on breast cancer cell lines. In particular, we reduced the levels of these two proteins in two breast cancer cell lines and measured the levels of cancer stem cell markers (see Materials and Methods). CD44+/CD24- cells are suggested to have cancer stem cell phenotype, although CD44+/CD24+ cells do possess cancer stem cell features.52-54 MCF-7 is a luminal cell line with 15% CD44+/CD24+ cells. Despite inefficient knockdown of SF3B1 (Fig. 8A), CD44+/CD24+ cells were reproducibly reduced upon SF3B1 knockdown (Fig. 8B). By contrast, SF3B1 knockdown cells displayed elevated levels of CD24 compared with control luciferase siRNA transfected cells (Fig. 8C). Similar results were obtained upon knockdown of PRPF8 in these cells (Fig. 8A-C). Interestingly, SF3B1 or PRPF8 knockdown in MDA-MB-231 cell line, which represents mesenchymal stem like triple negative breast cancer,55 had no effect on CD44+/CD24- status (Fig. 8). Thus, activity of SF3B1 and PRPF8 in breast cancer may be subtype-specific. Our multiple attempts to obtain MCF-7 cells with significant knockdown of SF3B1 were not successful although the same siRNA was effective in reducing SF3B1 in MDA-MB-231 cells. Inability to reduce SF3B1 in MCF-7 suggests the requirement of SF3B1 for survival of these cells. Consistent with this possibility, recent studies have demonstrated SF3B1 mutation (K700E) in breast cancer, preferentially in estrogen receptor positive breast cancer, to be a driver mutation.56 MCF-7 cells are estrogen receptor positive and are dependent on estrogen for survival.57 Overall, our results suggest breast cancer subtype-specific function of SF3B1 and PRPF8 (Fig. 8).
Figure 8.
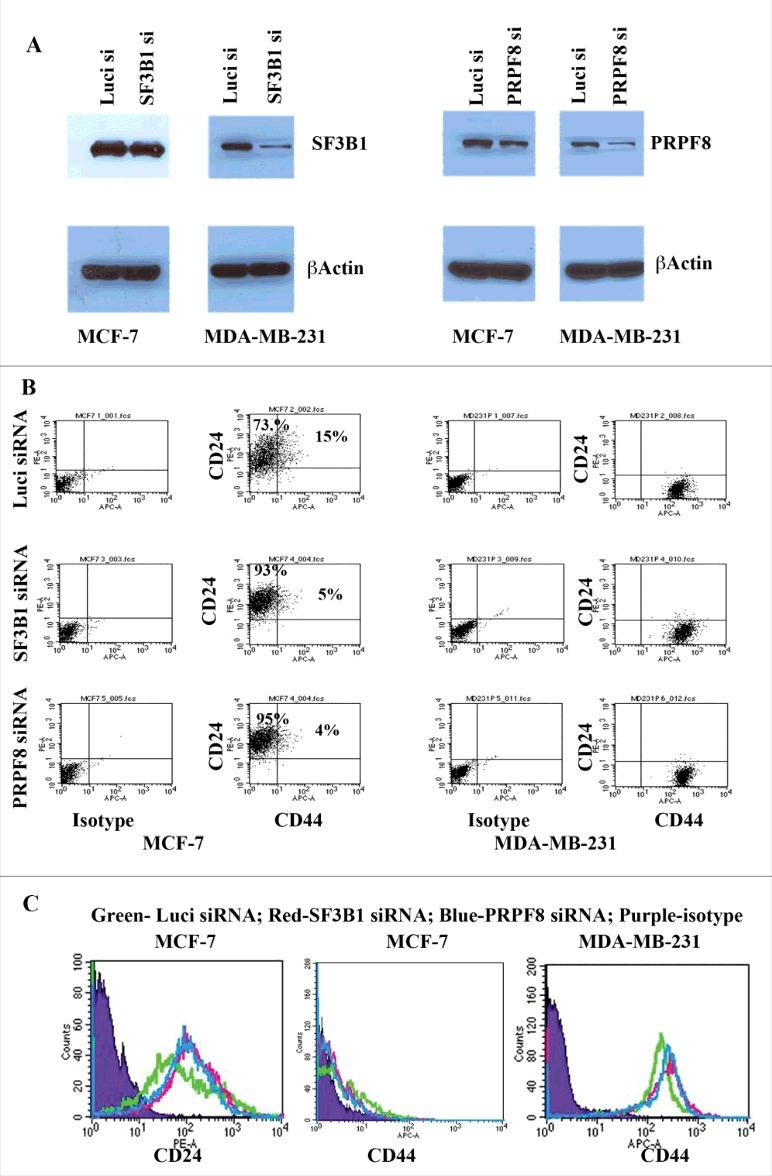
SF3B1 and PRPF8 alter CD44/CD24 profile in MCF-7 but not MDA-MB-231 cells. (A) siRNA-mediated knockdown of SF3B1 and PRPF8 in MCF-7 and MDA-MB-231 cells. Same blots were re-probed for β-Actin as a loading control. (B) CD44/CD24 staining pattern in control siRNA, SF3B1 siRNA and PRPF8 siRNA treated cells. Isotype controls were used to generate quadrants. SF3B1 and PRPF8 knockdown reduced the number of CD44+/CD24+ cells in MCF-7 cells but had no effect on MDA-MB-231 cells, despite much higher knockdown in these cells. MDA-MB-231 cells are predominantly CD44+/CD24-. (C). The effect of SF3B1 and PRPF8 on CD24 and CD44 protein levels. SF3B1 and PRPF8 knockdown increased CD24 but did not significantly effect CD44 levels in MCF-7 cells. SF3B1 and PRPF8 knockdown in MDA-MB-231 cells had no effect on CD44 expression.
Discussion
RNA Binding proteins (RBPs) are a class of proteins crucial in orchestrating several events of the post transcriptional regulation (PTR). Dysregulation of these proteins has been implicated in several disorders including cancer although the causes of such dysregulation is poorly understood. In this study, we delineate the mutational landscape of ∼1300 RNA-Binding proteins across 26 cancer types. Our computational analysis revealed that RBPs have an average of ∼3 mutations per Mb across 26 cancers and enabled the identification of 281 RBPs to be enriched for mutations in at least one cancer type. Among these, genes encoding for EPPK1, KMT2C, AHNAK and PLEC were found to be enriched for mutations in at least 10 cancers suggesting common players in mediating cancer phenotypes in different tissues. GC content and exome length were not found to play a major role in contributing to the mutational frequency of RBPs in majority of the studied cancers. However, it is possible to speculate that other properties such as high expression, prevalent in RBPs,4-6 can result in a tendency for genes to be clustered in specific regions of the genome, are likely to be in ‘open chromatin’ regions or related to cancer pathways/functions, leading to their specific high mutation rates. Hence, as the repertoire of RBPs and their genomic organization principles are increasingly studied, such contributions to the mutational landscape of RBPs will become clear across a broad range of cancer types. Our analyses also revealed that RBPs enriched for mutations in atleast one cancer type were seen to be undergoing frequent Frameshift and Inframe deletions, missense, nonsense and silent mutations when compared to those that are not enriched, revealing the abundance of these variant types in mutated RBPs as significant contributor for malfunction in cancer genomes. Functional analysis of the RBPs which are significantly mutated, revealed the enrichment of pathways related to apoptosis, splicing and translation. Additionally, we identified more than 200 RBPs that are candidate drivers in at least one cancer type. These drivers based on the impact of non-synonymous mutations on the function of RBPs included AHNAK and SYNE1 which were found to be significantly mutated in at least 12 and 8 cancer types respectively. We show that the presence of non-synonymous mutations correlate with change in the RNA levels of a significant fraction of driver RBPs (15% of the drivers), when cancer samples are grouped by the presence of mutations in an RBP irrespective of the cancer type. Also, protein-protein interaction network analysis of the driver genes identified in at least two cancer types revealed the presence of a cluster of mutated proteins involved in the spliceosomal machinery, suggesting a plausible mechanism for tumorogenesis. Knockdown of pan-cancer drivers such as SF3B1 and PRPF8 in breast cancer cell lines MCF7 and MDA-MB-231 using siRNAs, revealed cancer subtype-specific effects. Our knock down experiments indicated that deletion of either of these RBPs resulted in MCF7 cells, which are estrogen receptor positive, to exhibit reduced stem cell features. In contrast, MDA-MB-231 cells, which represent mesenchymal stem like triple negative breast cancer, did not exhibit any change in stem cell characteristics suggesting the cancer subtype specific effects imparted due to the alternations in the levels of driver RBPs. These observations suggest the need to account for tumor variability, due to the presence of multiple cell populations in a given tumor sample, in developing better cancer systems biology models which can account for clonal evolution of cancer genomes.58,59 Although, current depth of sequencing and level of annotation from TCGA datasets doesn't readily permit such high resolution analyses to dissect the prevalent clones, future studies focused on single cell sequencing of tumors should be able to dissect the clonal origin of the mutations for driver RBPs identified here. Network analysis of the driver RBPs in the protein interaction of RBPs clearly revealed higher network centrality measures suggesting the prominent positions they hold in the RNP network. These observations suggest that driver RBPs which are highly connected in the protein interaction network could contribute to the disruption in the formation of RNP complexes thereby effecting the post-transcriptional networks they control. To identify cancer-specific sub-networks which are likely to represent RNP complexes which are effected in different cancers, we employed the hotnet framework and identified several potential RNP complexes which are mutated in four different cancer types. Our results suggest that although RBP drivers might be common between cancers, their downstream RNP mutational hotspots could be very different thereby leading different post-transcriptional network changes across cancer types. This analysis should form a foundation to help us uncover the mutational spectrum of RBPs and their wiring dynamics in different cancer types thereby leading to dysregulation of post-transcriptional regulatory networks and also emphasizes the potential of various proteins of the splicesomal machinery as possible drug targets in cancer.
Materials and methods
Datasets used in the study
a. RNA binding proteins
We catalogued a set of 1344 genes encoding for RBPs in the human genome of which 1298 had mutation data. These consist of RBPs identified in recent experimental screens, including Castello et al.,60 Baltz et al.,61 Ray et al.,62 human orthologs of RBPs identified in mouse embryonic stem cells by Kwon et al.,63 and those reported in RBPDB.64 Complete dataset of RBPs is available as Table S1 and from READDB.65
b. Mutation data
Somatic mutation calls were downloaded as MAF Files from the Broad Firehouse for 26 cancer types: Adrenocorticol carcinoma (ACC), Bladder Urothelial carcinoma (BLCA), Breast invasive carcinoma (BRCA), Cervival squamous cell carcinoma an endocervical adenocarcinoma (CESC), Colon adenocarcinoma (COAD), Gliobastoma multiforme (GBM), Head and neck squamous cell carcinoma (HNSC), Kidney Chromophobe (KICH), Kidney renal cell carcinoma (KIRC), Kidney renal papillary cell carcinoma (KIRP), Acute myeloid leukemia (LAML), Lower grade glioma (LGG), Liver hepatocellular carcinoma (LIHC), Lung adenocarcinoma (LUAD), Lung squamous cell carcinoma (LUSC), Ovarian serous cystadenocarcinoma (OV), Pancreatic adenocarcinoma (PAAD), Pheochromocytoma and Paraganglioma (PCPG), Prostrate adenocarcinoma (PRAD), Rectum adenocarcinoma (READ), Skin cutaneous Melanoma (SKCM), Stomach adenocarcinoma (STAD), Thyroid carcinoma (THCA), Uterine corpus endometrial carcinoma (UCEC), Uterine carcinosarcoma (UCS), Uveal Melanoma (UVM). Mutations that were called using NCBI Build 36 were converted to build 37 using the NCBI lift over. The list of cancer types and their corresponding downloaded filenames are available as Table S1.
Mutation frequency
The overall mutation frequency of a gene in a given cancer type is calculated as shown in Fig. 1A. To summarize, the mutation frequency is calculated by normalizing the total number of mutations in a given gene by its exome length and the number of patients in the cancer type being analyzed. The obtained value is multiplied by 106 to estimate the mutation frequency per Mb. The Pearson correlation of mutation frequencies between different cancers was estimated using the “rcorr” function in the Hmisc package in R. Hierarchical clustering of mutational frequencies was performed using GENE-E (http://www.broadinstitute.org/cancer/software/GENE-E).
Identification of genes enriched in mutations (GEMs), analysis of gene sets and variant types
We calculated the significance of observing the total mutations in a gene, given its exome length against the whole genome as a background using Fisher's exact test (see Fig. 1B). The obtained p-values are corrected by Benjamini- Hochberg based FDR correction. Genes with corrected p < 0.01 and odds ratio < 1 were classified as Genes Enriched in Mutations (GEMs) in a given cancer. P-value and the odds ratio were calculated using the fisher.test function in R.
Functional analysis of gene sets was carried out using ClueGO, a cytoscape plugin used to identify and visualize functionally related clusters.66 Pathway annotations available in Reactome database67 were used to carry out the functional analysis. Clusters identified at p < 0.01 were used for interpretation.
For the analysis based on the variant types, we initially categorized RBPs in each cancer as GEMs and non-GEMs based on the above mentioned criteria. We then obtained the mutation frequency of these genes in each cancer type for nine different variant classes viz – Inframe deletion, Inframe insertion, Frameshift deletion, Frameshift Insertion, Missense mutation, Nonsense mutation, Nonstop mutation, Silent and Splice Site mutations. Variants were classified into the above mentioned categories based on the annotations provided in the downloaded MAF files. Also, mutation frequency of a gene for each of these variant types in a given cancer is calculated as shown in Fig. 1A (See “Mutation Frequency” section in Materials and Methods). Upon obtaining the mutation frequencies in each cancer type for all the variant classes, we pooled the mutational frequencies of RBPs enriched for mutations across the cancers into one bin named as GEM-RBPs (Fig. 3C) and mutation frequencies of RBPs that are not enriched for mutations in any cancer are labelled as NonGEM-RBPs in Fig. 3C. The significance of the differences between these two groups was calculated using Wilcox test.
Identifying candidate driver RBPs
We computed the bias of RBPs towards the accumulation of somatic mutations of high functional impact (FM bias) to identify potential driver genes among them, across each cohort of cancers. To this end, we first employed the IntOGen-FM pipeline,68 on each of the 26 cohorts of cancer samples used in this study. The pipeline first obtained three predicted functional impact scores for each of SIFT, PPH2 and Mutation Assessor algorithms19-21 for the somatic mutations observed in all genes across cancer samples of each cohort. We then used the OncodriveFM approach39 to compute the FM bias of RBPs. Somatic mutations in all genes were taken into account by OncodriveFM to compute the background functional impact of each RBP, and to correct FM bias p-values for multiple testing. The analysis of the 26 cohorts was carried out using expression filters and mutational thresholds as described in.68 In each cohort, RBPs identified at q-value < 0.01 were identified as candidate drivers.
Expression analysis
The cancer expression data was downloaded from The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov/tcga). TCGA provides multi-level data (clinical, genome sequencing, microarray, RNA sequencing etc.) procured from a number of institutions, from a variety of patients, for over 30 cancers. In this study, we collected RNAseq V2.0 data for more than 6000 patients spanning 26 cancers, by downloading the RNA expression levels for all the genes across the cancer cohorts by selecting the Level 3 from the data portal of the TCGA data access site. For a given gene predicted as a driver in a particular cancer, we divide the patient cohort into two groups – Mutated and Non-mutated samples corresponding to that gene. Mutated samples constitute patients with non-synonymous mutations in a given gene of interest. On the contrary, Non-mutated samples constitute those without the mutations. By using the TCGA sample barcodes we cross mapped between the mutation and expression data for each patient in each cohort. The significance of difference in the expression levels between these sample groups is calculated using the Wilcox-test. Genes which exhibited a significant difference in expression levels between the two groups (p < 0.01) were considered for further analysis, under the notion that these non-synonymous mutations in drivers are contributing to the changes in expression levels.
Functional interaction network analysis
RBPs predicted to be drivers in at least two cancers were used for analyzing the functional interaction networks. We used Cytoscape FI plugin69 to map the driver genes onto the interaction network. We allowed the presence of linker genes to expand the network of the genes. We then grouped the genes into enriched functions using the annotations available in the Reactome as a background. Complexes with more than five components and FDR corrected p < 1E-03 were identified to be significant and were highlighted in Fig. 6.
Network properties of candidate drivers
We constructed a network of 12299 interactions among 1247 RBPs using the data obtained from BioGRID for human proteins.70 For each node, network properties like Degree, Betweenness and Closeness were calculated using the built in functions in igraph (See http://cneurocvs.rmki.kfki.hu/igraph/ and http://www.r-project.org). Further, genes were categorized as drivers – if they are predicted to be candidate drivers in at least two cancer types and the remaining RBPs are termed nondrivers. Statistical significance was estimated using Wilcox test.
Identifying cancer specific subnetworks among candidate RBP drivers
We employed the HotNet2 (HotNet diffusion-oriented subnetworks) algorithm71 to identify subnetworks in a given network among the candidate drivers identified in SKCM, LUAD, COAD and STAD. We limited our analysis to these cancers based on the number of RBP drivers predicted (>40). To this end, we constructed a network constituting 12299 interactions among 1247 RBPs using BioGRID.70 We then performed permutations of the above network to generate 100 random networks. Subsequently, for each cancer type, every node in the protein-protein interaction network is given a “heatscore” as input to the algorithm that is identical to the q-value obtained from the above analysis (See Section titled “Identifying candidate driver RBPs”). Using these input parameters, the HotNet2 algorithm was run independently on all the four cancers to identify significant subnetworks. Further, the subnetworks identified at specific threshold delta values were considered for the analysis – SKCM (delta = 7.19E-06), LUAD (delta = 4.21E-06), COAD (delta = 2.47E-06), STAD (delta = 2.92E-06).
Cell lines, siRNA transfection, western blotting and flow cytometry
MCF-7 and MDA-MB-231 cells were maintained in minimum essential media (MEM) with 10% fetal calf serum and Penicillin/Streptomycin. Cells were transfected with 60 nM control luciferase siRNA, siRNA against SF3B1 (Ambion Cat# 16708A, Assay ID:19939) or PRPF8 (Ambion Cat# 16708A Assay ID: 241490) using Lipofectamine reagent (Invitrogen). SF3B1 and PRPF8 protein levels were measured by western blotting four days after siRNA transfection as described previously.54 CD44/CD24 staining and flow cytometry was also performed four days after transfection as described previously.54 Antibodies against SF3B1 (cat# 14434S, Cell Signaling), PRPF8 (Cat#Ab190347, Abcam), CD24 (Cat#555428, BD Biosciences) and CD44 (Cat#559942, BD Biosciences) were used as per instructions from manufacturers.
Supplementary Material
Funding Statement
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM123314 to SCJ. This work is also supported by a grant from the Susan G. Komen for the Cure (SAC110025 to HN).
Conflict of interest
The authors have no competing interests to declare.
Acknowledgments
The authors would like to thank members of the Janga Lab for critically reading a previous version of the manuscript and providing helpful comments. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank Nuria Lopez-Bigas for her support during the study. We thank Manjushree Anjanappa and IU Simon Cancer Center Flow Cytometry core for help with flow cytometry analysis.
References
- 1.Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–45. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–86. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janga SC. From specific to global analysis of posttranscriptional regulation in eukaryotes: posttranscriptional regulatory networks. Brief Funct Genomics. 2012;11:505–21. doi: 10.1093/bfgp/els046. [DOI] [PubMed] [Google Scholar]
- 4.Neelamraju Y, Hashemikhabir S, Janga SC. The human RBPome: From genes and proteins to human disease. J Proteomics. 2015;127(Pt A):61–70. doi: 10.1016/j.jprot.2015.04.031. [DOI] [PubMed] [Google Scholar]
- 5.Kechavarzi B, Janga SC. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol. 2014;15:R14. doi: 10.1186/gb-2014-15-1-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mittal N, Roy N, Babu MM, Janga SC. Dissecting the expression dynamics of RNA-binding proteins in posttranscriptional regulatory networks. Proc Natl Acad Sci U S A. 2009;106:20300–5. doi: 10.1073/pnas.0906940106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lukong KE, Chang KW, Khandjian EW, Richard S. RNA-binding proteins in human genetic disease. Trends Genet. 2008;24:416–25. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Kim MY, Hur J, Jeong S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Rep. 2009;42:125–30. doi: 10.5483/BMBRep.2009.42.3.125. [DOI] [PubMed] [Google Scholar]
- 9.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254–66. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- 10.Wurth L. Versatility of RNA-Binding Proteins in Cancer. Comp Funct Genomics. 2012;2012:178525. doi: 10.1155/2012/178525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matter N, Herrlich P, Konig H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature. 2002;420:691–5. doi: 10.1038/nature01153. [DOI] [PubMed] [Google Scholar]
- 12.Kawahara H, Imai T, Imataka H, Tsujimoto M, Matsumoto K, Okano H. Neural RNA-binding protein Musashi1 inhibits translation initiation by competing with eIF4G for PABP. J Cell Biol. 2008;181:639–53. doi: 10.1083/jcb.200708004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okano H, Kawahara H, Toriya M, Nakao K, Shibata S, Imai T. Function of RNA-binding protein Musashi-1 in stem cells. Exp Cell Res. 2005;306:349–56. doi: 10.1016/j.yexcr.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 14.Han W, Xin Z, Zhao Z, Bao W, Lin X, Yin B, Zhao J, Yuan J, Qiang B, Peng X. RNA-binding protein PCBP2 modulates glioma growth by regulating FHL3. J Clin Invest. 2013;123:2103–18. doi: 10.1172/JCI61820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castello A, Fischer B, Hentze MW, Preiss T. RNA-binding proteins in Mendelian disease. Trends Genet. 2013;29:318–27. doi: 10.1016/j.tig.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al.. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977–83. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bechara EG, Sebestyen E, Bernardis I, Eyras E, Valcarcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell. 2013;52:720–33. doi: 10.1016/j.molcel.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 21.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, et al.. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–6. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson D, Charoensawan V, Kummerfeld SK, Teichmann SA. DBD–taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res. 2008;36:D88–92. doi: 10.1093/nar/gkm964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–2. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez-Perez A, Jene-Sanz A, Lopez-Bigas N. The mutational landscape of chromatin regulatory factors across 4,623 tumor samples. Genome Biol. 2013;14:r106. doi: 10.1186/gb-2013-14-9-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiche G. Role of plectin in cytoskeleton organization and dynamics. J Cell Sci. 1998;111(Pt 17):2477–86. [DOI] [PubMed] [Google Scholar]
- 27.Bausch D, Thomas S, Mino-Kenudson M, Fernandez-del CC, Bauer TW, Williams M, Warshaw AL, Thayer SP, Kelly KA. Plectin-1 as a novel biomarker for pancreatic cancer. Clin Cancer Res. 2011;17:302–9. doi: 10.1158/1078-0432.CCR-10-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katada K, Tomonaga T, Satoh M, Matsushita K, Tonoike Y, Kodera Y, Hanazawa T, Nomura F, Okamoto Y. Plectin promotes migration and invasion of cancer cells and is a novel prognostic marker for head and neck squamous cell carcinoma. J Proteomics. 2012;75:1803–15. doi: 10.1016/j.jprot.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida T, Shiraki N, Baba H, Goto M, Fujiwara S, Kume K, Kume S. Expression patterns of epiplakin1 in pancreas, pancreatic cancer and regenerating pancreas. Genes Cells. 2008;13:667–78. doi: 10.1111/j.1365-2443.2008.01196.x. [DOI] [PubMed] [Google Scholar]
- 30.Guo X, Hao Y, Kamilijiang M, Hasimu A, Yuan J, Wu G, Reyimu H, Kadeer N, Abudula A. Potential predictive plasma biomarkers for cervical cancer by 2D-DIGE proteomics and Ingenuity Pathway Analysis. Tumour Biol. 2015;36:1711–20. doi: 10.1007/s13277-014-2772-5. [DOI] [PubMed] [Google Scholar]
- 31.Roth JR. Frameshift mutations. Annu Rev Genet. 1974;8:319–46. doi: 10.1146/annurev.ge.08.120174.001535. [DOI] [PubMed] [Google Scholar]
- 32.Challis D, Antunes L, Garrison E, Banks E, Evani US, Muzny D, Poplin R, Gibbs RA, Marth G, Yu F. The distribution and mutagenesis of short coding INDELs from 1,128 whole exomes. BMC Genomics. 2015;16:143. doi: 10.1186/s12864-015-1333-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 34.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 35.Hamby SE, Thomas NS, Cooper DN, Chuzhanova N. A meta-analysis of single base-pair substitutions in translational termination codons (‘nonstop’ mutations) that cause human inherited disease. Hum Genomics. 2011;5:241–64. doi: 10.1186/1479-7364-5-4-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng S, Kim H, Verhaak RG. Silent mutations make some noise. Cell. 2014;156:1129–31. doi: 10.1016/j.cell.2014.02.037. [DOI] [PubMed] [Google Scholar]
- 37.Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28:150–8. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 38.Green RE, Lewis BP, Hillman RT, Blanchette M, Lareau LF, Garnett AT, Rio DC, Brenner SE. Widespread predicted nonsense-mediated mRNA decay of alternatively-spliced transcripts of human normal and disease genes. Bioinformatics. 2003;19(Suppl 1):i118–21. doi: 10.1093/bioinformatics/btg1015. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012;40:e169. doi: 10.1093/nar/gks743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee IH, Sohn M, Lim HJ, Yoon S, Oh H, Shin S, Shin JH, Oh SH, Kim J, Lee DK, et al.. Ahnak functions as a tumor suppressor via modulation of TGFbeta/Smad signaling pathway. Oncogene. 2014;33:4675–84. doi: 10.1038/onc.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shankar J, Messenberg A, Chan J, Underhill TM, Foster LJ, Nabi IR. Pseudopodial actin dynamics control epithelial-mesenchymal transition in metastatic cancer cells. Cancer Res. 2010;70:3780–90. doi: 10.1158/0008-5472.CAN-09-4439. [DOI] [PubMed] [Google Scholar]
- 42.Shtivelman E, Cohen FE, Bishop JM. A human gene (AHNAK) encoding an unusually large protein with a 1.2-microns polyionic rod structure. Proc Natl Acad Sci U S A. 1992;89:5472–6. doi: 10.1073/pnas.89.12.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belgrader P, Maquat LE. Nonsense but not missense mutations can decrease the abundance of nuclear mRNA for the mouse major urinary protein, while both types of mutations can facilitate exon skipping. Mol Cell Biol. 1994;14:6326–36. doi: 10.1128/MCB.14.9.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14:447–59. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 45.Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, et al.. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3:1122–9. doi: 10.1158/2159-8290.CD-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al.. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44:53–7. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. doi: 10.1182/blood-2013-01-480970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bradham C, McClay DR. p38 MAPK in development and cancer. Cell Cycle. 2006;5:824–8. doi: 10.4161/cc.5.8.2685. [DOI] [PubMed] [Google Scholar]
- 49.Budak G, Srivastava R, Janga SC. Seten: A tool for systematic identification and comparison of processes, phenotypes and diseases associated with RNA-binding proteins from condition-specific CLIP-seq profiles. RNA. 2017;23(6):836–46. doi: 10.1261/rna.059089.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 51.Wang E, Zaman N, McGee S, Milanese JS, Masoudi-Nejad A, O'Connor-McCourt M. Predictive genomics: a cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin Cancer Biol. 2015;30:4–12. doi: 10.1016/j.semcancer.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 52.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R Jr, Badve S, Nakshatri H. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006;8:R59. doi: 10.1186/bcr1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhat-Nakshatri P, Appaiah H, Ballas C, Pick-Franke P, Goulet R Jr, Badve S, Srour EF, Nakshatri H. SLUG/SNAI2 and tumor necrosis factor generate breast cells with CD44+/CD24- phenotype. BMC Cancer. 2010;10:411. doi: 10.1186/1471-2407-10-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maguire SL, Leonidou A, Wai P, Marchio C, Ng CK, Sapino A, Salomon AV, Reis-Filho JS, Weigelt B, Natrajan RC. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol. 2015;235:571–80. doi: 10.1002/path.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al.. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang E, Zou J, Zaman N, Beitel LK, Trifiro M, Paliouras M. Cancer systems biology in the genome sequencing era: part 1, dissecting and modeling of tumor clones and their networks. Semin Cancer Biol. 2013;23:279–85. doi: 10.1016/j.semcancer.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 59.Wang E, Zou J, Zaman N, Beitel LK, Trifiro M, Paliouras M. Cancer systems biology in the genome sequencing era: part 2, evolutionary dynamics of tumor clonal networks and drug resistance. Semin Cancer Biol. 2013;23:286–92. doi: 10.1016/j.semcancer.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 60.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al.. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 61.Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al.. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46:674–90. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 62.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al.. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–7. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kwon SC, Yi H, Eichelbaum K, Fohr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, Kim VN. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013;20:1122–30. doi: 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- 64.Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011;39:D301–8. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hashemikhabir S, Neelamraju Y, Janga SC. Database of RNA binding protein expression and disease dynamics (READ DB). Database (Oxford). 2015;2015:bav072. doi: 10.1093/database/bav072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–3. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Croft D, O'Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, et al.. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–7. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, Santos A, Lopez-Bigas N. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013;10:1081–2. doi: 10.1038/nmeth.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010;11:R53. doi: 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–9. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leiserson MD, Vandin F, Wu HT, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al.. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 2015;47:106–14. doi: 10.1038/ng.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.