

Escherichia coli has been engineered to produce chlorophyll.
Abstract
Chlorophylls are essential cofactors for photosynthesis, which sustains global food chains and oxygen production. Billions of tons of chlorophylls are synthesized annually, yet full understanding of chlorophyll biosynthesis has been hindered by the lack of characterization of the Mg–protoporphyrin IX monomethyl ester oxidative cyclase step, which confers the distinctive green color of these pigments. We demonstrate cyclase activity using heterologously expressed enzyme. Next, we assemble a genetic module that encodes the complete chlorophyll biosynthetic pathway and show that it functions in Escherichia coli. Expression of 12 genes converts endogenous protoporphyrin IX into chlorophyll a, turning E. coli cells green. Our results delineate a minimum set of enzymes required to make chlorophyll and establish a platform for engineering photosynthesis in a heterotrophic model organism.
INTRODUCTION
Chlorophylls (Chls) underpin photosynthesis, which generates the oxygen that supports all complex life on Earth and the reducing potential to fix carbon dioxide as carbohydrates, the ultimate source of all the organic compounds required for life on Earth. The annual production of Chls, on land and in the oceans, is on the scale of billions of tons, yet the enzyme components of the biosynthesis pathway have neither been fully determined nor assembled to define the minimal set of genes required to make Chl. The Chl biosynthetic pathway (Fig. 1A) is a branch of tetrapyrrole biosynthesis, and it begins with protoporphyrin IX (PPIX), which is also the precursor for heme biosynthesis. Most life forms, including those able to photosynthesize, make PPIX and convert it to heme by inserting Fe2+ into the porphyrin macrocycle. Heme is a cofactor for respiratory proteins, and it is also converted into bilins, linear tetrapyrroles that are used as light-harvesting pigments in cyanobacteria.
Fig. 1. Assembly of the Chl biosynthesis pathway in Escherichia coli.
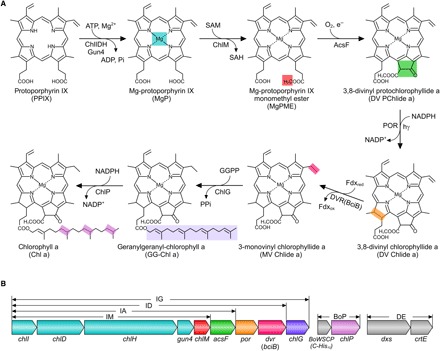
(A) Overall reactions from PPIX to Chl a catalyzed by the enzymes introduced to E. coli. The insertion of Fe2+ into PPIX (not shown) creates a biosynthetic branchpoint (not shown) that yields heme. Colored shading denotes the chemical change(s) at each step. ATP, adenosine triphosphate; ADP, adenosine diphosphate; SAM, S-adenosine-l-methionine; SAH, S-adenosyl-l-homocysteine; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of NADP+; Pi, inorganic phosphate; PPi, inorganic pyrophosphate. (B) Arrangement and relative size of each gene in the constructed plasmids. The chlI, chlD, chlH, gun4, chlM, acsF, dvr (bciB), and chlG genes were consecutively cloned into a modified pET3a vector with a single T7 promoter upstream of chlI and a ribosome-binding site upstream of each gene using the link-and-lock method (see fig. S2 for details) (40). Colors for genes correspond to those used in (A). Plasmid constructs and gene contents are IM (chlI–chlM), IA (chlI–acsF), ID (chlI–dvr), IG (chlI–chlG), BoP (BoWSCP and chlP), and DE (dxs and crtE). BoP is a pACYCDuet1-based plasmid containing a sequence encoding the BoWSCP protein with a C-terminal His10 tag (10) and the Synechocystis chlP gene. DE is a pCOLADuet1-based plasmid containing the E. coli dxs gene and the Rvi. gelatinosus crtE gene.
Chl biosynthesis is initiated when the magnesium chelatase enzyme complex (ChlIDH, Gun4) inserts Mg2+ into PPIX, so the Mg2+/Fe2+ Chl/heme branchpoint in photosynthetic bacteria, algae, and plants requires fine control to ensure that the correct amounts of hemes, bilins, and Chls are produced (1). Following the production of Mg-PPIX (MgP), six more enzymatic steps culminate in the production of Chl a, the ubiquitous pigment of oxygenic photosynthesis (see Fig. 1A). The MgP methyltransferase (ChlM) produces the substrate for the MgP monomethyl ester (MgPME) cyclase (AcsF), which produces 3,8-divinyl protochlorophyllide a (DV PChlide a); this intermediate has acquired a fifth ring that imparts the green color characteristic of Chls. Protochlorophyllide oxidoreductase (POR) produces 3,8-divinyl chlorophyllide a (DV Chlide a), which is reduced by divinyl reductase (DVR) and then esterified with geranylgeranyl pyrophosphate (GGPP) by Chl synthase (ChlG) to produce GG–Chl a. Finally, the reduction of the GG “tail,” catalyzed by GG reductase (ChlP), completes the pathway, and Chl a is produced. At this point, the pigment is hydrophobic and enters the membrane-bound assembly pathway for photosynthetic complexes, where it engages with the Sec/YidC-assembly machinery that coordinates the cotranslational insertion of nascent photosystem polypeptides with pigment production (2). Many steps of the Chl biosynthesis pathway have been studied individually in mutant strains of phototrophs, and some of the early steps have been characterized to define kinetic parameters (3), but it is not known whether these enzymes are sufficient to assemble the pathway and convert PPIX to Chl a. Apart from the global significance of Chl biosynthesis, the bottom-up construction of this pathway represents the first step in reprogramming a heterotrophic bacterium for growth under a variety of predetermined conditions of light intensity and wavelength. This metabolic engineering, which would also require genes encoding photosynthetic apoproteins, not only addresses important biological problems such as the concept of the minimal amount of genetic information required for photosynthetic life but also lays the groundwork for engineering cell factories with light-powered metabolism.
RESULTS AND DISCUSSION
In vivo cyclase activity of heterologously expressed AcsF
From the outset, the major obstacle for constructing a complete Chl biosynthetic pathway was the cyclase step, the activity of which has never been demonstrated using a recombinant enzyme. There are two mechanistically unrelated cyclases: an oxygen-sensitive, radical-SAM enzyme with a [4Fe-4S] cluster encoded by the bchE gene in most anoxygenic phototrophic bacteria (4) and an oxygen-dependent di-iron enzyme in some purple proteobacteria, as well as cyanobacteria, algae, and higher plants (5). We previously delineated three distinct classes of the oxygen-dependent cyclase in terms of subunit composition (5); one class comprises only one known subunit, AcsF, and the other two cyclases require an additional subunit, either BciE or Ycf54, for activity. Studies on barley mutants (6) indicated that additional subunits were required to form an active cyclase complex, and substantial effort had been made to identify a missing component. To verify that AcsF is the sole cyclase component, we transferred the acsF gene (7) from one purple bacterial phototroph, Rubrivivax (Rvi.) gelatinosus, to another that lacks the oxygen-dependent cyclase, Rhodobacter (Rba.) capsulatus. The bchE gene encoding the oxygen-sensitive cyclase was then deleted from the recipient (fig. S1A), as was the ccoP gene encoding a subunit of the oxygen scavenging cbb3 oxidase in Rba. capsulatus (fig. S1B). This latter manipulation should increase the intracellular oxygen concentration and allow the foreign AcsF cyclase to function (8). Bacteriochlorophyll biosynthesis and assembly of photosynthetic complexes were abolished in the ΔbchEΔccoP mutant, which, when harboring the pBB[acsF] plasmid-containing Rvi. gelatinosus acsF, regained nearly wild-type levels of pigmentation (Fig. 2, A and B).
Fig. 2. In vivo cyclase activity of heterologously expressed AcsF.
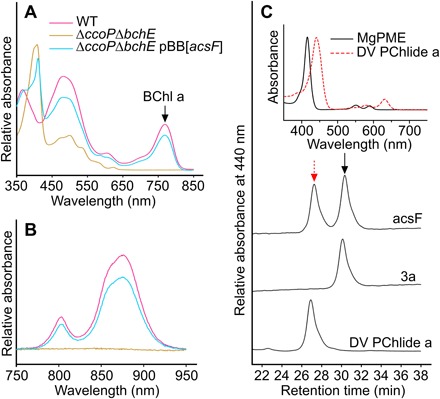
(A and B) The cyclase activity is linked to the (A) production of bacteriochlorophyll a (BChl a) and (B) assembly of photosynthetic complexes in Rba. capsulatus. (C) HPLC analysis of pigment methanol extracts from in vivo cyclase assays conducted with E. coli strains harboring either the pET3a (3a) or pET3a-acsF (acsF) plasmid. E. coli cultures were grown to an optical density at 600 nm (OD600) of 0.4 to 0.6 and then induced with isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated with MgPME.
To demonstrate AcsF activity in a nonphotosynthetic bacterium, we conducted an in vivo cyclase assay with an E. coli strain expressing the Rvi. gelatinosus acsF gene. E. coli cultures were fed MgPME, the cyclase substrate, which can diffuse across the lipid bilayer of E. coli membranes because of its hydrophobic nature; analysis of pigments by high-performance liquid chromatography (HPLC) showed that DV PChlide a, the product of the cyclase, was detected in the strain expressing acsF and was absent in the control strain (Fig. 2C). The in vivo conversion of MgPME into DV PChlide a in E. coli confirms that the Rvi. gelatinosus AcsF alone is a functional cyclase, requiring no additional subunit for activity; thus, it is a highly suitable candidate for assembling a Chl biosynthesis pathway in a heterotrophic bacterium, using the minimum number of components.
Assembly of the Chl biosynthesis pathway in E. coli
After demonstrating the in vivo cyclase activity of the Rvi. gelatinosus AcsF, we tested the known components of Chl biosynthesis by engineering the whole pathway (Fig. 1A) in the nonpigmented enteric bacterium E. coli. Genes encoding enzymes that mediate the six enzymatic steps of the Chl biosynthesis pathway were assembled into one operon (Fig. 1B), starting from the conversion of PPIX to MgP. We used the genes for Chl biosynthesis from the cyanobacterium Synechocystis sp. PCC 6803 (hereafter referred to as Synechocystis), except for the cyclase-encoding gene acsF, which was from Rvi. gelatinosus. The Rvi. gelatinosus crtE gene, encoding the GGPP synthase, was also included to modify the nonmevalonate pathway in E. coli to produce GGPP, a substrate for Chl synthase. To ensure sufficient isopentenyl PP for GGPP production, we overexpressed the native E. coli dxs gene, encoding one of the rate-limiting steps of the nonmevalonate pathway (9). A sequence encoding the Brassica oleracea (var. gemmifera) water-soluble Chl protein (BoWSCP) (10) was included, in an attempt to sequester the hydrophobic and phototoxic Chl molecules produced by this engineered pathway. All genes used are listed in table S1. The progressively larger constructs depicted in Fig. 1B represent an increasingly complete pathway and contain genes from chlI to chlM, then to acsF, to dvr, and finally to chlG; they were named IM, IA, ID, and IG, respectively. In addition, the Synechocystis cyclase-encoding genes cycI and ycf54 were cloned into the IM plasmid, resulting in the IM-cycI-ycf54 construct (fig. S3).
The E. coli C43(DE3) strain (11), a strain effective for expression of toxic proteins, was used as the host. Single transformations of E. coli C43(DE3) yielded the IM, IA, ID, IG, and IM-cycI-ycf54 strains; two and three sequential transformations were conducted to obtain the DE/IG and DE/BoP/IG strains, respectively. The resulting strains were assayed for their capacities to synthesize Chl intermediates at various stages and the final product, Chl a (see Materials and Methods). Pigments were extracted from the harvested E. coli cells using methanol and analyzed by HPLC with elution profiles monitored by absorbance at 416, 440, and 665 nm (Fig. 3, B to E; see figs. S3 to S5 for additional controls).
Fig. 3. Pigment accumulation in E. coli strains expressing Chl biosynthesis genes.

(A) Photographs of cell pellets of the E. coli control strain accumulating PPIX (3a) and the Chl a–producing strain (DE/BoP/IG) grown with supplementation of δ-aminolevulinic acid (ALA). Pigment production in described E. coli strains was analyzed by HPLC. m/z, mass/charge ratio. (B) Accumulation of MgPME in the IM strain monitored by absorbance at 416 nm. (C) Accumulation of DV PChlide a in the IA strain monitored by absorbance at 440 nm. (D) Light-dependent production of MV Chlide a in the ID strain monitored by absorbance at 440 nm (shown in black) and 665 nm (shown in blue). POR was activated by treatment with 5 μmol photons m−2 s−1 light at the latter stage of the incubation. The small MV Chlide a peak detected in the dark sample is due to unavoidable light exposure during experimental procedures. DV and MV pigments are differentiated by positions of Soret bands in the absorption spectra. (E) Accumulation of GG–Chl a in the DE/IG strain and of Chl a in the DE/BoP/IG strain monitored by absorbance at 665 nm. The production of authentic Chl a was further confirmed by LC-MS with the acquired mass spectrum shown in the inset (see fig. S6 for details). The pigment accumulated in the IG strain was not assigned because of lack of an appropriate pigment standard.
Control strain 3a, containing the empty pET3a vector, did not accumulate any Chl intermediates (Fig. 3). MgPME was produced in the IM strain (Fig. 3B), and the IA strain accumulated DV PChlide a (Fig. 3C), as did the IM-cycI-ycf54 strain containing the cycI and ycf54 genes (12–15) from the cyanobacterium Synechocystis (fig. S3). On the basis of retention times and positions of Soret absorption bands, the pigments in the ID strain were identified as monovinyl (MV) PChlide a and MV Chlide a (Fig. 3D). The trace level of MV Chlide a in the dark sample was likely produced by unavoidable light exposure during experimental procedures. The increased level of MV Chlide a and substantial decrease in MV PChlide a upon light treatment demonstrate that the ID strain harbors an active light-dependent POR enzyme and show that we have built a biosynthetic pathway that converts PPIX to MV Chlide a. Further biosynthetic steps (Fig. 3E) do not alter the absorption properties of the pigment, so HPLC was used to assess the completion of the pathway, which relies on the availability of GGPP to esterify MV Chlide a. The IG strain accumulated a new pigment (retention time, 46.9 min; Fig. 3E), which is much more hydrophobic than MV Chlide a (retention time, 20.2 min) and likely to be Chl a esterified with a farnesyl moiety; the immediate precursor of GGPP, farnesyl PP, is produced in E. coli, and Chl synthase has been reported to be able to use farnesyl PP as a substrate (16). The combination of the DE and IG plasmids enables E. coli to produce GG–Chl a. Three successive reductions of the GG tail, catalyzed by the GG reductase ChlP, complete the Chl pathway. The noticeable green color of the DE/BoP/IG strain and identification of Chl a by HPLC and liquid chromatography–mass spectrometry (LC-MS; Fig. 3E and fig. S6) demonstrate the successful assembly of the Chl biosynthetic pathway in E. coli. We estimate that the productivity of Chl a in the DE/BoP/IG strain under the tested conditions was ~1000 molecules per cell, equivalent to a concentration of ~1 μM in E. coli cells.
Finally, the pigmentation of the engineered E. coli cells was imaged at the single-cell level by structured illumination microscopy (SIM) (17) and fluorescence spectroscopy. Figure 4A demonstrates that newly synthesized Chl a molecules are exclusively localized in the native cytoplasmic membrane [with an in vivo emission maximum at 676 nm (Fig. 4H)] rather than in the cytosolic recombinant BoWSCP (see fig. S7 for immunodetection of the protein), a natural scavenger of Chls (18). In plants, cyanobacteria, and algae, the destination of Chls is a series of transmembrane multisubunit complexes, the assembly pathways of which are under active investigation in many laboratories (19–21). BoWSCP was included here because it represents a relatively simple, cytosolic target for binding newly synthesized Chls. Previously, we have shown that the phototrophic bacterium Rba. sphaeroides, which normally makes bacteriochlorophyll, can be engineered to synthesize Chl a, which was subsequently assembled in vivo into the BoWSCP (10). Although this protein is synthesized by E. coli (fig. S7), evidence for Chl binding was inconclusive because of the low levels of BoWSCP synthesized. The factors that determine in vivo assembly of Chl into water-soluble and membrane-bound complexes in E. coli require further study.
Fig. 4. SIM and fluorescence emission spectroscopy on the cellular distribution of Chl a in E. coli.
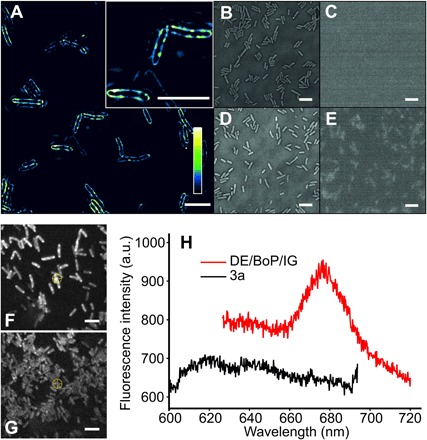
(A) SIM of the ALA-fed E. coli DE/BoP/IG cells. The color bar shows the color map of the image. The inset image shows the cellular distribution of Chl a in more detail. Scale bars, 5 μm. E. coli 3a cells with or without ALA feeding showed zero or limited fluorescence. (B to E) Bright-field and epifluorescence images of the E. coli 3a cells without (B and C) and with ALA feeding (D and E). Scale bars, 5 μm. (F and G) Fluorescence images of the ALA-fed E. coli (F) DE/BoP/IG and (G) 3a cells immobilized on an agar gel pad. Scale bars, 5 μm. (H) Fluorescence emission spectra recorded from individual E. coli DE/BoP/IG cells [marked with a front sight symbol in (G)]. a.u., arbitrary units.
Concluding remarks
This research completes our understanding of how the tetrapyrrole pigments of life, namely, siroheme (22), hemes (23, 24), bilins (25, 26), vitamin B12 (27), coenzyme F430 (28, 29), and now Chls, are synthesized from a common template. By identifying the elusive oxidative ring cyclase step that confers the green color to Chl, we have been able to assemble a cyclase gene, together with 11 others, to form a Chl biosynthetic pathway in E. coli. The PPIX substrate for this new pathway is furnished by the host’s tetrapyrrole metabolism, and the widespread availability of PPIX in most forms of life could provide the basis for installing Chl biosynthesis in a variety of heterotrophic organisms. This work forms the basis for performing in vitro cyclase assays with defined components and will enable molecular biologists to reprogram the cellular energetics of various platform organisms to include solar energy capture, commencing a new era of light-powered synthetic biology.
MATERIALS AND METHODS
Bacterial strains and plasmids
Bacterial strains and plasmids used in this study are listed in table S2. Primers used for plasmid construction and screening mutants are listed in table S3. E. coli strains were grown in LB medium at 37°C. If required, antibiotics were added at 30 μg ml−1 for kanamycin, 100 μg ml−1 for ampicillin, and 34 μg ml−1 for chloramphenicol. Rvi. gelatinosus strains were grown in polypeptone-yeast extract-sodium succinate (PYS) medium (30) at 30°C. Synechocystis strains were grown in blue-green 11 (BG-11) medium (31) buffered with 10 mM N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (pH 8.3, adjusted by KOH) at 30°C with continuous illumination. Glucose was added at 5 mM if required. The Rba. capsulatus strain (SB1003) (32), resistant to rifampicin, was obtained from C. Bauer (Indiana University) and grown in mineral-peptone-yeast extract (MPYE) medium (33) at 30°C. If required, antibiotics were added at 30 μg ml−1 for kanamycin and 20 μg ml−1 for rifampicin.
The allelic exchange suicide vector pK18mobsacB (34) was used to construct marker-free in-frame deletion mutant of Rba. capsulatus. For construction of the pK18ΔbchE plasmid, the upstream and downstream regions (approximately 500 base pairs) of the bchE gene were polymerase chain reaction (PCR)–amplified from Rba. capsulatus genomic DNA with primers bchEUpXbaIF/bchEUpR and bchEDownF/bchEDownHindIIIR, respectively. The two PCR products were fused by overlap extension PCR, digested with Xba I and Hind III, and ligated into the digested pK18mobsacB vector. The pK18ΔccoP plasmid was constructed in the same manner with the relevant primers. For construction of the pBB[acsF] plasmid, two parts of the acsF gene were PCR-amplified from Rvi. gelatinosus genomic DNA with the acsFBglIIF/acsFremoveBglIIR and acsFremoveBglIIF/acsFNotIR primers. The two PCR products were fused by overlap extension PCR, digested with Bgl II and Not I, and ligated into the digested pBBRBB-Ppuf843–1200 vector (35). For construction of the DE plasmid, the crtE gene was amplified by PCR from Rvi. gelatinosus genomic DNA with the crtENdeIF/crtEXhoIR primers, digested with Nde I and Xho I, and ligated into the pCOLADuet1 vector to obtain pCOLADuet1-crtE. Three parts of the E. coli dxs gene were amplified by PCR from a plasmid containing the gene with the dxsNcoIF/dxsremoveHindIII1R, dxsremoveHindIII1F/dxsremoveHindIII2R, and dxsremoveHindIII2F/dxsHindIIIR primers. The three PCR products were fused by overlap extension PCR, digested with Nco I and Hind III, and ligated into the digested pCOLADuet1-crtE to obtain the DE plasmid. For construction of the BoP plasmid, the chlP gene was PCR-amplified from Synechocystis genomic DNA with chlPNdeIF/chlPXhoIR primers, digested with Nde I and Xho I, and ligated into the digested pACYCDuet1 vector to obtain the pACYCDuet1-chlP plasmid. The sequence encoding the BoWSCP-His10 was cut from the pIND4[WSCP] plasmid (10) using Nco I and Hind III and ligated into the digested pACYCDuet1-chlP to obtain the BoP plasmid.
The Synechocystis chlI, chlD, and chlH genes were subcloned from the pET9a-SynI, pET9a-SynD, and pET9a-SynH plasmids (36), respectively. The Hind III and Xba I sites in the chlI gene were removed by site-directed mutagenesis with the chlIremoveHindIIIF/chlIremoveHindIIIR and chlIremoveXbaIF/chlIremoveXbaIR primers. An Spe I site was introduced to pET9a-SynI (Hind III and Xba I sites removed), pET9a-SynD, and pET9a-SynH by site-directed mutagenesis with the primers pETaddSpeIF/pETaddSpeIR. Then, the chlI, chlD, and chlH genes were excised from the pET9a constructs with Nde I and Spe I and ligated into the Spe I–digested pET3a vector. The Synechocystis gun4 gene was PCR-amplified from Synechocystis genomic DNA with the gun4NdeIF/gun4SpeIR primers, digested with Nde I and Spe I, and ligated into the digested Spe I–added pET3a vector. Then, the Hind III and Xba I sites in the gun4 gene were removed by site-directed mutagenesis with the gun4removeHindIIIF/gun4removeHindIIIR and gun4removeXbaIF/gun4removeXbaIR primers. The Synechocystis chlM, por, dvr, chlG, chlP, cycI, and ycf54 genes were PCR-amplified from Synechocystis genomic DNA with the relevant geneNdeIF/geneSpeIBamHIR primers, digested with Nde I and BamH I, and ligated into the digested pET3a vector. The Hind III site in the por gene and the Spe I site in the dvr gene were subsequently removed by site-directed mutagenesis with the porremoveHindIIIF/porremoveHindIIIR and dvrremoveSpeIF/dvrremoveSpeIR primers, respectively. The acsF gene was PCR-amplified from Rvi. gelatinosus genomic DNA with the acsFNdeIF/acsFSpeIBamHIR primers, digested with Nde I and BamH I, and ligated into the digested pET3a vector. Then, genes were cut from the pET3a constructs and adjoined one by one in the described order using the link-and-lock method, as depicted in fig. S2. Plasmids were sequenced by GATC Biotech.
Construction and phenotypic analysis of Rba. capsulatus mutants
The deletion plasmids pK18ΔbchE and pK18ΔbchE were transformed into the E. coli S17-1 strain (37), and transformants were selected on LB agar with kanamycin (30 μg ml−1). A single colony from the plate was inoculated to 5 ml of LB medium with kanamycin (30 μg ml−1) and grown at 37°C for 24 hours. E. coli cells harvested from 3 ml of the culture were resuspended in 50 μl of LB medium and mixed with Rba. capsulatus cells, which were harvested from 30 ml of liquid culture and resuspended in 100 μl of LB medium. The mating mixture was spotted onto solid LB medium and incubated at 30°C overnight. The mixture was streaked out onto MPYE agar supplemented with kanamycin (30 μg ml−1) and rifampicin (20 μg ml−1). The obtained kanamycin-resistant transconjugants were subcultured three times in nonselective MPYE medium to allow a second homologous recombination. Then, the culture was diluted and spread onto MPYE agar with 10% (w/v) sucrose. Colonies were then replica-plated onto MPYE plates containing 10% (w/v) sucrose, containing and lacking kanamycin (30 μg ml−1). The desired mutants were screened from the kanamycin-sensitive and sucrose-resistant colonies by PCR with the geneScreenF/geneScreenR primers. The expression plasmid pBB[acsF] was conjugated into Rba. capsulatus via E. coli S17-1 using the same method as described above. Transconjugants harboring the plasmid were selected on MPYE agar with kanamycin (30 μg ml−1) and rifampicin (20 μg ml−1).
For phenotypic analysis, Rba. capsulatus strains were grown in 10 ml of MPYE medium in 50-ml Falcon tubes with shaking at 230 rpm. Cells were harvested and resuspended in 1 ml of 60% (w/v) sucrose before recording absorption spectra of whole cells. Pigments were extracted from cells standardized by OD680 with methanol. The absorption spectra of whole cells and pigment extracts were recorded on a Cary 60 UV-Vis spectrophotometer, whereas those for whole cells were normalized and corrected for light scattering, as described previously (38).
In vivo enzymatic assay of cyclase in E. coli
E. coli C43(DE3) was transformed with either the pET3a or the pET3a-acsF plasmid and selected on LB agar with ampicillin (100 μg ml−1). For in vivo assays, a single colony was used to inoculate 10 ml of LB medium with ampicillin (100 μg ml−1) and grown overnight at 37°C, with shaking at 220 rpm. The next day, 30 to 50 μl of the resulting culture were used to inoculate 10 ml of LB medium with ampicillin (100 μg ml−1) and grown as above until the OD600 reached 0.4 to 0.6. IPTG was added at 0.5 mM to induce gene expression. Meanwhile, purified MgPME dissolved in methanol was directly added to cultures. The cultures were incubated for a further 24 hours at 30°C, with shaking at 175 rpm, before pigments were extracted from the cells.
Synthetic production of Chl a in E. coli
E. coli C43(DE3) was transformed with the pET3a-based plasmids containing Chl biosynthesis genes and selected on LB agar with ampicillin (100 μg ml−1). For the DE/IG strain, two sequential transformations were conducted, and the second transformation was selected on LB agar with ampicillin (100 μg ml−1) and kanamycin (30 μg ml−1). For the DE/BoP/IG strain, three sequential transformations were conducted, and the third transformation was selected on LB agar with ampicillin (100 μg ml−1), kanamycin (30 μg ml−1), and chloramphenicol (34 μg ml−1). A single colony was used to inoculate liquid medium and cultured as above, except that, at the point of induction, ALA and Mg2+ (equimolar mixture of MgCl2 and MgSO4) were also added at 10 mM. Cultures requiring light activation were illuminated with 5 μmol photons m−2 s−1 light for the final 4 hours.
Pigment extraction
E. coli cells were harvested from liquid cultures and washed in 25 mM Hepes buffer (pH 7.4). Pigments were extracted with an excess of methanol by vigorous shaking using a Mini-Beadbeater (BioSpec), incubated on ice for 10 min, and centrifuged at 16,000g for 5 min at 4°C. The resulting supernatant containing extracted pigments was transferred to a new tube and analyzed immediately or vacuum-dried at 30°C using a Concentrator plus (Eppendorf) and stored at −20°C for future analysis. GG–Chl a was extracted from a Synechocystis ΔchlP mutant (10) using the same method as described above. MgPME was extracted from a Rvi. gelatinosus ΔbchEΔacsF mutant (5), which excreted the pigment into the growth medium as granules. Cells were harvested from liquid culture and washed in ultrapure water, and the pellet was suspended in methanol with gentle shaking to facilitate dissolution of the pigment into the solvent. MgPME solution was collected from the colored supernatant after centrifugation at 5000g at 4°C for 10 min.
High-performance liquid chromatography
A methanolic pigment solution, either freshly extracted or reconstituted from dried samples, was analyzed on an Agilent 1200 HPLC system equipped with a diode array detector and a fluorescence detector. A published method (39) with some modifications was used for separation of Chl a and its precursors. The pigment solution was loaded onto a Sigma-Aldrich Discovery C18 reversed-phase column (particle size, 5 μm; 250 mm × 4.6 mm). Solvent A was methanol/500 mM ammonium acetate (30/70, v/v). Solvent B was methanol. Pigment species were eluted at 40°C at a flow rate of 1 ml min−1 with a linear gradient of 65 to 75% solvent B over 35 min, followed by column wash with 100% solvent B for 15 min. The eluates were monitored by absorbance at 416, 440, and 665 nm.
Liquid chromatography–mass spectrometry
An Agilent 1200 HPLC system coupled to a 6410 QQQ mass spectrometer was used for confirmation of production of authentic Chl a. Data were collected and analyzed with MassHunter Software. Pigments were initially separated by reversed-phase HPLC using an identical program to that described above. The elution of Chl a from the column was monitored by checking absorbance at 665 nm. The assignment of m/z to eluates was performed using in-line electrospray ionization in positive mode monitoring m/z between 800 and 1000.
Structured illumination microscopy
E. coli cells were pelleted and washed with phosphate-buffered saline and resuspended to an appropriate density for microscopy. Cell suspensions were incubated on an agarose pad for 30 min and mounted in VECTASHIELD Antifade Medium (Vector Laboratories). Samples were imaged on a DeltaVision OMX V4 microscope with the Blaze 3D structured illumination module equipped with a 60× oil objective [numerical aperture (NA), 1.42]. Illumination was performed with a 642-nm laser with the emitted light collected through a 683/40 band-pass filter. For each SIM image, nine axial planes were captured with a spacing of 125 nm, and the data were reconstructed with the SoftWoRx 6.0 software package (GE Healthcare).
Fluorescence spectroscopy
Resuspended frozen E. coli cells (20 μl) were spotted onto LB agar and left until the liquid was absorbed into the agar. The cell spot was cut out of the agar and flipped over onto a glass-bottomed petri dish (WillCo Wells, GWSt-3522). The petri dish was then mounted in the microscope sample holder for imaging. Fluorescence imaging and emission spectral measurements were performed on a home-built imager equipped with a spectrometer (Acton SP2558, Princeton Instruments) and an electron-multiplying charge-coupled device camera (ProEM 512, Princeton Instruments). Dual-sample illumination was provided by a 470-nm light-emitting diode light source (M470L2, ThorLabs) to provide Köhler illumination for wide-field imaging and a 485-nm pulsed laser (LDH-D-C-485, PicoQuant) for a critical illumination when recording the emission spectra of individual cells. The laser beam was focused on the sample surface to a diffraction-limited spot using a 100× objective (PlanFluorite; NA, 1.4; oil immersion, Olympus), illuminating only a portion of the individual cell. Fluorescence signal was filtered through a 495-nm dichroic mirror (FF495-Di03, Semrock) and a 594-nm long-pass filter (BLP01-594R-25, Semrock). The emission spectra were recorded using a diffraction grating of 150 g mm−1 with an 800-nm blaze, resulting in a wavelength range of 114 nm. The central wavelength was always selected to match the peak emission wavelength of the sample.
Supplementary Material
Acknowledgments
We thank C. Bauer (Indiana University) for the gift of the Rba. capsulatus SB1003 strain, M. Proctor for providing the DV PChlide a and DV Chlide a standards, A. Hitchcock for providing the Chl a standard, and M. Radle (Pennsylvania State University) for assistance with LC-MS data collection. Funding: C.N.H. acknowledges financial support from Advanced Award 338895 from the European Research Council. C.N.H., G.E.C., and A.A.B. acknowledge funding from the Biotechnology and Biological Sciences Research Council (award number BB/M000265/1). D.P.C. is supported by the European Commission (Marie Skłodowska-Curie Global Fellowship 660652). S.F.H.B. was supported by a University of Sheffield 2022 Futures Studentship. D.A.B. acknowledges funding by NSF grant MCB-1613022 and Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy (DOE) grant DE-FG02-94ER20137. Research in the laboratories of C.N.H. and D.A.B. was also conducted under the auspices of the Photosynthetic Antenna Research Center, an Energy Frontier Research Center funded by the DOE, Office of Science, Office of Basic Energy Sciences under award number DE-SC 0001035. The SIM imaging was performed at the University of Sheffield Wolfson Light Microscopy Facility and was partly funded by Medical Research Council grant MR/K015753/1. S.H. was supported by a doctoral studentship from the University of Sheffield. Author contributions: G.E.C., S.H., and A.A.B. made the plasmid constructs. G.E.C. made the Rba. capsulatus mutants and conducted in vivo E. coli assays and pigment analyses. D.P.C. provided the LC-MS data and contributed ideas concerning interpretation of the data and the manuscript. S.F.H.B. provided the SIM data. C.V. provided the fluorescence emission spectroscopy data. D.A.B. contributed ideas concerning experimental evidence, the manuscript, and support for D.P.C. G.E.C. and C.N.H. designed the experiments, interpreted the data, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaaq1407/DC1
fig. S1. Deletions of the bchE and ccoP genes in Rba. capsulatus.
fig. S2. Diagram of the link-and-lock method for plasmid construction.
fig. S3. The production of DV PChlide a in the IA and IM-cycI-ycf54 strains.
fig. S4. The light-dependent production of MV Chlide a in the ID strain.
fig. S5. The production of GG–Chl a in the DE/IG strain and of Chl a in the DE/BoP/IG strain.
fig. S6. Verification of the production of Chl a in E. coli by LC-MS.
fig. S7. Western blot analysis of the BoWSCP-His10 expression in the DE/BoP/IG strain.
table S1. List of genes used to assemble the Chl biosynthesis pathway in E. coli.
table S2. Strains and plasmids described in this study.
table S3. Oligonucleotide primers used in this study.
REFERENCES AND NOTES
- 1.Brzezowski P., Richter A. S., Grimm B., Regulation and function of tetrapyrrole biosynthesis in plants and algae. Biochim. Biophys. Acta 1847, 968–985 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Chidgey J. W., Linhartová M., Komenda J., Jackson P. J., Dickman M. J., Canniffe D. P., Koník P., Pilný J., Hunter C. N., Sobotka R., A cyanobacterial chlorophyll synthase-HliD complex associates with the Ycf39 protein and the YidC/Alb3 insertase. Plant Cell 26, 1267–1279 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Y. Fujita, H. Yamakawa, Biochemistry of chlorophyll biosynthesis in photosynthetic prokaryotes, in Modern Topics in the Phototrophic Prokaryotes, P. Hallenbeck, Ed. (Springer, 2017), pp. 67–122. [Google Scholar]
- 4.Gough S. P., Petersen B. O., Duus J. Ø., Anaerobic chlorophyll isocyclic ring formation in Rhodobacter capsulatus requires a cobalamin cofactor. Proc. Natl. Acad. Sci. U.S.A. 97, 6908–6913 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen G. E., Canniffe D. P., Hunter C. N., Three classes of oxygen-dependent cyclase involved in chlorophyll and bacteriochlorophyll biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 114, 6280–6285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rzeznicka K., Walker C. J., Westergren T., Kannangara C. G., von Wettstein D., Merchant S., Gough S. P., Hansson M., Xantha-l encodes a membrane subunit of the aerobic Mg-protoporphyrin IX monomethyl ester cyclase involved in chlorophyll biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 102, 5886–5891(2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinta V., Picaud M., Reiss-Husson F., Astier C., Rubrivivax gelatinosus acsF (previously orf358) codes for a conserved, putative binuclear-iron-cluster-containing protein involved in aerobic oxidative cyclization of Mg-protoporphyrin IX monomethylester. J. Bacteriol. 184, 746–753 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen G. E., Canniffe D. P., Martin E. C., Hunter C. N., Absence of the cbb3 terminal oxidase reveals an active oxygen-dependent cyclase involved in bacteriochlorophyll biosynthesis in Rhodobacter sphaeroides. J. Bacteriol. 198, 2056–2063 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim S.-W., Keasling J. D., Metabolic engineering of the nonmevalonate isopentenyl diphosphate synthesis pathway in Escherichia coli enhances lycopene production. Biotechnol. Bioeng. 72, 408–415 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Hitchcock A., Jackson P. J., Chidgey J. W., Dickman M. J., Hunter C. N., Canniffe D. P., Biosynthesis of chlorophyll a in a purple bacterial phototroph and assembly into a plant chlorophyll–protein complex. ACS Synth. Biol. 5, 948–954 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Miroux B., Walker J. E., Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Minamizaki K., Mizoguchi T., Goto T., Tamiaki H., Fujita Y., Identification of two homologous genes, chlAI and chlAII, that are differentially involved in isocyclic ring formation of chlorophyll a in the cyanobacterium Synechocystis sp. PCC 6803. J. Biol. Chem. 283, 2684–2692 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Peter E., Salinas A., Wallner T., Jeske D., Dienst D., Wilde A., Grimm B., Differential requirement of two homologous proteins encoded by sll1214 and sll1874 for the reaction of Mg protoporphyrin monomethylester oxidative cyclase under aerobic and micro-oxic growth conditions. Biochim. Biophys. Acta 1787, 1458–1467 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Hollingshead S., Kopečná J., Jackson P. J., Canniffe D. P., Davison P. A., Dickman M. J., Sobotka R., Hunter C. N., Conserved chloroplast open-reading frame ycf54 is required for activity of the magnesium protoporphyrin monomethylester oxidative cyclase in Synechocystis PCC 6803. J. Biol. Chem. 287, 27823–27833 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hollingshead S., Kopečná J., Armstrong D. R., Bucinska L., Jackson P. J., Chen G. E., Dickman M. J., Williamson M. P., Sobotka R., Hunter C. N., Synthesis of chlorophyll-binding proteins in a fully segregated Δycf54 strain of the cyanobacterium Synechocystis PCC 6803. Front. Plant Sci. 7, 292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rüdiger W., Benz J., Guthoff C., Detection and partial characterization of activity of chlorophyll synthetase in etioplast membranes. Eur. J. Biochem. 109, 193–200 (1980). [DOI] [PubMed] [Google Scholar]
- 17.Gustafsson M. G., Shao L., Carlton P. M., Wang C. J. R., Golubovskaya I. N., Cande W. Z., Agard D. A., Sedat J. W., Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 94, 4957–4970 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satoh H., Uchida A., Nakayama K., Okada M., Water-soluble chlorophyll protein in Brassicaceae plants is a stress-induced chlorophyll-binding protein. Plant Cell Physiol. 42, 906–911 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Nickelsen J., Rengstl B., Photosystem II assembly: From cyanobacteria to plants. Annu. Rev. Plant Biol. 64, 609–635 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Yang H., Liu J., Wen X., Lu C., Molecular mechanism of photosystem I assembly in oxygenic organisms. Biochim. Biophys. Acta 1847, 838–848 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Eaton-Rye J. J., Sobotka R., Assembly of the photosystem II membrane-protein complex of oxygenic photosynthesis. Front. Plant Sci. 8, 884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raux E., Leech H. K., Beck R., Schubert H. L., Santander P. J., Roessner C. A., Scott A. I., Martens J. H., Jahn D., Thermes C., Rambach A., Warren M. J., Identification and functional analysis of enzymes required for precorrin-2 dehydrogenation and metal ion insertion in the biosynthesis of sirohaem and cobalamin in Bacillus megaterium. Biochem. J. 370, 505–516 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinemann I. U., Jahn M., Jahn D., The biochemistry of heme biosynthesis. Arch. Biochem. Biophys. 474, 238–251 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Layer G., Reichelt J., Jahn D., Heinz D. W., Structure and function of enzymes in heme biosynthesis. Protein Sci. 19, 1137–1161 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.S. I. Beale, Biosynthesis of Open-Chain Tetrapyrroles in Plants, Algae, and Cyanobacteria, in Ciba Foundation Symposium 180-The Biosynthesis of the Tetrapyrrole Pigments, D. J. Chadwick, K. Ackrill, Eds. (Wiley, 1994), pp. 156–176. [DOI] [PubMed] [Google Scholar]
- 26.Alvey R. M., Biswas A., Schluchter W. M., Bryant D. A., Attachment of noncognate chromophores to CpcA of Synechocystis sp. PCC 6803 and Synechococcus sp. PCC 7002 by heterologous expression in Escherichia coli. Biochemistry 50, 4890–4902 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Warren M. J., Raux E., Schubert H. L., Escalante-Semerena J. C., The biosynthesis of adenosylcobalamin (vitamin B12). Nat. Prod. Rep. 19, 390–412 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Zheng K., Ngo P. D., Owens V. L., Yang X.-P., Mansoorabadi S. O., The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science 354, 339–342 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Moore S. J., Sowa S. T., Schuchardt C., Deery E., Lawrence A. D., Ramos J. V., Billig S., Birkemeyer C., Chivers P. T., Howard M. J., Rigby S. E. J., Layer G., Warren M. J., Elucidation of the biosynthesis of the methane catalyst coenzyme F430. Nature 543, 78–82 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagashima K. V., Shimada K., Matsuura K., Shortcut of the photosynthetic electron transfer in a mutant lacking the reaction center-bound cytochrome subunit by gene disruption in a purple bacterium, Rubrivivax gelatinosus. FEBS Lett. 385, 209–213 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Rippka R., Deruelles J., Waterbury J. B., Herdman M., Stanier R. Y., Generic assignments, strain histories and properties of pure cultures of cyanobacteria. Microbiology 111, 1–61 (1979). [Google Scholar]
- 32.Yen H.-C., Marrs B., Map of genes for carotenoid and bacteriochlorophyll biosynthesis in Rhodopseudomonas capsulata. J. Bacteriol. 126, 619–629 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koch H.-G., Hwang O., Daldal F., Isolation and characterization of Rhodobacter capsulatus mutants affected in cytochrome cbb3 oxidase activity. J. Bacteriol. 180, 969–978 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schäfer A., Tauch A., Jäger W., Kalinowski J., Thierbach G., Pühler A., Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: Selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145, 69–73 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Tikh I. B., Held M., Schmidt-Dannert C., BioBrick™ compatible vector system for protein expression in Rhodobacter sphaeroides. Appl. Microbiol. Biotechnol. 98, 3111–3119 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Jensen P. E., Gibson L. C., Henningsen K. W., Hunter C. N., Expression of the chlI, chlD, and chlH genes from the cyanobacterium Synechocystis PCC6803 in Escherichia coli and demonstration that the three cognate proteins are required for magnesium-protoporphyrin chelatase activity. J. Biol. Chem. 271, 16662–16667 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Simon R., Priefer U., Pühler A., A broad host range mobilization system for in vivo genetic engineering: Transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1, 784–791 (1983). [Google Scholar]
- 38.Swainsbury D. J., Scheidelaar S., van Grondelle R., Killian J. A., Jones M. R., Bacterial reaction centers purified with styrene maleic acid copolymer retain native membrane functional properties and display enhanced stability. Angew. Chem. Int. Ed. 53, 11803–11807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sobotka R., Tichy M., Wilde A., Hunter C. N., Functional assignments for the carboxyl-terminal domains of the ferrochelatase from Synechocystis PCC 6803: The CAB domain plays a regulatory role, and region II is essential for catalysis. Plant Physiol. 155, 1735–1747 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGoldrick H. M., Roessner C. A., Raux E., Lawrence A. D., McLean K. J., Munro A. W., Santabarbara S., Rigby S. E. J., Heathcote P., Scott A. I., Warren M. J., Identification and characterization of a novel vitamin B12 (cobalamin) biosynthetic enzyme (CobZ) from Rhodobacter capsulatus, containing flavin, heme, and Fe-S cofactors. J. Biol. Chem. 280, 1086–1094 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaaq1407/DC1
fig. S1. Deletions of the bchE and ccoP genes in Rba. capsulatus.
fig. S2. Diagram of the link-and-lock method for plasmid construction.
fig. S3. The production of DV PChlide a in the IA and IM-cycI-ycf54 strains.
fig. S4. The light-dependent production of MV Chlide a in the ID strain.
fig. S5. The production of GG–Chl a in the DE/IG strain and of Chl a in the DE/BoP/IG strain.
fig. S6. Verification of the production of Chl a in E. coli by LC-MS.
fig. S7. Western blot analysis of the BoWSCP-His10 expression in the DE/BoP/IG strain.
table S1. List of genes used to assemble the Chl biosynthesis pathway in E. coli.
table S2. Strains and plasmids described in this study.
table S3. Oligonucleotide primers used in this study.