

Abstract
In lipoxygenases, the topologically conserved C-terminal domain catalyzes the oxidation of polyunsaturated fatty acids, generating an assortment of biologically relevant signaling mediators. Plant and animal lipoxygenases also contain a 100-150-amino acid N-terminal C2-like domain that has been implicated in interactions with isolated fatty acids and at the phospholipid bilayer. These interactions may lead to increased substrate availability and contribute to the regulation of active-site catalysis. Because of a lack of structural information, a molecular understanding of this lipid-protein interaction remains unresolved. Herein, we employed hydrogen–deuterium exchange MS (HDXMS) to spatially resolve changes in protein conformation upon interaction of soybean lipoxygenase with a fatty acid surrogate, oleyl sulfate (OS), previously shown to act at a site separate from the substrate-binding site. Specific, OS-induced conformational changes are detected both at the N-terminal domain and within the substrate portal nearly 30 Å away. Combining previously measured kinetic properties in the presence of OS with its impact on the Kd for linoleic acid substrate binding, we conclude that OS binding brings about an increase in rate constants for both the ingress and egress of substrate. We discuss the role of OS-induced changes in protein flexibility in the context of changes in the mechanism of substrate acquisition.
Keywords: allosteric regulation, enzyme inhibitor, fatty acid, hydrogen exchange mass spectrometry, inhibition mechanism, SLO, allostery, hydrogen-deuterium exchange (HDX), lipoxygenase
Introduction
Lipoxygenases, widely represented among plants, animals, fungi, and bacteria, catalyze the C–H abstraction from long-chain polyunsaturated fatty acids in the process of generating a diverse repertoire of (per)oxidized products that play important roles in various signaling pathways (1, 2). Select eicosanoid products from human enzymes have been implicated in a number of inflammation-based diseases (examples include arthritis, asthma, atherosclerosis, and cancer) (3); lipoxygenases also represent a “neglected frontier for regulation of chronic pain” (4). Because of the role of lipoxygenases in a wide range of signaling pathways, an understanding of allosteric regulation of active-site chemistry is fundamental to a mechanistic framework of enzyme reactivity as well as the success of future rational drug design efforts.
Lipoxygenases are structurally characterized by a 500–700-amino acid (predominantly α-helical) C-terminal domain (Fig. 1). The C-terminal domain contains the catalytic metallocofactor, typically a ferric hydroxide that is responsible for catalysis (Fig. 2). Although there is considerable variability in sequence and loop features, the structural topology of this domain is fairly conserved; however, a 100–150-amino acid N-terminal polycystin-1/lipoxygenase/α-toxin (PLAT)3 domain is only found in animals and plants (5–7). Of the plant lipoxygenases, soybean lipoxygenase-1 (SLO) has emerged as the paradigm of the family due to its native abundance, ease of purification, and structural stability. Because SLO and human lipoxygenases catalyze similar reactions and share similar structural elements, SLO is often related to the molecular enzymology of human enzymes as well.
Figure 1.
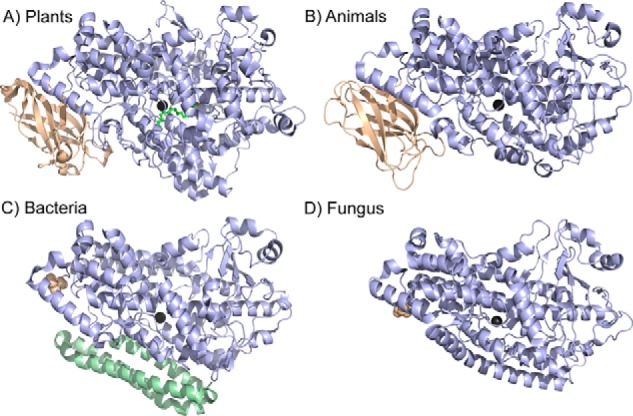
Structural comparison of lipoxygenases from various kingdoms. In A, SLO was selected as the representative for plant lipoxygenases, whereas in B, the human 5-LO isoenzyme is shown. The structures correspond to the Protein Data Bank entries 3PZW (A), 3O8Y (B), 4G32 (C), and 5FNO (D). The structurally conserved lipoxygenase fold of the catalytic domain is colored light blue; the catalytic metal is shown as dark gray in all cases. For both plants and animals, the N-terminal PLAT domain is colored in wheat (A and B). Bacterial and fungal enzymes do not contain a PLAT domain, but the resolved N-terminal amino acid is shown as wheat spheres. In C, the bacterial enzymes have additional α helices, as colored in pale green.
Figure 2.

Mechanism of lipid peroxidation by lipoxygenase. Left, reaction of model enzyme, soybean lipoxygenase, with substrate linoleic acid as an example. The metallocofactor, often a ferric hydroxide, although a manganese center is employed by fungal enzymes (63), abstracts a hydrogen atom from polyunsaturated fatty acids, in a regio- and stereospecific manner, through a proton-coupled electron transfer (PCET) mechanism (49). Molecular oxygen inserts into the delocalized radical intermediates, forming the product upon reverse proton-coupled electron transfer. The carbon numbering of the LA substrate is shown for reference. Relevant microscopic rate constant, k2, associated with the rate-determining C–H abstraction, is labeled. The enzyme-substrate and enzyme-LA radical complexes are labeled as ES′ and ES•, respectively. Right, the structures of substrates, as well as the allosteric effectors OA and OS, are shown for reference. The reactive carbon for each substrate is designated by an asterisk.
Whereas the function of the C-terminal domain is well established, the role for the N-terminal domain has not been fully elucidated. The N-terminal PLAT domain is structurally reminiscent of C2 domains in proteins that have been established to bind calcium and associate with the lipid membrane bilayer for cell signaling (8, 9). Studies on the human lipoxygenases have implicated the role of this PLAT domain in Ca2+-mediated phospholipid membrane binding (10–14).
In the case of plant enzyme SLO, calcium does not regulate catalysis or mediate membrane binding (15); instead, isolated fatty acids, such as the substrate linoleic acid (LA) at high concentrations, are postulated to associate at a remote site, presumably within the protein's N terminus, and alter the interaction of substrate within the catalytic center (16–18). Effectors such as oleyl sulfate (OS) and mixed inhibitors, such as oleic acid (OA), have been shown to stimulate the same allosteric behavior (18, 19), suggesting that LA, OA, and OS (and perhaps the phospholipid membrane (16)) may all interact at (or near) a remote allosteric site. Biochemical analysis of the product feedback inhibition found in humans, although lacking in SLO, has further supported the presence of an allosteric site for isolated lipid effector interactions within or near the PLAT domain (20, 21). Thus, although an ongoing recognized hypothesis is that the N-terminal domain of lipoxygenase interacts with lipid effectors to promote changes in substrate binding within the C-terminal catalytic domain, a network for communication between the allosteric and active sites has yet to be resolved.
To extend the molecular framework for lipid-induced allostery in lipoxygenases, we present HDXMS experiments with SLO in the absence and presence of the effector, OS. HDXMS has emerged as an incisive tool to describe networks of allosteric communication in proteins and to offer molecular insight into the mechanism of enzyme inhibition and/or activation (22–27). In the context of remote binding effects that alter lipoxygenase behavior, we find that SLO may serve as a tractable system, due to its structural stability and previously well-characterized kinetic impact from effectors such as OS. Further, we recently reported conditions in which HDXMS is capable of spatially resolving remote changes in SLO flexibility that correlate with the introduction of catalysis-impairing active-site mutations (28). Consistent with evidence from lipid interaction studies on SLO and human lipoxygenases (for examples, see Refs. 16 and 21), this current HDXMS study supports a localization of OS, as a lipid surrogate, at the N terminus (within the PLAT domain). Further, six non-overlapping peptides within the catalytic domain display a significant increase in the observed rate of deuterium exchange upon OS addition. This change in apparent rates of HDX supports a model in which lipid interactions at the N terminus are linked to the destabilization of a group of α helices stemming from the N terminus to the substrate entrance portal. Integrating prior kinetic analyses of OS inhibition (18) with measurement of the impact of OS on the Kd for linoleic acid substrate has enabled us to estimate the degree to which rates for LA binding and release are increased, implicating a shift in an internal equilibrium constant describing open and closed ES complexes and attributed to the aforementioned helical destabilization. Taken together, the data highlight the ability of HDXMS to interrogate the molecular mechanism of long-range allosteric effects in lipoxygenases.
Results
The conditions of the described HDXMS experiments are at temperatures ranging from 10 to 40 °C and effective pD 8 (see “Experimental procedures” for details), where the rate-limiting step for kcat has been shown to involve the chemical C–H bond cleavage (29) and HDX has been shown (28) to proceed via the EX-2 exchange mechanism (30, 31). EX-2 conditions permit a temperature-dependent analysis of regional conformational flexibility (28, 32, 33). To streamline data analysis, 45 non-overlapping peptides (Fig. 3) were selected from the nearly 300 available for this 839-amino acid protein, providing 63 and 89% sequence coverage of the N- and C-terminal domains, respectively. For HDXMS experiments with OS, the peptide sequence map is identical to that for SLO in the absence of effector. HDXMS was performed for both sets of peptides as a function of time (10 s to 4 h) and temperature (10, 20, 30, and 40 °C); previous controls showed that wildtype SLO is structurally stable, contains nearly complete iron occupancy, and maintains high activity (>90%) during the course of these incubations (28). For each peptide, the percentage of deuterium exchange (or incorporation) was determined by calculating the difference between the mass of the peptide before and after a given incubation time; the data in the presented HDXMS traces (Figs. 4 and 6 and Fig. S1) have been corrected for peptide-specific back-exchange during work-up.
Figure 3.

Primary sequence coverage map for the defined non-overlapping peptides in SLO. The color code corresponds to that for Fig. 1. Wheat, the N-terminal β-barrel, “PLAT” domain is represented by residues 1–144; light blue, the catalytic domain comprises residues 145–839.
Figure 4.
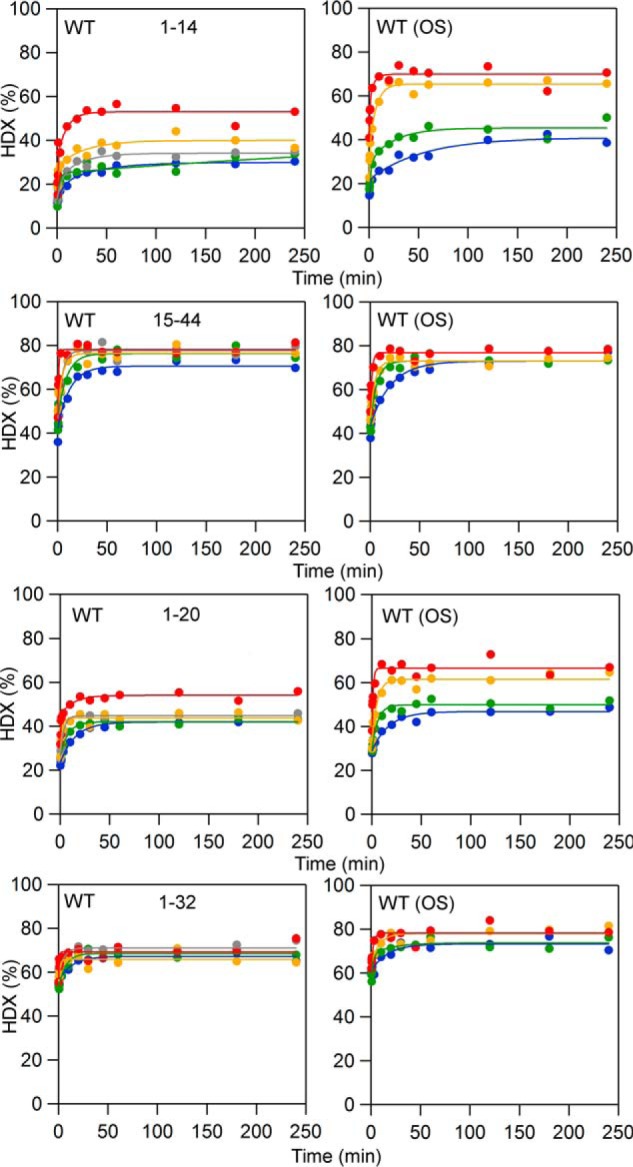
HDXMS traces showing peptides that exhibit increased extent of exchange at longer time points in the presence of OS. Blue, 10 °C; green, 20 °C; yellow, 30 °C; red, 40 °C. The data are shown in dots; the fits to the data are solid lines.
Figure 6.

Comparative time-dependent HDXMS traces, collected at 10 °C, for select peptides whose rates of exchange are faster with OS. The color coding of the HDXMS traces represents SLO in the absence of OS (black) and in the presence of OS (red). Middle, a partial structure of the catalytic domain is shown with a focus on the peptides affected by the presence of OS. The color coding of the peptides refers to their unique HDXMS behaviors and represents residues 239–256 and 257–273 (salmon), 297–305 and 306–316 (wheat), 317–334 (pale yellow), and 751–761 (pale green). The number of the N-terminal amino acid of each peptide, affected by OS, is shown in the structure for reference. Insets, the HDXMS traces are reproduced but are plotted with the x axis in logarithmic scale.
Two sets of experiments were conducted, both on wildtype SLO, in which one set of samples was incubated in the presence of 10 μm OS and another set was incubated without OS. Although OA can act as an allosteric effector (17), we chose to focus on OS to preclude competition between OA and LA at the substrate-binding site. As shown previously, OS does not compete with substrate binding and exclusively targets an alternate site in SLO (19). The concentration of OS was chosen to be saturating (Kd ∼0.6 μm) while remaining below the critical micelle concentration (≥18 μm) (18). A thorough inspection of the impact of OS on HDXMS, using pattern recognition (28), revealed that for the majority (38 of the 45 peptides), protein flexibility was unaffected by OS (see Fig. S1 for the complete set of HDXMS traces). Among the remaining peptides, two regionally defined behaviors emerged from this data analysis, with OS observed either to increase the extent of exchange at the N terminus (Fig. 4) or to increase the rate of exchange in the C-terminal domain (see Fig. 6 and Table 1).
Table 1.
Apparent average rates (min−1) of exchange, kHDX, from fits to HDXMS data (at 10 °C) for peptides affected by OS
Apparent average rate constants were determined from one- or two-exponential fits to the data within the dynamic range of the measurement. The rates are represented within one S.D. from the exponential fits to the data. For complete rate constant analysis across all temperatures, see Table S1; Arrhenius plots are shown in Fig. 7 for reference.
| Peptide | SLO (without OS) | SLO (with OS) |
|---|---|---|
| 1–14 | 0.066 ± 0.008 | 0.06 ± 0.01 |
| 239–256 | 0.3 ± 0.1 | 2.1 ± 0.9 |
| 257–273 | 0.11 ± 0.07 | 2.8 ± 0.9 |
| 297–305 | 0.10 ± 0.02 | 1.0 ± 0.2 |
| 306–316 | 0.14 ± 0.04 | 0.9 ± 0.1 |
| 317–334 | 0.009 ± 0.004 | 0.08 ± 0.02 |
| 751–761 | 0.06 ± 0.02 | 1.1 ± 0.4 |
Conformational change in the N-terminal PLAT domain
We first searched for changes in the extent of the deuterium exchange (at 4 h) in the presence of OS with the expectation of observing a canonical “protection” effect (i.e. decreased HDX) due to formation of a complex of OS with protein. In contrast, at all temperatures examined, OS displayed a significantly increased extent of exchange at a single, N-terminal peptide, 1–14 (Fig. 4). The difference in percent exchange, between SLO/OS and SLO alone, shows its largest impact at elevated temperatures (30 and 40 °C). Based on the percent exchange and number of exchangeable amide linkages (13 for this peptide), we calculate that OS induces a perturbation to 1–3 amide hydrogen bonds (depending on temperature) within the N-terminal peptide, 1–14. Comparison with the next sequential peptide, 15–44, showed no noticeable change to the extent or rate of exchange. We further examined peptides that overlap with 1–14, namely 1–20 and 1–32 (Fig. 4). As anticipated, the extents of exchange are increased in the presence of OS. The differences in percent exchange are smaller for these longer peptides, although the number of altered amide hydrogen bonds remains the same (i.e. 1–3, depending on temperature). These results suggest that OS binds to or in the vicinity of residues 1–14 and introduces a distinct change in conformation within the SLO N-terminal domain (localized around peptide 1–14). Our inability to detect any protection from HDX by OS at its actual binding site is probably due to relatively poor peptide coverage within the N-terminal region of protein, only 63% compared with 89% coverage within the C-terminal catalytic domain.
From analysis of the X-ray–derived structure (Fig. 5A), the first six amino acids (MFSAGH) could not be modeled, suggesting a high degree of structural mobility in crystallo. The rest of this peptide sequence is predominantly hydrophobic, aside from two lysine residues (KIKGTVVL), and forms β-sheet secondary structure. As noted above, further localization of the OS interaction was not feasible because peptides within the β-barrel that sandwich the peptide 1–14 through interstrand hydrogen bonding are not covered (Fig. 5A, gray schematic) in our HDXMS measurements. Attempts by our group to soak crystals or co-crystalize with OS were unsuccessful; no electron density could be assigned to OS (or even OA) at the N terminus or anywhere else in the protein structure.
Figure 5.

Structure of the N-terminal domain and the putative OS-binding site in SLO. The primary structure of peptide 1–14 is shown in A as yellow sticks for reference; the first six amino acids (MFSAGH) are not resolved in this high-resolution structure (1.4 Å). The N-terminal and C-terminal domains are colored wheat and light blue, respectively. Peptides not covered by HDXMS experiments in the PLAT domain are colored gray. B, space-filled model of the PLAT domain from the SLO structure. A loop 68–79 is colored in pale green and is located spatially over the putative binding site for OS and provides a relatively hydrophobic pocket for fatty acid binding. OS association in this pocket could trigger a conformational change in which the loop would close over the pocket.
It is possible that the negative charge on the sulfate group of OS alone is sufficient to interact with one or both of the above highlighted lysine residues to elicit a conformational change. However, these residues are not conserved among plant and animal lipoxygenases, and OS has been shown to bind with comparable affinity to the human 15-lipoxygenase (18). Further, previous stopped-flow kinetic experiments with palmitoyl sulfate, a compound structurally similar to OS with 2 fewer carbons, did not exhibit a high specificity for the allosteric site (Ki ∼140 μm, compared with 0.6 μm for OS) (19). Both oleic (18:1) and linoleic (18:2) acid also bind at the allosteric site (Kd ≥ 20 μm), although not nearly as tightly as OS (18). The length of the molecule and the structure of the headgroup probably play a role in the tight association of OS with the allosteric site.
Destabilization of C-terminal α helices
The second revealing pattern in the data is the observation of six non-overlapping SLO peptides whose apparent rate of exchange is increased upon incubation with OS (Fig. 6). HDX is typically regarded as a multiexponential process with slow, intermediate, and fast kinetic exchange rate constants. Whereas many reports generally use three exponentials to fit data within the dynamic range, we have found, using various multiexponential fitting functions, that the SLO HDXMS traces can be accurately modeled with two or even one exponential kinetic process without over- or underfitting data. In the case of peptides with two exponentials, the reported apparent rate constants represent their weighted average. A complete comparison of apparent rate constants can be found in Table S1.
Comparing rate constants across all temperatures revealed three peptides with rates of exchange increased by at least 3-fold: 297–305, 306–316, and 317–334 (cf. Table S1). The apparent rates of exchange for these peptides were previously shown to be sensitive to active-site mutations (28). A more thorough inspection of rate constants showed that three additional peptides exhibit altered rates, but only at lower incubation temperatures: 239–256, 257–273, and 751–761 (Table 1). These peptides have previously been characterized as having rapid exchange approaching nearly full extent. Indeed, at elevated temperatures (30 and 40 °C), the exchange rates for the latter three peptides (Table S1) are rapid and comparable for SLO in the presence and absence of effector (≥3 min−1). However, at 10 °C, the exchange process for the SLO protein in the absence of effector is slowed, sufficiently to be monitored versus time; thus, we focus on the HDXMS results at 10 °C, where the largest exchange differences are observed. In the presence of OS, a substantial increase in the apparent rate of exchange (by ≥8-fold) is observed for all six peptides at 10 °C (Fig. 6 and Table 1). Peptide 257–273 (and perhaps 751–761) is the most impacted by the presence of OS. Overall, the increase in the rates of exchange for all peptides shown in Fig. 6 is consistent with a destabilization of their secondary structure (34).
Besides the peptide 317–334, which is modeled primarily as a loop structural element in the X-ray structure, the remaining five C-terminal peptides affected by OS are α helices (Fig. 6). Our collective HDXMS data support a model of OS binding at the N-terminal PLAT domain that triggers the destabilization of these α helices in the catalytic domain. Previously, Tatulian et al. (16) provided evidence from the vibrational shifts in the peptidyl amide I vibrational modes that linoleic acid induces destabilization of α helices (i.e. to a more open structure). It has been shown that at elevated concentrations, substrate LA and mixed inhibitor, OA, associate at both an allosteric site and the active site in SLO. As stated above, OS exclusively interacts at the allosteric site of SLO and alters the binding of LA at the active site. Our HDXMS results, when combined with studies of OS binding (18, 19), support the location of an allosteric site for SLO in the PLAT domain that leads to destabilization of α helices within the catalytic domain.
Assessment of the rates of HDX for the peptides affected by OS as a function of the four temperatures studied here further revealed striking changes in the apparent activation energies for exchange; the Arrhenius plots are displayed in Fig. 7. A change in the Arrhenius slope is related to an altered activation energy of exchange. For EX-2 conditions, the observed rate of exchange (kHDX) will represent the product between the equilibrium constant for opening (kopen) and closing (kclose) processes, Kop = kopen/kclose, and the intrinsic chemical exchange rate, kint: kHDX = Kop × kint (30, 31, 35). The latter term represents the exchange rate on a fully exposed amide bond and depends on inductive effects from side-chain composition. In the absence of OS (Fig. 7, black dots/lines), the apparent activation energies of HDX for peptides shown (15 ± 2 < EaHDX < 20 ± 2 kcal/mol) are within error of the enthalpy for the intrinsic exchange (ΔH‡int = 17 kcal/mol) (64).
Figure 7.
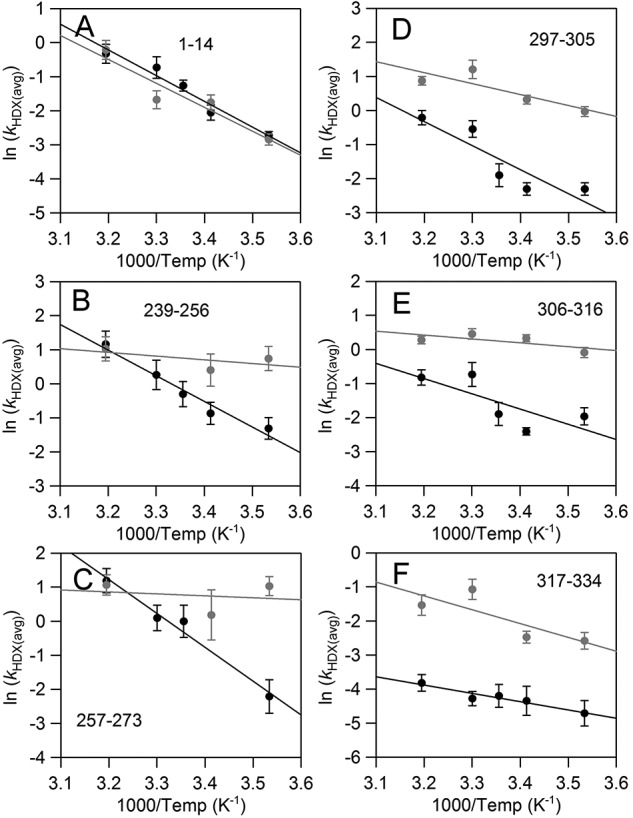
Arrhenius-like plots of the weighted average exchange rates, ln(kHDX(avg)), for SLO in the absence (black) or presence (gray) of OS. Apparent rates of HDXMS were determined from fits to the traces in Fig. S1. All rate constants can be found in Table S1.
Upon OS addition, the first clear trend is that the exchange rates for N-terminal peptide, 1–14, are unaffected across all temperatures (Fig. 7A). The same is true for all other N-terminal domain peptides (Table S1). The second feature to emerge is a drastic change in the Arrhenius slopes for 239–256 and 257–273 (Fig. 7, B and C). For these two peptides in the presence of OS, EaHDX(avg) values are considerably reduced; apparent activation energies of exchange decreased from the intrinsic value of 17 kcal/mol, requiring negative values for Kop that can be interpreted in the context of increased disorder within this region upon remote binding of OS (36, 37). For the remaining three peptides affected by OS, 297–305, 306–316, and 317–334 (Fig. 7, D–F), Arrhenius slopes are seen to be only modestly altered by OS when compared with the behavior of 239–256 and 257–273. The temperature dependence of the exchange rates for peptide 317–334 is perhaps the least affected. This behavior is in contrast to a previous observation from active-site mutational impact, in which this loop displayed a linear relationship between the enthalpic barrier for HDX and the corresponding barrier for catalytic PCET (28). An analysis of the temperature dependence of 751–761 has been omitted, as reliable rate constants were not obtainable at temperatures above 20 °C.
We point out that analyses of HDXMS traces at a single temperature, for example 30 °C in which most of the kcat, Km, and Dkcat kinetic parameters are often reported for SLO, would have revealed only changes to the loop region including peptides within the 297–334 segment. The impact on conformational flexibility at peptides 239–256, 257–273, and 751–761 is masked at this temperature because the exchange rates are practically identical for both conditions interrogated here (absence versus presence of OS). Whereas peptides including loop 317–334 and those spatially adjacent (297–305 and 306–316) are seen to have slightly impacted temperature dependences for the apparent rate constants, the change in the temperature dependence is most pronounced for 239–256 and 257–273. These results illustrate the advantage of performing HDXMS experiments at multiple temperatures.
HDXMS experiments conducted with OA (collected at 30 °C and with 45 μm OA) display trends similar to those for OS at the single temperature analyzed (Fig. S2). OA has also been shown to bind at the active site (38); however, OA is poorly reactive with ferric SLO (39) and non-reactive with the ferrous state of the enzyme (40) studied here. Whereas OA is expected to interact at both the allosteric and active sites, HDXMS traces at 30 °C for OA and OS are nearly indistinguishable (Fig. S2). Most notably, OA causes a similar conformational change at the N-terminal peptide, 1–14; these data are consistent with association of lipids, both OS and OA, at the proposed allosteric site located within the N-terminal PLAT domain. The observation of similar HDXMS behavior between free and substrate-bound protein has been documented previously for other systems (41, 42). HDXMS experiments with SLO were not performed in the presence of substrate, LA, because of the complicated results that were expected from structural alterations occurring during the catalytic cycle.
Discussion
HDXMS is proving to be a vital tool for the interrogation of protein flexibility associated with long-range allosteric communication, capable of detecting subtle changes in backbone mobility that are often invisible with X-ray crystallography. This work represents the HDXMS interrogation of lipid-induced allostery in lipoxygenase. Here, two distinct patterns of HDX have emerged from interactions of the lipid-like allosteric effector, OS, with the model plant enzyme, SLO. First, in the N-terminal PLAT domain, found only in humans and plants, OS induces a significant increase in the overall extent of HDX at long incubation times, consistent with the additional exchange of 1–3 amide hydrogens. We interpret this effect in the context of a conformational change, associated with OS binding nearby. Further, six peptides in the catalytic domain, where substrate binds and is oxidized, are seen to undergo dramatic increases in flexibility, supporting destabilization of their secondary structure.
Typically, protein ligand–inhibitor interactions show patterns of rates and/or extents of HDX that are reduced from the native protein (35, 43, 44). This canonical ligand-induced behavior is often attributed to enhanced intramolecular hydrogen bonding (i.e. structural stabilization). Protection of amide hydrogen bonds from exchange, as read out in HDXMS experiments as decreased exchange (extent and/or rates), is widely accepted as a hallmark for protein-protein and protein-ligand interactions. However, protein-ligand binding can exhibit enhanced rates and/or extents, as documented previously (for examples, see Refs. 45–47), and these patterns are characteristic of a different type of HDX behavior (48). This latter type of HDX behavior in which ligand binding increases HDX rates has been suggested to arise when the ligand binds following (or followed by)4 a protein structural change (48). Such may be the case for OS association at the SLO PLAT domain, where the presence of this effector shifts a protein conformational equilibrium toward a more solvent-exposed state within both the N terminus and catalytic domains.
The previously characterized kinetic ramifications from OS binding provide a mechanistic framework for the HDXMS results presented herein (18, 50). Select kinetic parameters for the SLO reactions on substrate LA in the absence and presence of OS are summarized below in Table 2. Importantly, an increase in the Dkcat/Km was shown upon the addition of OS; this has been linked to an increased microscopic rate constant for the substrate off rate, koff, by 60-fold compared with the absence of OS (see Table 2 for details). From the estimated off rates, the relative substrate on rates, kon, can be determined from the dissociation constants, Kd = koff/kon. Because C–H bond cleavage is fully rate-limiting for deuterated substrate (29), Kd is equivalent to the Michaelis–Menten constant for the deuterated substrate (i.e. Km (D) = Kd) measured herein. Estimated kon values, reported in Table 2, are seen to be accelerated by ∼12-fold in the presence of OS. These microscopic rate constants suggest that substrate diffusion is no longer partially rate-limiting under these conditions. This effect is corroborated by the demonstrated elimination of the solvent viscosity dependence ((kcat/Km°)/(kcat/Km)) in the presence of OS (see Table 2) (18). We note that the summarized kinetic properties of SLO (Table 2) represent a pH value slightly different from that of the HDXMS analyses (pH of 9 versus pD 8, respectively). From previous studies, the kcat and Dkcat parameters are seen to be identical at pH 8 and 9, whereas kcat/Km undergoes only a modest change; further, the value of Dkcat/Km at pH 9 is within experimental error of the value at pH 8 (29). For these reasons, it has been possible to quantify the kinetic impact of OS at pH 9, where the majority of kinetic data have been collected for SLO, and relate its behavior to HDXMS at pD 8, conditions where EX2 behavior has been demonstrated (28).
Table 2.
SLO kinetic parameters and microscopic rate constants in the absence and presence of OS
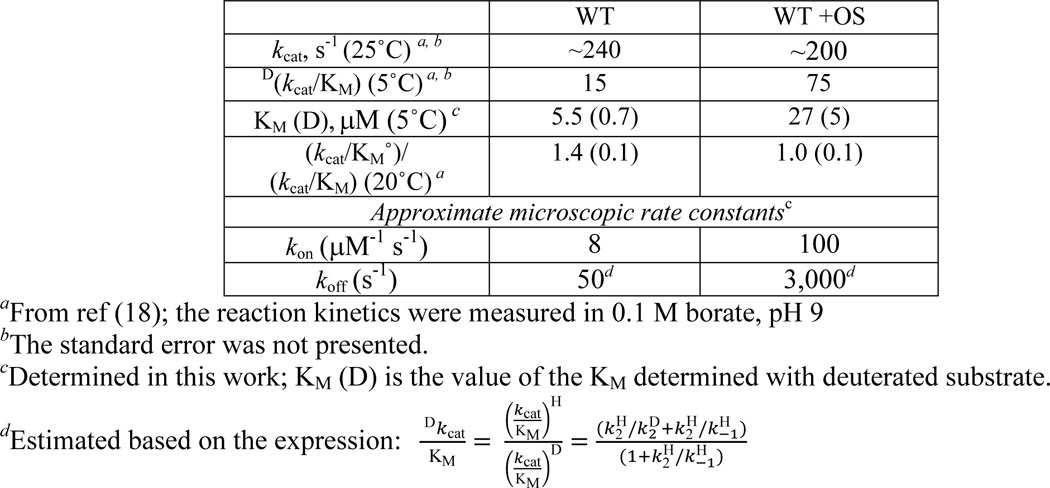
To explain the altered kinetics for substrate binding, in which koff is increased to a larger extent than kon, we propose a two-step mechanism for productive enzyme-substrate complex formation with an internal reorganization ES⇄ES′ associated with the equilibrium constant, Keq (Fig. 8). In the native system without OS, Keq favors ES′ so that the rate-determining steps for kcat/Km include k2 (chemistry) and substrate diffusional control. In the presence of OS, substrate diffusion no longer (significantly) contributes to the rate-determining steps (i.e. there is no viscogen dependence, (kcat/Km°)/(kcat/Km) = 1). Because kon and koff are both dependent on Keq (i.e. kon = k1[S] × Keq; koff = k−1 × 1/Keq; see Fig. 8) and OS addition impacts the off rate 5 times more than the on rate, the data imply that Keq is reduced in the presence of OS, shifting this internal equilibrium toward a “more open” form (denoted as ES).
Figure 8.

Schematic representing the impact of effector addition upon the reaction coordinate (A) and proposed mechanism (B) in soybean lipoxygenase chemistry. In A, the solid line represents the reaction coordinate (up to the first irreversible step, k2) for SLO reaction with LA in the absence of effector. The dashed line represents the changes to the steps in the reaction coordinate in the presence of OS determined from kinetic analysis. The addition of effector, OS, causes a relative reduced barrier for the first binding step and a change in the internal reorganization. Keq refers to this proposed internal reorganization from the initial enzyme-substrate complex (ES) to ES′ structure, with the latter representing the productive binding for chemistry. The substrate on rate (kon = k1[S] × Keq) and off rate (koff = k−1 × 1/Keq) are both a product of this internal equilibrium constant.
We propose that the enhanced substrate on and off rates may be facilitated by the destabilization of peptide 257–273, which is anticipated to line the substrate portal in SLO (51, 52). This putative entrance for substrate is located over 30 Å from the N-terminal peptide, 1–14, whose OS-induced conformational change is linked to enhanced substrate on and off rates (Fig. 9A). This raises the question: What is the link between the OS-induced conformational change at the N-terminal PLAT domain and the destabilization of α helices in the catalytic domain? A conserved cation-π interaction between Trp-130 and Arg-242 (Fig. 9, A and B; SLO numbering) has been previously proposed to serve as a mediator of Ca2+ activation in mammalian enzymes (13). Further, the homologous tryptophan in the human 5-lipoxygenase lies within a segment (FPCYRW102) that has been speculated to interact with the phospholipid membrane and an activating protein, coactosin-like protein, and elicit conformational allostery (53, 54). The HDXMS-identified network of allosteric communication in SLO, stemming from the PLAT domain to the substrate binding portal, may also be facilitated by this structural feature.
Figure 9.
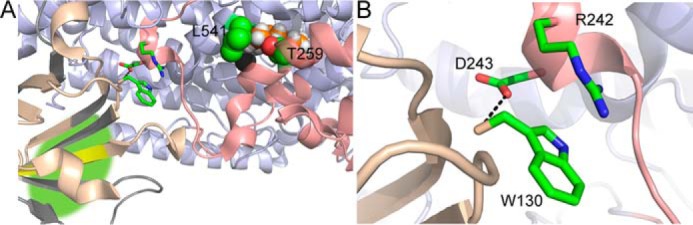
Structure of SLO depicting the peptides affected by interactions with allosteric effector, OS. The SLO model (Protein Data Bank code 3PZW) is colored pale blue. Peptides, whose backbone HDX properties are impacted by interactions with OS, are colored as follows. Yellow, increased extent of exchange (not rate); salmon, increased rate of exchange. A, proposed communication network for OS-mediated allostery includes the conserved cation-π interaction (green sticks). The iron cofactor is represented as a dark gray sphere. The modeled substrate is depicted in spheres (orange, carbon; white, hydrogen). Side chains Thr-259 and Leu-541 at the substrate entrance are displayed in spheres for reference. The green highlighted area suggests the binding site for OS. B, a focused view of the cation-π interaction between Trp-130 and Arg-242. The conserved carboxylate, Asp-243, also forms hydrogen bonds to Trp-130 backbone.
The current HDXMS and kinetic study was carried out at pH values elevated in relation to physiological conditions. The rationale for the conditions used here was to develop a molecular framework for the regulation of substrate diffusion by allosteric effectors. The key finding from the current work is the implied localization of lipid (surrogate) binding interactions at the N-terminal PLAT domain together with long-range allostery that arises from the lipoxygenase-lipid interactions. Beyond the present results, the most substantiated evidence to implicate the association of N-terminal PLAT domains with the free lipids or the lipid membranes can be attributed to N-terminal deletions. Examples from select plant and mammalian lipoxygenases suggest that removal of the PLAT domain impairs stability and catalytic proficiency and affects allosteric regulation (see Refs. 21, 55, and 56 and references within).
At the time of submission of this work, an HDXMS study of human 15-lipoxygenase, 15-LOX-2, appeared in the literature (14). This study showed little impact of either Ca2+ or lipid nanodiscs on HDXMS, whereas substrate was shown to yield a pattern of protection within the protein overall. Important differences between that study and the current work are the reduced coverage of the protein sequence for 15-LOX-2 in relation to SLO and the focus in the 15-LOX-2 study on a single temperature and different time regimes. As we show herein, changes in temperature can have a dramatic impact on observed patterns of HDXMS (Figs. 4 and 7), expanding our capacity to infer mechanistic features.
It should be noted that whereas Ca2+ triggers translocation of human LOX to the phospholipid membrane, calcium does not regulate membrane binding for SLO (15). Human enzymes also exhibit product feedback inhibition, whereas SLO does not. Together with the fact that SLO is 20% larger than human LOXs, the data suggest that 15-LOX-2 and SLO behave as different model enzymes.
To conclude, an important feature of the presented work is the focus on the interactions of SLO with OS, a molecule that neither turns over with SLO nor binds at the active site. These properties have allowed us to isolate the allosteric properties of a lipid effector as monitored by HDX and steady-state kinetics. The enhanced HDX observed for the SLO PLAT domain provides strong support for OS binding to the N terminus of SLO, whereas detailed analyses of HDXMS at multiple temperatures have uncovered a long-range interaction between the remote PLAT domain and the substrate binding portal of SLO. We believe that these HDXMS results provide the first robust structural evidence for the interaction of lipids at the N terminus of an intact SLO, with a resulting communication of dynamical effects to the catalytic domain.
Experimental procedures
Materials
Buffers, salts, and OA were obtained at the highest commercial purity available. Dideuterated linoleic acid, 11,11-2H2-LA(D2-LA), was synthesized as described (57). OS was synthesized from OA, as described previously (18). 1H NMR spectra of final product (CD3OD): δ = 5.25 (m, 2H), 3.90 (t, 2H), 1.95 (m, 4H), 1.57 (m, 2H), 1.23 (m, 22H), 0.81 (t, 3H). HRMS, calculated: 347.2256; observed: 347.2252.
SLO protein expression and purification
SLO was expressed using the T7-7 plasmid in BL21(DE3) Codon Plus RIL cells and purified as described previously (58). Purity was assessed by SDS-PAGE to be 90%. LC-MS (electrospray ionization) observed: 94,411 Da, calculated: 94,411. Iron analysis using the ferrozine assay resulted in 0.9 ± 0.05 Fe/SLO. The quality of the enzyme preparation was also confirmed by kinetic assays. Before freezing purified aliquots, the protein was dialyzed in 0.1 m borate, pH 9, and concentrated to 100 μm. Steady-state kinetic measurements with D2-LA were performed (as described previously (58)) in triplicate with 0.1 m borate, pH 9.0, buffer in the presence and absence of effector, OS.
HDX sample preparation
Purified aliquots of wildtype SLO were thawed and diluted to 50 μm. Protein aliquots (5 μl) from this stock sample were diluted 10-fold with 45 μl of D2O buffer (10 mm HEPES, pD 7.4; corrected as pD = pHread + 0.4 (59)) to a pD of 8. Controls indicate that the presence of 10% H2O does not alter this value appreciably. For the interrogation of OS (or OA), the purified oils were first dissolved in MeOD at 50 mm concentrations. These samples were then diluted in D2O buffer and added to SLO to a final OS concentration of 10 μm or a final OA concentration of 45 μm. (The order of addition did not impact HDXMS behavior, as the addition of OA or OS to SLO in H2O buffer, 1 min before mixing with the D2O buffer, had no notable effect.) The concentrations for OA and OS were selected based on previously determined Ki values of 22–36 μm (18, 60) and 0.6 μm, respectively. OA concentration was limited due to the critical micelle concentration, measured to be in the range of 55–70 μm, using a fluorescence approach with 1,6-diphenyl-1,3,5-hexatriene as described previously (61).
HDX samples were prepared randomly at a specified temperature (temperatures studied: 10, 20, 30, and 40 °C; bath stability ±0.1 °C), using a NESLAB RTE-111 water bath, for 14 time points (0, 10, 30, 45, 60, 180, 600, 1200, 1800, 2700, 3600, 7200, 10,800, and 14,400 s) for each temperature studied. Samples were randomized and collected over multiple days to avoid systematic error. Upon completion of the designated incubation time, samples were rapidly cooled (−20 °C bath) and acid-quenched (to pH 2.4, confirmed with pH electrode, with 0.32 m citric acid stock solution at 0 °C). SLO-derived peptides were generated from 2.5-min incubations with pre-exchanged, 150-μl immobilized pepsin (G-biosciences) after the addition of buffered (pH 2.4) guanidine HCl (∼0.5 m final concentration). The peptide fragments were removed from the immobilized pepsin via short (10 s) centrifugations using spin cups (0.45-μm cellulose acetate; Pierce) and then flash-frozen immediately in 250-μl MS tube inserts using liquid N2. Samples were stored at −80 °C until data collection.
Liquid chromatography–mass spectrometry for hydrogen–deuterium exchange measurements
Peptide assignments (from the digestion in water) were previously determined for WT SLO (28). Deuterated, pepsin-digested samples of SLO from hydrogen–deuterium exchange experiments were analyzed using a 1200 series liquid chromatograph (LC; Agilent) that was connected in-line with an LTQ Orbitrap XL mass spectrometer (Thermo). The LC was equipped with a reversed-phase analytical column (Viva C8, 30-mm length × 1.0-mm inner diameter, 5-μm particles; Restek) and guard precolumn (C8, Restek). Solvent A was 99.9% water, 0.1% formic acid, and solvent B was 99.9% acetonitrile, 0.1% formic acid (v/v). Each sample was thawed immediately before injection onto the column. The elution program consisted of a linear gradient from 5 to 10% B over 1 min, a linear gradient to 40% B over 5 min, a linear gradient to 100% B over 4 min, isocratic conditions at 100% B for 3 min, a linear gradient to 5% B over 0.5 min, and isocratic conditions at 5% B for 5.5 min, at a flow rate of 300 μl/min. The column compartment was maintained at 4 °C and lined with towels to absorb atmospheric moisture condensation. The column exit was connected to the electrospray ionization source of the mass spectrometer using PEEK tubing (0.005-inch inner diameter × -inch outer diameter; Agilent). Mass spectra were acquired in the positive ion mode over the range m/z = 350–1800 using the Orbitrap mass analyzer, in profile format, with a mass resolution setting of 100,000 (at m/z = 400). Data acquisition was controlled using Xcalibur software (version 2.0.7, Thermo).
HDX data analysis
Mass spectral data acquired for HDX measurements were analyzed using the software HDX WorkBench (62). The percentage of deuterium incorporation was calculated for each of these peptides, taking into account the number of amide linkages (excluding proline residues) and the calculated number of deuterons incorporated. The values were normalized for 100% D2O and corrected for peptide-specific back-exchange, as determined previously (28). The data were plotted as deuterium exchange versus time using Igor Pro software. The rates and extents of exchange (at 4 h) were determined from one- or two-exponential fits to the analyzed time-resolved HDX data.
Author contributions
A. R. O. and J. P. K. designed and analyzed all experiments. A. R. O. and A. T. I. performed all experiments. A. R. O. and J. P. K. wrote the paper. All authors have approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Shenshen Hu for helpful discussions. We also thank Dr. Svetlana Pakhomova and Prof. Marcia Newcomer for attempts to co-crystalize the SLO-OS complex. The QB3/Chemistry Mass Spectrometry Facility at UC Berkeley receives support from National Institutes of Health Grant 1S10OD020062-01.
This work was supported by the National Institutes of Health Grants GM113432 (to A. R. O.) and GM118117 (to J. P. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1 and S2.
It is important to note here that the exact order of these events (binding of molecules induces conformational change, or conformational change induces ligand binding) is difficult to clarify because the HDXMS experiment as assessed here is a thermally averaged measurement under EX-2 conditions. The data are consistent with a conformational change that occurs with ligand (i.e. lipid) binding.
- PLAT
- polycystin-1/lipoxygenase/α-toxin
- HDX
- hydrogen–deuterium exchange
- HDXMS
- hydrogen–deuterium exchange mass spectrometry
- SLO
- soybean lipoxygenase
- OS
- oleyl sulfate
- OA
- oleic acid
- LA
- linoleic acid; kcat is the maximal rate at substrate saturation; Dkcat is the ratio of kcat for protio substrate to the kcat for deuterio substrate.
References
- 1. Porta H., and Rocha-Sosa M. (2002) Plant lipoxygenases: physiological and molecular features. Plant Physiol. 130, 15–21 10.1104/pp.010787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Feussner I., and Wasternack C. (2002) The lipoxygenase pathway. Annu. Rev. Plant Biol. 53, 275–297 10.1146/annurev.arplant.53.100301.135248 [DOI] [PubMed] [Google Scholar]
- 3. Haeggström J. Z., and Funk C. D. (2011) Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev. 111, 5866–5898 10.1021/cr200246d [DOI] [PubMed] [Google Scholar]
- 4. Whitehouse M. W., and Rainsford K. D. (2006) Lipoxygenase inhibition: the neglected frontier for regulating chronic inflammation and pain. Inflammanopharmacology 14, 99–102 10.1007/s10787-006-1523-7 [DOI] [PubMed] [Google Scholar]
- 5. Andreou A., and Feussner I. (2009) Lipoxygenases: structure and reaction mechanism. Phytochemistry 70, 1501–1510 10.1016/j.phytochem.2009.05.008 [DOI] [PubMed] [Google Scholar]
- 6. Ivanov I., Heydeck D., Hofheinz K., Roffeis J., O'Donnell V. B., Kuhn H., and Walther M. (2010) Molecular enzymology of lipoxygenases. Arch. Biochem. Biophys. 503, 161–174 10.1016/j.abb.2010.08.016 [DOI] [PubMed] [Google Scholar]
- 7. Newcomer M. E., and Brash A. R. (2015) The structural basis for specificity in lipoxygenase catalysis. Protein Sci. 24, 298–309 10.1002/pro.2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nalefski E. A., and Falke J. J. (1996) The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 5, 2375–2390 10.1002/pro.5560051201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang D., and Aravind L. (2010) Identification of novel families and classification of the C2 domain superfamily elucidate the origin and evolution of membrane targeting activities in eukaryotes. Gene 469, 18–30 10.1016/j.gene.2010.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hammarberg T., Provost P., Persson B., and Rådmark O. (2000) The N-terminal domain of 5-lipoxygenase binds calcium and mediates calcium stimulation of enzyme activity. J. Biol. Chem. 275, 38787–38793 10.1074/jbc.M006136200 [DOI] [PubMed] [Google Scholar]
- 11. Kulkarni S., Das S., Funk C. D., Murray D., and Cho W. (2002) Molecular basis of the specific subcellular localization of the C2-like domain of 5-lipoxygenase. J. Biol. Chem. 277, 13167–13174 10.1074/jbc.M112393200 [DOI] [PubMed] [Google Scholar]
- 12. Oldham M. L., Brash A. R., and Newcomer M. E. (2005) Insights from the X-ray crystal structure of coral 8R-lipoxygenase: calcium activation via a C2-like domain and a structural basis for product chirality. J. Biol. Chem. 280, 39545–39552 10.1074/jbc.M506675200 [DOI] [PubMed] [Google Scholar]
- 13. Eek P., Järving R., Järving I., Gilbert N. C., Newcomer M. E., and Samel N. (2012) Structure of a calcium-dependent 11R-lipoxygenase suggests a mechanism for Ca2+ regulation. J. Biol. Chem. 287, 22377–22386 10.1074/jbc.M112.343285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Droege K. D., Keithly M. E., Sanders C. R., Armstrong R. N., and Thompson M. K. (2017) Structural dynamics of 15-lipoxygenase-2 via hydrogen–deuterium exchange. Biochemistry 56, 5065–5074 10.1021/acs.biochem.7b00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Restrepo F., Snyder H. E., and Zimmerman G. L. (1973) Calcium activation of soybean lipoxygenase. J. Food Sci. 38, 779–782 10.1111/j.1365-2621.1973.tb02074.x [DOI] [Google Scholar]
- 16. Tatulian S. A., Steczko J., and Minor W. (1998) Uncovering a calcium-regulated membrane-binding mechanism for soybean lipoxygenase-1. Biochemistry 37, 15481–15490 10.1021/bi981062t [DOI] [PubMed] [Google Scholar]
- 17. Lewis E. R., Johansen E., and Holman T. R. (1999) Large competitive kinetic isotope effects in human 15-lipoxygenase catalysis measured by a novel HPLC method. J. Am. Chem. Soc. 121, 1395–1396 10.1021/ja983083g [DOI] [Google Scholar]
- 18. Mogul R., Johansen E., and Holman T. R. (2000) Oleyl sulfate reveals allosteric inhibition of soybean lipoxygenase-1 and human 15-lipoxygenase. Biochemistry 39, 4801–4807 10.1021/bi992805t [DOI] [PubMed] [Google Scholar]
- 19. Ruddat V. C., Whitman S., Holman T. R., and Bernasconi C. F. (2003) Stopped-flow kinetic investigations of the activation of soybean lipoxygenase-1 and the influence of inhibitors on the allosteric site. Biochemistry 42, 4172–4178 10.1021/bi020698o [DOI] [PubMed] [Google Scholar]
- 20. Wecksler A. T., Kenyon V., Garcia N. K., Deschamps J. D., van der Donk W. A., and Holman T. R. (2009) Kinetic and structural investigations of the allosteric site in human epithelial 15-lipoxygenase-2. Biochemistry 48, 8721–8730 10.1021/bi9009242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joshi N., Hoobler E. K., Perry S., Diaz G., Fox B., and Holman T. R. (2013) Kinetic and structural investigations into the allosteric and pH effect on the substrate specificity of human epithelial 15-lipoxygenase-2. Biochemistry 52, 8026–8035 10.1021/bi4010649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoofnagle A. N., Resing K. A., Goldsmith E. J., and Ahn N. G. (2001) Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. U.S.A. 98, 956–961 10.1073/pnas.98.3.956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anand G. S., Hughes C. A., Jones J. M., Taylor S. S., and Komives E. A. (2002) Amide H/2H exchange reveals communication between the cAMP and catalytic subunit-binding sites in the R1α subunit of protein kinase A. J. Mol. Biol. 323, 377–386 10.1016/S0022-2836(02)00919-1 [DOI] [PubMed] [Google Scholar]
- 24. Yang J., Garrod S. M., Deal M. S., Anand G. S., Woods V. L. Jr., and Taylor S. (2005) Allosteric network of cAMP-dependent protein kinase revealed by mutation Tyr204 in the P+1 loop. J. Mol. Biol. 346, 191–201 10.1016/j.jmb.2004.11.030 [DOI] [PubMed] [Google Scholar]
- 25. Rand K. D., Jørgensen T. J., Olsen O. H., Persson E., Jensen O. N., Stennicke H. R., and Andersen M. D. (2006) Allosteric activation of coagulation factor VIIa visualized by hydrogen exchange. J. Biol. Chem. 281, 23018–23024 10.1074/jbc.M602968200 [DOI] [PubMed] [Google Scholar]
- 26. Underbakke E. S., Iavarone A. T., Chalmers M. J., Pascal B. D., Novick S., Griffin P. R., and Marletta M. A. (2014) Nitric oxide-induced conformational changes in soluble guanylate cyclase. Structure 22, 602–611 10.1016/j.str.2014.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deredge D., Li J., Johnson K. A., and Wintrode P. L. (2016) Hydrogen/deuterium exchange kinetics demonstrate long range allosteric effects of thumb site 2 inhibitors of hepatitis C viral RNA-dependent RNA polymerase. J. Biol. Chem. 291, 10078–10088 10.1074/jbc.M115.708370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Offenbacher A. R., Hu S., Poss E. M., Carr C. A. M., Scouras A. D., Prigozhin D. M., Iavarone A. T., Palla A., Alber T., Fraser J. S., and Klinman J. P. (2017) Hydrogen deuterium exchange of lipoxygenase uncovers a relationship between distal, solvent exposed protein motions and the thermal activation barrier for catalytic proton-coupled electron tunneling. ACS Cent. Sci. 3, 570–579 10.1021/acscentsci.7b00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glickman M. H., and Klinman J. P. (1995) Nature of the rate-limiting steps in the soybean lipoxygenase-1 reaction. Biochemistry 34, 14077–14092 10.1021/bi00043a013 [DOI] [PubMed] [Google Scholar]
- 30. Hoofnagle A. N., Resing K. A., and Ahn N. G. (2003) Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 32, 1–25 10.1146/annurev.biophys.32.110601.142417 [DOI] [PubMed] [Google Scholar]
- 31. Englander S. W. (2006) Hydrogen exchange and mass spectrometry: A historical perspective. J. Am. Soc. Mass Spectrom. 17, 1481–1489 10.1016/j.jasms.2006.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang Z.-X., Lee T., Resing K. A., Ahn N. G., and Klinman J. P. (2004) Thermal-activated protein mobility and its correlation with catalysis in thermophilic alcohol dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 101, 9556–9561 10.1073/pnas.0403337101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oyeyemi O. A., Sours K. M., Lee T., Resing K. A., Ahn N. G., and Klinman J. P. (2010) Temperature dependence of protein motions in a thermophilic dihydrofolate reductase and its relationship to catalytic efficiency. Proc. Natl. Acad. Sci. U.S.A. 107, 10074–10079 10.1073/pnas.1003678107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hvidt A., and Nielsen S. O. (1966) Hydrogen exchange in proteins. Adv. Protein Chem. 21, 287–386 10.1016/S0065-3233(08)60129-1 [DOI] [PubMed] [Google Scholar]
- 35. Wales T. E., and Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 10.1002/mas.20064 [DOI] [PubMed] [Google Scholar]
- 36. Englander S. W., and Kallenbach N. R. (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16, 521–655 [DOI] [PubMed] [Google Scholar]
- 37. Oliveberg M., Tan Y. J., and Fersht A. R. (1995) Negative activation enthalpies in the kinetics of protein folding. Proc. Natl. Acad. Sci. U.S.A. 92, 8926–8929 10.1073/pnas.92.19.8926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van der Hijdt L. M., Schilstra M. J., Feiters M. C., Nolting H.-F., Hermes C., Veldink G. A., and Vliegenthart J. F. (1995) Changes in the iron coordination sphere of Fe(II) lipoxygenase-1 from soybean upon binding of linoleate or oleate. Eur. J. Biochem. 231, 186–191 10.1111/j.1432-1033.1995.tb20685.x [DOI] [PubMed] [Google Scholar]
- 39. Clapp C. H., Strulson M., Rodriguez P. C., Lo R., and Novak M. J. (2006) Oxygenation of monounsaturated fatty acids by soybean lipoxygenase-1: evidence for transient hydroperoxide formation. Biochemistry 45, 15884–15892 10.1021/bi0619425 [DOI] [PubMed] [Google Scholar]
- 40. Clapp C. H., Senchak S. E., Stover T. J., Potter T. C., Findeis P. M., and Novak M. J. (2001) Soybean lipoxygenase-mediated oxygenation of monounsaturated fatty acids to enones. J. Am. Chem. Soc. 123, 747–748 10.1021/ja0032071 [DOI] [PubMed] [Google Scholar]
- 41. Liu Y.-H., and Konermann L. (2006) Enzyme conformational dynamics during catalysis and in the “resting state” monitored by hydrogen/deuterium exchange mass spectrometry. FEBS Lett. 580, 5137–5142 10.1016/j.febslet.2006.08.042 [DOI] [PubMed] [Google Scholar]
- 42. Liu Y.-H., and Konermann L. (2008) Conformational dynamics of free and catalytically active thermolysin are indistinguishable by hydrogen/deuterium exchange mass spectrometry. Biochemistry 47, 6342–6351 10.1021/bi800463q [DOI] [PubMed] [Google Scholar]
- 43. Shi Z., Resing K. A., and Ahn N. G. (2006) Networks for the allosteric control of protein kinases. Curr. Opin. Struct. Biol. 16, 686–692 10.1016/j.sbi.2006.10.011 [DOI] [PubMed] [Google Scholar]
- 44. Konermann L., Pan J., and Liu Y.-H. (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 40, 1224–1234 10.1039/C0CS00113A [DOI] [PubMed] [Google Scholar]
- 45. Lee T., Hoofnagle A. N., Kabuyama Y., Stroud J., Min X., Goldsmith E. J., Chen L., Resing K. A., and Ahn N. G. (2004) Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell 14, 43–55 10.1016/S1097-2765(04)00161-3 [DOI] [PubMed] [Google Scholar]
- 46. Burke J. E., Babakhani A., Gorfe A. A., Kokotos G., Li S., Woods V. L. Jr., McCammon J. A., and Dennis E. A. (2009) Location of inhibitors bound to group IVA phospholipase A2 determined by molecular dynamics and deuterium exchange mass spectrometry. J. Am. Chem. Soc. 131, 8083–8091 10.1021/ja900098y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sowole M. A., and Konermann L. (2014) Effects of protein-ligand interactions on hydrogen/deuterium exchange kinetics: conanonical and noncanonical scenarios. Anal. Chem. 86, 6715–6722 10.1021/ac501849n [DOI] [PubMed] [Google Scholar]
- 48. Konermann L., Rodriguez A. D., and Sowole M. A. (2014) Type 1 and type 2 scenarios in hydrogen exchange mass spectrometry studies on protein-ligand complexes. Analyst 139, 6078–6087 10.1039/C4AN01307G [DOI] [PubMed] [Google Scholar]
- 49. Hatcher E., Soudackov A. V., and Hammes-Schiffer S. (2004) Proton-coupled electron transfer in soybean lipoxygenase. J. Am. Chem. Soc. 126, 5763–5775 10.1021/ja039606o [DOI] [PubMed] [Google Scholar]
- 50. Wecksler A. T., Garcia N. K., and Holman T. R. (2009) Substrate specificity effects of lipoxygenase products and inhibitors on soybean lipoxygenase-1. Bioorg. Med. Chem. 17, 6534–6539 10.1016/j.bmc.2009.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Minor W., Steczko J., Stec B., Otwinowski Z., Bolin J. T., Walter R., and Axelrod B. (1996) Crystal structure of soybean lipoxygenase L-1 at 1.4 Å resolution. Biochemistry 35, 10687–10701 10.1021/bi960576u [DOI] [PubMed] [Google Scholar]
- 52. Gaffney B. J., Bradshaw M. D., Frausto S. D., Wu F., Freed J. H., and Borbat P. (2012) Locating a lipid at the portal to the lipoxygenase active site. Biophys. J. 103, 2134–2144 10.1016/j.bpj.2012.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Allard J. B., and Brock T. G. (2005) Structural organization of the regulatory domain of human 5-lipoxygenase. Curr. Protein Pept. Sci. 6, 125–131 10.2174/1389203053545417 [DOI] [PubMed] [Google Scholar]
- 54. Esser J., Rakonjac M., Hofmann B., Fischer L., Provost P., Schneider G., Steinhilber D., Samuelsson B., and Rådmark O. (2009) Coactosin-like protein functions as a stabilizing chaperone for 5-lipoxygenase: a role for tryptophan 102. Biochem. J. 425, 265–274 10.1042/BJ20090856 [DOI] [PubMed] [Google Scholar]
- 55. Di Venere A., Salucci M. L., van Zadelhoff G., Veldink G., Mei G., Rosato N., Finazzi-Agrò A., and Maccarrone M. (2003) Structure-to-function relationship of mini-lipoxygenase, a 60-kDa fragment of soybean lipoxygenase-1 with lower stability but higher enzymatic activity. J. Biol. Chem. 278, 18281–18288 10.1074/jbc.M212122200 [DOI] [PubMed] [Google Scholar]
- 56. Walther M., Hofheinz K., Vogel R., Roffeis J., and Kühn H. C. (2011) The N-terminal β-barrel domain of mammalian lipoxygenases including mouse t-lipoxygenase is not essential for catlytic activity and membrane binding but exhibits regulatory functions. Arch. Biochem. Biophys. 516, 1–9 10.1016/j.abb.2011.09.004 [DOI] [PubMed] [Google Scholar]
- 57. Offenbacher A. R., Zhu H., and Klinman J. P. (2016) Synthesis of site-specifically 13C labeled linoleic acids. Tetrahedron Lett. 57, 4537–4540 10.1016/j.tetlet.2016.08.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hu S., Sharma S. C., Scouras A. D., Soudackov A. V., Carr C. A. M., Hammes-Schiffer S., Alber T., and Klinman J. P. (2014) Extremely elevated room-temperature kinetic isotope effects quantify the critical role of barrier width in enzymatic C–H activation. J. Am. Chem. Soc. 136, 8157–8160 10.1021/ja502726s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schowen K. B., and Schowen R. L. (1982) Solvent isotope effects on enzyme systems. Methods Enzymol. 87, 551–606 10.1016/S0076-6879(82)87031-6 [DOI] [PubMed] [Google Scholar]
- 60. Van der Heijdt L. M., Feiters M. C., Navaratnam S., Nolting H.-F., Hermes C., Veldink G. A., and Vliegenthart J. F. (1992) X-ray absorption spectroscopy of soybean lipoxygenase-1. Eur. J. Biochem. 207, 793–802 10.1111/j.1432-1033.1992.tb17110.x [DOI] [PubMed] [Google Scholar]
- 61. Chattopadhyay A., and London E. (1984) Fluorimetric determination of critical micelle concentration avoiding interference from detergent charge. Anal. Biochem. 139, 408–412 10.1016/0003-2697(84)90026-5 [DOI] [PubMed] [Google Scholar]
- 62. Pascal B. D., Willis S., Lauer J. L., Landgraf R. R., West G. M., Marciano D., Novick S., Goswami D., Chalmers M. J., and Griffin P. R. (2012) HDX Workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 23, 1512–1521 10.1007/s13361-012-0419-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wennman A., Oliw E. H., Karkehabadi S., and Chen Y. (2016) Crystal structure of manganese lipoxygenase of the rice blast fungus Magnaporthe oryzae. J. Biol. Chem. 291, 8130–8139 10.1074/jbc.M115.707380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bai Y., Milne J. S., Mayne L., and Englander S. W. (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86 10.1002/prot.340170110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.