

Abstract

The bromodomain and extra-terminal (BET) family proteins, consisting of BRD2, BRD3, BRD4, and testis-specific BRDT members, are epigenetic “readers” and play a key role in the regulation of gene transcription. BET proteins are considered to be attractive therapeutic targets for cancer and other human diseases. Recently, heterobifunctional small-molecule BET degraders have been designed based upon the proteolysis targeting chimera (PROTAC) concept to induce BET protein degradation. Herein, we present our design, synthesis, and evaluation of a new class of PROTAC BET degraders. One of the most promising compounds, 23, effectively degrades BRD4 protein at concentrations as low as 30 pM in the RS4;11 leukemia cell line, achieves an IC50 value of 51 pM in inhibition of RS4;11 cell growth and induces rapid tumor regression in vivo against RS4;11 xenograft tumors. These data establish that compound 23 (BETd-260/ZBC260) is a highly potent and efficacious BET degrader.
Introduction
Bromodomain-containing proteins are epigenetic “readers”. By binding to acetylated lysine residues on the histone tails, bromodomain-containing proteins play a key role in regulation of gene transcription.1 Among bromodomain-containing proteins, the bromodomain and extra-terminal domain (BET) family of proteins, consisting of BRD2, BRD3, BRD4, and testis-specific BRDT members, have emerged as exciting new therapeutic targets for cancer and other human diseases.2−6 The discovery of 1 ((+)-JQ-1) as the first potent and selective BET inhibitor (Figure 1) has greatly promoted investigations of BET proteins as new therapeutic targets in human cancers and other diseases.7 Several BET inhibitors such as 2 (OTX015) and 3 (I-BET762) (Figure 1) have been advanced into clinical development.8−13 Recently, preliminary clinical data have provided an important clinical proof-of-concept that BET inhibition has therapeutic potential for the treatment of certain forms of human cancer, including NUT (nuclear protein in testis) midline carcinoma, multiple myeloma, and acute myeloid leukemia (AML).7,10,11,14 Preclinical studies have also suggested that BET inhibitors may have therapeutic potential for the treatment of other human cancers, as well as other human diseases and conditions.15−19
Figure 1.

Chemical structures of representative BET inhibitors 1, 2, 3, 7, 8 and three representative previously reported BET degraders 4, 5, and 6.
Small-molecule BET inhibitors are designed to bind the BET bromodomains and to block the interaction of BET proteins with acetylated lysine residues on histone tails to regulate gene transcription. In addition to small-molecule BET inhibitors, a new approach has recently been developed to target BET proteins for degradation based upon the proteolysis targeting chimera (PROTAC) concept.20 In this approach, a heterobifunctional (chimeric) molecule is designed to contain a BET inhibitor, which binds to BET proteins, another small-molecule ligand, which binds to an E3 ubiquitin ligase complex, and a linker to tether these two ligands together.21,22 A number of BET degraders have been reported, including 4 (dBET1),235 (ARV-771),246 (ARV-825),25 and MZ126 (Figure 1). Compounds 4 and 6 were designed using 1 or 2, two closely related BET inhibitors, and thalidomide, which is a ligand for cereblon, a component of the Cullin4A ubiquitin ligase complex.23,25 In comparison, BET degrader 5 was designed using 1 for the BET inhibitor portion and a ligand for the von Hippel–Lindau E3 ubiquitin ligase.24 These BET degraders have been shown to efficiently induce BET protein degradation and to be more potent in inhibition of cancer cell growth and in induction of apoptosis than their corresponding BET inhibitors. Compound 4 is more effective in inhibition of tumor growth in vivo than 1 in an acute leukemia model in mice,23 and compound 5 achieves effective inhibition of tumor growth in a castration-resistance prostate cancer xenograft model in mice.24 Collectively, these studies provide evidence that small-molecule degraders of BET proteins may have a promising therapeutic potential for the treatment of human cancers and potentially other diseases and conditions.
Recently, our laboratory reported the discovery of azacarbazoles as a new class of potent and selective BET bromodomain inhibitors.27 In the present study, we report the discovery of a new class of small-molecule BET degraders designed based upon our azacarbazole-based BET inhibitors and thalidomide/lenalidomide as ligands for cereblon/Cullin4A. Through extensive optimization of the linker region, we have obtained a series of highly potent BET degraders. Among these, compound 23 (BETd-260) is capable of inducing degradation of BRD2, BRD3, and BRD4 proteins at 30–100 pM in the RS4;11 leukemia cells. Compound 23 achieves an IC50 value of 51 pM in inhibition of the RS4;11 cell growth and induces rapid tumor regression of the RS4;11 xenograft tumors with no signs of toxicity in mice. Compound 23 is a highly potent and efficacious BET degrader and warrants extensive evaluation for the treatment of human cancers and other diseases.
Results and Discussion
Starting from our previously reported BET inhibitor 7(27) (Figure 1), we performed further optimization for this class of BET inhibitors and identified 8 (HJB97) as a high-affinity BET inhibitor. In our FP-based competitive binding assays, 8 binds to BRD2, BRD3, and BRD4 with high affinities (Ki < 1 nM) and is >10 times more potent than 1 or 2 (Table 1). Compound 8 also potently inhibits cell growth in RS4;11 and MOLM-13 acute leukemia cell lines, which are known to be sensitive to BET inhibitors. Hence, compound 8 is a potent BET inhibitor and was employed in our design of BET degraders.
Table 1. Binding Affinities of BET Inhibitors.

IC50 values were obtained from three separate experiments.
Upon the basis of the cocrystal structure of BRD4 BD2 complexed with 7 (PDB code 4Z93), we modeled the structure of BRD4 BD2 complexed with 8 (Figure 2). Our modeled structure showed that the 2-carboxamide group attached to the [6,5,6] tricyclic system in 8 is exposed to solvent, making it a suitable site for tethering to thalidomide/lenalidomide for the design of potential PROTAC degraders of BET proteins (Figure 2). Accordingly, we designed and synthesized 9 as a potential BET degrader using 8 for the BET inhibitor portion, thalidomide as the cereblon ligand, and the same linker that was used in 4. In a cell growth assay, the BET degrader 9 has an IC50 value of 4.3 nM in the RS4;11 acute leukemia cell line and is 6 times more potent than the corresponding BET inhibitor 8 (Table 2). Western blotting analysis showed that 9, at concentrations as low as 3–10 nM, is effective in decreasing the level of BRD2, BRD3, and BRD4 proteins in the RS4;11 cells whereas the BET inhibitor 8 at both 100 and 300 nM fails to decrease the level of BRD2–4 proteins (Figure 3). Two previously reported BET degraders, 4 and 6, also effectively decrease the level of BRD2–4 proteins in the RS4;11 cell line (Figure 3). Consistent with its testis-specific expression, BRDT protein was not detected in the RS4;11 cells. We concluded that 9 is a promising BET degrader for further optimization.
Figure 2.

(A) Cocrystal structure of BRD4 BD2 complexed with 7 (green, PDB code 4Z93). (B) Modeled structure of BRD4 BD2 complexed with 8 (yellow). Water molecules are shown as red spheres. Hydrogen bonds are depicted in dashed lines. Residues in BRD4 BD2 close to the proposed linkage site of 8 are labeled in red.
Table 2. Optimization of Linker Length and Composition.


IC50 values were obtained from three independent experiments.
Figure 3.
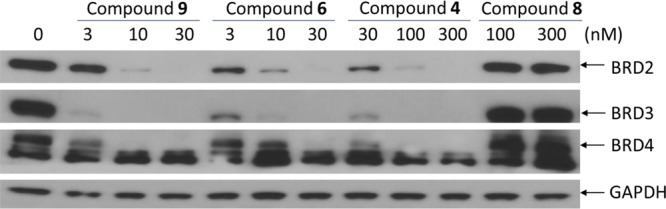
Western blotting analysis of BRD2, BRD3, and BRD4 proteins in RS4;11 cells treated with BET degraders 4, 6, and 9 and BET inhibitor 8. RS4;11 cells were treated for 3 h with each individual compound at indicated concentrations, and proteins were probed by specific antibodies. GAPDH was used as the loading control.
We next explored the length and the composition of the linker in 9. Replacing the oxygen atom in 9 with an amino group resulted in 10, whose cell growth inhibitory activity is 2 times better than that of 9. Conversion of the amide group (-CO-NH-) in 9 to an ethylene group (-CH2-CH2-) generated 11, which has an IC50 value of 0.48 nM in inhibition of cell growth in the RS4;11 cell line and is thus 10 times more potent than 9. Similar conversion of the amide group in 10 to an ethylene group yielded 12 which, with an IC50 value of 0.20 nM in inhibition of RS4;11 cell growth, is 10 times more potent than 10. These data demonstrate that both the length and the composition of the linker have a considerable influence on the cellular potencies of the resulting BET degraders.
We next sought to determine the optimal linker length in 12 for cellular potencies by shortening the linker by one methylene group progressively, which resulted in compounds 13–17 (Table 2). Compound 13 with a linker one methylene group shorter than that in 12 is 3 times less potent than 12. However, compound 14 with two methylene groups shorter than 12 in the linker achieves an IC50 value of 0.14 nM in inhibition of RS4;11 cell growth and is slightly more potent than 12. Shortening the linker in 14 by one additional methylene group resulted in 15, which is ∼3 times less potent than 14 in inhibition of RS4;11 cell growth, and further shortening the linker in 15 by one methylene or ethylene group yielded 16 or 17, respectively, which is 5 and 10 times less potent than 15, respectively, in inhibition of RS4;11 cell growth. These data demonstrate that for achieving the most potent cell growth inhibition activity, an optimal linker length should comprise -(CH2)4–7NH- in these BET degraders, as is shown in compounds 12–15. When the linker becomes too short as in, for examples, compounds 16 and 17, the cellular potency is greatly decreased.
To investigate whether the SAR results obtained in the RS4;11 cell line are valid in a different cell line, we evaluated this series of compounds together with 4 and 6 as control compounds for their cell growth inhibitory activity in the MOLM-13 leukemia cell line, which harbors a mixed lineage leukemia protein 1 (MLL1) fusion gene and is also responsive to BET inhibitors in our previous study.27 The data obtained are summarized in Table 2. In general, the IC50 values obtained for all these BET degraders in the MOLM-13 cell line are 5–10 times higher than those obtained in the RS4;11 cell line. Compounds 6 and 4 have IC50 values of 18.2 nM and 657 nM, respectively, in inhibition of MOLM-13 cell growth and are 5.5 and 8.3 times less potent than those in the RS4;11 cell line. We obtained a very similar SAR result for BET degraders 9–17 in the MOLM-13 cell line when compared to that in the RS4;11 cell line. Compounds 12 and 14 are two of the most potent BET degraders in this series with IC50 values of 1.2 nM and 2.1 nM, respectively, in inhibition of MOLM-13 cell growth.
Having determined the optimal linker length, we next performed further modifications of the linker composition in compound 14 (Table 3). Replacement of the NH group in 14 with a methylene group yielded 18, which with an IC50 value of 0.17 nM in inhibition of RS4;11 cell growth is as potent as 14. Replacement of one methylene group in 18 with an oxygen atom generated 19, which has an IC50 value of 0.42 nM in inhibition of RS4;11 cell growth and is 2 times less potent than 18. Shortening the linker in 18 by one methylene group resulted in 20 which, with an IC50 value of 0.14 nM in inhibition of RS4;11 cell growth, is equipotent to 14. Thus, modifications of the linker compositions identified compounds 18 and 20 as two very potent BET degraders with subnanomolar IC50 values in inhibition of RS4;11 cell growth.
Table 3. Further Optimization of the Linker and Phthalimide Moiety.


IC50 values were obtained from three independent experiments.
In all of the above synthesized compounds, we employed thalidomide as the ligand for cereblon. Lenalidomide has been developed as a second generation thalidomide analogue for the treatment of multiple myeloma and myelodysplastic syndromes.28 Although thalidomide and lenalidomide have similar binding affinities to cereblon,29 they may have different cell permeability and/or pharmacokinetic profiles, and thus we investigated the effect of replacing thalidomide with lenalidomide in three potent BET degraders 18, 19, and 20. This effort resulted in 21, 22, and 23, which achieve IC50 values of 0.037 nM, 0.90 nM, and 0.051 nM, respectively, in inhibition of RS4;11 cell growth. Hence, 21 and 23 are 2–3 times more potent than 18 and 20. Interestingly, in contrast to 21 and 23, compound 22 has an IC50 value of 0.9 nM in inhibition of RS4;11 cell growth and is thus 2 times less potent than 19. To further confirm the significance of the linker length, we synthesized compounds 24 and 25 with one methylene or ethylene group shorter than that in 23. Compounds 24 and 25 have IC50 values of 0.98 nM and 9.6 nM in inhibition of RS4;11 cell growth, respectively, and are therefore 19 and 188 times less potent than 23.
To further investigate the mechanism of action for this class of BET degraders, we have designed two control compounds based upon 23. It has been shown that installation of a methyl group on the amino group in the lenalidomide moiety blocks the binding of thalidomide analogues to cereblon.25,29 Accordingly, we installed a methyl group on the amino group in the lenalidomide moiety, which resulted in 26 (Table 4). Compound 26 has an IC50 value of 35.1 nM in inhibition of RS4;11 cell growth and is therefore >600 times less potent than 23, but the IC50 is similar to that obtained for the BET inhibitor 8. Furthermore, 26 fails to induce degradation of BRD2, BRD3, and BRD4 proteins in the RS4;11 cell line, indicating that it inhibits cell growth by acting not as a BET degrader but as a BET inhibitor (Figure S1 in Supporting Information). We synthesized compound 27 based upon a less potent BET inhibitor 28, which binds to BRD2, BRD3, and BRD4 proteins with affinities of >100 times less potent than that of 8 (Table 1). Compound 27 has an IC50 value of >1000 nM and is thus >10 000 and >400 times less potent than 23 in RS4;11 and MOLM13 cell growth inhibition assays, respectively (Table 4). These data indicate that in order to achieve potent cell growth inhibition, a BET degrader must be able to bind to both BET proteins and to the ubiquitin ligase complex, which is consistent with their PROTAC design and the expected mechanism of action.
Table 4. Investigation of the Effect of BET Protein and Cereblon Binding on Cellular Potencies of BET Degraders.


IC50 values were obtained from three independent experiments.
We examined the ability of four representative BET degraders (21 and 23–25) to induce BET degradation in the RS4;11 cell line, and the data are shown in Figure 4. All these four compounds can effectively and potently induce degradation of BET proteins in a dose-dependent manner after a 3 h treatment, and their potencies in reducing the levels of BRD2–4 proteins correlate well with their cellular potencies in inhibition of cell growth in the RS4;11 cell line. At concentrations as low as 10 nM, 25 effectively decreases the level of BRD3 and BRD4 proteins but is less potent and effective in decreasing in the level of BRD2 protein. Compound 24 is very effective in reducing the level of BRD2 and BRD4 at concentrations as low as 3 nM and the level of BRD3 at 1 nM. The two most potent BET degraders, 21 and 23, are highly effective in decreasing the level of BRD2 and BRD4 proteins at concentrations as low as 0.3 nM and decreasing the level of BRD3 at 0.1 nM with a 3 h treatment. Therefore, 21 and 23 are extremely potent degraders of BET proteins.
Figure 4.

Western blotting analysis of BRD2, BRD3, and BRD4 proteins in RS4;11 cells treated with compounds 21 and 23–25. RS4;11 cells were treated for 3 h with individual compounds at indicated concentrations, and proteins were probed by specific antibodies. GAPDH was used as the loading control.
We further investigated the potency of compound 23 in inducing BET protein degradation in the RS4;11 cells with a 24 h treatment, with compound 8 included as a control (Figure 5). Compound 23 is capable of effectively reducing the level of BRD2 and BRD3 proteins at concentrations as low as 0.1 nM and the level of BRD4 protein at a concentration of 0.03 nM. In comparison, the BET inhibitor 8 has no effect on the level of BRD2–4 proteins at all concentrations tested (30–1000 nM).
Figure 5.
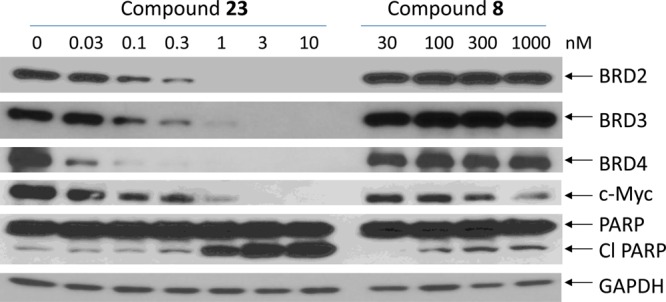
Western blotting analysis of BRD2, BRD3, and BRD4 proteins, as well as c-Myc and PARP in RS4;11 cells treated with BET degrader 23 and BET inhibitor 8. RS4;11 cells were treated for 24 h with individual compounds at indicated concentrations, and proteins were probed by specific antibodies. GAPDH was used as the loading control.
We examined the effects of the BET degrader 23 and the BET inhibitor 8 in the RS4;11 cell line on the level of c-Myc (Figure 5), a protein known to be down-regulated by both BET inhibitors and degraders.25,30 Consistent with its extremely high potency in inducing BET protein degradation, 23 effectively down-regulates the level of c-Myc at concentrations as low as 0.1 nM. In comparison, the BET inhibitor 8 can also effectively down-regulate the level of c-Myc but at concentrations of 300–1000 nM in the RS4;11 cell line. Hence, the BET degrader 23 is >1000 times more potent than the BET inhibitor 8 in down-regulation of c-Myc protein in the RS4;11 cell line.
The mechanism of action of BET protein degradation by 23 was further investigated (Figure 6). Addition of the BET inhibitor 8 effectively blocks the degradation of BRD2, BRD3, and BRD4 proteins induced by 23 (Figure 6A), further confirming that the degradation of BET proteins by 23 requires its binding to BET proteins. Similarly, addition of lenalidomide also effectively blocks the degradation induced by 23 for all three BET proteins (Figure 6B), clearly indicating that degradation of BET proteins by 23 is cereblon-dependent. The proteasome inhibitor MG-132 and the NEDD8-activating enzyme (NAE) inhibitor MLN4924 also completely block the degradation of BET proteins by 23 (Figure 6C), indicating that BET protein degradation by 23 depends upon proteasome and NAE. These mechanistic data constitute clear evidence that 23 is a bona fide and highly potent BET degrader.
Figure 6.
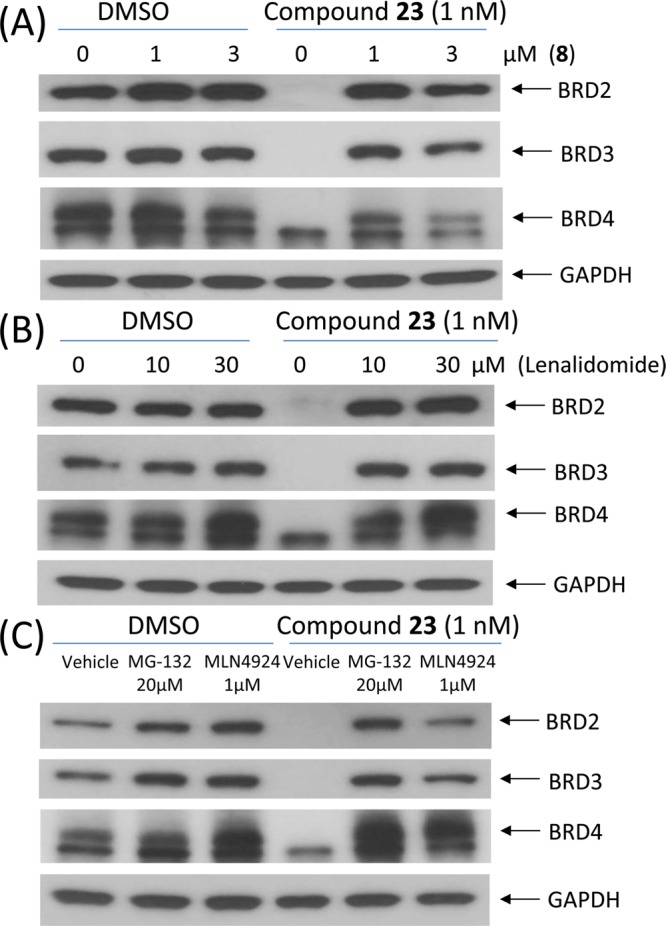
Western blotting analysis of BRD2, BRD3, and BRD4 proteins after a 2 h pretreatment with 8 (A), lenalidomide (B), or a proteasome inhibitor MG-132 or a NEDD8-activating enzyme (NAE) inhibitor MLN4924 (C), followed by a 3 h treatment with 23 at 1 nM in RS4;11 cells.
We employed flow cytometry analysis to investigate the ability of the BET degrader 23 and the BET inhibitor 8 to induce cell cycle arrest and apoptosis in the RS4;11 and MOLM-13 cell lines (Figure 7). Both compounds were found to effectively induce cell cycle arrest in a dose-dependent manner in both cell lines, but they have very different potencies. While the BET inhibitor 8 is effective at 30 nM in the RS4;11 cell line in inducing cell cycle arrest, the BET degrader 23 has a strong effect at concentrations as low as 0.3 nM. In the MOLM-13 cell line, 8 is effective at 30–100 nM in inducing cell cycle arrest while 23 is very effective at 1–3 nM. We also observed a sharp contrast in their ability to induce apoptosis. While the BET degrader 23 induces robust apoptosis in both RS4;11 and MOLM-13 cell lines at 3–10 nM concentrations with a 24 h treatment, the BET inhibitor 8 is ineffective in both cell lines at concentrations as high as 300 nM.
Figure 7.

Induction of cell cycle arrest and apoptosis by BET degrader 23 and BET inhibitor 8 in the RS4;11 and MOLM-13 cell lines by flow cytometry analysis.
We tested the antitumor activity of 23 in the RS4;11 xenograft model in mice, and the results are shown in Figure 8. Dosing of the mice bearing RS4;11 xenograft tumors with 5 mg/kg of 23 intravenously every other day, three times a week for 3 weeks, achieved rapid tumor regression with a maximum of >90% regression observed. There was no animal weight loss or other signs of toxicity in mice treated with compound 23, and the animal weight gained in the vehicle control group of mice was attributed largely to rapid tumor growth. Therefore, the in vivo data firmly establish that 23 has highly efficacious antitumor activity at a well-tolerated dose-schedule in mice.
Figure 8.

BET degrader 23 induces complete tumor regression in the xenograft model of human RS4;11 tumor cells with minimal body weight change.
To gain a better understanding of the strong antitumor activity of 23 and its mechanism of action in vivo, we performed a pharmacodynamics (PD) analysis in SCID mice bearing the RS4;11 xenograft tumors (Figure 9). In this experiment, mice bearing one or two RS4;11 xenograft tumors were administered a single dose of 23 at 5 mg/kg intravenously and were sacrificed at 1, 3, 6, and 24 h time-points after the treatment. Mice treated with vehicle control were sacrificed at the 6 h time point. Western blotting analysis was performed to probe the level of BRD2, BRD3, and BRD4 proteins, as well as c-Myc, caspase-3, and PARP proteins in the tumor tissue. Our PD data (Figure 9) clearly show that a single dose of 23 was highly effective, dramatically reducing the level of BRD2, BRD3, and BRD4 proteins in the RS4;11 tumor tissue, starting from 1 h with the effect persisting for >24 h. The level of c-Myc was strongly down-regulated, with the effect persisting for at least 6 h. Robust cleavage of PARP and caspase-3 was observed, starting from the 3 h time-point and with a peak effect at the 6 h time point, indicating strong apoptosis induction by 23. Therefore, our PD analysis clearly demonstrates that a single dose of 23 is sufficient to induce near complete degradation of BRD2, BRD3, and BRD4 proteins for >24 h, accompanied by robust cleavage of PARP and caspase-3, and strong down-regulation of c-Myc protein.
Figure 9.
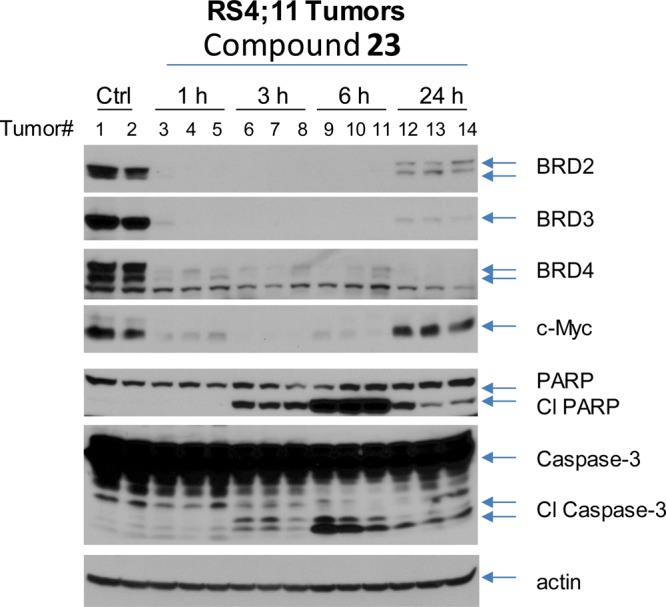
Pharmacodynamic analysis of compound 23 in RS4;11 xenograft tumor tissue. SCID mice bearing RS4;11 tumors were treated with a single intravenous dose of 23 at 5 mg/kg. Mice were sacrificed at 1, 3, 6, and 24 h time-points after administration with compound 23 or at 6 h with vehicle control, and tumors were harvested from mice for Western blotting analysis of BRD2, BRD3, BRD4, c-Myc, PARP and cleaved PARP (Cl PARP), and caspase-3 and cleaved caspase-3 (Cl caspase-3). Actin was used as the loading control. Two mice were used for each time-point with each mouse bearing either one or two tumors.
We analyzed concentrations of 23 in plasma and also in RS4;11 tumors in the same mice used in the PD experiment (Table 5). Our data showed that despite its relatively large size (MW = 798.8), 23 can effectively penetrate the RS4;11 xenograft tumor tissue and has a concentration of 166.3, 98.5, and 35.8 ng/g in the tumor at 1, 3, and 6 h time-points, respectively, exceeding the drug concentrations needed for effective BET degradation shown in our in vitro experiments (Figures 4 and 5). The drug concentration, however, was undetectable after 24 h. Together with the PD data, our PK data indicate that a single dose of 23 at 5 mg/kg achieves a sufficient exposure in the tumor tissue for effective induction of BET protein degradation for over 24 h.
Table 5. Analysis of Concentrations of Compound 23 in Plasma and RS4;11 Tumor Tissuea.
| concn
in plasma (ng/mL) |
concn
in tumor (ng/g) |
|||||||
|---|---|---|---|---|---|---|---|---|
| time-point (h) | individual mice | mean | individual tumor | mean | SD | |||
| 1 | 422 | 362 | 392 | 135.5 | 123 | 240.5 | 166.3 | 64.5 |
| 3 | 58 | 76.1 | 67.1 | 86 | 89 | 120.5 | 98.5 | 19.1 |
| 6 | 5.5 | 7.4 | 6.4 | 39.4 | 31.9 | 36.2 | 35.8 | 3.8 |
| 24 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | |
Mice bearing RS4;11 xenograft tumors were administered with a single dose of compound 23 at 5 mg/kg intravenously. Mice were sacrificed at 1, 3, 6, and 24 h time-points after administration with compound 23, and plasma and tumor tissue were collected for analysis. Two mice were used for each time-point with each mouse bearing either one or two tumors (three tumors for each time point).
We also evaluated the metabolism of compound 23 in mouse liver microsomes. The major metabolite was proposed to be mono- or dihydroxylated product occurring in the alkyl chain in the linker. In addition, cleavage of the C(sp3)–C(sp2) bond between the linker and cereblon binding ligand and N-deethylation of the pyrazole moiety are other alternative metabolic pathways (for detailed information, see Figure S5).
Chemistry
The synthesis of compounds 8 and 28 is outlined in Scheme 1. Briefly, 3-cyclopropyl-3-oxopropanenitrile (29) and ethylhydrazine oxalate (30) were heated in EtOH to generate 31. Compound 32 was prepared as described, and intermediate 33 was obtained from 32 in four steps.31 Coupling of 31 with 33 followed by hydrolysis of the methyl ester produced the key intermediate 34. Amidation of 34 with MeNH2 in the presence of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIPEA) at room temperature in dimethylformamide (DMF) gave the target compound 8. Compound 36 was formed by dehalogenation of 33 in the presence of Pd/C and H2, followed by hydrolysis. Acyl chloride formation followed by amidation of 36 generated 28.
Scheme 1. Compounds 8 and 28.

Reaction conditions: (a) EtOH, reflux; (b) NCCOOEt, 4 N HCl in dioxane; (c) 10% NaOH (aq), EtOH, reflux; (d) conc H2SO4, MeOH, reflux; (e) POCl3, reflux; (f) Pd2(dba)3, BINAP, K3PO4, toluene, reflux; (g) LiOH, THF, rt; (h) NH2Me, HATU, DIPEA, DMF, rt; (i) Pd/C, H2, DMF, 60 °C; (j) LiOH, THF, rt; (k) 2 drops of DMF, SOCl2, reflux; (l) methylamine hydrochloride, THF, rt.
The synthesis of compound 9 is shown in Scheme 2. Condensation of 3-hydroxyphthalic anhydride (37) with 3-aminopiperidine-2,6-dione hydrochloride (38) afforded the intermediate 39. Alkylation of 39 with tert-butyl bromoacetate generated 40, which was subjected to trifluoroacetic acid-promoted ester hydrolysis. Amide condensation and Boc-deprotection reactions afforded 41. Condensation of 41 with 34 under the same conditions described for the synthesis of 8 gave compound 9.
Scheme 2. Compound 9.

Reaction conditions: (a) TEA, toluene, reflux; (b) tert-butyl bromoacetate, KI, KHCO3, DMF, 60 °C; (c) TFA, rt; (d) N-Boc-1,4-butanediamine, HATU, DIPEA, DMF, rt; (e) TFA, DCM, rt; (f) HATU, DIPEA, DMF, rt.
The synthetic route to compound 10 is shown in Scheme 3. Compound 43 was obtained by treatment of 3-nitrophthalic anhydride (42) with benzyl bromide, followed by reduction of the nitro group with stannous chloride dihydrate and alkylation with tert-butyl bromoacetate. Removal of the benzyl groups of 43 followed by amide condensation resulted in 44. Compound 10 was prepared from 44 by the same procedure as was used for the synthesis of 9.
Scheme 3. Compound 10.

Reaction conditions: (a) TsOH·H2O, BnOH, 100 °C; BnBr, NaHCO3, DMF, 100 °C; (b) SnCl2·2H2O, EtOAc, 50 °C; (c) tert-butyl bromoacetate, DIPEA, DMF, 90 °C; (d) Pd/C, H2, EtOH, rt; (e) 38, pyridine, 110 °C; (f) TFA, rt; (g) N-Boc-1,4-butanediamine, HATU, DIPEA, DMF, rt; (h) TFA, DCM, rt; (i) HATU, DIPEA, DMF, rt.
As shown in Scheme 4, compound 11 was synthesized starting from the intermediate 39. Alkylation of the hydroxyl group of 39 with 7-bromo-1-heptanol gave 46. Compound 47 was obtained by conversion of the hydroxyl group to an amine group in 46 through three steps. Condensation of 47 with 34 using previously described methods afforded compound 11.
Scheme 4. Compound 11.

Reaction conditions: (a) 7-bromoheptan-1-ol, NaHCO3, KI, DMF, 80 °C; (b) MsCl, TEA, DCM; (c) NaN3, DMF, 80 °C; (d) PPh3, THF/H2O, rt; (e) HATU, DIPEA, DMF, rt.
Compounds 12–17 were synthesized according to the route shown in Scheme 5. Compound 49 was prepared from 48 by the strategy used in the preparation of 39. Compounds 50a–f were formed by substitution reaction of 49 with different length of mono-Boc protected alkyl diamines. Boc-deprotection with TFA in DCM afforded the intermediates 51a–f, which were subsequently condensed with 34 to generate compounds 12–17.
Scheme 5. Compounds 12–17.

Reaction conditions: (a) NaOAc, AcOH, reflux; (b) DIPEA, DMF, 80 °C; (c) TFA, DCM, rt; (d) HATU, DIPEA, DMF, rt.
The synthesis of compounds 18 and 20 is shown in Scheme 6. Sonogashira coupling of 53, which was prepared in a manner similar to that used to produce 49, with different terminal alkyne-containing linker substrates gave 54a,b. Reduction of the alkyne group in 54a,b resulted in 55a,b, whose Boc group was removed in TFA/DCM to form 56a,b. Compounds 18 and 20 were obtained by condensation of 56a,b with 34 using the same method as was used for 9.
Scheme 6. Compounds 18 and 20.

Reaction conditions: (a) NaOAc, AcOH; (b) tert-butyl pent-4-yn-1-ylcarbamate or tert-butyl hex-5-yn-1-ylcarbamate, Pd(PPh3)2Cl2, CuI, DMF/TEA, 80 °C; (c) Pd/C, H2, EtOH, rt; (d) TFA, DCM, rt; (e) HATU, DIPEA, DMF, rt.
Synthesis of compounds 19 and 22 is shown in Scheme 7. Bromination of 57 with N-bromosuccinimide (NBS) and benzoyl peroxide (BPO) gave 58, which was heated with 38 and triethylamine (TEA) in MeCN to generate 59. Compounds 19 and 22 were prepared starting from 53 and 59, respectively, in the same way as 53 was converted to 18 and 20.
Scheme 7. Compounds 19 and 22.

Reaction conditions: (a) NBS, BPO, benzene, reflux; (b) TEA, MeCN, reflux; (c) tert-butyl (2-(prop-2-yn-1-yloxy)ethyl)carbamate, Pd(PPh3)2Cl2, CuI, DMF/TEA, 80 °C; (d) Pd/C, H2, EtOH, rt; (e) TFA, DCM, rt; (f) HATU, DIPEA, DMF, rt.
Starting from 59, compounds 21, 23–25, and 27 were prepared using the procedure outlined in Scheme 8, which is similar to the procedure used in the preparation of 22 from 59.
Scheme 8. Compounds 21, 23–25, and 27.

Reaction conditions: (a) tert-butyl prop-2-yn-1-ylcarbamate or tert-butyl but-3-yn-1-ylcarbamate or tert-butyl pent-4-yn-1-ylcarbamate or tert-butyl hex-5-yn-1-ylcarbamate, Pd(PPh3)2Cl2, CuI, DMF/TEA, 80 °C; (b) Pd/C, H2, EtOH, rt; (c) TFA, DCM, rt; (d) HATU, DIPEA, DMF, rt.
Finally, the synthesis of compound 26 is outlined in Scheme 9. Methylation of 59 with MeI yielded 63. Compound 26 was prepared from 63 by a process similar to that used for the synthesis of 18 and 20 from 53.
Scheme 9. Compound 26.

Reaction conditions: (a) MeI, K2CO3, DMF, 60 °C; (b) tert-butyl pent-4-yn-1-ylcarbamate, Pd(PPh3)2Cl2, CuI, DMF/TEA, 80 °C; (c) Pd/C, H2, EtOH, rt; (d) TFA, DCM, rt; (e) HATU, DIPEA, DMF, rt.
Conclusion
Upon the basis of a novel, azacarbazole-containing potent BET inhibitor and thalidomide/lenalidomide, we have designed a new class of PROTAC small-molecule degraders of BET proteins. Through extensive optimization of the linker length and composition, we have obtained a number of highly potent small-molecule BET protein degraders as exemplified by compound 23. Compound 23 effectively induces degradation of the BET proteins BRD2, BRD3, and BRD4 at concentrations as low as 0.1–0.3 nM with a 3 h treatment in the RS4;11 acute leukemia cell line and is capable of inducing degradation of BRD4 protein at concentrations as low as 30 pM with a 24 h treatment. Compound 23 achieves an IC50 value of 51 pM and 2.3 nM in inhibition of cell growth in the RS4;11 and MOLM-13 acute leukemia cell lines, respectively. Our mechanistic investigation firmly establishes that compound 23 is a bona fide PROTAC BET degrader, whose activity requires that 23 binds to BET proteins and the cereblon/Cullin4A E3 ubiquitin ligase complex. Compound 23 effectively induces both cell cycle arrest and apoptosis in RS4;11 and MOLM-13 acute leukemia cell lines at subnanomolar to low nanomolar concentrations. Significantly, 23 achieves >90% tumor regression in the RS4;11 xenograft model in mice at a well-tolerated dose-schedule. Our PD analysis showed that a single, intravenous dose of 23 is capable of inducing profound degradation of BET proteins in the tumor tissue with effect persisting for >24 h, accompanied by robust cleavage of PARP and caspase-3 and strong down-regulation of c-Myc. In addition to its potent anticancer activity against acute leukemia cells, our recent study also showed that compound 23 is very potent and efficacious against triple-negative human breast cancer in vitro and in vivo.32 Collectively, our data demonstrate that 23 is a highly potent, efficacious, and promising BET degrader and warrants further evaluation as a potential new therapy for the treatment of human acute leukemia and other types of human cancer.
Experimental Section
Chemistry. General Experiment and Information
Unless otherwise noted, all purchased reagents were used as received without further purification. 1H NMR and 13C NMR spectra were recorded on a Bruker Advance 400 or 300 MHz spectrometer. 1H NMR spectra were reported in parts per million (ppm) downfield from tetramethylsilane (TMS). All 13C NMR spectra were reported in ppm and obtained with 1H decoupling. In reported spectral data, the format (δ) chemical shift (multiplicity, J values in Hz, integration) was used with the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. MS analyses were carried out with a Waters UPLC–mass spectrometer. The final compounds were all purified by C18 reverse phase preparative HPLC column with solvent A (0.1% TFA in H2O) and solvent B (0.1% TFA in MeCN) as eluents. The purity of all the final compounds was confirmed to be >95% purity by UPLC–MS or UPLC.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-N-methyl-9H-pyrimido[4,5-b]indole-2-carboxamide (8)
Ethylhydrazine oxalate (30, 41.0 g, 0.27 mmol, 1.2 equiv) in 200 mL of EtOH was added to a stirred solution of 3-cyclopropyl-3-oxopropanenitrile (29, 25.0 g, 0.23 mol, 1.0 equiv). The resulting solution was heated to reflux for 12 h. After cooling to room temperature, most of the solvent was evaporated. Workup was performed with EtOAc and saturated brine solution. The combined organic layer was dried over anhydrous Na2SO4. After filtration and concentration, the residue was purified by flash column chromatography with hexane/EtOAc to afford the desired compound 31 as a slightly yellow solid (22.6 g, 65% yield). 1H NMR (400 MHz, CDCl3) 5.07 (s, 1H), 3.82 (q, J = 7.2 Hz, 2H), 3.55 (s, 2H, NH2), 1.78–1.71 (m, 1H), 1.28 (t, J = 7.2 Hz, 3H), 0.80–0.75 (m, 2H), 0.57–0.54 (m, 2H). UPLC–MS calculated for C8H14N3 [M + H]+: 152.12, found 152.09.
Compound 32, Prepared in Three Steps Following a Published Method32
Compound 32 (13.16 g, 40 mmol, 1.0 equiv) and ethyl cyanoformate (19.76 mL, 0.2 mol, 5.0 equiv) were added to a round-bottom flask at room temperature. A 4.0 M hydrogen chloride solution in dioxane (90 mL) was added, and the reaction mixture was refluxed at 82 °C for 12 h. The reaction was then cooled to room temperature, and the solvents were removed on a rotary evaporator. To this crude mixture, 10% NaOH aqueous solution (120 mL) and EtOH (100 mL) were added, and the solution was heated to reflux for 6 h. The volatile components were then removed on a rotary evaporator, and the aqueous residue was acidified with 2 N aqueous HCl. The product was allowed to precipitate at 0 °C. Filtration of the mixture furnished pure 7-(3,5-dimethylisoxazol-4-yl)-4-hydroxy-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxylic acid as a yellow solid (11.34 g, 80% yield). UPLC–MS calculated for C17H15N4O5 [M + H]+: 355.10, found 355.45.
Concentrated sulfuric acid (15 mL) was added to a solution of 7-(3,5-dimethylisoxazol-4-yl)-4-hydroxy-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxylic acid (11.34 g, 32 mmol) in EtOH (300 mL) in a round-bottom flask. The mixture was heated to reflux for 12 h. The volatile components were removed on a rotary evaporator. Then 400 mL of EtOAc was added. The product was allowed to precipitate. Filtration of the mixture furnished pure methyl 7-(3,5-dimethylisoxazol-4-yl)-4-hydroxy-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxylate as a solid (10.61 g, 90% yield). 1H NMR (300 MHz, CD3OD) δ (ppm) 7.86 (s, 1H), 7.39 (s, 1H), 4.07 (s, 3H), 3.93 (s, 3H), 2.34 (s, 3H), 2.17 (s, 3H).
Methyl 7-(3,5-dimethylisoxazol-4-yl)-4-hydroxy-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxylate (10.61 g, 28.8 mmol) and POCl3 (200 mL) were stirred in a round-bottom flask at 90 °C for 12 h, then cooled to room temperature, and the volatile components were removed on a rotary evaporator. EtOAc (200 mL) was added, and the pH was adjusted to 8 using aqueous NaOH solution. Filtration of the mixture furnished compound 33 as a yellow solid (7.80 g, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.81 (s, 1H), 7.55 (s, 1H), 3.92 (s, 3H), 3.90 (s, 3H), 2.33 (s, 3H), 2.12 (s, 3H). UPLC–MS calculated for C18H16ClN4O4 [M + H]+: 387.09, found 387.18.
Pd2(dba)3 (183 mg, 0.2 mmol, 0.1 equiv), racemic BINAP (249 mg, 0.4 mmol, 0.2 equiv), and K3PO4 (1.70 g, 8.0 mmol, 4.0 equiv) were added sequentially to a solution of compound 33 (774 mg, 2.0 mmol, 1.0 equiv) in toluene (25 mL). The solution was purged and refilled with nitrogen three times before compound 31 (604 mg, 4.0 mmol, 2.0 equiv) was added. The solution was purged and refilled with nitrogen again. The resulting solution was heated to 110 °C and stirred for 12 h. After cooling to room temperature, the solution was filtered through Celite and concentrated. The residue was purified by flash column chromatography with DCM/MeOH to afford methyl 4-((3-cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxylate as a yellow solid (702 mg, 70% yield). 1H NMR (400 MHz, DMSO-d6) 12.15 (s, 1H), 12.05 (s, 1H), 8.06 (s, 1H), 7.56 (s, 1H), 7.22 (s, 1H), 3.99 (q, J = 7.2 Hz, 2H), 3.79 (s, 3H), 3.13 (s, 3H), 2.24 (s, 3H), 2.05 (s, 3H), 1.78–1.71 (m, 1H), 1.14 (t, J = 7.2 Hz, 3H), 0.80–0.75 (m, 2H), 0.57–0.54 (m, 2H).
LiOH (101 mg, 4.2 mmol, 3.0 equiv) was added to a solution of methyl 4-((3-cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxylate (702 mg, 1.4 mmol, 1.0 equiv) in THF (20 mL). The solution was stirred at room temperature for 12 h. After concentration, the residue was purified by preparative HPLC to afford the desired compound 34 as a yellow solid (545 mg, 80% yield).
HATU (27 mg, 0.07 mmol, 1.4 equiv), DIPEA (27 μL, 0.15 mmol, 3.0 equiv), and methylamine (2.0 M in THF 0.1 mL, 0.20 mmol, 4.0 equiv) were added sequentially to a stirred solution of compound 34 (24 mg, 0.05 mmol, 1.0 equiv) in DMF (2.0 mL). The solution was stirred at room temperature for 2 h. Preparative HPLC afforded the pure product (8) as a slightly yellow solid (17 mg, 68% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.17 (s, 1H), 9.27 (s, 1H), 8.21 (s, 1H), 7.43 (s, 1H), 7.34 (s, 1H), 5.92 (s, 1H), 3.96 (q, J = 7.2 Hz, 2H), 3.81 (s, 3H), 2.81 (d, J = 4.8 Hz, 3H), 2.30 (s, 3H), 2.09 (s, 3H), 1.92–1.85 (m, 1H), 1.32 (t, J = 7.2 Hz, 3H), 0.87–0.82 (m, 2H), 0.64–0.60 (m, 2H). UPLC–MS calculated for C26H29N8O3 [M + H]+: 501.24, found 501.22. Purity, 99.0%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(4-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)butyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (9)
In a round-bottom flask, 3-hydroxyphthalic anhydride 37 (1.0 g, 6.09 mmol, 1.0 equiv) and 3-aminoperidine-2,6-dione hydrochloride 38 (1.0 g, 6.09 mmol, 1.0 equiv) were mixed in toluene (50 mL). TEA (0.93 mL, 6.7 mmol, 1.1 equiv) was added. The resulting reaction mixture was heated to reflux for 12 h with Dean–Stark trap equipment. After cooling to ambient temperature, evaporation of most of the solvent afforded a crude product, which was purified by flash column chromatography with DCM/MeOH to obtain the desired product 39 as a slightly yellow solid (1.5 g, 90% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 11.16 (s, 1H), 11.08 (s, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.32 (d, J = 7.2 Hz, 1H), 7.25 (d, J = 8.4 Hz, 1H), 5.07 (dd, J = 12.8 Hz, J = 5.2 Hz, 1H), 2.93–2.84 (m, 1H), 2.61–2.46 (m, 1H), 2.05–2.01 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ (ppm) 173.28, 170.48, 167.50, 166.30, 155.99, 136.83, 133.60, 124.05, 114.82, 114.72, 49.11, 31.43, 22.51.
In a round-bottom flask, compound 39 (1.5 g, 5.5 mmol, 1.0 equiv) was dissolved in DMF (10 mL). KI (91 mg, 0.55 mmol, 0.1 equiv) and KHCO3 (826 mg, 8.25 mmol, 1.5 equiv) were added to the stirred solution. Then tert-butyl bromoacetate (0.98 mL, 6.6 mmol, 1.2 equiv) was added dropwise. The resulting mixture was stirred at 60 °C for 12 h. After normal workup with EtOAc and saturated brine, the combined organic layer was dried over Na2SO4. After filtration and evaporation, the residue was purified by flash column chromatography with DCM/MeOH to get the intermediate 40 as a white solid (1.7 g, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 11.13 (s, 1H), 7.80 (t, J = 8.0 Hz, 1H), 7.48 (d, J = 7.2 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 5.13 (dd, J = 12.8 Hz, J = 5.2 Hz, 1H), 4.97 (s, 2H), 2.97–2.85 (m, 1H), 2.65–2.52 (m, 2H), 2.14–2.03 (m, 1H), 1.43 (s, 9H).
The intermediate 40 was dissolved in TFA (8.0 mL). The reaction mixture was stirred at room temperature for 2 h. After evaporation of the solvent, the residue was freeze-dried on a lyophilizer to afford a white solid, which was used in the following steps without further purification. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 13.16 (s, 1H), 11.11 (s, 1H), 7.80 (t, J = 8.0 Hz, 1H), 7.48 (d, J = 7.2 Hz, 1H), 7.40 (d, J = 8.8 Hz, 1H), 5.11 (dd, J = 12.8 Hz, J = 5.2 Hz, 1H), 4.99 (s, 2H), 2.95–2.86 (m, 1H), 2.63–2.48 (m, 2H), 2.08–2.03 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ (ppm) 173.26, 170.38, 169.97, 167.21, 165.64, 155.61, 137.23, 133.73, 120.35, 116.80, 116.23, 65.48, 49.27, 31.42, 22.44. UPLC–MS calculated for C15H13N2O7 [M + H]+: 333.07, found 333.17.
In a round-bottom flask, the intermediate obtained from the previous step (99.7 mg, 0.3 mmol, 1.0 equiv) was dissolved in anhydrous DMF (2 mL). N-Boc-1,4-butanediamine (68 mg, 0.36 mmol, 1.2 equiv), HATU (137 mg, 0.36 mmol, 1.2 equiv), and DIPEA (157 μL, 0.9 mmol, 3.0 equiv) were added sequentially. The reaction mixture was stirred at room temperature for 2 h and then purified by HPLC to get the product as a slightly yellow solid (128 mg, 85% yield) which was dissolved in DCM (2.0 mL) and TFA (1.0 mL). After stirring for 1 h, the solvent was evaporated and the residue 41 was used as a stored solution (0.1 M in DMF) in the next step without further purification.
HATU (27 mg, 0.07 mmol, 1.4 equiv), DIPEA (27 μL, 0.15 mmol, 3.0 equiv), and the amine intermediate 41 (0.1 M in DMF 0.7 mL, 0.07 mmol, 1.4 equiv) were added sequentially to a stirred solution of compound 34 (24 mg, 0.05 mmol, 1.0 equiv) in DMF (2.0 mL). The solution was then stirred at room temperature for 2 h. Preparative HPLC purification afforded the pure product (9) as a slightly yellow solid (27 mg, 63% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.21 (s, 1H), 11.10 (s, 1H), 9.30 (s, 1H), 8.23 (t, J = 6.0 Hz, 1H), 8.00 (t, J = 6.0 Hz, 1H), 7.81 (t, J = 7.6 Hz, 1H), 7.47 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.34 (s, 1H), 5.93 (s, 1H), 5.11 (dd, J = 12.8 Hz, J = 5.2 Hz, 1H), 4.78 (s, 2H), 3.95 (q, J = 7.2 Hz, 2H), 3.82 (s, 3H), 3.30–3.25 (m, 2H), 3.22–3.16 (m, 2H), 2.89–2.85 (m, 1H), 2.60–2.45 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 2.05–2.01 (m, 1H), 1.90–1.85 (m, 1H), 1.51–1.49 (m, 4H), 1.31 (t, J = 7.2 Hz, 3H), 0.86–0.81 (m, 2H), 0.63–0.60 (m, 2H). UPLC–MS calculated for C44H46N11O9 [M + H]+: 872.35, found 872.37. Purity, >99.0%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(4-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)acetamido)butyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (10)
In a round-bottom flask, 3-nitrophthalic anhydride 42 (5.79 g, 30 mmol, 1.0 equiv) and p-toluenesulfonic acid monohydrate (571 mg, 3 mmol, 0.1 equiv) were mixed in benzyl alcohol (20 mL). The mixture was heated to 100 °C and stirred for 12 h. After cooling to room temperature, benzyl bromide (7.1 mL, 45 mmol, 1.5 equiv), KI (498 mg, 3 mmol, 0.1 equiv), KHCO3 (9.0 g, 90 mmol, 3.0 equiv), and DMF (25 mL) were added. The mixture was heated to 100 °C and stirred for 6 h. After cooling to room temperature, the solvent was evaporated and the mixture was then poured into H2O (300 mL). The solution was extracted with EtOAc. The combined organic layers were washed with brine and dried over anhydrous Na2SO4. After filtration and evaporation, the crude residue was purified by flash column chromatography with hexane/EtOAc to give the intermediate dibenzyl 3-nitrophthalate as a slightly yellow solid (9.4 g, 80% yield).
In a round-bottom flask, dibenzyl 3-nitrophthalate (9.4 g, 24 mmol, 1.0 equiv) was dissolved in EtOAc (100 mL). Then stannous chloride dihydrate (11.3 g, 50 mmol, 2.08 equiv) was added portionwise to the reaction mixture. The resulting reaction mixture was heated to 50 °C and stirred for 12 h. Then NaOH (aq) was added to the reaction mixture to quench the reaction. The reaction mixture was filtered through Celite and washed with EtOAc. The filtrate was extracted with EtOAc and brine. The combined organic layer was dried over anhydrous Na2SO4. After filtration and evaporation, the crude residue was purified by flash column chromatography with hexane/EtOAc to give dibenzyl 3-aminophthalate as a slightly yellow solid (7.8 g, 90% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.45–7.36 (m, 10H), 7.22 (t, J = 8.4 Hz, 1H), 6.96 (d, J = 7.2 Hz, 1H), 6.76 (d, J = 7.6 Hz, 1H), 5.36 (s, 2H, NH2), 5.26 (s, 2H), 5.10 (s, 2H).
In a round-bottom flask, dibenzyl 3-aminophthalate (2.0 g, 5.54 mmol, 1.0 equiv) and KI (100 mg, 0.56 mmol, 0.1 equiv) were combined with anhydrous DMF (10 mL). tert-Butyl bromoacetate (2.4 mL, 16.6 mmol, 3.0 equiv) and DIPEA (4.8 mL, 27.7 mmol, 5.0 equiv) were added to the reaction mixture which was then heated to 90 °C and stirred for 12 h. After cooling to room temperature, most of the solvent was evaporated and the residue was purified by column chromatography with hexane/EtOAc to give compound 43 as a slightly yellow solid (1.05 g, 40% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.40–7.25 (m, 11H), 6.87 (d, J = 7.2 Hz, 1H), 6.65 (d, J = 8.4 Hz, 1H), 5.20 (s, 2H), 4.95 (s, 2H), 3.88 (d, J = 4.8 Hz, 2H), 1.52 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm) 169.16, 168.86, 167.61, 148.44, 135.78, 135.66, 135.42, 132.82, 128.72, 128.62, 128.58, 128.40, 128.22, 128.16, 116.54, 113.89, 111.32, 82.38, 67.32, 66.97, 46.07, 28.27.
In a round-bottom flask, compound 43 (1.0 g, 2.1 mmol) was dissolved in EtOH (20 mL). 100 mg of Pd/C (10 wt %) was added under N2. The flask was purged and refilled with H2 three times. Then the reaction mixture was stirred at room temperature under 1 atm of H2. Once the starting material disappeared, as judged by TLC, the mixture was filtered through Celite and washed with EtOH. After evaporation of the solvent, 3-aminopiperidine-2,6-dione hydrochloride 38 (380 mg, 2.31 mmol, 1.1 equiv) and pyridine (20 mL) were added. The reaction mixture was heated to 110 °C and stirred overnight. After cooling to room temperature, the solvent was evaporated as much as possible and the residue was poured into H2O. After extraction three times with EtOAc, the combined organic layer was washed with brine and dried over anhydrous Na2SO4. After filtration and evaporation, the crude residue was purified by flash column chromatography with DCM/MeOH to give compound 44 as a yellow solid (325 mg, 40% yield).
TFA (2.0 mL) was added to compound 44, obtained as described above. The intermediate (2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)glycine was obtained by evaporation of the solvent without further purification. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.91 (s, 1H, COOH), 11.10 (s, 1H, NH), 7.59 (t, J = 7.6 Hz, 1H), 7.08 (d, J = 6.8 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.86 (t, J = 5.6 Hz, 1H, NH), 5.08 (dd, J = 13.2 Hz, J = 5.6 Hz, 1H), 4.12 (d, J = 5.2 Hz, 2H), 2.94–2.85 (m, 1H), 2.63–2.49 (m, 2H), 2.09–2.07 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ (ppm) 173.28, 171.90, 170.52, 169.26, 167.75, 146.28, 136.60, 132.48, 118.18, 111.54, 110.11, 60.22, 49.07, 31.46, 22.61. UPLC–MS calculated for C15H14N3O6 [M + 1]+: 332.09, found 332.00.
Following the procedure for the synthesis of compound 41, compound 45 (250 mg, 0.76 mmol) was synthesized from the intermediate (2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)glycine.
HATU (27 mg, 0.07 mmol, 1.4 equiv), DIPEA (27 μL, 0.15 mmol, 3.0 equiv), and compound 45 (28 mg, 0.07 mmol, 1.4 equiv) were added sequentially to a stirred solution of compound 34 (24 mg, 0.05 mmol, 1.0 equiv) in DMF (2.0 mL). The solution was stirred at room temperature for 2 h. Preparative HPLC purification afforded the pure product (10) as a slightly yellow solid (32 mg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.32 (s, 1H), 11.09 (s, 1H), 9.41 (s, 1H), 8.26 (t, J = 5.6 Hz, 1H), 8.19 (t, J = 5.6 Hz, 1H), 7.58 (t, J = 8.0 Hz, 1H), 7.48 (s, 1H), 7.41 (s, 1H), 7.03 (d, J = 6.8 Hz, 1H), 6.87 (d, J = 8.8 Hz, 1H), 5.94 (s, 1H), 5.07 (dd, J = 12.8 Hz, J = 5.6 Hz, 1H), 3.97 (q, J = 7.2 Hz, 2H), 3.94 (s, 2H), 3.82 (s, 3H), 3.29–3.26 (m, 2H), 3.15–3.12 (m, 2H), 2.93–2.84 (m, 1H), 2.61–2.45 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 2.05–2.01 (m, 1H), 1.92–1.85 (m, 1H), 1.51–1.45 (m, 4H), 1.31 (t, J = 7.2 Hz, 3H), 0.87–0.82 (m, 2H), 0.64–0.60 (m, 2H). UPLC–MS calculated for C44H47N12O8 [M + H]+: 871.36, found 871.39. Purity, 91.4%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)heptyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (11)
KI (33 mg, 0.2 mmol, 0.1 equiv), NaHCO3 (336 mg, 4.0 mmol, 2.0 equiv), and 7-bromo-1-heptanol (468 mg, 2.4 mmol, 1.2 equiv) were added sequentially to a solution of compound 39 (548 mg, 2.0 mmol, 1.0 equiv) in DMF (10 mL). The resulting solution was heated to 60 °C and stirred for 12 h. After cooling to room temperature, the solution was filtered through Celite and concentrated. The residue was purified by preparative HPLC to afford 46 as a colorless solid (589 mg, 76% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 11.10 (s, 1H), 7.80 (t, J = 7.80 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.44 (d, J = 7.2 Hz, 1H), 5.09 (dd, J = 12.8 Hz, J = 5.2 Hz, 1H), 4.20 (t, J = 6.4 Hz, 3H), 3.39 (t, J = 6.8 Hz, 2H), 2.90–2.86 (m, 1H), 2.62–2.50 (m, 2H), 2.08–2.02 (m, 1H), 1.78–1.72 (m, 2H), 1.48–1.29 (m, 8H); 13C NMR (100 MHz, DMSO-d6) δ (ppm) 173.24, 170.42, 167.32, 165.77, 156.50, 137.47, 133.72, 120.23, 116.69, 115.57, 69.27, 61.16, 49.22, 32.94, 31.43, 29.07, 28.89, 25.94, 25.82, 22.48. UPLC–MS calculated for C20H25N2O6 [M + H]+: 389.17, found 389.18.
MsCl (0.14 mL, 1.70 mmol, 1.2 equiv) and TEA (0.30 mL, 2.13 mmol, 1.5 equiv) were added sequentially to a solution of compound 46 (589 mg, 1.42 mmol, 1.0 equiv) in DCM (10 mL) at 0 °C. The resulting solution was stirred at 0 °C for 2 h. Then the solvent was evaporated to afford the crude residue. Sodium azide (277 mg, 4.25 mmol, 3.0 equiv) was added to the above residue in DMF (6 mL). The solution was stirred at 70 °C for 2 h and then filtered through Celite. The residue was purified by preparative HPLC to afford the desired product as a white solid (386 mg, 66% yield in two steps). 10% Pd/C (30 mg) was added to the above compound in EtOH (10 mL). The flask was purged and refilled with H2 three times. The solution was stirred at room temperature under H2 for 12 h. Filtration through Celite removed the Pd/C, and then the solution was concentrated and the residue was purified by preparative HPLC to afford the desired compound 47 as a colorless oil (72 mg, 20% yield).
HATU (27 mg, 0.07 mmol, 1.4 equiv), DIPEA (27 μL, 0.15 mmol, 3.0 equiv), and the amine 47 (27.1 mg, 0.07 mmol, 1.4 equiv) were added sequentially to a stirred solution of the intermediate 34 (24 mg, 0.05 mmol, 1.0 equiv) in DMF (2.0 mL). The solution was stirred at room temperature for 2 h. Preparative HPLC purification afforded the pure product 11 as a slightly yellow solid (19 mg, 45% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.30 (s, 1H), 11.08 (s, 1H), 9.28 (s, 1H), 8.16 (t, J = 5.6 Hz, 1H), 7.79 (t, J = 8.0 Hz, 1H), 7.52 (s, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.34 (s, 1H), 5.94 (s, 1H), 5.07 (dd, J = 12.8 Hz, J = 5.6 Hz, 1H), 4.21 (t, J = 6.4 Hz, 1H), 3.94 (q, J = 7.2 Hz, 2H), 3.83 (s, 3H), 3.29–3.24 (m, 2H), 2.91–2.82 (m, 1H), 2.59–2.44 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 2.05–1.99 (m, 1H), 1.91–1.84 (m, 1H), 1.81–1.74 (m, 2H), 1.53–1.47 (m, 4H), 1.41–1.30 (m, 7H), 0.86–0.81 (m, 2H), 0.64–0.60 (m, 2H). UPLC–MS calculated for C45H49N10O8 [M + H]+: 857.37, found 857.35. Purity, 98.2%.
General Procedure for Synthesis of Compounds 12–17
In a round-bottom flask, 3-fluorophthalic anhydride 48 (6.64 g, 40 mmol, 1.0 equiv), 3-aminopiperidine-2,6-dione hydrochloride 38 (6.58 g, 40 mmol, 1.0 equiv), and sodium acetate (3.94 g, 48 mmol, 1.2 equiv) were mixed in AcOH (120 mL). The resulting reaction mixture was heated to reflux at 140 °C for 12 h. After cooling to room temperature, most of the AcOH was evaporated and the residue was purified by flash column chromatography with DCM/MeOH to obtain compound 49 as a slightly yellow solid (9.7 g, 88% yield). UPLC–MS calculated for C13H10FN2O4 [M + H]+: 277.06, found 277.02. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 11.15 (s, 1H), 7.98–7.93 (m, 1H), 7.80–7.72 (m, 2H), 5.17 (dd, J = 13.2 Hz, J = 5.2 Hz, 1H), 2.95–2.86 (m, 1H), 2.64–2.47 (m, 2H), 2.10–2.06 (m, 1H).
Mono-Boc protected alkyl diamines (1.13 mmol, 1.1 equiv) of different lengths were added to a stirred solution of compound 49 (285 mg, 1.03 mmol, 1.0 equiv) in DMF (6.0 mL) and DIPEA (0.36 mL, 2.06 mmol, 2.0 equiv). The reaction mixture was stirred at 90 °C for 12 h. Then the mixture was cooled to room temperature, poured into H2O, and extracted twice with EtOAc. The combined organic layer was washed with brine, dried over anhydrous Na2SO4. After filtration and evaporation, the crude residue was purified by preparative HPLC to give the intermediate (50a–f) which was dissolved in DCM (4.0 mL) and TFA (2.0 mL). After stirring for 1 h, the solvent was evaporated to give the crude product 51a–f, which was used in the next step without further purification.
HATU (27 mg, 0.07 mmol, 1.4 equiv), DIPEA (27 μL, 0.15 mmol, 3.0 equiv), and the amine intermediate 51a–f (0.07 mmol, 1.4 equiv) were added sequentially to a stirred solution of compound 34 (24 mg, 0.05 mmol, 1.0 equiv) in DMF (2.0 mL). The solution was stirred at room temperature for 2 h. Preparative HPLC purification afforded the pure product 12–17.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)heptyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (12)
1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.30 (s, 1H), 11.08 (s, 1H), 9.31 (s, 1H), 8.17 (s, 1H), 7.58–7.54 (m, 2H), 7.39 (s, 1H), 7.08 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 6.8 Hz, 1H), 6.52 (s, 1H), 5.95 (s, 1H), 5.04 (dd, J = 12.8 Hz, J = 5.6 Hz, 1H), 3.95 (q, J = 7.2 Hz, 2H), 3.83 (s, 3H), 3.31–3.25 (m, 4H), 2.92–2.83 (m, 1H), 2.59–2.49 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 2.05–2.00 (m, 1H), 1.90–1.85 (m, 1H), 1.61–1.56 (m, 2H), 1.54–1.49 (m, 2H), 1.45–1.28 (m, 6H), 1.30 (t, J = 7.2 Hz, 3H), 0.87–0.82 (m, 2H), 0.65–0.61 (m, 2H). UPLC–MS calculated for C45H50N11O7 [M + H]+: 856.39, found 856.39. Purity, 97.9%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (13)
1H NMR (400 MHz, methanol-d4) δ 7.54 (s, 1H), 7.46 (s, 1H), 7.39–7.36 (m, 1H), 7.02 (d, J = 7.4 Hz, 1H), 6.93 (d, J = 7.1 Hz, 1H), 5.49 (s, 1H), 5.05–4.98 (m, 1H), 4.24–4.17 (q, J = 7.3 Hz, 2H), 3.98 (s, 2H), 3.89 (s, 3H), 3.37–3.22 (m, 6H), 3.00 (s, 2H), 2.88–2.84 (m, 2H), 2.68–2.64 (m, 2H), 2.34 (s, 3H), 2.17 (s, 3H), 1.98–1.91 (m, 1H), 1.73–1.68 (m, 2H), 1.48 (t, J = 7.3 Hz, 3H), 1.15–1.05 (m, 2H), 0.89–0.81 (m, 2H). UPLC–MS calculated for C44H47N11O7 [M + H]+: 842.37, found 842.36. UPLC analysis (10 min from 10% to 100% (MeCN/H2O containing 0.1% CF3CO2H)): retention time, 5.21 min; peak area, 98.5%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (14)
1H NMR (400 MHz, MeOD-d4) δ 7.51–7.42 (m, 2H), 7.38 (s, 1H), 6.98 (d, J = 8.9 Hz, 1H), 6.88 (d, J = 7.0 Hz, 1H), 6.08 (s, 1H), 4.93–4.97 (m, 1H), 4.15 (q, J = 7.0 Hz, 2H), 3.88 (s, 3H), 3.52–3.48 (m, 2H), 3.35 (s, 4H), 2.74–2.53 (m, 2H), 2.34 (s, 3H), 2.17 (s, 3H), 2.05–1.93 (m, 4H), 1.78–1.64 (m, 2H), 1.42 (t, J = 7.0 Hz, 3H), 1.40–1.34 (m, 1H), 1.08–0.96 (m, 2H), 0.80–0.73 (m, 2H). UPLC–MS calculated for C43H45N11O7 [M + H]+: 828.35, found 828.35. UPLC analysis (10 min from 10% to 100% (MeCN/H2O containing 0.1% CF3CO2H)): retention time, 4.80 min; peak area, 99.4%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (15)
1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.29 (s, 1H), 11.08 (s, 1H), 9.36 (s, 1H), 8.31 (t, J = 5.6 Hz, 1H), 7.56 (t, J = 7.2 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.01 (d, J = 6.8 Hz, 1H), 6.58 (s, 1H), 5.96 (s, 1H), 5.05 (dd, J = 12.8 Hz, J = 5.2 Hz, 1H), 3.97 (q, J = 7.2 Hz, 2H), 3.83 (s, 3H), 3.40–3.30 (m, 4H), 2.92–2.83 (m, 1H), 2.60–2.49 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 2.07–2.01 (m, 1H), 1.91–1.84 (m, 1H), 1.65–1.55 (m, 4H), 1.31 (t, J = 7.2 Hz, 3H), 0.86–0.81 (m, 2H), 0.64–0.60 (m, 2H). UPLC–MS calculated for C42H44N11O7 [M + H]+: 814.34, found 814.37. Purity, 89.9%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (16)
1H NMR (400 MHz, MeOD-d4) δ 7.55–7.49 (m, 2H), 7.39 (s, 1H), 7.04 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 7.1 Hz, 1H), 4.89–4.81 (m, 1H), 4.18 (q, J = 7.3 Hz, 2H), 3.89 (s, 3H), 3.70–3.56 (m, 2H), 3.52–3.42 (m, 2H), 3.35 (d, J = 1.4 Hz, 2H), 2.34 (s, 3H), 2.17 (s, 3H), 2.05–2.01 (m, 4H), 1.88–1.79 (m, 1H), 1.44 (t, J = 7.3 Hz, 3H), 1.09–1.01 (m, 2H), 0.82–0.73 (m, 2H). UPLC–MS calculated for C41H41N11O7 [M + H]+: 800.32, found 800.23. UPLC analysis (10 min from 10% to 100% (MeCN/H2O containing 0.1% CF3CO2H)): retention time, 4.43 min; peak area, 98.0%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (17)
1H NMR (400 MHz, methanol-d4) δ 7.55–7.48 (m, 1H), 7.46 (s, 1H), 7.33–7.27 (m, 1H), 7.20 (d, J = 8.6 Hz, 1H), 7.01 (d, J = 7.1 Hz, 1H), 6.16 (s, 1H), 5.05–4.95 (m, 1H), 4.20 (q, J = 7.2 Hz, 2H), 3.87 (s, 3H), 3.72–3.70 (m, 2H), 3.66–3.62 (m, 2H), 2.83–2.73 (m, 1H), 2.71–2.58 (m, 2H), 2.33 (s, 3H), 2.16 (s, 3H), 1.99–1.97 (m, 1H), 1.46 (t, J = 7.2 Hz, 3H), 1.11–1.02 (m, 2H), 0.83–0.79 (m, 2H). UPLC–MS (ESI+): m/z calculated for C40H39N11O7 [M + H]+: 786.30, found 786.18. UPLC analysis (10 min from 10% to 100% (MeCN/H2O containing 0.1% CF3CO2H)): retention time, 4.37 min; peak area, 97.4%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(6-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)hexyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (18)
In a round-bottom flask, 3-bromophthalic anhydride 52 (2.27 g, 10.0 mmol, 1.0 equiv), 3-aminopiperidine-2,6-dione hydrochloride 38 (1.81 g, 11 mmol, 1.1 equiv), and sodium acetate (0.98 g, 12 mmol, 1.2 equiv) were mixed in acetic acid (30 mL). The resulting reaction mixture was heated to reflux at 140 °C for 12 h. After cooling to room temperature, most of the AcOH was evaporated and the residue was purified by flash column chromatography with DCM/MeOH to give compound 53 as a purple solid (2.70 g, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.94 (d, J = 7.2 Hz, 1H), 7.78 (t, J = 7.6 Hz, 1H), 5.18 (dd, J = 12.8 Hz, J = 5.6 Hz, 1H), 2.95–2.86 (m, 1H), 2.65–2.48 (m, 2H), 2.11–2.05 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 173.21, 170.18, 166.07, 165.65, 139.66, 136.73, 134.15, 129.20, 123.31, 118.11, 49.65, 31.39, 22.30. UPLC–MS calculated for C13H10BrN2O4 [M + H]+: 336.98, found 336.95.
In a round-bottom flask, compound 53 (674 mg, 2.0 mmol, 1.0 equiv) and tert-butyl hex-5-yn-1-ylcarbamate (934 mg, 4.0 mmol, 2.0 equiv) were added to a solution of CuI (76 mg, 0.4 mmol, 0.2 equiv) and Pd(PPh3)2Cl2 (140 mg, 0.2 mmol, 0.1 equiv) in DMF (10 mL). The solution was purged and refilled with nitrogen three times. Then trimethylamine (5.0 mL) was added. The solution was purged and refilled with nitrogen again. The mixture was stirred at 70 °C for 3 h under Ar. The solution was cooled to room temperature and filtered through Celite. The residue was purified by flash column chromatography to yield compound 54b as a slightly yellow solid (653 mg, 72% yield). UPLC–MS calculated for C24H27N3NaO6 [M + Na]+ = 476.18, found 476.18.
10% Pd/C (60 mg) was added to a solution of compound 54b (653 mg, 1.44 mmol, 1.0 equiv) in EtOH (10 mL) under N2. The solution was purged and refilled with hydrogen three times. The solution was stirred at room temperature for 12 h. After filtration, the solvent was evaporated to give the crude residue 55b, which was dissolved in DCM (4.0 mL) and TFA (2.0 mL). The reaction mixture was stirred at room temperature for 1 h. Preparative HPLC purification afforded compound 56b as a colorless oil (411 mg, 80% yield in two steps).
HATU (27 mg, 0.07 mmol, 1.4 equiv), DIPEA (27 μL, 0.15 mmol, 3.0 equiv), and 56b (25 mg, 0.07 mmol, 1.4 equiv) were added sequentially to a stirred solution of compound 34 (24 mg, 0.05 mmol, 1.0 equiv) in DMF (2.0 mL). The solution was stirred at room temperature for 2 h. Preparative HPLC purification afforded the pure product 18 as a slightly yellow solid (29 mg, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.25 (s, 1H), 11.10 (s, 1H), 9.29 (s, 1H), 8.15 (t, J = 5.6 Hz, 1H), 7.78–7.69 (m, 3H), 7.58 (s, 1H), 7.37 (s, 1H), 5.94 (s, 1H), 5.12 (dd, J = 13.2 Hz, J = 5.2 Hz, 1H), 3.94 (q, J = 7.2 Hz, 2H), 3.83 (s, 3H), 3.28–3.23 (m, 2H), 3.05 (t, J = 8.0 Hz, 2H), 2.93–2.83 (m, 1H), 2.61–2.49 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 2.07–2.03 (m, 1H), 1.89–1.82 (m, 1H), 1.66–1.60 (m, 2H), 1.53–1.47 (m, 2H), 1.36–1.27 (m, 7H), 0.84–0.79 (m, 2H), 0.63–0.59 (m, 2H). UPLC–MS calculated for C44H47N10O7 [M + H]+: 827.36, found 827.28. Purity, 98.7%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(2-(3-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)propoxy)ethyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (19)
Following the procedure for the synthesis of compound 18 from the intermediate 53, compound 19 was obtained with tert-butyl (2-(prop-2-yn-1-yloxy)ethyl)carbamate as the linker instead of t-butyl hex-5-yn-1-ylcarbamate. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.24 (s, 1H), 11.10 (s, 1H), 9.27 (s, 1H), 8.23 (t, J = 5.6 Hz, 1H), 7.73–7.68 (m, 3H), 7.57 (s, 1H), 7.35 (s, 1H), 5.91 (s, 1H), 5.11 (dd, J = 12.8 Hz, J = 5.2 Hz, 1H), 3.94 (q, J = 7.2 Hz, 2H), 3.83 (s, 3H), 3.50–3.43 (m, 6H), 3.09 (t, J = 7.6 Hz, 2H), 2.92–2.82 (m, 1H), 2.61–2.46 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 2.05–2.02 (m, 2H), 1.93–1.82 (m, 3H), 1.29 (t, J = 7.2 Hz, 3H), 0.84–0.79 (m, 2H), 0.63–0.59 (m, 2H). UPLC–MS calculated for C43H45N10O8 [M + H]+: 829.34, found 829.41. Purity, >99.0%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(5-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)pentyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (20)
Following the procedure used in the synthesis of compound 18 from the intermediate 53, compound 20 was obtained with tert-butyl pent-4-yn-1-ylcarbamate as the linker instead of tert-butyl hex-5-yn-1-ylcarbamate. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.22 (s, 1H), 11.10 (s, 1H), 9.29 (s, 1H), 8.19 (t, J = 5.6 Hz, 1H), 7.76–7.71 (m, 3H), 7.54 (s, 1H), 7.35 (s, 1H), 5.93 (s, 1H), 5.12 (dd, J = 13.2 Hz, J = 5.2 Hz, 1H), 3.95 (q, J = 7.2 Hz, 2H), 3.82 (s, 3H), 3.29–3.24 (m, 2H), 3.06 (t, J = 8.0 Hz, 2H), 2.92–2.83 (m, 1H), 2.61–2.55 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 2.05–2.00 (m, 1H), 1.90–1.85 (m, 1H), 1.67–1.61 (m, 2H), 1.57–1.52 (m, 2H), 1.41–1.35 (m, 2H), 1.31 (t, J = 7.2 Hz, 3H), 0.85–0.81 (m, 2H), 0.63–0.59 (m, 2H). UPLC–MS calculated for C43H44N10O7 [M + H]+: 812.34, found 813.33. Purity, 95.3%.
General Procedure for Synthesis of Compounds 21–25 and 27
NBS (17.09 g, 96 mmol, 1.2 equiv) and BPO (1.938 g, 8.0 mmol, 0.1 equiv) were added to a stirred solution of methyl 3-bromo-2-methylbenzoate 57 (18.33 g, 80 mmol, 1.0 equiv) in benzene (150 mL). The solution was heated at reflux for 6 h. After cooling to room temperature, the solvent was evaporated and the residue was purified by flash column chromatography with hexane/EtOAc to afford the intermediate 58 (22.17 g, 90% yield) as a slightly yellow solid.
Compound 38 (14.48 g, 88 mmol, 1.22 equiv) and TEA (13.38 mL, 96 mmol, 1.33 equiv) were added to a stirred solution of compound 58 (22.17 g, 72 mmol, 1.0 equiv) in MeCN (150 mL). The solution was stirred at 80 °C for 12 h and then cooled to room temperature, and most of the solvent was evaporated. EtOAc (200 mL) and H2O (200 mL) were added to the residue and the solution was filtered to afford the crude product as a purple solid 59 (17.4 g, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 11.00 (s, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 7.6 Hz, 1H), 7.51 (t, J = 8.0 Hz, 1H), 5.15 (dd, J = 13.2 Hz, J = 5.2 Hz, 1H), 4.42 (d, J = 17.6 Hz, 1H), 4.27 (d, J = 17.6 Hz, 1H), 2.96–2.87 (m, 1H), 2.62–2.42 (m, 2H), 2.07–2.00 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ (ppm) 173.30, 171.32, 167.69, 142.58, 135.10, 134.41, 130.98, 122.96, 117.79, 52.22, 48.46, 31.66, 22.74. UPLC–MS calculated for C13H12BrN2O3 [M + H]+: 323.00, found 322.90.
Following the procedures used to prepare compound 18 from the intermediate 53, compounds 21–25 and 27 with different alkynyl chains were obtained as indicated in Schemes 8 and 9.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(6-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)hexyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (21)
1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.25 (s, 1H), 10.97 (s, 1H), 9.31 (s, 1H), 8.16 (t, J = 5.2 Hz, 1H), 7.60–7.21 (m, 5H), 5.95 (s, 1H), 5.13 (dd, J = 13.2 Hz, J = 4.8 Hz, 1H), 4.48 (d, J = 17.2 Hz, 1H), 4.32 (d, J = 17.2 Hz, 1H), 3.95 (q, J = 7.2 Hz, 2H), 3.83 (s, 3H), 3.29–3.24 (m, 2H), 2.94–2.87 (m, 1H), 2.73–2.45 (m, 4H), 2.30 (s, 3H), 2.10 (s, 3H), 2.03–1.98 (m, 1H), 1.89–1.82 (m, 1H), 1.66–1.62 (m, 2H), 1.52–1.48 (m, 2H), 1.36–1.27 (m, 7H), 0.84–0.79 (m, 2H), 0.63–0.59 (m, 2H). UPLC–MS calculated for C44H49N10O6 [M + H]+: 813.38, found 813.29. Purity, 95.3%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(2-(3-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)propoxy)ethyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (22)
1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.23 (s, 1H), 10.96 (s, 1H), 9.27 (s, 1H), 8.23 (t, J = 5.2 Hz, 1H), 7.57–7.41 (m, 4H), 7.35 (s, 1H), 5.90 (s, 1H), 5.10 (dd, J = 13.2 Hz, J = 5.2 Hz, 1H), 4.47 (d, J = 17.2 Hz, 1H), 4.32 (d, J = 17.2 Hz, 1H), 3.93 (q, J = 7.2 Hz, 2H), 3.83 (s, 3H), 3.50–3.40 (m, 6H), 2.88–2.84 (m, 1H), 2.71 (t, J = 7.6 Hz, 2H), 2.67–2.43 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 1.98–1.81 (m, 4H), 1.29 (t, J = 7.2 Hz, 3H), 0.83–0.78 (m, 2H), 0.62–0.58 (m, 2H). UPLC–MS calculated for C43H47N10O7 [M + H]+: 815.36, found 815.26. Purity, >99.0%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (23)
See Figures S2–S4 in Supporting Information. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.21 (s, 1H), 10.98 (s, 1H), 9.29 (s, 1H), 8.18 (t, J = 5.2 Hz, 1H), 7.57 (d, J = 6.8 Hz, 2H), 7.49–7.43 (m, 2H), 7.35 (s, 1H), 5.93 (s, 1H), 5.13 (dd, J = 13.2 Hz, J = 5.2 Hz, 1H), 4.49 (d, J = 17.2 Hz, 1H), 4.33 (d, J = 17.2 Hz, 1H), 3.95 (q, J = 7.2 Hz, 2H), 3.83 (s, 3H), 3.29–3.24 (m, 2H), 2.96–2.87 (m, 1H), 2.70–2.44 (m, 4H), 2.30 (s, 3H), 2.10 (s, 3H), 2.03–2.00 (m, 1H), 1.88–1.85 (m, 1H), 1.67–1.65 (m, 2H), 1.58–1.54 (m, 2H), 1.39–1.24 (m, 5H), 0.84–0.82 (m, 2H), 0.63–0.59 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ (ppm) 172.87, 171.04, 168.37, 165.48, 162.96, 159.17, 156.48, 155.10, 154.07, 152.42, 152.20, 140.49, 137.46, 136.47, 131.88, 131.56, 131.43, 128.26, 120.60, 119.04, 117.43, 113.85, 113.41, 104.51, 98.35, 97.22, 56.01, 51.55, 46.24, 42.41, 31.16, 29.00, 28.80, 26.30, 22.51, 14.86, 11.28, 10.34, 9.58, 7.59. UPLC–MS calculated for C43H47N10O6 [M + H]+: 799.37, found 799.19. Purity, 98.9%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)butyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (24)
1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.22 (s, 1H), 10.96 (s, 1H), 9.29 (s, 1H), 8.23 (t, J = 6.0 Hz, 1H), 7.58–7.44 (m, 4H), 7.36 (s, 1H), 5.92 (s, 1H), 5.11 (dd, J = 13.2 Hz, J = 5.2 Hz, 1H), 4.47 (d, J = 17.2 Hz, 1H), 4.32 (d, J = 17.2 Hz, 1H), 3.94 (q, J = 7.2 Hz, 2H), 3.83 (s, 3H), 3.35–3.32 (m, 2H), 2.94–2.84 (m, 1H), 2.69 (t, J = 7.6 Hz, 2H), 2.56–2.35 (m, 2H), 2.30 (s, 3H), 2.10 (s, 3H), 2.01–1.96 (m, 1H), 1.87–1.80 (m, 1H), 1.67–1.54 (m, 4H), 1.29 (t, J = 7.2 Hz, 3H), 0.84–0.79 (m, 2H), 0.62–0.58 (m, 2H). UPLC–MS calculated for C42H45N10O6 [M + H]+: 785.35, found 785.32. Purity, 94.2%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-N-(3-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)propyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (25)
1H NMR (400 MHz, MeOD-d4) δ (ppm) 7.59 (d, J = 7.2 Hz, 1H), 7.50–7.43 (m, 3H), 7.25 (s, 1H), 5.98 (s, 1H), 5.09 (dd, J = 13.2 Hz, J = 4.8 Hz, 1H), 4.48 (d, J = 17.2 Hz, 1H), 4.42 (d, J = 17.2 Hz, 1H), 4.13 (q, J = 6.8 Hz, 2H), 3.86 (s, 3H), 3.50–3.40 (m, 2H), 2.83–2.61 (m, 4H), 2.45–2.25 (m, 4H), 2.25–1.91 (m, 7H), 1.43 (t, J = 7.2 Hz, 3H), 0.92–0.89 (m, 2H), 0.68–0.66 (m, 2H). UPLC–MS calculated for C41H43N10O6 [M + H]+: 771.34, found 771.35. Purity, >99.0%.
4-((3-Cyclopropyl-1-ethyl-1H-pyrazol-5-yl)amino)-7-(3,5-dimethylisoxazol-4-yl)-6-methoxy-N-(5-(2-(1-methyl-2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-9H-pyrimido[4,5-b]indole-2-carboxamide (26)
K2CO3 (332 mg, 2.4 mmol, 1.2 equiv) and MeI (0.19 mL, 3.0 mmol, 1.5 equiv) were added to a solution of compound 59 (646 mg, 2.0 mmol, 1.0 equiv) in DMF (5.0 mL). The solution was stirred at 60 °C for 3 h. Preparative HPLC purification afforded the desired compound 63 as a white solid (485 mg, 72% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.86 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 7.2 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 5.22 (dd, J = 13.2 Hz, J = 5.2 Hz, 1H), 4.41 (d, J = 17.6 Hz, 1H), 4.26 (d, J = 17.6 Hz, 1H), 3.06–2.95 (m, 4H), 2.79–2.73 (m, 1H), 2.52–2.41 (m, 1H), 2.07–2.01 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ (ppm) 172.33, 170.94, 167.73, 142.60, 135.11, 134.38, 130.96, 122.99, 117.79, 52.69, 48.41, 31.80, 27.06, 22.05. UPLC–MS calculated for C14H14BrN2O3 [M + H]+: 337.02, found 336.98.
Following the procedures used for the conversion of the intermediates 53–18, compound 26 was obtained from the intermediate 63. 1H NMR (400 MHz, MeOD-d4) δ (ppm) 7.56–7.54 (m, 2H), 7.43–7.36 (m, 3H), 6.20 (s, 1H), 5.12 (dd, J = 13.6 Hz, J = 5.2 Hz, 1H), 4.47 (d, J = 17.2 Hz, 1H), 4.41 (d, J = 17.2 Hz, 1H), 4.22 (q, J = 6.8 Hz, 2H), 3.89 (s, 3H), 3.50–3.40 (m, 2H), 3.07 (s, 3H), 2.90–2.81 (m, 2H), 2.72–2.61 (m, 2H), 2.51–2.40 (m, 1H), 2.32 (s, 3H), 2.16 (s, 3H), 2.13–2.09 (m, 1H), 2.03–1.99 (m, 1H), 1.70–1.66 (m, 4H), 1.48–1.44 (m, 5H), 1.10–1.05 (m, 2H), 0.83–0.81 (m, 2H). UPLC–MS calculated for C44H49N10O6 [M + H]+: 813.38, found 813.34. Purity, >99.0%.
7-(3,5-Dimethylisoxazol-4-yl)-N-(5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-6-methoxy-9H-pyrimido[4,5-b]indole-2-carboxamide (27)
1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.57 (s, 1H), 10.97 (s, 1H), 9.60 (s, 1H), 9.01 (t, J = 6.0 Hz, 1H), 8.12 (s, 1H), 7.56–7.54 (m, 2H), 7.52 (s, 1H), 7.48–7.41 (m, 2H), 5.13 (dd, J = 13.2 Hz, J = 5.2 Hz, 1H), 4.48 (d, J = 17.2 Hz, 1H), 4.32 (d, J = 17.2 Hz, 1H), 3.91 (s, 3H), 3.38–3.33 (m, 2H), 2.96–2.87 (m, 1H), 2.68–2.57 (m, 3H), 2.51–2.42 (m, 1H), 2.32 (s, 3H), 2.12 (s, 3H), 2.07–1.99 (m, 1H), 1.69–1.60 (m, 4H), 1.44–1.36 (m, 2H). UPLC–MS calculated for C35H36N7O6 [M + H]+: 650.27, found 650.26. Purity, >99.0%.
7-(3,5-Dimethylisoxazol-4-yl)-6-methoxy-N-methyl-9H-pyrimido[4,5-b]indole-2-carboxamide (28)
10% Pd/C (20 mg) was added to a stirred solution of compound 33 (200 mg, 0.52 mmol, 1.0 equiv) in 10 mL of DMF under N2. The flask was purged and refilled with H2 three times. The solution was then stirred at 60 °C for 12 h. After cooling to room temperature, the solution was filtered through Celite. After concentration, the residue crude intermediate 35 was suspended in THF (30 mL), and LiOH (38 mg, 1.55 mmol, 3.0 equiv) was added. The resulting solution was stirred at room temperature for 12 h. After concentration, the residue was purified by preparative HPLC to afford the compound 36 as a yellow solid (52 mg, 30% yield for two steps). UPLC–MS calculated for C17H15N4O4 [M + H]+: 339.11, found 339.08.
Compound 36 (150 mg, 0.44 mmol) was suspended in thionyl chloride (0.64 mL, 8.8 mmol), and two drops of dry DMF were added. The mixture was stirred at reflux for 2 h. Excess thionyl chloride was removed in vacuo, and the resulting solid was dried. The resulting acyl chloride was used without further purification. Dry THF (15 mL) was added to the acyl chloride. The mixture was cooled to 0 °C, and subsequently, methylamine hydrochloride (45 mg, 0.67 mmol) was added. After stirring at room temperature for 2 h, the mixture was evaporated and purified by preparative HPLC to get 61.8 mg of compound 28 with a yield of 40%. ESI-MS calculated for C18H17N5O3 [M + H]+ = 52.13. Obtained: 352.17. 1H NMR (400 MHz, MeOD-d4) δ 9.55 (s, 1H), 8.03 (s, 1H), 7.54 (s, 1H), 3.96 (s, 3H), 2.35 (s, 3H), 2.18 (s, 3H), 1.31 (t, J = 7.3 Hz, 3H). UPLC analysis (10 min from 10% to 100% (MeCN/H2O containing 0.1% CF3CO2H)): retention time, 2.70 min; peak area, 98.12%.
Determination of Biochemical Binding Affinities to BET Proteins
Binding affinities of BET inhibitors to BRD2 (BD1 and BD2 proteins), BRD3 (BD1 and BD2 proteins), BRD4 (BD1 and BD2 proteins) were determined using our established fluorescence-polarization binding assays as described previously.27
Cell Growth Inhibition, Apoptosis Analysis, and Western Blotting
The human acute leukemia RS4;11 cell line (CRL-1873) was purchased from the American Type Culture Collection, and the human acute leukemia MOLM-13 cell line was purchased from the DSMZ German cell bank (ACC554). In all experiments, RS4;11 and MOLM-13 human leukemia cells were used within three months of thawing fresh vials. RS4;11 and MOLM-13 cells were cultured in RPMI 1640 media supplemented with 10% FBS and 1% penicillin–streptomycin at 37 °C in a humidified atmosphere containing 5% CO2 in air.
In cell growth experiments, cells were seeded in 96-well cell culture plates at a density of 10000−20000 cells/well in 100 μL of culture medium. Each compound tested was serially diluted in the appropriate medium, and 100 μL of the diluted solution containing the tested compound was added to the appropriate wells of the cell plate. After addition of the tested compound, the cells were incubated for 4 days at 37 °C in an atmosphere of 5% CO2. Cell growth was evaluated by a lactate dehydrogenase-based WST-8 assay (Dojindo Molecular Technologies) using a Tecan Infinite M1000 multimode microplate reader (Tecan, Morrisville, NC). The WST-8 reagent was added to the plate, incubated for at least 1 h, and read at 450 nm. The readings were normalized to the DMSO-treated cells, and the IC50 was calculated by nonlinear regression analysis using GraphPad Prism 6 software.
Flow cytometry was used to analyze effects of the drugs on cell cycle (propidium iodide staining) and apoptosis (annexin V and propidium iodide staining). Cell were treated with compounds at the indicated concentrations for 24 h, collected, stained, and analyzed by flow cytometry.
For Western blot analysis, 2 × 106 cells/well were treated with compounds at the indicated concentrations for various times. Cells were collected and lysed in RIPA buffer containing protease inhibitors. An amount of 20 μg of lysate was run in each lane of a PAGE–SDS and blotted into PVDF membranes. Antibodies for immunoblotting were BRD2, BRD3, and BRD4 purchased from Bethyl Laboratories (Montgomery, TX, USA), c-Myc from Cell Signaling Technology (Danvers, MA, USA), and GAPDH from Santa Cruz Biotechnologies (Dallas, TX, USA).
Efficacy and Pharmacodynamics Studies in the RS4;11 Xenograft Model in Mice
All animal experiments were done under the guidelines of the University of Michigan Committee for Use and Care of Animals and using an approved animal protocol (PRO00005315, PI, Shaomeng Wang).
To develop xenograft tumors, 5 × 106 RS4;11 cells with 50% Matrigel were injected subcutaneously on the dorsal side of severe combined immunodeficient (SCID) mice, obtained from Charles River, one tumor per mouse. When tumors reached ∼100 mm3, mice were randomly assigned to treatment and vehicle control groups. Animals were monitored daily for any signs of toxicity and weighed 2–3 times per week during the treatment and weighed at least weekly after the treatment ended. Tumor size was measured 2–3 times per week by electronic calipers during the treatment period and at least weekly after the treatment was ended. Tumor volume was calculated as V = LW2/2, where L is the length and W is the width of the tumor.
For pharmacodynamic analysis, resected RS4;11 xenograft tumor tissues were ground into powder in liquid nitrogen and lysed in lysis buffer (1% CHAPS, 150 mM NaCl, 20 mM Tris-HCl, 1 mM. EDTA, 1 mM EGTA, and COMPLETE proteinase inhibitor (Roche)). Whole tumor lysates were separated on 4–20% Novex gels. The separated proteins were transferred to a polyvinylidene difluoride membrane for immunoblotting. The following antibodies were used: rabbit polyclonal antibodies for BRD2 (A302-583A), BRD3 (A302-368A), and BRD4 (A301-985A100) from Bethyl Laboratories; c-Myc (D84C12), PARP (46D11), and caspase-3 (8G10) from Cell Signaling Technology, and actin goat polyclonal antibody from Santa Cruz Biotechnology. The secondary antibody used was horseradish peroxidase conjugated goat anti-rabbit (Thermo Scientific). The BIO-RAD Clarity Western Enhanced Chemiluminescence Substrates and HyBlot Chemiluminescence film were used for signal development and detection using a SRX-101A tabletop processor (Konica Minolta).
Determination of Drug Concentrations in Plasma and RS4;11 Tumor Tissue
Pharmacokinetics of compound 23 was determined in female SCID C.B-17 mice bearing RS4;11 tumors following a single intravenous dose of 5 mg/kg. Compound 23 was dissolved in the vehicle containing 20% (v/v) polyethylene glycol 400, 6% (v/v) Cremophor EL and 74% (v/v) PBS (20% PCP). Mice were sacrificed at 1, 3, 6, and 24 h after drug treatment and at 6 h after vehicle treatment, followed by collection of blood samples (300 μL) and tumor samples. Blood samples were centrifuged at 15 000 rpm for 10 min, then the supernatant plasma was saved for analysis. Isolated tumor samples were immediately frozen and ground with a mortar and pestle in liquid nitrogen. All plasma and tumor samples were stored at −80 °C prior to analysis.
Plasma and tumor concentrations of compound 23 were determined by a LC–MS/MS method developed and validated for this study. The LC–MS/MS method, consisting of a Shimadzu HPLC system and chromatographic separation of tested compound, was achieved using a Waters XBridge-C18 column (5 cm × 2.1 mm, 3.5 μm). An AB Sciex QTrap 5500 mass spectrometer equipped with an electrospray ionization source (Applied Biosystems, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. The mobile phases were 0.1% formic acid in purified water (A) and 0.1% formic acid in acetonitrile (B). The gradient (B) was held at 10% (0–0.3 min), increased to 95% at 0.7 min, then held at isocratic 95% B for 2.3 min and then immediately stepped back down to 10% for 2 min re-equilibration. The flow rate was set at 0.4 mL/min.
In Vitro Metabolite Identification in Mouse Liver Microsomes
Compound 23 (10 μM) was incubated with mouse liver microsomes (MLM) and β-NADPH at 37 °C for 20 and 40 min. The reaction was quenched by adding a 3-fold volume of ice-cold acetonitrile. The mixture was centrifuged at 15 000 rpm for 10 min, and the supernatant was saved under −80 °C for analysis. The negative control samples were prepared by a similar procedure without NADPH or using boiled microsomes. The general approach for metabolite identification using AB Sciex QTrap 5500 mass spectrometer involves the following steps: (1) Obtain a product ion spectrum of the parent compound to establish fragmentation. (2) Interpret the spectrum to identify major fragment ion and possible neutral loss. (3) Collect spectra of samples using both established precursor ion scan and neutral loss scan. EMS scan of both control and samples were acquired also. (4) Run product ion scans and MRM scans for all possible metabolite identified from step 3 plus expected metabolite. (5) Interpret the spectrum of the metabolites and determine the structure with their logical fragmentation pattern.
Acknowledgments
This study is supported in part by funding from the Breast Cancer Research Foundation (to S.W.), the University of Michigan Comprehensive Cancer Center Strategic Fund for Breast Cancer (to S.W.), the University of Michigan Comprehensive Cancer Center Core Grant from the National Cancer Institute, NIH (Grant P30CA046592), and Medsyn Biopharma.
Glossary
Abbreviations Used
- AML
acute myeloid leukemia
- BD1
bromodomain 1
- BD2
bromodomain 2
- BET
bromodomain and extra-terminal domain
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthalene
- BRD2
bromodomain-containing protein 2
- BRD3
bromodomain-containing protein 3
- BRD4
bromodomain-containing protein 4
- BRDT
bromodomain testis-specific protein
- FP
fluorescence polarization
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- MLL1
mixed lineage leukemia protein 1
- MRM
multiple reaction monitoring
- NUT
nuclear protein in testis
- PBS
phosphate buffered saline
- PD
pharmacodynamics
- PK
pharmacokinetics
- PROTAC
proteolysis targeting chimera
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01816.
Western Blotting analysis of BRD2, BRD3, and BRD4 proteins in RS4;11 cells treated with compounds 8, 23, and 26; 1H NMR spectrum for compound 23; 13C NMR spectrum for compound 23; UPLC–MS results for compound 23PDF)
Coordinates information for structure representation (PDB)
Molecular formula strings and some data (CSV)
Author Contributions
∇ B.Z., J.H., F.X., Z.C., L.B., E.F.-S., and M.L. contributed equally.
The authors declare the following competing financial interest(s): S. Wang, B. Zhou, F. Xu, J. Hu, L. Bai, C.-Y. Yang, D. McEachern, and S. Przybranowski are co-inventors for BET degraders disclosed in this study. The University of Michigan has filed a number of patent applications related to these BET degraders, which have been licensed to Medsyn Biopharma. S. Wang is a co-founder, stock holder, and consultant for Medsyn Biopharma.
Supplementary Material
References
- Patel D. J.; Wang Z. Readout of epigenetic modifications. Annu. Rev. Biochem. 2013, 82, 81–118. 10.1146/annurev-biochem-072711-165700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina A. C.; Denis G. V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Kouzarides T.; Huntly B. J. P. Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367, 647–657. 10.1056/NEJMra1112635. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Muller S.; Filippakopoulos P.; Knapp S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.-W. Small molecule bromodomain Inhibitors: extending the druggable genome. Prog. Med. Chem. 2012, 51, 1–55. 10.1016/B978-0-12-396493-9.00001-7. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boi M.; Gaudio E.; Bonetti P.; Kwee I.; Bernasconi E.; Tarantelli C.; Rinaldi A.; Testoni M.; Cascione L.; Ponzoni M.; Mensah A. A.; Stathis A.; Stussi G.; Riveiro M. E.; Herait P.; Inghirami G.; Cvitkovic E.; Zucca E.; Bertoni F. The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 2015, 21, 1628–1638. 10.1158/1078-0432.CCR-14-1561. [DOI] [PubMed] [Google Scholar]
- Berthon C.; Raffoux E.; Thomas X.; Vey N.; Gomez-Roca C.; Yee K.; Taussig D. C.; Rezai K.; Roumier C.; Herait P.; Kahatt C.; Quesnel B.; Michallet M.; Recher C.; Lokiec F.; Preudhomme C.; Dombret H. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, 186–195. 10.1016/S2352-3026(15)00247-1. [DOI] [PubMed] [Google Scholar]
- Odore E.; Lokiec F.; Cvitkovic E.; Bekradda M.; Herait P.; Bourdel F.; Kahatt C.; Raffoux E.; Stathis A.; Thieblemont C.; Quesnel B.; Cunningham D.; Riveiro M. E.; Rezaï K. Phase I population pharmacokinetic assessment of the oral bromodomain Inhibitor OTX015 in patients with haematologic malignancies. Clin. Pharmacokinet. 2016, 55, 397–405. 10.1007/s40262-015-0327-6. [DOI] [PubMed] [Google Scholar]
- Coudé M.-M.; Braun T.; Berrou J.; Dupont M.; Bertrand S.; Masse A.; Raffoux E.; Itzykson R.; Delord M.; Riveiro M. E.; Herait P.; Baruchel A.; Dombret H.; Gardin C. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. 10.18632/oncotarget.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C. W.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O.; Gosmini R.; Toum J.; Clement C. A.; Barnathan M.; Brusq J. M.; Mordaunt J. E.; Grimes R. M.; Crowe M.; Pineau O.; Ajakane M.; Daugan A.; Jeffrey P.; Cutler L.; Haynes A. C.; Smithers N. N.; Chung C. W.; Bamborough P.; Uings I. J.; Lewis A.; Witherington J.; Parr N.; Prinjha R. K.; Nicodeme E. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 2013, 56, 7501–7515. 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- Stathis A.; Zucca E.; Bekradda M.; Gomez-Roca C.; Delord J. P.; de La Motte Rouge T.; Uro-Coste E.; de Braud F.; Pelosi G.; French C. A. Clinical response of carcinomas harboring the BRD4-NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discovery 2016, 6, 492–500. 10.1158/2159-8290.CD-15-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaidos A.; Caputo V.; Gouvedenou K.; Liu B.; Marigo I.; Chaudhry M. S.; Rotolo A.; Tough D. F.; Smithers N. N.; Bassil A. K.; Chapman T. D.; Harker N. R.; Barbash O.; Tummino P.; Al-Mahdi N.; Haynes A. C.; Cutler L.; Le B.; Rahemtulla A.; Roberts I.; Kleijnen M.; Witherington J. J.; Parr N. J.; Prinjha R. K.; Karadimitris A. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood 2014, 123, 697–705. 10.1182/blood-2013-01-478420. [DOI] [PubMed] [Google Scholar]
- Banerjee C.; Archin N.; Michaels D.; Belkina A. C.; Denis G. V.; Bradner J.; Sebastiani P.; Margolis D. M.; Montano M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukocyte Biol. 2012, 92, 1147–1154. 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Gaiha G. D.; John S. P.; Pertel T.; Chin C. R.; Gao G.; Qu H.; Walker B. D.; Elledge S. J.; Brass A. L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816. 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis G. V. Bromodomain coactivators in cancer, obesity, type 2 diabetes, and inflammation. Discovery Med. 2010, 10, 489–499. [PMC free article] [PubMed] [Google Scholar]
- Matzuk M. M.; McKeown M. R.; Filippakopoulos P.; Li Q.; Ma L.; Agno J. E.; Lemieux M. E.; Picaud S.; Yu R. N.; Qi J.; Knapp S.; Bradner J. E. Small-molecule inhibition of BRDT for male contraception. Cell 2012, 150, 673–684. 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure M.; Crews C. M. Small-molecule PROTACS: new approaches to protein degradation. Angew. Chem., Int. Ed. 2016, 55, 1966–1973. 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Lu J.; Qian Y.; Altieri M.; Gordon D.; Rossi A. M. K.; Wang J.; Chen X.; Dong H.; Siu K.; Winkler J. D.; Crew A. P.; Crews C. M.; Coleman K. G. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 7124–7129. 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran X.; Zhao Y.; Liu L.; Bai L.; Yang C.-Y.; Zhou B.; Meagher J. L.; Chinnaswamy K.; Stuckey J. A.; Wang S. Structure-based design of γ-carboline analogues as potent and specific BET bromodomain inhibitors. J. Med. Chem. 2015, 58, 4927–4939. 10.1021/acs.jmedchem.5b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldis J. B.; Knight R.; Hussein M.; Chopra R.; Muller G. A review of the history, properties, and use of the immunomodulatory compound lenalidomide. Ann. N. Y. Acad. Sci. 2011, 1222, 76–82. 10.1111/j.1749-6632.2011.05974.x. [DOI] [PubMed] [Google Scholar]
- Fischer E. S.; Bohm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E. J.; Harper J. W.; Jenkins J. L.; Thoma N. H. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Ran X.; Zhao Y.; Yang C.-Y.; Liu L.; Bai L.; McEachern D.; Stuckey J.; Meagher J. L.; Sun D.; Li X.; Zhou B.; Karatas H.; Luo R.; Chinnaiyan A.; Asangani I. A.. Preparation of pyridoindole, pyridazinoindole and pyrimidinoindole compounds as BET bromodomain inhibitors. US 20140256706 A1, September 11, 2014.
- Bai L.; Zhou B.; Yang C.-Y.; Ji J.; McEachern D.; Przybranowski S.; Jiang H.; Hu J.; Xu F.; Zhao Y.; Liu L.; Fernandez-Salas E.; Xu J.; Dou Y.; Wen B.; Sun D.; Meagher J. L.; Stuckey J.; Hayes D. F.; Li S.; Ellis M. J.; Wang S.. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017, DOI: 10.1158/0008-5472.CAN-16-2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.