

ABSTRACT
The lungs are in direct contact with the environment through the tubular structure that constitutes the airway. Starting from the nasal orifice, the airway is exposed to foreign particles including infectious agents, allergens, and other substances that can damage the airways. Therefore, the airway must have a functional epithelial barrier both in the upper and lower airways to protect against these threats. As with the skin, it is likely that the pathogenesis of respiratory diseases is a consequence of epithelial barrier defects in these airways. The characteristics of this system, starting from the beginning of life and extending into maturing and aging, determine the prognosis of respiratory diseases. In this article, we discuss the pathogenesis, clinical phenotype, and prognosis of respiratory diseases from newborns to adulthood in the context of epithelial barrier function and dysfunction.
KEYWORDS: airway, barrier, dysfunction, epithelium, respiratory disease
Introduction
The human lungs contain a vast amount of epithelial tissue which, like the skin, is continually in contact with the external environment.1 The surface area of the adult respiratory system is about 80–100 m2, which is about half the area of a tennis court.2 The nose, combined with paranasal sinuses, has a surface area of about 100–200 cm2 in adults.3 Every minute, about 6–12 liters of air is inhaled, which results in the exposure of an extensive amount of epithelial tissue to harmful environmental agents and infectious organisms.1 In a healthy nose, 80–90% of inhaled minute particles (e.g. 10 μm) are captured by the nasal mucosal surface and transported by the mucociliary mechanism to the pharynx, where they are either swallowed or expelled by coughing.3 This supports the claim that the nasal epithelium is exposed to all environmental agents including infectious agents (such as viruses, bacteria, and fungi), allergens, and air contaminants before the bronchial epithelium, thus protecting the lower airway.3,4 However, some infectious agents, allergens, and air contaminants may still reach the lower airway. Therefore, both the upper and lower airways must have a functionally adequate epithelial barrier to ensure our survival. The pathogenesis of respiratory tract diseases is likely a result of imbalances between the microbiome and the innate immunity on both sides of the airway epithelium barrier.5 The characteristics of this system, starting from the beginning of life and extending into maturing and aging, determine the prognosis of respiratory diseases.
Embryology, histology and functional aspects of the epithelium in the upper and lower airways
During embryonic development, the upper airway originates from the pharyngeal arc. The pharyngeal arc is formed both by the outer layer of ectodermal tissue that surrounds the mesenchymal tissue nucleus and by the endodermal epithelium layer that lines the inside.6 In the embryonic stage, the respiratory endoderm develops from a small cell cluster located in the ventral anterior stomodeum. Epithelial cells of the primitive stomodeum cover the inner surface of the surrounding mesenchyme and bifurcate to form the proximal structures of tracheobronchial tree.7 Prior to this proximal-distal pattern, the pulmonary endoderm expresses numerous transcriptional regulators including Nkx2.1, Gata6, and Foxa1/2. The proximal pulmonary epithelium is formed from endoderm precursors which express Sox2 and differentiate into the ciliary, secretory, and basal cells lines during development. More distally, endoderm precursors expressing Sox9 / Id2 produce critical alveolar epithelium cell lines known as type I and II alveolar epithelial cells (AEC I/II).7
The respiratory epithelial and peri-epithelial complex consists of various cell types including ciliary, mucous, basal, and Clara cells in nearly all airways and bronchi, and of type I and type II cells in the alveoli.8 The nasal epithelium comprises 3 cell types: basal cells, goblet cells, and ciliary and nonciliary columnar cells. Although the nasal and bronchial mucosa have similar features such as the presence of pseudostratified epithelium with ciliary and columnar cells over a basement membrane, they generally show different features at the submucosal level. While large vascular structures predominate in the nasal mucosa, the bronchial airways are characterized by the muscular structures surrounding the airways.9
The respiratory epithelium constitutes a physical barrier between the outer and inner environments, and the sinonasal mucosa is crucial in protecting the interior.10,11 Epithelial cells are connected to their neighbors by cell-cell junctions, including tight junctions (TJs), adherens junctions (AJs), gap junctions, and desmosomes, which are the central components of this physical barrier (Fig. 1).3,12,13,14,15 The epithelial cell, also an element of innate immunity, acts as a classic physical barrier as well as prevents the development of inflammation by excreting cytokines and other mediators against harmful agents (viruses, bacteria, allergens, etc.), thus playing an important role in inflammation formation and the secondary anatomic and pathological changes.16 Furthermore, the excreted mucus contains antimicrobial molecules (defensin, bacteridisin, etc.), antiproteases, and antioxidants, and enables the capture and expulsion of inhaled foreign particles via mucociliary movements.1,17
Figure 1.
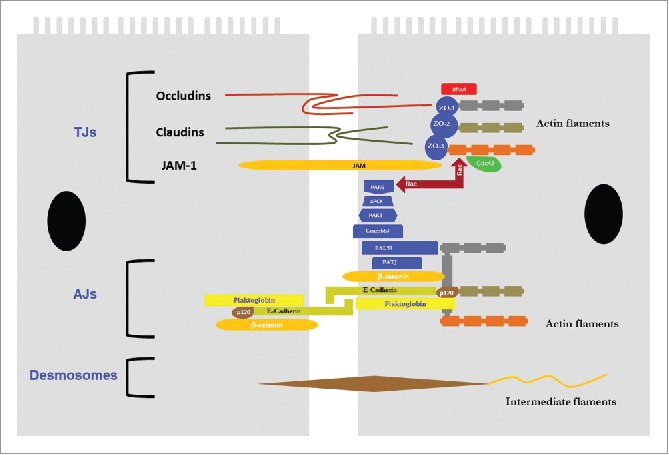
Junction components that form the barrier between adjacent epithelial cells in the airway. TJs, Tight junctions; AJs, Adherens junctions; JAM, Junctional adhesion molecule; ZO, Zonula occludens.
Functional importance of the airway epithelial barrier in health and disease
Barrier function of the epithelium
The respiratory epithelium, consisting of ciliary columnar cells, goblet cells, and Clara cells, forms a continuous and fairly regular lining of the airway lumen and protects it against inhaled environmental agents including aeroallergens, contaminants, and pathogens.6
Three main ultrastructural components are required to maintain the integrity of the airway epithelium. Epithelial barrier function is maintained by proper cell-cell connections among TJs (zonula occludens, ZO), AJs (zonula adherens), and desmosomes (macula adherens). The transmembrane proteins forming these tight junctions are connected to components of the cytoskeleton (Fig. 1).8,15,17,18,19
Tight junctions
Tight junctions, which constitute the most important part of the physical barrier function of epithelium, determine paracellular permeability.20,21 TJs are situated at the apicolateral boundary of epithelial cells to establish the major barrier to paracellular ion and fluid traffic between the inner and outer environments. TJs occlude the apical surface of the epithelial cells, thus separating the apical and basolateral regions. Approximately 40 different proteins have been identified as TJ components, and new junctional components and protein interactions are continually being discovered. This vast constellation of components includes the main integral TJ proteins claudin, occludin, and junction adhesion molecule (JAM)-1; many peripheral membrane proteins such as scaffold PDZ-containing proteins ZO-1, ZO-2, ZO-3, and multi-PDZ domain protein-1 (MUPP1); membrane-associated guanylate kinase with inverted orientation-1 (MAGI)-1, MAGI-2, MAGI-3; cell polarity molecules ASIP/PAR-3, PAR-6, PALS-1, and PALS-1-associated tight junction (PATJ); and non-PDZ proteins, cingulin, symplekin, ZONAB, GEF-H1, aPKC, PP2A, Rab3b, Rab13, PTEN, and 7H6.21–25 The configuration of these proteins within the structure of TJs is such that JAM, claudin, and occludin are in transmembrane orientation.
The major junctional complex proteins include 3 TJ families. Firstly, claudin has 27 different types and determines calcium-free cell adhesion, tight junction fiber formation, and intercellular ion selectivity.26 Claudins may be barrier-promoting or barrier-disrupting (or “leaky”). For example, claudin-1, the founding family member, is necessary and sufficient for junction formation. Mice with claudin-1 deficiency die immediately after birth and suffer extreme transepidermal water loss. In contrast, claudin-2 is an example of a leaky claudin which is associated with interleukin-13 (IL-13)-induced STAT6-dependent increase in intestinal permeability.27 It is found in the intestines of patients with increased epithelium permeability or inflammatory bowel diseases such as Crohn's disease and ulcerative colitis.28 Human pulmonary epithelial cell lines NCI-H441 and A549 to determine their similarity to primary human alveolar type II cells. NCI-H441 cells exhibited high expression of barrier-promoting claudin-3, -4, -5 while A549 cells exhibited high expression of leaky claudin-2.29
The second group of TJ proteins is the tight junction-associated marvel protein (TAMP) family, which has 3 members: occludin, tricellulin, and MARVELD3. Contrary to claudins, TAMP family members are not required for normal epithelium development and barrier function. However, they seem to play a role in barrier regulation during inflammation. The intracytoplasmic tails of occludin and other TAMP family members are subject to numerous post-translational modifications which are thought to affect interactions with scaffold components, signaling molecules, and the actin cytoskeleton.27 Occludin, the first identified integral membrane protein of TJs, is most commonly expressed in the basolateral surface of epithelial cells and is the most reliable immunohistochemical TJ marker.21 Overexpression of occludin increases barrier function, which is exhibited as an increase in transepithelial electrical resistance (TER) in mammal epithelial cells.30 Tricellulin was first identified at sites of tricellular epithelial contact, and is also shown to have barrier function.31 Less is known about the pulmonary expression or function of tricellulin and MARVELD3, and more research is needed concerning airway inflammation and the role of epithelial occludin and the TAMP family in asthma.27
The third respiratory TJ group is an immunoglobulin-like family whose most notable members are the JAM and coxsackie adenovirus receptor (CAR). These TJ components are receptors for several important viruses.32 Respiratory epithelium cells express a large number of CAR isoforms that promote the entry of viral particles.33 In respiratory epithelial cells, binding of Coxsackievirus and adenovirus to CAR causes TJ disruption and also reduces transepithelial resistance.1 A preliminary study showed that the binding of adenovirus to CAR resulted in the fragmentation of junctional complexes and increased epithelial permeability.33 Therefore, junction dysfunction may represent a strategy used by viruses to increase replication.33 JAMs (JAM-A, -B, -C, -4) are immunoglobulin superfamily proteins expressed in the cell junctions of epithelial and endothelial cells as well as on the surfaces of leukocytes, thrombocytes, and erythrocytes. JAM creates homophilic and heterophilic adhesion.22 TJs are important for various cellular processes including leukocyte transfer, thrombocyte activation, angiogenesis, and adenovirus binding.22 The available evidence indicates that JAM-A dimerization is required for functional regulation of the barrier.21
TJs and AJs are linked to cytoplasmic proteins and attach to the actin cytoskeleton to form a “cytosolic plaque.” ZO-1, ZO-2 and ZO-3 are members of the membrane-associated guanylate kinase (MAGUK) family, also called scaffolding proteins. They link directly to occludin and claudin at one end and bind to actin fibers at the other end, thus playing a direct role in the formation of barrier function.22,34
Adherens junctions
AJs are adherence structures that surround the cell and create an intercellular space of 25–35 nm. They are located directly beneath the TJs and consist of 2 basic adhesive parts, referred to as the nectin-afadin and E-cadherin-catenin complexes. Nectin forms the primary structure for both AJs and TJs, whereas afadin, to which it is attached, is connected to actin filaments.22 The degradation of actin filaments results in the functional and morphological disruption of TJ barriers.15 E-cadherin and β-catenin are important for adhesive function and regulate intercellular motility.22
E-cadherin is a calcium-dependent adhesion molecule expressed on epithelial cells and essentially mediates homophilic cell-cell adhesion. There is evidence concerning the effects of E-cadherin-mediated normal homophilic junctions on the integrity of TJs between epithelial cells. E-cadherin consists of an extracellular domain in the form of calcium-dependent homotypic adherence between epithelial cells and a highly conserved cytoplasmic tail. This structural cytoplasmic domain is stabilized on the membrane by means of p120 catenin, β-catenin, and α-catenin proteins, thus connecting to microtubule and actin cytoskeleton.35,36 E-cadherin is believed to be necessary for the maintenance of cell structure and other TJ structures. When E-cadherin is not properly expressed in the epidermis, TJ proteins ZO-1, occludin, and claudin are delocalized and TJs become distorted.15 The presence of low calcium concentration and blocking antibodies against E-cadherin causes fragmentation of not only E-cadherin itself but also ZO-1 and actin filaments. Therefore, the functional activity and normal expression of E-cadherin are important for ensuring the continuity of TJs between epithelial cells and the continuity of normal intercellular barrier function in respiratory epithelial cells.15 In addition, E-cadherin also modulates the activity of epidermal growth factor receptor (EGFR) and β-catenin to regulate cell proliferation and differentiation. Under normal conditions, E-cadherin prevents the liberation of the respiratory epithelium by binding EGFR at AJs, therefore inhibiting EGFR activation.37 Both separation of E-cadherin from the AJ complex or reduced expression of E-cadherin may occur during epithelial-mesenchymal transition (EMT), which can result in EGFR dissociating from the AJ and being redistributed, where it may be easily activated by EGF ligands.38 Free cytoplasmic β-catenin may translocate to the nucleus in the presence of Wingless integration zone (Wnt) ligands and initiate the transcription of target genes involved in embryonic development and pulmonary adult homeostasis.37
While inducing E-cadherin expression suppresses nuclear factor kappa β (NF-κ β) activation, in cases of E-cadherin-mediated cell-cell adhesion loss, NF-κ β signal is stimulated as in cancerous cell lines. Thus the balance between E-cadherin expression and NF-κ β activation results in the epithelial cell becoming a tolerogenic and proinflammatory/immunogenic phenotype.15
Desmosomes and hemidesmosomes
Desmosomes are E-cadherin-mediated cell-cell adhesion structures which provide mechanical support to the tissue. They are located predominantly along the lateral surfaces of columnar epithelial cells, particularly toward the basal surface, and establish connections between columnar and basal cells. Columnar cells do not establish adhesive connections to the basement membrane, but connect to the basement membrane via desmosomal attachments. Hemidesmosomes connect basal cells to the extracellular matrix in the basement membrane. They primarily include integrins. Besides adhesion, they may also play a role in cell signaling pathways.15
Cell polarization in airway epithelium
The formation of apicolateral junctional complexes in airway epithelium is closely related to cell polarization. Recent gene sequencing studies have demonstrated the expression of 2 polarity complexes in respiratory epithelial cells, the Crumbs (CRB) complex and partitioning defective (PAR) complex.39 The CRB complex comprises integral membrane protein Crumbs3 and scaffolding proteins, Lin 7 1 (PALS1) associated protein, and PALS1 associated tight junction protein (PATJ).1,27 Polarity proteins are important for proper morphogenesis and for the maintenance of apical-basolateral polarization.27 Reduced Crumbs3 expression delays the formation of TJ and cilia. The consumption of PALS1 leads to PATJ loss, disruption of cell polarity, reduction of TER, and alteration of E-cadherin traffic.1 Coronavirus envelope protein E is bound to PALS1, and the expression of protein E delays TJ formation in MDCK cells.1 The central components of the PAR complex are scaffolding proteins PAR3, PAR6, and atypical protein kinase C; however, the molecular actions of the PAR complex in respiratory epithelium polarity are not known.1
Stress response of the epithelium: Intracellular signaling by e-cadherin and epithelial mesenchymal transition
Cytokines and chemokines excreted by respiratory epithelial cells activate and regulate the function of immune cells and structural cells, and also act as chemoattractants. Epithelial cells also express many cytokine and chemokine receptors whose activation can influence epithelial functions. IL-4 and IL-3, cytokines associated with Th2-mediated immune response, and the epithelium-derived cytokines IL-25, IL-33, and Thymic stromal lymphopoietin (TSLP) have crucial roles in the pathogenesis of atopic asthma, allergic rhinitis (AR), and chronic rhinosinusitis (CRS).10,11,17,40,41,42,43
Another important role of the epithelium lies in its relation to the underlying mesenchyme, and cytokine and growth factors are excreted as a result of the connection between these 2 structures; this superstructure is called the epithelial-mesenchymal trophic unit (EMTU).44 Epithelial mesenchymal transition (EMT) is a process in which epithelial cells lose their epithelial character and gain mesenchymal properties. The main factors that induce EMT are tyrosine kinase receptors for the TGF-β superfamily which are activated by growth factors and canonical Wnt/β-catenin signals, which may regulate EMT in the respiratory tract or lungs (Fig. 2).37 EMT has a complex molecular basis and occurs during both embryogenesis and epithelial neoplasia.45 It results in the downregulation of E-cadherin. Disruption of the functional activity and normal expression of E-cadherin leads to the functional and morphological degradation of TJ barriers.15 This disruption of epithelial barrier function plays a key role in increasing the access of inhaled allergens to antigen-presenting cells in the submucosa and stimulating Th2-mediated immune response and EMT.18,27,46,47
Figure 2.
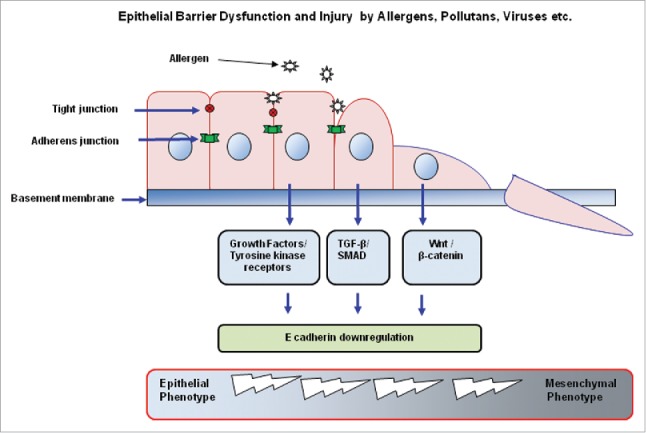
The main features and elements of epithelial-mesenchymal transition. TGF, Transforming growth factor; SMAD, Sma and mad related family; Wnt, Wingless / integrated.
When the E-cadherin-β-catenin structure is disrupted, β-catenin released from its cytosolic segment is freed and translocates to the nucleus, where it activates the Wnt/β-catenin signaling pathway; this process is involved in tissue remodeling.46,48 The available data indicate that EMT is a characteristic of the epithelial cells in asthma and is highly likely to contribute to airway remodeling.49 Interestingly, it has been shown in vitro that house dust mite (HDM) extracts, particularly combined with TGF-β1, induce EMT properties in 16HBE epithelial cells.46 In another study, chronic HDM administration in mice resulted in E-cadherin and occludin loss and airway remodeling.50 Recently, Hupin et al. demonstrated the properties of EMT in chronic rhinosinusitis without nasal polyps (CRSsNP) and with nasal polyps (CRSwNP) patients.10,51 In summary, further research must be conducted to understand how EMT and other changes in epithelial differentiation affect the properties of the respiratory epithelium barrier.
Rhinitis, asthma, and chronic obstructive pulmonary disease: The role of epithelial barrier dysfunction (origins in childhood, morbidity, epidemiology)
Allergic rhinitis
AR refers to nasal mucosal inflammation caused by IgE-mediated response to inhaled allergens. It causes symptoms such as nasal congestion and bleeding, sneezing, and nose itchiness in 30% of the Western population.52 It has been reported that in lower airway diseases (for example, asthma, bronchiolitis obliterans, chronic obstructive pulmonary disease and cystic fibrosis), the pseudostratified respiratory epithelium is seriously damaged and has to be restored to perform its defensive functions. The same results were found in the sinonasal epithelial cells obtained from CRSwNP and AR patients.41,53 A healthy nasal epithelial barrier is known to be critical for protection against environmental factors, including allergens.54,55 Other epithelial barrier indicators such as filaggrin (FLG), a member of the epidermal differentiation complex on chromosome 1q21, are reported to have polymorphisms and loss-of-function mutations associated with atopic dermatitis and AR.53 It is well known that some inhaled allergens are proteinases that directly cause epithelial barrier dysfunction. It has been shown that blocking protease allergens reduces allergic response in AR.41 Similar to the signs of epithelial damage seen in asthma, the pathological findings in AR include increased respiratory epithelial barrier permeability and higher sensitivity to oxidants caused by the overproduction of mucus and inadequate innate immune response to viral infections in the respiratory tract.56 Evidence shows that defective and dysfunctional epithelial TJs and AJs are, along with nasal polyps, a common feature of allergic rhinitis and chronic rhinosinusitis. This leaky epithelium is accompanied by reduced expression of occludin and claudin-4 in nasal biopsies.12,57 Leaky epithelium may cause deeper penetration of inhaled allergens, bacteria, and viruses into the subepithelial zone, facilitating antibody intake and activating epithelial signaling and innate adaptive immune responses.58,59 House dust mites, pollens, and allergens such as fungi with cysteine proteinase activity may degrade occludin or may directly disrupt TJ integrity.52,60 CRSwNP patients exhibit a defective epithelial barrier, mostly caused by secreted proinflammatory cytokines such as IFN-gamma, IL-4, and oncostatin M.60 It has yet to be determined why the epithelial barrier is defective in CRSwNP. It is possible that the epithelial cells are naturally abnormal in CRSwNP.The association between asthma and CRSwNP has been more extensively described.55 Most asthma patients (88%) have sinonasal inflammation, at least radiographically.55
Asthma
Asthma is a chronic inflammatory airway disease accompanied by structural changes involving various cellular components and cells, particularly mast cells, eosinophils, T cells, macrophages, and epithelial cells.61 Asthma patients suffer from wheezing, expectorating, varying degrees of air flow restriction, and extreme sensitivity of the airway to environmental bronchospasmogenic stimuli.62
As a barrier against the external environment, the bronchial epithelium plays a key role in the gene-environment interaction in asthma.63,64 Numerous studies have shown that the respiratory epithelium is disrupted in asthma.20,56,62 It has also been reported that the bronchial epithelial cells of asthma patients exhibit antimicrobial response defects.62 Biopsy studies conducted in children suggest that structural changes in the respiratory epithelium may occur before the onset of airway inflammation in the early phase of asthma pathogenesis.65 It has been argued that the structural and functional abnormalities in respiratory epithelium cause abnormal signaling to the immune system and structural cells, and lead to restructuring, inflammation, and oversensitivity of the allergic airway.62
Airway inflammation patterns seem to be similar in all clinical forms of asthma (allergic, nonallergic, and aspirin-induced) and in all age groups.61 Asthmatic inflammation is characterized by the dominance of Th2 cells and Th2-mediated chemokine and cytokine responses. Asthmatic inflammation forms according to factors determined by the individual's genetics and the environment in which they live.44 It has been determined that the immune system is Th2-dominant in utero and in the early neonatal period. The general view according to the hygiene theory is that infections and endotoxin exposure early in life incline the immune system toward Th1, while fewer encounters with microorganisms tilt it toward Th2 dominance.22,66 In developed countries, the frequency of atopy in children and adolescents may be up to 40%, but only one-third of these children develop asthma. The exact mechanisms by which epithelial high-sensitivity gene expression brings about a functionally modified response to asthma aeroallergens remain unknown.44
Epithelial fragility and epithelial shedding are the characteristic features of asthma,15,43 with selective loss of columnar cells and epithelial damage.20 It is not exactly known why asthma patients are prone to epithelial fragility, but their epithelial damage may be caused by internal factors such as genetic polymorphism or external factors such as certain respiratory viruses, environmental contaminants including ozone, cigarette smoke, and allergens such as Dermatophagoides pteronyssinus 1 (Der p 1) and fungal allergens.20,67,68
In vivo and in vitro studies have shown that epithelial barrier function is disrupted in asthma.44 Studies on the respiratory epithelium junctional proteins in asthma patients have repeatedly demonstrated reduced expression of ZO-1, E-cadherin, and occludin in the asthmatic epithelium both in vivo and in vitro, which indicates a major defect in adhesion mechanisms. This may make the respiratory epithelial cells of asthma patients more sensitive to allergens.35,36,62,69 In the early 1990s, ultrastructural studies showed that asthmatic patients had fewer intercellular adhesion junction complexes compared with nonasthmatic patients, but the underlying mechanisms of these structural differences remain to be clarified.18
Disruption of epithelial barrier function has presumed consequences such as easier penetration of inhaled allergens into respiratory tissue, increased allergen access to antigen-presenting cells in the submucosa, and greater extension of spasmogenic agonists to smooth muscles.18 Proposed mechanisms of epithelial barrier damage are via indirect suppression of junctional proteins due to the release of inflammatory mediators, or fragmentation directly related to allergens (Fig. 3).70 HDM, cockroaches, animal and fungal allergen components, protease-releasing environmental factors including some tree and plant pollens (such as Olea europaea, Dactylis glomerata), and protease-releasing eosinophils and neutrophils may degrade ZO-1 and E-cadherin molecules.36,58,71 In addition, proteases activate protease-activated receptors (PAR), which lead to proinflammatory gene transcription. PAR-2 receptors are expressed in the respiratory epithelium. The receptor itself causes signal activation that causes cytokine release, metalloproteinase activation, antigen-triggered signal transduction, and airway hyperreactivity.72 In addition to direct proteolytic breakdown, HDM, cockroach, and fungal extracts activate PAR-2. It has been shown that PAR-2 activation primarily causes loss of E-cadherin-dependent cell-cell adhesion in the human respiratory epithelium.15,73 However, it is not fully understood why allergens with protease activity induce structural modifications only in asthma patients and do not cause any changes in healthy epithelium.46,74
Figure 3.
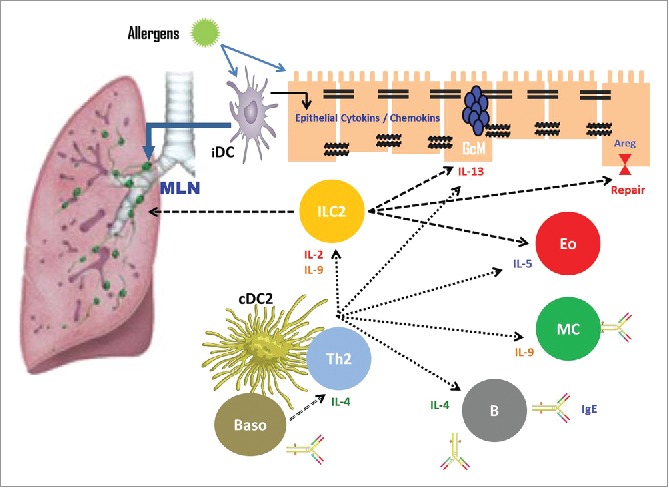
Indirect suppression of barrier proteins by inflammatory mediators in allergen-induced epithelial barrier damage. IL, Interleukin; ILC, Innate lymphoid cells; MLN, Mediastinal lymph nodes; MC, Macrophage cell; cDC, Conventional dendritic cell; Th2, T helper 2; B, B lymphocytes; Eo, Eosinophil; GCM, goblet-cell metaplasia.
Chronic obstructive pulmonary disease
Chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and mortality worldwide. COPD is usually defined by chronic airflow obstruction with a forced expired volume in one second/forced vital capacity (FEV1/FVC) ratio of < 0.7 following a bronchodilator challenge.75 It is generally considered to be a respiratory tract disease that affects smoking adults and is related to structural airway modifications (remodeling). Respiratory epithelium cells isolated from healthy smokers and COPD patients show reduced expression of occludin and claudins 1, 3, 7, and 8, which may contribute to barrier dysfunction seen in the subjects.39 As cigarette smoke causes an abnormal activation of canonical Wnt/β-catenin signaling, it is believed that exposure to chronic cigarette smoke reduces barrier function and facilitates invasion of the respiratory epithelium by environmental allergens, contaminants, and pathogens.76
Bronchopulmonary dysplasia and hyperoxia
Bronchopulmonary dysplasia (BPD) is one of the most serious complications of lung damage in preterm infants due to high oxygen exposure and mechanical ventilation. Children with BPD require long-term oxygen therapy after being discharged from the hospital.77 A hyperoxia-induced BPD model showed that BPD is accompanied by destruction of TJ structures in the lung, increased permeability of the pulmonary epithelial barrier, and a decline in the concentrations of certain TJ proteins.78 Destruction of the pulmonary epithelial TJ structures is a key event in the pathogenesis of pulmonary edema in BPD.77 It has been shown that AEC II also underwent EMT in addition to transdifferentiation in response to hyperoxia stimulation.77
Maturational, functional, and responsive role of the epithelium from wheezing to asthma and from asthma to chronic obstructive pulmonary disease (as a pediatric disease)
Early life events are important throughout the life of a patient, and airway diseases appear to originate very early in infancy/childhood. Wheezing syndromes are seen frequently in early childhood and some children with early onset wheezing syndromes will later develop asthma.79 There are 2 hypotheses regarding how respiratory tract diseases and asthma in childhood influence long-term pulmonary development and functioning: 1) an early viral infection alters lung development and switches the host to airway obstruction, or 2) lung development is altered in utero or in early life due to antenatal insults and predisposes the ‘primed host’ to early respiratory illnesses and subsequent chronic airway obstruction.75
Viral infection of the respiratory tract may further disrupt barrier function or may delay repolarization and differentiation. Exposure of respiratory epithelial cells to double-stranded RNA, rhinovirus (RV), or respiratory syncytial virus (RSV) infection induces a pronounced upregulation of TSLP expression.5 TSLP is an epithelial cell-derived interleukin (IL)-7-like cytokine that contributes to mucosal immunity induced by microbes. It is currently the focus of much attention because there may be a link between asthma and viral infections.71 At the same time, RV infection in the respiratory epithelium contributes to the Th2 immune response by triggering the release of epithelial cytokines IL-33 and IL-25.5,71 IL-33 is considered an alarm signal for epithelial damage and is released by the epithelium in response to triggers such as allergens or infectious agents.80 Similar to allergens, RV and RSV impact epithelium integrity and reduce expression of epithelial junction proteins. RV increases respiratory epithelium permeability via loss of ZO-1 from TJs and reduced transepithelial resistance.15,62 Similarly, RSV infection led to a significant reduction in TER and increase in permeability. Additionally it caused disruption of the epithelial apical junction complex and remodeling of the apical actin cytoskeleton.81 These findings may help explain why respiratory viruses exacerbate allergic inflammation despite evoking type-1 immune responses that should theoretically be able to balance type-2-mediated inflammation.62 These intercellular junctions also function as signaling platforms that regulate gene expression, cell reproduction, and differentiation.62 Wheezing respiratory diseases due to RV in the first 3 y of life is an important risk factor associated to subsequent asthma.82,83 Because airway remodeling begins in the same early life period, it is conceivable that RV infections may also play a role in inducing and advancing airway remodeling.82
Epigenetic factors can significantly affect gene transcription without altering the gene sequence.17,64 They influence gene activity and change the structural conformation and accessibility of genes, for example through DNA methylation or histone acetylation/deacetylation, without making any modifications in the DNA sequence.17 These alterations orchestrate a complex early-life reprogramming of immune T cell response, dendritic cell function, macrophage activation, and a breach of airway epithelial barrier that dictates asthma risk and severity in later life. Current knowledge on the epigenetic effects of tobacco smoke, microbial allergens, oxidants, airborne particulate matter, diesel exhaust particles, dietary methyl donors, and other nutritional factors, and dust mites is discussed.84 Many asthma genes recently identified in genome-wide association studies (GWAS) are expressed by respiratory structural cells, particularly the epithelium, and this suggests that the processes in the respiratory mucosal surface are critical for the development of asthma.40,62 It has been established that the IL-33 and IL-1 receptor-like 1 (IL1RL1)/IL18R1 loci are important in asthma development.43,85 The GABRIEL study demonstrated that polymorphisms in IL1RL1 and IL-33 are more strongly associated with early onset asthma (< 16 years).86 IL33-IL1RL1 pathway polymorphisms are related to specific wheezing phenotypes and particularly to mid-onset wheezing. The IL33-IL1RL1 pathway may act in wheezing and asthma development by means of allergic sensitization development during childhood.80 Gene association studies have established a connection between extreme airway sensitivity and the CDHR3 and PCDH1 genes, both of which encode proteins involved in epithelial barrier function.43 Similarly, the discovery that variants in cutaneous barrier genes such as FLG and SPINK5 lead to a predisposition for allergic disorders such as atopic dermatitis and asthma in the presence of eczema suggests that atopy may be the result of barrier disruption. These findings have changed the views about the pathogenesis of atopic dermatitis and suggest that the same mechanisms in asthma may apply.17,87 However, asthma is not determined entirely by genetic factors, but also by gene-environment interactions. In particular relation to asthma susceptibility, the exposure to specific environmental factors can be a key factor in the induction or suppression of asthma-related genes.64
Epidemiological findings provide ample evidence that antenatal programming, genes, and the environment all play etiological roles in chronic adult illnesses such as COPD.75 Five childhood risk factors (asthmatic mother and father, maternal smoking, childhood asthma, and airway infections) are strongly correlated with progressive reduction of pulmonary function and COPD. Factors adversely affecting pulmonary development include transgenerational (smoking grandmother), antenatal (exposure to tobacco and contaminants), and early childhood (exposure to tobacco and other contaminants, including pesticides) influences. All those factors also affect airway epithelial maturation and development, as alveolar development is now believed to continue through somatic growth and is adversely effected by early exposure to tobacco smoke. This results in further deterioration of the growing lungs and airway wall, including the epithelial layer. In early childhood, repeated epithelial damage and gene-environment interactions manifest as lower pulmonary function and airway obstruction in sensitive children.75 More than 30 y ago, Burrows suggested that disturbed early development of the lungs might underlie the susceptibility to COPD. Low birth weight and respiratory infections in infancy reduced lung function in adulthood. Consistent with this, death from COPD was associated with lower birth weight.88 Barker et al. concluded that COPD morbidity (reduced lung function) and mortality were more strongly influenced by respiratory infections in childhood than by cigarette smoking in their cross-sectional study.89
Genetic factors are also relevant, with genes important in lung development and early wheezing also being implicated in COPD.90 Further evidence suggesting that COPD has origins in early childhood was provided by the PIAMA and ALSPAC studies, which demonstrated associations between important COPD-associated genes.91 They found that at least 3 COPD genes were involved in lung growth and development and were involved in antenatal and early-life responses to tobacco smoke exposure.91 RAGE has been reported to be strongly associated with higher levels of soluble forms of this protein. RAGE expression is most abundant in the lung, and the intensity of expression in respiratory epithelial cells varies during lung morphogenesis. It has been shown to play a role in critical processes directly involved in perinatal transitioning of the embryonic lung into a mature functional organ.92 Moreover, these transient early wheeze (TEW)-related airway development anomalies have been shown to be associated with reduced lung function even when the symptoms disappear, and the condition continues through the rest of the childhood and adolescence. It is believed that TEW may be related to subsequent COPD development.91 In a cohort study following children aged 10–15 y for 50 years, Tagiyeva et al. demonstrated that childhood asthma was associated with increased risk for COPD, while childhood wheezy bronchitis increased the risk of COPD to a lesser degree.93
Therapeutic strategies to improve barrier dysfunction: A new and rational approach to the treatment of airway diseases
Asthma is a chronic inflammatory disease of the airways, which in the long-term results in remodeling that lead to irreversible demage of the airway wall.94 In addition, many viruses which cause lower respiratory infection lead to additional damage of the EMTU and treatment of viral infection does not have any restorative effect on the airway wall and epithelium-related microstructures. Then that barrier restoration might provide an option.
The respiratory epithelium constitutes a potential key target for inhaler therapies such as inhaled corticosteroids (ICS) and bronchodilators such as β-agonists and muscarinic antagonists. Various studies have shown that glucocorticoids play a fundamental role in the function and maintenance of cell–cell contact by inducing tight junction formation and increasing transepithelial resistance; however, their effects on the distribution and expression of AJ proteins (E-cadherin, β-catenin) are not completely clear. Treating bronchial epithelial cell lines Calu-3 and 16HBE with dexamethasone and fluticasone propionate stimulated expression of occludin and ZO-1, thus increasing epithelial barrier integrity. Bronchial biopsies obtained from asthma patients showed that ICS treatment reduced inflammation and increased the number of ciliary cells.37 Similarly, Yuksel et al. revealed increased levels of occludin, claudin, and JAM in a mouse model of asthma treated with steroids.95 In a study by Doerner et al., corticosteroid treatment did not substantially alter TGF-β1-mediated downregulation of E-cadherin mRNA.96 Song et al. demonstrated partial restoration of E-cadherin distribution in an experimental asthma model after pretreatment with dexamethasone.97 A recent study showed that barrier leakiness in asthmatic patients is induced by Th2 cells, IL-4, and IL-13 and histone deacetylases (HDAC) activity. The inhibition of endogenous HDAC activity reconstitutes defective barrier by increasing TJ expression.98 In a recent randomized, controlled study, it was reported that ICS may also reduce the EMT properties in the bronchial tissue of COPD patients.99 Recent findings in primary bronchial epithelial cells have shown that ICS is protective against cigarette smoke-induced barrier dysfunction.100 Inhaled β-agonists have a broad bronchodilator effect on airway smooth muscle but also act as bronchodilators by means of AECs expressing β-adrenoreceptors.37,101 Cytokine release and adhesion molecule expression by stimulated human AECs are downregulated by salmeterol.37 EMT could be inhibited by muscarinic antagonists.102
Despite available treatment options such as intranasal steroids (INS), antihistamines, leukotriene receptor antagonists, and immunotherapy, 20% of patients with AR do not respond favorably to treatment.52 In the complex interaction between human nasal epithelial cells and environmental pathogens, host factors play an important role in the severity and progression of disease and response to pharmacologic therapies.52 The cause of nonresponse to treatment is multifactorial, with barrier dysfunction being one of these factors. A dysfunctional epithelium barrier increases the intake of allergens and exogen particles, which lead to further activation of mast cells and nerve fibers. Therefore, research on epithelial barrier and TJ function in AR patients is very important.52 Fluticasone propionate increased the barrier integrity of nasal epithelial cells both in healthy controls and in allergic rhinitis patients with HDM allergy.52
Conclusions
In summary, potential novel treatment strategies may provide new information and new therapeutic targets for the restoration of disrupted epithelial barrier function, especially for asthma. For asthma, drugs for improving Caveolin-1 function, improving steroid response or inhibiting pathways like TGF-β/Smad3 signaling which are involved in EMT may be promising. Blocking the epithelial cytokines TSLP, IL-33, and IL-25 may be a powerful tool for limiting Th2 responses.
References
- [1].Ganesan S, Comstock AT, Sajjan US. Barrier function of airway tract epithelium. Tissue Barriers. 2013;1(4):e24997. doi: 10.4161/tisb.24997. PMID:24665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Colebatch HJ, Ng CK. Estimating alveolar surface area during life. Respir Physiol. 1992;88(1-2):163-70. doi: 10.1016/0034-5687(92)90037-W. PMID:1626135. [DOI] [PubMed] [Google Scholar]
- [3].Wang DY, Li Y, Yan Y, Li C, Shi L. Upper airway stem cells: understanding the nose and role for future cell therapy. Curr Allergy Asthma Rep. 2015;15(1):490. doi: 10.1007/s11882-014-0490-0. PMID:25430951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yu F, Zhao X, Li C, Li Y, Yan Y, Shi L, Gordon BR, Wang DY. Airway stem cells: review of potential impact on understanding of upper airway diseases. Laryngoscope. 2012;122(7):1463-9. doi: 10.1002/lary.23320. PMID:22555982. [DOI] [PubMed] [Google Scholar]
- [5].Smits HH, van der Vlugt LE, von Mutius E, Hiemstra PS. Childhood allergies and asthma: New insights on environmental exposures and local immunity at the lung barrier. Curr Opin Immunol. 2016;42:41-7. doi: 10.1016/j.coi.2016.05.009. PMID:27254380. [DOI] [PubMed] [Google Scholar]
- [6].Pohunek P. Development, structure and function of the upper airways. Paediatr Respir Rev. 2004;5(1):2-8. doi: 10.1016/j.prrv.2003.09.002. PMID:15222948. [DOI] [PubMed] [Google Scholar]
- [7].Swarr DT, Morrisey EE. Lung endoderm morphogenesis: gasping for form and function. Annu Rev Cell Dev Biol. 2015;31:553-73. doi: 10.1146/annurev-cellbio-100814-125249. PMID:26359777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tam A, Wadsworth S, Dorscheid D, Man SF, Sin DD. The airway epithelium: more than just a structural barrier. Ther Adv Respir Dis. 2011; 5: 255-73. doi: 10.1177/1753465810396539. PMID:21372121. [DOI] [PubMed] [Google Scholar]
- [9].Bourdin A, Gras D, Vachier I, Chanez P. Upper airway x 1: allergic rhinitis and asthma: united disease through epithelial cells. Thorax. 2009;64:999-1004. doi: 10.1136/thx.2008.112862. PMID:19864543. [DOI] [PubMed] [Google Scholar]
- [10].Khalmuratova R, Park JW, Shin HW. Immune Cell Responses and Mucosal Barrier Disruptions in Chronic Rhinosinusitis. Immune Netw. 2017;17(1):60-7. doi: 10.4110/in.2017.17.1.60. PMID:28261021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lambrecht BN, Hammad H.. Allergens and the airway epithelium response: gateway to allergic sensitization. J Allergy Clin Immunol. 2014;134(3):499-507. doi: 10.1016/j.jaci.2014.06.036. PMID:25171864. [DOI] [PubMed] [Google Scholar]
- [12].Soyka MB, Wawrzyniak P, Eiwegger T, Holzmann D, Treis A, Wanke K, Kast JI, Akdis CA. Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-γ and IL-4. J Allergy Clin Immunol. 2012;130(5):1087-1096.e10. doi: 10.1016/j.jaci.2012.05.052. PMID:22840853. [DOI] [PubMed] [Google Scholar]
- [13].Nomura K, Obata K, Keira T, Miyata R, Hirakawa S, Takano K, Kohno T, Sawada N, Himi T, Kojima T. Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells.Respir Res. 2014 Feb 18;15:21. doi: 10.1186/1465-9921-15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang N, Van Crombruggen K, Gevaert E, Bachert C. Barrier function of the nasal mucosa in health and type-2 biased airway diseases. Allergy. 2016;71(3):295-307. doi: 10.1111/all.12809. PMID:26606240. [DOI] [PubMed] [Google Scholar]
- [15].Nawijn MC, Hackett TL, Postma DS, van Oosterhout AJ, Heijink IH. E-cadherin: gatekeeper of airway mucosa and allergic sensitization. Trends Immunol. 2011; 32: 248-55. doi: 10.1016/j.it.2011.03.004. PMID:21493142. [DOI] [PubMed] [Google Scholar]
- [16].Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev. 2011; 242: 186-204. doi: 10.1111/j.1600-065X.2011.01033.x. PMID:21682746. [DOI] [PubMed] [Google Scholar]
- [17].Loxham M, Davies DE, Blume C. Epithelial function and dysfunction in asthma. Clin Exp Allergy. 2014;44:1299-313. doi: 10.1111/cea.12309. PMID:24661647. [DOI] [PubMed] [Google Scholar]
- [18].Knight DA, Stick SM, Hackett TL. Defective function at the epithelial junction: a novel therapeutic frontier in asthma? J Allergy Clin Immunol. 2011;128:557-8. doi: 10.1016/j.jaci.2011.07.031. PMID:21878242. [DOI] [PubMed] [Google Scholar]
- [19].Saatian B, Rezaee F, Desando S, Emo J, Chapman T, Knowlden S, Georas SN. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers. 2013;1(2):e24333. doi: 10.4161/tisb.24333. PMID:24665390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Swindle EJ, Collins JE, Davies DE. Breakdown in epithelial barrier function in patients with asthma: identification of novel therapeutic approaches. J Allergy Clin Immunol. 2009; 124: 23-34. doi: 10.1016/j.jaci.2009.05.037. PMID:19560576. [DOI] [PubMed] [Google Scholar]
- [21].Kojima T, Go M, Takano K, Kurose M, Ohkuni T, Koizumi J, Kamekura R, Ogasawara N, Masaki T, Fuchimoto J, et al.. Regulation of tight junctions in upper airway epithelium. Biomed Res Int. 2013;2013:947072. doi: 10.1155/2013/947072. PMID:23509817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 2007;127: 2525-32. doi: 10.1038/sj.jid.5700865. PMID:17934504. [DOI] [PubMed] [Google Scholar]
- [23].Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286(6):C1213-28. doi: 10.1152/ajpcell.00558.2003. PMID:15151915. [DOI] [PubMed] [Google Scholar]
- [24].Sawada N, Murata M, Kikuchi K, Osanai M, Tobioka H, Kojima T, Chiba H. Tight junctions and human diseases. Med Electron Microsc. 2003;36(3):147-56. doi: 10.1007/s00795-003-0219-y. PMID:14505058. [DOI] [PubMed] [Google Scholar]
- [25].Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2(4):285-93. doi: 10.1038/35067088. PMID:11283726. [DOI] [PubMed] [Google Scholar]
- [26].Schlingmann B, Molina SA, Koval M. Claudins: Gatekeepers of lung epithelial function. Semin Cell Dev Biol. 2015;42:47-57. doi: 10.1016/j.semcdb. PMID:25951797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol. 2014;134:509-20. doi: 10.1016/j.jaci.2014. PMID:25085341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Steelant B, Seys SF, Boeckxstaens G, Akdis CA, Ceuppens JL, Hellings PW. Restoring airway epithelial barrier dysfunction: a new therapeutic challenge in allergic airway disease. Rhinology. 2016;54(3):195-205. doi: 10.4193/Rhin15.376. PMID:27316042. [DOI] [PubMed] [Google Scholar]
- [29].Ren H, Birch NP, Suresh V. An Optimised Human Cell Culture Model for Alveolar Epithelial Transport. PLoS One. 2016 Oct 25;11(10):e0165225. doi: 10.1371/journal.pone.0165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Takano K, Kojima T, Sawada N, Himi T. Role of tight junctions in signal transduction: an update. EXCLI J. 2014;13:1145-62. eCollection 2014. PMCID: PMC4464418 PMID:26417329. [PMC free article] [PubMed] [Google Scholar]
- [31].Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 2005;171(6):939-45.doi: 10.1083/jcb.200510043. PMID:16365161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Excoffon KJ, Guglielmi KM, Wetzel JD, Gansemer ND, Campbell JA, Dermody TS, Zabner J. Reovirus preferentially infects the basolateral surface and is released from the apical surface of polarized human respiratory epithelial cells. J Infect Dis. 2008;197(8):1189-97. doi: 10.1086/529515. PMID:18419529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Walters RW, Freimuth P, Moninger TO, Ganske I, Zabner J, Welsh MJ. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell. 2002;110(6):789-99. doi: 10.1016/S0092-8674(02)00912-1. PMID:12297051. [DOI] [PubMed] [Google Scholar]
- [34].Rezaee F, Georas SN. Breaking Barriers: New Insights Into Airway Epithelial Barrier Function in Health and Disease. Am J Respir Cell Mol Biol. 2014;50(5):857-69. doi: 10.1165/rcmb.2013-0541RT. PMID:24467704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Goto Y, Uchida Y, Nomura A, Sakamoto T, Ishii Y, Morishima Y, Masuyama K, Sekizawa K.. Dislocation of E-cadherin in the airway epithelium during an antigen-induced asthmatic response. Am J Respir Cell Mol Biol. 2000;23:712-8. doi: 10.1165/ajrcmb.23.6.4031. PMID:11104722. [DOI] [PubMed] [Google Scholar]
- [36].de Boer WI, Sharma HS, Baelemans SM, Hoogsteden HC, Lambrecht BN, Braunstahl GJ. Altered expression of epithelial junctional proteins in atopic asthma: possible role in inflammation. Can J Physiol Pharmacol. 2008;86:105-12. doi: 10.1139/y08-004. PMID:18418437. [DOI] [PubMed] [Google Scholar]
- [37].Gohy ST, Hupin C, Pilette C, Ladjemi MZ. Chronic inflammatory airway diseases:the central role of the epithelium revisited. Clin Exp Allergy. 2016;46(4):529-42. doi: 10.1111/cea.12712. PMID:27021118. [DOI] [PubMed] [Google Scholar]
- [38].Wendt MK, Smith JA, Schiemann WP. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010;29:6485-98. doi: 10.1038/onc.2010.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici-Barel Y, Salit J, Harvey BG, Crystal RG. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68(5):877-92. doi: 10.1007/s00018-010-0500-x. PMID:20820852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tsicopoulos A, de Nadai P, Glineur C. Environmental and genetic contribution in airway epithelial barrier in asthma pathogenesis. Curr Opin Allergy Clin Immunol. 2013;13(5):495-9. doi: 10.1097/ACI. PMID:23945177. [DOI] [PubMed] [Google Scholar]
- [41].Suzuki M, Itoh M, Ohta N, Nakamura Y, Moriyama A, Matsumoto T, Ohashi T, Murakami S. Blocking of protease allergens with inhibitors reduces allergic responses in allergic rhinitis and other allergic diseases. Acta Otolaryngol. 2006;126(7):746-51. doi: 10.1080/00016480500475625. PMID:16803715. [DOI] [PubMed] [Google Scholar]
- [42].Kim DW, Cho SH. Emerging Endotypes of Chronic Rhinosinusitis and Its Application to Precision Medicine. Allergy Asthma Immunol Res. 2017;9(4):299-306. doi: 10.4168/aair.2017.9.4.299. PMID:28497916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Davies DE. Epithelial barrier function and immunity in asthma. Ann Am Thorac Soc. 2014 Dec;11 Suppl 5:S244-51. doi: 10.1513/AnnalsATS.201407-304AW. [DOI] [PubMed] [Google Scholar]
- [44].Holgate ST. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev. 2011;242:205-19. doi: 10.1111/j.1600-065X.2011.01030.x. PMID:21682747. [DOI] [PubMed] [Google Scholar]
- [45].Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347-76. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- [46].Heijink IH, Postma DS, Noordhoek JA, Broekema M, Kapus A. House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am J Respir Cell Mol Biol. 2010;42:69-79. doi: 10.1165/rcmb.2008-0449OC. PMID:19372245. [DOI] [PubMed] [Google Scholar]
- [47].Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax 2014;69:760-5. doi: 10.1136/thoraxjnl-2013-204608. PMID:24334519. [DOI] [PubMed] [Google Scholar]
- [48].Lampugnani MG. Endothelial cell-to-cell junctions: adhesion and signaling in physiology and pathology. Cold Spring Harb Perspect Med. 2012;2(10). doi: 10.1101/cshperspect.a006528. PMID:23028127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol. 2012;12:53-9. doi: 10.1097/ACI.0b013e32834ec6eb. PMID:22217512. [DOI] [PubMed] [Google Scholar]
- [50].Johnson JR, Roos A, Berg T, Nord M, Fuxe J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS One. 2011; 6:e16175. doi: 10.1371/journal.pone.0016175. PMID:21283768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hupin C, Gohy S, Bouzin C, Lecocq M, Polette M, Pilette C. Features of mesenchymal transition in the airway epithelium from chronic rhinosinusitis. Allergy. 2014;69(11):1540-9. doi: 10.1111/all.12503. PMID:25104359. [DOI] [PubMed] [Google Scholar]
- [52].Steelant B, Farré R, Wawrzyniak P, Belmans J, Dekimpe E, Vanheel H, Van Gerven L, Kortekaas Krohn I, Bullens DM, Ceuppens JL, et al.. Impaired barrier function in patients with house dust mite-induced allergic rhinitis is accompanied by decreased occludin and zonula occludens-1expression. J Allergy Clin Immunol. 2016;137(4):1043-53.e1-5. doi: 10.1016/j.jaci.2015.10.050. PMID:26846377. [DOI] [PubMed] [Google Scholar]
- [53].Portelli MA, Hodge E, Sayers I. Genetic risk factors for the development of allergic disease identified by genome-wide association. Clin Exp Allergy. 2015;45(1):21-31. doi: 10.1111/cea.12327. PMID:24766371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Toppila-Salmi S, Renkonen J, Joenväärä S, Mattila P, Renkonen R. Allergen interactions with epithelium. Curr Opin Allergy Clin Immunol. 2011;11(1):29-32. doi: 10.1097/ACI.0b013e328342319e. PMID:21150436. [DOI] [PubMed] [Google Scholar]
- [55].Stevens WW, Schleimer RP, Kern RC. Chronic Rhinosinusitis with Nasal Polyps. J Allergy Clin Immunol Pract. 2016;4(4):565-72. doi: 10.1016/j.jaip.2016.04.012. PMID:27393770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Grainge CL, Davies DE. Epithelial injury and repair in airways diseases. Chest. 2013;144(6):1906-12. doi: 10.1378/chest.12-1944. PMID:24297122. [DOI] [PubMed] [Google Scholar]
- [57].Meng J, Zhou P, Liu Y, Liu F, Yi X, Liu S, Holtappels G, Bachert C, Zhang N. The development of nasal polyp disease involves early nasal mucosal inflammation and remodelling. PLoS One. 2013;8(12):e82373. doi: 10.1371/journal.pone.0082373. PMID:24340021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Vinhas R, Cortes L, Cardoso I, Mendes VM, Manadas B, Todo-Bom A, Pires E, Veríssimo P. Pollen proteases compromise the airway epithelial barrier through degradation of transmembrane adhesion proteins and lung bioactive peptides. Allergy. 2011;66(8):1088-98. doi: 10.1111/j.1398-9995.2011.02598.x. PMID:21480927. [DOI] [PubMed] [Google Scholar]
- [59].Tai HY, Tam MF, Chou H, Peng HJ, Su SN, Perng DW, Shen HD. Pen ch 13 allergen induces secretion of mediators and degradation of occludin protein of human lung epithelial cells. Allergy. 2006;61(3):382-8. doi: 10.1111/j.1398-9995.2005.00958.x. PMID:16436150. [DOI] [PubMed] [Google Scholar]
- [60].Pothoven KL, Norton JE, Hulse KE, Suh LA, Carter RG, Rocci EL, Harris KE, Shintani-Smith S, Conley DB, Chandra RK, et al.. Oncostatin M promotes mucosal epithelial barrier dysfunction, and its expression is increased in patients with eosinophilic mucosal disease. J Allergy Clin Immunol. 2015;136(3):737-746.e4. doi: 10.1016/j.jaci.2015.01.043. PMID:25840724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].2014. GINA REPORT, Global strategy for asthma management and prevention. Updated December 2011. Available from: http://www.ginasthma.org/GINA-Report,-Global-Strategy-for-Asthma-Management-and-Prevention. [Google Scholar]
- [62].Heijink IH, Nawijn MC, Hackett TL. Airway epithelial barrier function regulates the pathogenesis of allergic asthma. Clin Exp Allergy. 2014;44(5):620-30. doi: 10.1111/cea.12296. PMID:24612268. [DOI] [PubMed] [Google Scholar]
- [63].Blume C, Swindle EJ, Gilles S, Traidl-Hoffmann C, Davies DE. Low molecular weight components of pollen alter bronchial epithelial barrier functions. Tissue Barriers. 2015;3:e1062316. doi: 10.1080/15476286.2015. PMID:26451347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Moheimani F, Hsu AC, Reid AT, Williams T, Kicic A, Stick SM, Hansbro PM, Wark PA, Knight DA. The genetic and epigenetic landscapes of the epithelium in asthma. Respir Res. 2016;17(1):119. doi: 10.1186/s12931-016-0434-4. PMID:27658857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Barbato A, Turato G, Baraldo S, Bazzan E, Calabrese F, Panizzolo C, et al.. Epithelial damage and angiogenesis in the airways of children with asthma. Am J Respir Crit Care Med. 2006;174:975-981. doi: 10.1164/rccm.200602-189OC. PMID:16917118. [DOI] [PubMed] [Google Scholar]
- [66].Effros RM, Nagaraj H. Asthma: new developments concerning immune mechanisms, diagnosis and treatment. Curr Opin Pulm Med. 2007;13:37-43. doi: 10.1097/MCP.0b013e3280108757. PMID:17133123. [DOI] [PubMed] [Google Scholar]
- [67].Proud D. Biology of Epithelial Cells In: Adkinson NF, Bochner BS, Busse WW, editors. Middleton's Allergy: Principles and Practice. 7th edition. China: Mosby Elsevier Publishers; 2009. p. 373-86. [Google Scholar]
- [68].Blume C, Swindle EJ, Dennison P, Jayasekera NP, Dudley S, Monk P, Behrendt H, Schmidt-Weber CB, Holgate ST, Howarth PH, et al.. Barrier responses of human bronchial epithelial cells to grass pollen exposure. Eur Respir J. 2013;42(1):87-97. doi: 10.1183/09031936.00075612. PMID:23143548. [DOI] [PubMed] [Google Scholar]
- [69].Yuksel H, Turkeli A, Taneli F, Horasan GD, Kanik ET, Kizilkaya M, Gozukara C, Yilmaz O. E-cadherin as an epithelial barrier protein in exhaled breath condensate. J Breath Res. 2014;8(4):046006. doi: 10.1088/1752-7155/8/4/046006. PMID:25379974. [DOI] [PubMed] [Google Scholar]
- [70].Hammad H, Lambrecht BN. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity. 2015;43(1):29-40. doi: 10.1016/j.immuni.2015.07.007. PMID:26200011. [DOI] [PubMed] [Google Scholar]
- [71].Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18:673-83. doi: 10.1038/nm.2731. PMID:22561831. [DOI] [PubMed] [Google Scholar]
- [72].Winter MC, Shasby SS, Ries DR, Shasby DM. PAR2 activation interrupts E-cadherin adhesion and compromises the airway epithelial barrier: protective effect of beta-agonists. Am J Physiol Lung Cell Mol Physiol. 2006;291:628-35. doi: 10.1152/ajplung.00046.2006. [DOI] [PubMed] [Google Scholar]
- [73].Heijink IH, van Oosterhout A, Kapus A. Epidermal growth factor receptor signalling contributes to house dust mite-induced epithelial barrier dysfunction. Eur Respir J. 2010; 36: 1016-26. doi: 10.1183/09031936.00125809. PMID:20351035. [DOI] [PubMed] [Google Scholar]
- [74].Gershwin LJ. Effects of allergenic extracts on airway epithelium. Curr Allergy Asthma Rep. 2007;7:357-62. doi: 10.1007/s11882-007-0054-7. PMID:17697644. [DOI] [PubMed] [Google Scholar]
- [75].Narang I, Bush A. Early origins of chronic obstructive pulmonary disease. Semin Fetal Neonatal Med. 2012;17(2):112-8. doi: 10.1016/S0140-6736(14)60446-3. PMID:22265926. [DOI] [PubMed] [Google Scholar]
- [76].Khan EM, Lanir R, Danielson AR, Goldkorn T. Epidermal growth factor receptor exposed to cigarette smoke is aberrantly activated and undergoes perinuclear trafficking. FASEB J 2008; 22:910-7. doi: 10.1096/fj.06-7729com. PMID:17971399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Xu S, Xue X, You K, Fu J. Caveolin-1 regulates the expression of tight junction proteins during hyperoxia-induced pulmonary epithelial barrier breakdown. Respir Res. 2016;17(1):50. doi: 10.1186/s12931-016-0364-1. PMID:27176222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].You K, Xu X, Fu J, Xu S, Yue X, Yu Z, Xue X. Hyperoxia disrupts pulmonary epithelial barrier in newborn rats via the deterioration of occludin and ZO-1. Respir Res. 2012;13:36. doi: 10.1186/1465-9921-13-36. PMID:22559818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Martinez FD. The connection between early life wheezing and subsequent asthma:The viral march. Allergol Immunopathol (Madr). 2009;37(5):249-51. doi: 10.1016/j.aller.2009.06.008. PMID:19875225. [DOI] [PubMed] [Google Scholar]
- [80].Savenije OE, Mahachie John JM, Granell R, Kerkhof M, Dijk FN, de Jongste JC, Smit HA, Brunekreef B, Postma DS, Van Steen K, et al.. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J Allergy Clin Immunol. 2014;134(1):170-7. doi: 10.1016/j.jaci.2013.12.1080. PMID:24568840. [DOI] [PubMed] [Google Scholar]
- [81].Rezaee F, DeSando SA, Ivanov AI, Chapman TJ, Knowlden SA, Beck LA, Georas SN. Sustained protein kinase D activation mediates respiratory syncytial virus-induced airway barrier disruption. J Virol. 2013t;87:11088-95. doi: 10.1128/JVI.01573-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Shelfoon C, Shariff S, Traves SL, Kooi C, Leigh R, Proud D. Chemokine release from human rhinovirus-infected airway epithelial cells promotes fibroblast migration. J Allergy Clin Immunol. 2016;138(1):114-122.e4. doi: 10.1016/j.jaci.2015.12.1308. PMID:26883463. [DOI] [PubMed] [Google Scholar]
- [83].Holgate ST, Arshad HS, Roberts GC, Howarth PH, Thurner P, Davies DE. A new look at the pathogenesis of asthma. Clin Sci (Lond). 2009;118(7):439-50. doi: 10.1042/CS20090474. PMID:20025610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ho SM. Environmental epigenetics of asthma: an update. J Allergy Clin Immunol. 2010;126(3):453-65. doi: 10.1016/j.jaci.2010.07.030. PMID:20816181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Grotenboer NS, Ketelaar ME, Koppelman GH, Nawijn MC. Decoding asthma: Translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J Allergy Clin Immunol 2013;131:856-65. doi: 10.1016/j.jaci.2012.11.028. PMID:23380221. [DOI] [PubMed] [Google Scholar]
- [86].Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, von Mutius E, Farrall M, Lathrop M, Cookson WO, GABRIEL Consortium . A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211-21. doi: 10.1056/NEJMoa0906312. PMID:20860503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Roth HM, Wadsworth SJ, Kahn M, Knight DA. The airway epithelium in asthma: developmental issues that scar the airways for life? Pulm Pharmacol Ther. 2012Dec;25(6):420-6[62].doi: 10.1016/j.pupt.2012.09.004.. [DOI] [PubMed] [Google Scholar]
- [88].Burrows B, Knudson RJ, Lebowitz MD. The relationship of childhood respiratory illness to adult obstructive airway disease. Am Rev Respir Dis 1977;115:751e60. doi: 10.1164/arrd.1977.115.5.751. [DOI] [PubMed] [Google Scholar]
- [89].Barker DJ, Godfrey KM, Fall C, Osmond C, Winter PD, Shaheen SO. Relation of birth weight and childhood respiratory infection to adult lung function anddeath from chronic obstructive airways disease. BMJ 1991;303:671e5. doi: 10.1136/bmj.303.6804.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Bush A. Lung Development and Aging. Ann Am Thorac Soc. 2016 Dec;13(Supplement_5):S438-S446. doi: 10.1513/AnnalsATS.201602-112AW. [DOI] [PubMed] [Google Scholar]
- [91].Kerkhof M, Boezen HM, Granell R, Wijga AH, Brunekreef B, Smit HA, et al.. Transient early wheeze and lung function in early childhood associated with chronic obstructive pulmonary disease genes. J Allergy Clin Immunol. 2014 Jan;133(1):68-76.e1-4. doi: 10.1016/j.jaci.2013.06.004. [DOI] [PubMed] [Google Scholar]
- [92].Reynolds PR, Kasteler SD, Cosio MG, Sturrock A, Huecksteadt T, Hoidal JR. RAGE: Developmental expression and positive feedback regulation by egr-1 during cigarette smoke exposure in pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1094-101. doi: 10.1152/ajplung.00318.2007. PMID:18390831. [DOI] [PubMed] [Google Scholar]
- [93].Tagiyeva N, Devereux G, Fielding S, Turner S, Douglas G. Outcomes of childhood asthma and wheezy bronchitis: a 50-year cohort study. Am J Respir Crit Care Med. 2016;193:23-30. doi: 10.1164/rccm.201505-0870OC. PMID:26351837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yuksel H, Yilmaz O, Karaman M, Bagriyanik HA, Firinci F, Kiray M, Turkeli A, Karaman O. Role of vascular endothelial growth factor antagonism on airway remodeling in asthma. Ann Allergy Asthma Immunol. 2013;110(3):150-5. doi: 10.1016/j.anai.2012.12.015. PMID:23548522. [DOI] [PubMed] [Google Scholar]
- [95].Yuksel H, Yilmaz O, Karaman M, Firinci F, Turkeli A, Kanik ET, Inan S. Vascular endothelial growth factor antagonism restores epithelial barrier dysfunction via affecting zonula occludens proteins. Exp Ther Med. 2015;10(1):362-8. doi: 10.3892/etm.2015.2502 PMID:26170963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. 2009 Oct 27;10:100. doi: 10.1186/1465-9921-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Song J, Zhao H, Dong H, Zhang D, Zou M, Tang H, Liu L, Liang Z, Lv Y, Zou F, et al.. Mechanism of E-cadherin redistribution in bronchial airway epithelial cells in a TDI-induced asthma model. Toxicol Lett. 2013;220(1):8-14. doi: 10.1016/j.toxlet.2013.03.033. PMID:23566898. [DOI] [PubMed] [Google Scholar]
- [98].Wawrzyniak P, Wawrzyniak M, Wanke K, Sokolowska M, Bendelja K, Rückert B, Globinska A, Jakiela B, Kast JI, Idzko M, et al.. Regulation of bronchial epithelial barrier integrity by type 2 cytokines and histonedeacetylases in asthmatic patients. J Allergy Clin Immunol. 2017;139(1):93-103. doi: 10.1016/j.jaci.2016.03.050. PMID:27312821. [DOI] [PubMed] [Google Scholar]
- [99].Sohal SS, Soltani A, Reid D, Ward C, Wills KE, Muller HK, Walters EH. A randomized controlled trial of inhaled corticosteroids (ICS) on markers of epithelial-mesenchymal transition (EMT) in large airway samples in COPD: an exploratory proof of concept study. Int J Chron Obstruct Pulmon Dis. 2014;9:533-42. doi: 10.2147/COPD.S63911. PMID:24920891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Heijink I, van Oosterhout A, Kliphuis N, Jonker M, Hoffmann R, Telenga E, Klooster K, Slebos DJ, ten Hacken N, Postma D, et al.. Oxidant-induced corticosteroid unresponsiveness in human bronchial epithelial cells. Thorax. 2014;69(1):5-13. doi: 10.1136/thoraxjnl-2013-203520. PMID:23980116. [DOI] [PubMed] [Google Scholar]
- [101].Yilmaz O, Yuksel H. Where does current and future pediatric asthma treatment stand? Remodeling and inflammation: Bird's eye view. Pediatr Pulmonol. 2016;51(12):1422-9. doi: 10.1002/ppul.23488. PMID:27233079. [DOI] [PubMed] [Google Scholar]
- [102].Yang K, Song Y, Tang YB, Xu ZP, Zhou W, Hou LN, Zhu L, Yu ZH, Chen HZ, Cui YY. mAChRs activation induces epithelial-mesenchymal transition on lung epithelial cells. BMC Pulm Med. 2014;14:53. doi: 10.1186/1471-2466-14-53. PMID:24678619. [DOI] [PMC free article] [PubMed] [Google Scholar]