

Abstract
Pancreatic-Derived Factor (PANDER, FAM3B) is a member of the FAM3 family of cytokine molecules that were initially described in 2002. PANDER expression is primarily localized to the endocrine pancreas and is secreted from both pancreatic α and β-cells. Initial characterization of PANDER revealed a potential role in pancreatic islet apoptosis. However, recent animal models have indicated PANDER functions as a hormone by regulating glucose levels via interaction with both the liver and the endocrine pancreas. An understanding of the function of PANDER can further the insight into the mechanisms of glucose regulation and potentially provide additional therapeutic targets for the treatment of diabetes. This review details the supporting data demonstrating PANDER has a biological function in glycemic regulation.
Keywords: PANDER, FAM3B, glucose regulation, islets
1. Introduction to PANDER: a novel endocrine hormone
PANcreatic-DERived factor (PANDER, FAM3B) was originally described in 2002 as a result of a search for novel cytokines. PANDER was primarily localized to the endocrine pancreas and identified in both α and β-cells. Initial characterization evaluating the impact of PANDER on islets revealed induction of apoptosis via intracellular viral delivery or exogenous recombinant protein application. Induction of islet apoptosis via PANDER is regulated via a caspase-3-dependent mechanism. Additional microarray analysis indicated PANDER induced activation of various cell death pathways via both caspase-3 and cyclin-dependent kinase inhibitor 1A (p21). These initial results presented a case for PANDER as a potential activator or cofactor in the induction of islet apoptosis with regard to type 1 diabetes. However, recent evidence has begun to mount indicating another biological role for PANDER in the regulation of glycemic levels via interaction with both the liver and the pancreas. Furthermore, additional studies have demonstrated that PANDER is significantly regulated by both glucose and insulin and further supports a biological role in glycemic regulation. The initial murky picture of PANDER has become clearer with the recent creation and characterization of PANDER animal models including both the knockout and acute overexpression model. These recent findings have warranted a comprehensive review of the studies surrounding PANDER and now significantly demonstrate that PANDER represents a novel hormone with pleiotropic effects on multiple tissues impacting euglycemia.
2. Identification of PANDER
PANDER is one of four members of the family with sequence similarity 3 (FAM3) of predicted cytokine-like proteins, discovered through a screen for new cytokines based on the characteristic secondary structure of a four-helix bundle with up-up-down-down topology that is stabilized by disulfide bonds formed between conserved cysteines [1]. Whereas primary sequence homology is very low among cytokines, this secondary structure is highly conserved [2] and present in many cytokines, including interleukin (IL)-2,−3, −4, −5, −6, −7, −9, −10, and −13, granulocyte-macrophage colony stimulating factor (GM-CSF) and growth hormone (GH).
To search for novel families that fit these parameters, the algorithm Ostensible Recognition of Folds (ORF), which predicts the likelihood that an amino acid in a primary sequence will be in an alpha helix (a), a beta sheet (b) or a coil (c), was employed [3]. Using probes derived from short chain cytokines such as IL-2, −4 and GM-CSF, the first member of the family with sequence similarity 3 member A (FAM3A), was discovered and later confirmed to have the four-helix bundle structure with four conserved cysteines. Subsequently, three putative genes were identified that had significant homology to FAM3A but not to any other genes. These were later named FAM3B, FAM3C and FAM3D and comprise the other members of the FAM3 family. All members have 224–235 amino acids, including 4 conserved cysteines and a predicted signal sequence. Although amino acid sequence homology is only 31.6–53.3% within the family, the members have no significant similarity with any other known cytokines.
FAM3B was later renamed Pancreatic-derived factor (PANDER) [4] due its robust expression in the endocrine pancreas. Human FAM3B shares 78% primary sequence homology with its murine homolog and also contains 235 amino acids with 4 conserved cysteine’s and secretion signal peptide.
3. Tissue-specific expression within the endocrine pancreas
Characterization of PANDER’s tissue distribution by northern blot analysis demonstrated both human and murine PANDER were very highly expressed in the pancreas and to a minimal extent in the small intestine followed by the least expression in the prostate [1]. A recent study detected PANDER mRNA in a broad range of tissues including muscle and liver [5], but this has yet to be confirmed by other groups. Quantitative real time PCR (q-PCR) analysis of various murine tissues further confirmed the pancreatic expression of PANDER [6]. Similarly, murine PANDER mRNA levels have been quantified in various pancreas-derived cell lines. Using muscle-derived C2C12 cells as a negative control, the α-cell derived cell line, α-TC3 had the highest level of PANDER mRNA compared to the β-cell derived β-TC3 cells [7, 8]. Similarly, in fluorescence-activated cell sorted murine pancreatic islet cells, PANDER mRNA expression was highest in the glucagon positive, α-cell enriched population, at levels 6-fold more than in the insulin positive, β-cell enriched population. PANDER mRNA was also detected in α-TC1-9 and α-TC1-6 α-cell lines and the MIN-6 β-cell line [9, 10]. However, despite relatively high PANDER expression in these cell lines, murine islets have mRNA levels at least 100-fold greater than in pancreatic-derived cell lines [7].
Immunohistochemical (IHC) staining for murine PANDER protein revealed specific localization to the islet of Langerhans of the endocrine pancreas with no expression in the exocrine pancreas. This was confirmed in both human and rat islets, in which immuno-fluorescence staining for PANDER overlapped significantly with both insulin positive β-cells and glucagon positive α-cells. PANDER protein was also detected in β-TC3 and α-TC3 cell lines [4, 8].
4. Localization within pancreatic α and β-cells
Analysis of PANDER’s sub-cellular localization within β-cells by immunogold electron microscopy and confocal microscopy indicated that PANDER was co-localized with insulin to secretory vesicles [8]. In αTC1-6 cells, PANDER was detected in the glucagon-negative granular cytosolic compartment including cytoplasmic granules, neuroendocrine vesicles, endoplasmic reticulum (ER), and the cis-Golgi [10].
5. Transcriptionally regulated expression
The murine PANDER gene consists of 8 exons spanning an approximate 32 kb region located on chromosome 16B-C4 [1]. Human chromosomal location for PANDER is syntenic to human chromosome 21q22. For murine PANDER, a single transcriptional start site located 520 bp upstream of the translational start codon has been identified [7]. The murine promoter contains several transcriptional binding sites for elements that are known to confer both pancreatic-islet specific and glucose-responsive expression similar to that of insulin [7]. In the region between −335 to +523 relative to the transcriptional start site there are binding sites for A and E box elements, hepatic nuclear factors (HNF) 1 and 4α, and signal transducer and activation of transcription (STAT) 3, 5, and 6. These A and E box elements were hypothesized to confer tissue specific PANDER expression by allowing the interaction of transcription factors such as Pancreatic/Duodenal Homeobox domain-1 (PDX-1) and β-cell E box Transactivator 2 (BETA2/NeuroD) respectively, which are important for pancreas development and conferring islet specific expression of many islet genes. In fibroblast cells, co-transfection of a construct that expresses luciferase under control of the PANDER promoter, with plasmids directing over expression of islet-specific transcription factors (TFs) MafA, BETA2/NeuroD and PDX-1 revealed that although all three TFs individually increased luciferase activity above control levels, PDX-1 had the greatest impact. Furthermore, PDX-1 bound to three A box sites within the PANDER promoter, and simultaneous ablation of two of these sites significantly abrogated PANDER promoter activity [11].
Besides conferring tissue specific expression, these binding motifs are also known to promote glucose responsive expression. Glucose robustly induced PANDER promoter activity in both β-TC3 cells and mouse islets in a dose-dependent manner, and PDX-1 may be involved in this glucose responsive regulation [11]. Wang et al. confirmed this glucose responsive regulation in MIN6 cells, in which glucose significantly increased PANDER transcription via a cAMP regulator element binding protein (CREB) -dependent mechanism [9]. Glucose-stimulated transcription occurred via either Ca2+/PKA/ERK1/2 or Ca2+/PKC/ pathway, and blockage of these pathways significantly reduced PANDER promoter activity and gene expression. No glucose responsive promoter activity was observed in α-TC3 cells despite high levels of PANDER mRNA [7]. In addition to glucose, a combination of the pro-inflammatory cytokines, interleukin-1β (IL-1β), tumor necrosis factor (TNFα), and interferon-γ (IFNγ) also robustly increased PANDER mRNA in β-TC3 cells and mouse islets [8] (Fig. 1). IFNγ alone produced a 3-fold increase that was both time- and dose-dependent.
Figure 1. Secretagogues inducing PANDER expression or secretion.
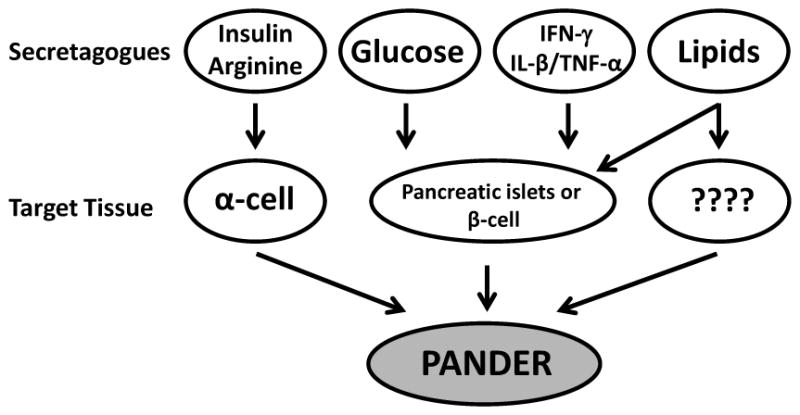
Various factors have been demonstrated to regulate PANDER. These include insulin, arginine, glucose, IFNγ, TNFα, IL-1β and lipids. Insulin and arginine have increased PANDER expression and secretion from the α-TC1-6 glucagonoma cell line. Glucose has been significantly demonstrated to increase PANDER expression and secretion from both isolated pancreatic islets and various pancreatic β-cell lines. IFNγ alone or in combination with TNFα and IL-1β induce PANDER mRNA expression in both primary islets and pancreatic β-cell lines. A single study has indicated that PANDER mRNA and protein levels are increased in the livers of high-fat diet-fed mice and insulin-resistant db/db mice. Concordantly, aged and fat mice have increased levels of serum PANDER as compared to lean control mice. However, given the fact that PANDER is expressed to some degree in other tissues (i.e. small intestine, prostate, and potentially liver) there is uncertainty as to the source of increased PANDER secretion in these high-fat models. Nonetheless, there is a preponderance of evidence that indicates that lipids are inducing an increase in PANDER expression potentially from multiple tissues.
6. Glucose and insulin regulated secretion
Like the other members of the FAM3 family, PANDER has a hydrophobic leader sequence on its N-terminus suggesting that it will be targeted to the ER and processed within cells for secretion. As previously mentioned, PANDER is localized within the ER and Golgi of αTC1-6 cells and in secretory vesicles in β cells. In CHO cells overexpressing human PANDER, PANDER was detected in the culture media and determined to be a monomer by analytical ultracentrifugation and non-reducing gel electrophoresis. Two N-termini were identified at either Glutamate 30 or Serine 46 [1]. Similarly, two N-termini were identified for murine PANDER expressed in Pichia Pastoris, Serine 46 and Alanine 55, a possible variant product from Pichia production. Cleavage at these two sites resulted in proteins of molecular weights 21 and 20 kDa respectively. In transfected β-TC3 cells or primary hepatocytes infected with PANDER adenovirus, 26 and 23 kDa products were detected in lysate and media respectively, presumably the full length and Glutamate 30-cleaved PANDER, potentially carrying posttranslational modifications due to expression in mammalian cells.
The localization of PANDER to insulin granules in β-cells suggested that PANDER might be co-secreted with insulin in response to glucose. In β cell lines, PANDER and insulin secretions were increased with glucose incubation in a dose- and time-dependent manner [9, 12] (Fig. 1). Potassium chloride, a potent insulin secretagogue, also caused a robust increase in PANDER secretion. Chronic glucose stimulation also significantly increased PANDER secretion from mouse islets overexpressing PANDER in β-cells. Therefore, it was concluded that PANDER secretion from β-cells is regulated in a manner similar to insulin secretion.
In α-cells, neither increasing nor decreasing glucose concentrations had any effect on PANDER secretion or content. This suggested that in the whole islets, the dominant source of secreted PANDER in response to glucose is the β-cell. However in αTC1-6 cells, arginine, a potent stimulator of glucagon secretion, increased PANDER secretion at concentrations ≥ 10 mM [10] (Fig. 1). Surprisingly, low insulin concentrations (17 or 50 nM) also stimulated PANDER release from α-cells more robustly than arginine. Studies performed by Carnegie et al. utilizing a C-terminal PANDER-FLAG fusion protein revealed PANDER localization to a glucagon-negative granular cytoplasmic compartment in α-TC1-6 cells. PANDER, if it is secreted in tandem with glucagon, is not likely to contribute to PANDER levels in the blood during stimulated insulin secretion when paracrine regulated glucagon release may be inhibited by insulin. Nonetheless, the nutrient and hormonal regulation of PANDER is consistent with a potential glucoregulatory function (Fig. 1).
7. Biological impact of PANDER in-vitro and in-vivo
To β or not to β?: Induction of β-cell apoptosis and impact on β-cell function
Initial assessment of a potential autocrine effect of PANDER demonstrated that islets and β-TC3 cells treated with recombinant PANDER (typically in the nanomolar range) or PANDER adenovirus (which expresses full length PANDER) had high levels of apoptotic and necrotic cell death compared to untreated cells [4, 13]. The increase in apoptosis was not accompanied by changes in the protein levels of apoptotic mediators such as Akt, STAT1, NF-κB, and Fas known to be important for cytokine-induced autoimmune β-cell death [13]. However, PANDER-induced apoptosis was mediated by caspase-3 cleavage, without any effect on intracellular Ca2+ or nitric oxide (NO) production suggesting the involvement of “noncanonical” pathways.
In a microarray study of PANDER-treated islets, 20% of the genes differentially expressed after 48 hrs were associated with cell death compared to 47% after 72 hrs of treatment [14]. Genes with the highest fold changes included cyclin-dependent kinase inhibitor 1A (CDKN1A/p21), insulin like growth factor binding protein (IGFBP4) and lipocalin 2 (LCN2). The anti-apoptotic gene CDKN1A/p21, downregulated 3.4 fold with PANDER treatment, appeared to be central to several apoptotic networks which included caspase-3 and 7. Cleaved caspase-3 protein levels were also increased in PANDER-treated β-TC3 cells with a subsequent decrease in CDKN1A, indicating that CDKN1A/caspase-3 network appears to be important for PANDER-induced β-cell death.
In addition to β-cell apoptosis, recombinant PANDER treatment decreased basal insulin secretion from β-TC3 cells but had no effect on glucose stimulated secretion or β-cell glucose metabolism [1]. Overexpression of PANDER in isolated murine islets via adenoviral delivery or recombinant protein application had no impact on glucose-stimulated insulin secretion (GSIS), yet insulin secretion was impaired in the presence of glucose with various insulin secretagogues such as carbachol (muscarinic agonist) or potassium chloride [13]. From these experiments, it was concluded that PANDER appears to impact the amplifying not initiating signals of GSIS suggesting potential defects in calcium signaling. Unfortunately, calcium signaling was not evaluated within islets overexpressing PANDER.
PANDER−/− (PANKO) mice did not display any abnormalities in pancreatic islet architecture, or expression of various genes involved in GSIS [6]. However, with regard to function, insulin secretion in response to arginine injection was blunted and pancreatic islets isolated from these mice showed diminished GSIS and abnormal Ca2+ response to glucose. The intracellular calcium response in PANKO islets differed from wild-type with the following observations: hyper-response of Ca2+ at low glucose (3 mM) conditions; lack of transient Ca2+ decrease immediately following high glucose (16 mM) stimulation attributed to uptake by the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) pumps; impaired amplitude of Ca2+ at high glucose condition, and blunted KCl response. The observed abnormal Ca2+ handling identified in the pancreatic islets may provide a causative mechanism for the observed impaired GSIS in-vitro and glucose intolerance observed in-vivo (Table 1). However, in mice overexpressing PANDER by adenoviral gene delivery with targeted hepatic PANDER overexpression, increasing circulating PANDER levels did not affect fractional insulin secretion or islet insulin content [15] (Table 1). These findings are discordant from the results in both the PANKO mice and isolated pancreatic islets overexpressing PANDER and this may be attributed to several reasons including different experimental models (KO versus liver-specific overexpression) or site and amount of PANDER overexpression (liver vs. pancreatic islets). Overall, the compilation of results indicate that PANDER is acting within the pancreatic islets to promote β-cell function and suggests a role for intracellular PANDER in the regulation or facilitation of insulin secretion (Fig. 2).
Table 1.
Phenotypic comparison of PANDER adenoviral and knockout mice
| Ad-PANDER | PANDER KO | |
|---|---|---|
| Fasting Blood Glucose | Elevated | No difference in mice on normal diet Lower in mice on HFD |
| Fasting Serum Metabolites | ↑ Insulin ↑ Corticosterone |
↑ Insulin ↑ Leptin |
| Glucose Tolerance | Impaired due to fasting hyperglycemia | Impaired due to impaired β-cell function |
| Hepatic Glucose Production by HE-clamp |
Not Evaluated | Decreased |
| Glucose Production from primary hepatocytes | Increased | Not Evaluated |
| Gluconeogenic gene expression (PEPCK and G6Pase) | Elevated | Decreased |
| β-cell cell function | Normal | Decreased |
Figure 2. Mechanism of PANDER action during the fasted and fed state in the regulation of euglycemia.
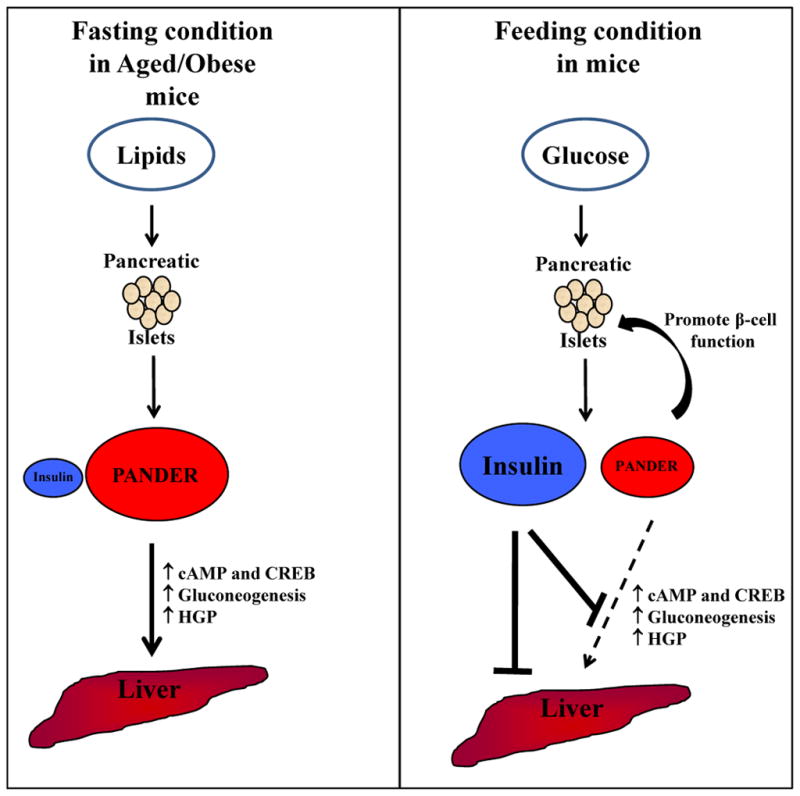
Evidence to date indicates that PANDER is a novel hormone that is counterregulatory to insulin. In addition, PANDER has been demonstrated to promote pancreatic β-cell function but not stimulate but rather enhance or facilitate insulin secretion. Multiple secretagogues have been identified that stimulate PANDER secretion from pancreatic islets including glucose, lipids, and insulin (for α-cells). Therefore, postprandial ingestion of these various nutrients and subsequent release of insulin would induce PANDER release.
In lean mice, PANDER levels are low during fasting but in age- or diet-induced obesity, PANDER levels are high in fasted mice possible due to a stimulatory effect of lipids on the β-cells (left panel). In the absence of high insulin levels, high circulating PANDER levels impact the liver to increase cAMP and pCREB with subsequent upregulation of critical gluconeogenic enzymes of G6Pase and PEPCK gene expression. This results in an overall increase in HGP and fasting hyperglycemia.
With refeeding, glucose stimulates both PANDER and insulin secretion from the β-cells (right panel). In the presence of high insulin levels and its strong suppressive effect on HGP, the effect of PANDER on the liver is blocked and HGP is shut down. Therefore, PANDER expression and secretion induced during postprandial conditions would be to strictly enhance β-cell function and calcium handling within the pancreatic islet itself. However, distal action in the liver would be serving a function counter-regulating insulin action.
8. PANDERing elsewhere - Regulation of hepatic glucose production
In a screen for extra-pancreatic targets of PANDER, specific binding of 125I-PANDER to liver membrane was detected [16] indicating that there might be a hepatic PANDER receptor. In HepG2 cells, a human hepatoma cell line, recombinant PANDER treatment resulted in an inhibition in insulin signaling as measured by decreased activation of the insulin receptor-β (IRβ), insulin receptor substrate-1 (IRS-1), phosphatidylinositol-3′-OH-kinase (PI3K), and Akt [16]. In primary murine hepatocytes, PANDER treatment increased cAMP and cAMP-responsive element binding protein (CREB) signaling [15]. These effects were proposed to have occurred via interaction with the unidentified hepatic PANDER receptor.
The effect of circulating PANDER on the liver appears to be important for regulating hepatic glucose production (HGP) during fasting. In mice, PANDER overexpression promotes hyperglycemia, compensatory hyperinsulinemia, decreased liver triglycerides, and elevated corticosterone levels only in fasted mice, [15] (Table 1), but does not affect ad libitum fed glucose levels or serum metabolites. Even the glucose intolerance observed in these mice was due to elevated fasting glucose levels. The increase in fasting glucose was accompanied by increased transcription of hepatic phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase). In vitro, PANDER-treated hepatocytes demonstrated elevated glucose production and gluconeogenic gene expression. Mechanistically, the effect on gluconeogenic transcription and glucose output appeared to be due to enhanced cAMP and CREB signaling after stimulation with forskolin or 8-bromocAMP and dexamethasone respectively and not to any impairment in insulin’s ability to suppress HGP.
Concordantly, PANKO mice displayed a decrease in HGP during a hyperinsulinemic-euglycemic clamp and decreased gluconeogenic gene expression in fasting PANKO mice compared to WT controls. Surprisingly, glucose levels were not altered in PANKO mice compared to controls. The compilation of data from both the animal models and in-vitro data indicate that during fasting, the liver represents a strong distal target for PANDER action with subsequent impact on hepatic gluconeogenesis and overall regulation of euglycemia (Fig. 2).
In addition to the extracellular actions we have observed, Li et al reported that PANDER is found in hepatocytes, with increased expression in steatotic livers of diabetic (db/db) and high fat diet (HFD, 45% fat by calories) fed mice. Furthermore, knockdown of PANDER within the liver attenuated hyperglycemia (and insulin resistance) in db/db mice. Concordantly, PANDER overexpression in liver promoted lipogenesis, lipid accumulation, and Forkhead box 1 (FOXO1) expression, Furthermore, overexpression of FOXO1 concordantly increased hepatic PANDER levels [5]. No information on PANDER secretion from the liver or the effect on circulating PANDER levels was reported.
9. PANDER, Lipids and Hyperglycemia
Presently, the only physiological condition under which we have observed chronically elevated circulating PANDER levels is in fat, aged C57Bl/6 mice [17]. Two striking differences in PANDER levels were observed in these mice compared to young, lean mice: (1) PANDER levels were significantly higher both during feeding and fasting and (2) serum levels in fat mice did not decrease with fasting as is seen in lean mice (levels are 2-fold lower with fasting), but remained high. These data suggest that there is a positive correlation between increased body fat and elevated PANDER levels. Furthermore, while ad libitum fed glucose levels were not significantly different between the two groups, after a 24-hr fast, fat mice were hyperglycemic compared to lean mice. Therefore, elevated fasting PANDER levels in these fat mice may be an important contributor to fasting hyperglycemia due to increased hepatic glucose production. While increased triglyceride storage may promote hyperglycemia [18–20], it is unlikely that PANDER is just an innocent bystander. For example, in mice overexpressing PANDER in the β-cell, there was an increased onset of HFD-induced glucose intolerance compared to WT controls [21].
Conversely, while there were no differences in fasted glucose levels in the PANKO mice on normal chow diet, after HFD feeding for 10 weeks, glucose levels following a 24- or 48-hr fast were significantly lower in PANKO mice [17, 22] and more similar to their normal diet (ND, 16.7% fat by calories) fed counterparts. This suggests that loss of PANDER may protect mice on a HFD from developing gross fasting hyperglycemia and that PANDER may be an important target for inhibiting HFD-induced fasting hyperglycemia.
Mechanistically, a positive effect of lipids on PANDER secretion is not implausible as free fatty acids (FFAs) promote insulin secretion. As PANDER is co-stored and -secreted with insulin, higher PANDER levels in fat mice may be due to a FFA-induced stimulation of insulin secretory vesicles. A similar effect has been observed for amylin, a peptide that is also localized to insulin secretory vesicles and co-secreted with insulin [23–25]. FFAs increase amylin mRNA expression and protein [26] and long-term HFD feeding in mice increases plasma amylin levels [27, 28]. Amylin secretion is also increased in obesity, and insulin resistance and decreased in patients with T2DM [29]. A similar mechanism might explain higher circulating PANDER levels in fat mice.
10. PANDER as a therapeutic target for diabetes
Type 2 diabetes is characterized by a progressive loss of glucose tolerance over several years due to three key abnormalities: impaired insulin secretion, impaired insulin action in liver, muscle, adipose, and abnormal splanchnic glucose metabolism [30–32]. Prior to the development of diabetes, many pre-diabetic patients present with impaired fasting glucose (fasting hyperglycemia) and/or impaired glucose tolerance (IGT) [33, 34], which are both risk factors for the development of T2DM [35]. Additionally, there is a strong correlation between the severity of fasting hyperglycemia in T2DM and increased HGP [36, 37]. As fasting hyperglycemia predicts unusually high postprandial glucose in humans [38], the development of drugs to treat this condition has strong clinical relevance.
Data generated from both of our PANDER animal models have revealed multiple metabolic abnormalities that are shared with the pathogenesis of type 2 diabetes including increased hepatic glucose output (Table 1). Therefore, the significance of PANDER is that it represents a potential molecule that may impact diabetes based on the effect that PANDER has on target tissues involved in glucose regulation.
It is interesting to note that in fed insulin sensitive mice there, does not appear to be an effect of PANDER on the liver to promote HGP. This is likely due to two reasons. 1) Insulin exerts a very strong suppressive effect on HGP even if PANDER is present and 2) PANDER enhances stimulated levels HGP but cannot itself directly increase HGP. Therefore in the fed state the liver does not appear to be PANDER’s primary target. However, in fasted mice, signals that act via cAMP and CREB dependent pathways increase gluconeogenic gene expression and HGP. Furthermore, low insulin levels during fasting do not inhibit these processes. When PANDER levels are high under these conditions, such as in aged, fat mice, then PANDER enhances cAMP and CREB signaling to promote fasting hyperglycemia. In addition, small interfering RNA (siRNA)-mediated knockdown of hepatic PANDER significantly attenuated steatosis, insulin resistance, and fasting levels in db/db mice [5]. Therefore, PANDER’s effect on the liver is revealed under the pathophysiological state of excess fat and insulin resistance.
One potentially attractive target for a novel class of type 2 diabetic compounds could be focused on the inhibition of PANDER hepatic signaling to block HGP and improve hepatic insulin sensitivity either at the receptor level or intracellularly. Furthermore, reducing circulating PANDER may also be beneficial as high PANDER levels may indirectly promote fasting hyperglycemia via corticosterone (the active glucocorticoid in mice) which is significantly higher in fasted mice overexpressing PANDER [15]. Likewise, many rodent models of T2DM are associated with elevated glucocorticoids levels [39] but antagonism of hepatic glucocorticoid signaling normalizes elevated HGP and improves glycemic control [40, 41].
Furthermore, conditions that perpetuate type 2 diabetes including hyperglycemia, and hyperlipidemia would all contribute to hyperPANDERemia since these conditions have previously been demonstrated to increase PANDER expression and would subsequently result in induction of increased HGP and glycemic levels (Fig. 3). Therefore, not only might therapeutic intervention of PANDER action decrease glycemic levels but also inhibit the further progression of type 2 diabetes.
Figure 3. PANDER and the potential progression of type 2 diabetes.
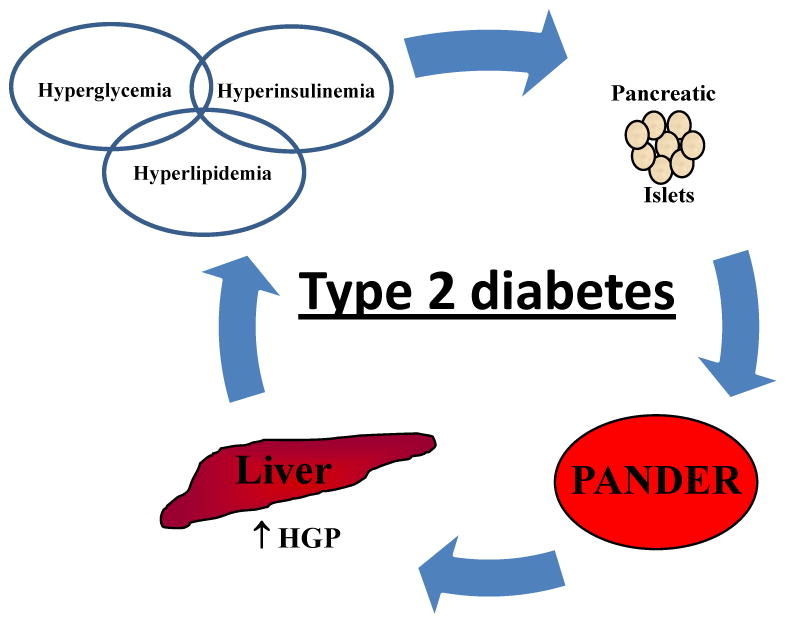
Type 2 diabetes is characterized by a progressive loss of glucose tolerance as a result of impaired insulin sensitivity. Several hallmarks of this disease include increased glycemic and insulin levels along with potential hyperlipidemia. Concordantly, both glucose and insulin have been identified to stimulate PANDER secretion from both pancreatic β and α-cells, respectively. In addition, PANDER sera levels are increased in aged and heavier mice. This condition may also stimulate PANDER levels during type 2 diabetes or metabolic syndrome and ultimately result in increased hepatic glucose production and increased glycemic levels. Therefore, one potential therapeutic option would be to inhibit PANDER hepatic action and improve glycemic control via inhibition of hepatic glucose output.
11. Concluding Remarks
In conclusion, as the field of PANDER research continually expands it has become more evident that PANDER represents a novel hormone that serves a biological function in the regulation of glycemic levels via interaction with both the endocrine pancreas and liver. PANDER is intimately coupled to insulin with regard to modification of both its action and secretion. Several of the causative parameters associated with both type 1 (increased cytokine expression) and type 2 (hyperlipidemia, hyperglycemia, and hyperinsulinemia) diabetes may increase circulating PANDER levels and perpetuate hyperglycemia and glucose intolerance. This may initiate or exacerbate progression of diabetes mellitus (Fig. 3) and therefore the inhibition of hepatic PANDER action may provide a therapeutic option to inhibit the further deterioration of glycemic control.
Highlights.
PANDER serves as a novel hormone regulating glycemic levels.
PANDER is positively regulated by glucose and insulin.
PANDER impacts pancreatic β-cell glucose-stimulated insulin secretion.
PANDER interacts with liver and increases hepatic glucose production.
High fat animal models demonstrate increased PANDER expression and hepatic action.
Acknowledgments
This work was supported by grant K01-DK070744 (to B.R.B.) from the NIDDK, National Institutes of Health and the Juvenile Diabetes Research Foundation.
Footnotes
DISCLOSURE STATEMENT: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhu Y, Xu G, Patel A, McLaughlin MM, Silverman C, Knecht K, Sweitzer S, Li X, McDonnell P, Mirabile R, et al. Cloning, expression, and initial characterization of a novel cytokine-like gene family. Genomics. 2002;80:144–150. doi: 10.1006/geno.2002.6816. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Meguid SS, Shieh HS, Smith WW, Dayringer HE, Violand BN, Bentle LA. Three-dimensional structure of a genetically engineered variant of porcine growth hormone. Proc Natl Acad Sci U S A. 1987;84:6434–6437. doi: 10.1073/pnas.84.18.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aurora R, Rose GD. Seeking an ancient enzyme in Methanococcus jannaschii using ORF, a program based on predicted secondary structure comparisons. Proc Natl Acad Sci U S A. 1998;95:2818–2823. doi: 10.1073/pnas.95.6.2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao X, Gao Z, Robert CE, Greene S, Xu G, Xu W, Bell E, Campbell D, Zhu Y, Young R, et al. Pancreatic-derived factor (FAM3B), a novel islet cytokine, induces apoptosis of insulin-secreting beta-cells. Diabetes. 2003;52:2296–2303. doi: 10.2337/diabetes.52.9.2296. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Chi Y, Wang C, Wu J, Yang H, Zhang D, Zhu Y, Wang N, Yang J, Guan Y. Pancreatic-derived factor promotes lipogenesis in the liver: Role of FOXO1 signaling pathway. Hepatology. doi: 10.1002/hep.24295. In-press. [DOI] [PubMed] [Google Scholar]
- 6.Robert-Cooperman CE, Carnegie JR, Wilson CG, Yang J, Cook JR, Wu J, Young RA, Wolf BA, Burkhardt BR. Targeted disruption of pancreatic-derived factor (PANDER, FAM3B) impairs pancreatic beta-cell function. Diabetes. 2010;59:2209–2218. doi: 10.2337/db09-1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burkhardt BR, Yang MC, Robert CE, Greene SR, McFadden KK, Yang J, Wu J, Gao Z, Wolf BA. Tissue-specific and glucose-responsive expression of the pancreatic derived factor (PANDER) promoter. Biochim Biophys Acta. 2005;1730:215–225. doi: 10.1016/j.bbaexp.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 8.Xu W, Gao Z, Wu J, Wolf BA. Interferon-gamma-induced regulation of the pancreatic derived cytokine FAM3B in islets and insulin-secreting betaTC3 cells. Mol Cell Endocrinol. 2005;240:74–81. doi: 10.1016/j.mce.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 9.Wang O, Cai K, Pang S, Wang T, Qi D, Zhu Q, Ni Z, Le Y. Mechanisms of glucose-induced expression of pancreatic-derived factor in pancreatic beta-cells. Endocrinology. 2008;149:672–680. doi: 10.1210/en.2007-0106. [DOI] [PubMed] [Google Scholar]
- 10.Carnegie JR, Robert-Cooperman CE, Wu J, Young RA, Wolf BA, Burkhardt BR. Characterization of the expression, localization, and secretion of PANDER in alpha-cells. Mol Cell Endocrinol. 2010;325:36–45. doi: 10.1016/j.mce.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burkhardt BR, Cook JR, Young RA, Wolf BA. PDX-1 interaction and regulation of the Pancreatic Derived Factor (PANDER, FAM3B) promoter. Biochim Biophys Acta. 2008;1779:645–651. doi: 10.1016/j.bbagrm.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Robert CE, Burkhardt BR, Young RA, Wu J, Gao Z, Wolf BA. Mechanisms of glucose-induced secretion of pancreatic-derived factor (PANDER or FAM3B) in pancreatic beta-cells. Diabetes. 2005;54:3217–3228. doi: 10.2337/diabetes.54.11.3217. [DOI] [PubMed] [Google Scholar]
- 13.Cao X, Yang J, Burkhardt BR, Gao Z, Wong RK, Greene SR, Wu J, Wolf BA. Effects of overexpression of pancreatic derived factor (FAM3B) in isolated mouse islets and insulin-secreting betaTC3 cells. Am J Physiol Endocrinol Metab. 2005;289:E543–550. doi: 10.1152/ajpendo.00113.2005. [DOI] [PubMed] [Google Scholar]
- 14.Burkhardt BR, Greene SR, White P, Wong RK, Brestelli JE, Yang J, Robert CE, Brusko TM, Wasserfall CH, Wu J, et al. PANDER-induced cell-death genetic networks in islets reveal central role for caspase-3 and cyclin-dependent kinase inhibitor 1A (p21) Gene. 2006;369:134–141. doi: 10.1016/j.gene.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 15.Wilson CG, Schupp M, Burkhardt BR, Wu J, Young RA, Wolf BA. Liver-specific overexpression of pancreatic-derived factor (PANDER) induces fasting hyperglycemia in mice. Endocrinology. 2010;151:5174–5184. doi: 10.1210/en.2010-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, Wang C, Li J, Burkhardt BR, Robert-Cooperman CE, Wilson C, Gao Z, Wolf BA. PANDER binds to the liver cell membrane and inhibits insulin signaling in HepG2 cells. FEBS Lett. 2009;583:3009–3015. doi: 10.1016/j.febslet.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Wilson CG. PhD Doctoral. University of Pennsylvania; Philadelphia: 2010. Characterization of the effect of Pancreatic Derived Factor (PANDER) on hepatic regulation of glucose homeostasis. [Google Scholar]
- 18.Sundaresan S, Vijayagopal P, Mills N, Imrhan V, Prasad C. A mouse model for nonalcoholic steatohepatitis. J Nutr Biochem. 2010 doi: 10.1016/j.jnutbio.2010.08.011. In-press. [DOI] [PubMed] [Google Scholar]
- 19.Varela GM, Antwi DA, Dhir R, Yin X, Singhal NS, Graham MJ, Crooke RM, Ahima RS. Inhibition of ADRP prevents diet-induced insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2008;295:G621–628. doi: 10.1152/ajpgi.90204.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorman T, Hope DC, Brownlie R, Yu A, Gill D, Lofvenmark J, Wedin M, Mayers RM, Snaith MR, Smith DM. Effect of high-fat diet on glucose homeostasis and gene expression in glucokinase knockout mice. Diabetes Obes Metab. 2008;10:885–897. doi: 10.1111/j.1463-1326.2007.00819.x. [DOI] [PubMed] [Google Scholar]
- 21.Robert CE, Wu J, Burkhardt BR, Wolf BA. Transgenic Mice Overexpressing the Novel Islet Specific Cytokine, PANDER, Exhibit Glucose Intolerance. Diabetes. 2005;54(Suppl 1):74-LB. (Abstract) [Google Scholar]
- 22.Robert-Cooperman CE, Wilson CG, Burkhardt BR. PANDER KO mice on high-fat diet are glucose intolerant yet resistant to fasting hyperglycemia and hyperinsulinemia. FEBS Lett. 585(9):1345–9. doi: 10.1016/j.febslet.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gasa R, Gomis R, Casamitjana R, Rivera F, Novials A. Glucose regulation of islet amyloid polypeptide gene expression in rat pancreatic islets. Am J Physiol. 1997;272:E543–549. doi: 10.1152/ajpendo.1997.272.4.E543. [DOI] [PubMed] [Google Scholar]
- 24.Gasa R, Gomis R, Casamitjana R, Novials A. Signals related to glucose metabolism regulate islet amyloid polypeptide (IAPP) gene expression in human pancreatic islets. Regul Pept. 1997;68:99–104. doi: 10.1016/s0167-0115(96)02109-x. [DOI] [PubMed] [Google Scholar]
- 25.Hartter E, Svoboda T, Ludvik B, Schuller M, Lell B, Kuenburg E, Brunnbauer M, Woloszczuk W, Prager R. Basal and stimulated plasma levels of pancreatic amylin indicate its co-secretion with insulin in humans. Diabetologia. 1991;34:52–54. doi: 10.1007/BF00404025. [DOI] [PubMed] [Google Scholar]
- 26.Qi D, Cai K, Wang O, Li Z, Chen J, Deng B, Qian L, Le Y. Fatty acids induce amylin expression and secretion by pancreatic {beta}-cells. Am J Physiol Endocrinol Metab. 2009;298:E99–E107. doi: 10.1152/ajpendo.00242.2009. [DOI] [PubMed] [Google Scholar]
- 27.Mulder H, Martensson H, Sundler F, Ahren B. Differential changes in islet amyloid polypeptide (amylin) and insulin mRNA expression after high-fat diet-induced insulin resistance in C57BL/6J mice. Metabolism. 2000;49:1518–1522. doi: 10.1053/meta.2000.18511. [DOI] [PubMed] [Google Scholar]
- 28.Westermark GT, Leckstrom A, Ma Z, Westermark P. Increased release of IAPP in response to long-term high fat intake in mice. Horm Metab Res. 1998;30:256–258. doi: 10.1055/s-2007-978878. [DOI] [PubMed] [Google Scholar]
- 29.Ludvik B, Lell B, Hartter E, Schnack C, Prager R. Decrease of stimulated amylin release precedes impairment of insulin secretion in type II diabetes. Diabetes. 1991;40:1615–1619. doi: 10.2337/diab.40.12.1615. [DOI] [PubMed] [Google Scholar]
- 30.Rutter GA. Diabetes: the importance of the liver. Curr Biol. 2000;10:R736–738. doi: 10.1016/s0960-9822(00)00737-5. [DOI] [PubMed] [Google Scholar]
- 31.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–1346. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- 32.Lin Y, Sun Z. Current views on type 2 diabetes. J Endocrinol. 204:1–11. doi: 10.1677/JOE-09-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Unwin N, Shaw J, Zimmet P, Alberti KG. Impaired glucose tolerance and impaired fasting glycaemia: the current status on definition and intervention. Diabet Med. 2002;19:708–723. doi: 10.1046/j.1464-5491.2002.00835.x. [DOI] [PubMed] [Google Scholar]
- 34.Abdul-Ghani MA, DeFronzo RA. Plasma glucose concentration and prediction of future risk of type 2 diabetes. Diabetes Care. 2009;32(Suppl 2):S194–198. doi: 10.2337/dc09-S309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdul-Ghani MA, Lyssenko V, Tuomi T, DeFronzo RA, Groop L. Fasting versus postload plasma glucose concentration and the risk for future type 2 diabetes: results from the Botnia Study. Diabetes Care. 2009;32:281–286. doi: 10.2337/dc08-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in non-insulin-dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism. 1989;38:387–395. doi: 10.1016/0026-0495(89)90129-7. [DOI] [PubMed] [Google Scholar]
- 37.Radziuk J, Pye S. Tracer-determined glucose fluxes in health and type 2 diabetes: basal conditions. Best Pract Res Clin Endocrinol Metab. 2003;17:323–342. doi: 10.1016/s1521-690x(03)00038-1. [DOI] [PubMed] [Google Scholar]
- 38.Carroll MF, Izard A, Riboni K, Burge MR, Schade DS. Fasting hyperglycemia predicts the magnitude of postprandial hyperglycemia: implications for diabetes therapy. Diabetes Care. 2002;25:1247–1248. doi: 10.2337/diacare.25.7.1247. [DOI] [PubMed] [Google Scholar]
- 39.Livingstone DE, Jones GC, Smith K, Jamieson PM, Andrew R, Kenyon CJ, Walker BR. Understanding the role of glucocorticoids in obesity: tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology. 2000;141:560–563. doi: 10.1210/endo.141.2.7297. [DOI] [PubMed] [Google Scholar]
- 40.Edgerton DS, Jacobson PB, Opgenorth TJ, Zinker B, Beno D, von Geldern T, Ohman L, Scott M, Neal D, Cherrington AD. Selective antagonism of the hepatic glucocorticoid receptor reduces hepatic glucose production. Metabolism. 2006;55:1255–1262. doi: 10.1016/j.metabol.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 41.Jacobson PB, von Geldern TW, Ohman L, Osterland M, Wang J, Zinker B, Wilcox D, Nguyen PT, Mika A, Fung S, et al. Hepatic glucocorticoid receptor antagonism is sufficient to reduce elevated hepatic glucose output and improve glucose control in animal models of type 2 diabetes. J Pharmacol Exp Ther. 2005;314:191–200. doi: 10.1124/jpet.104.081257. [DOI] [PubMed] [Google Scholar]