

ABSTRACT
Mismatch-repair deficiency (MMR-D) is closely linked to hypermutation and accordingly, high immunogenicity. MMR-D-related tumors thus constitute ideal vaccination targets for both therapeutic and prophylactic approaches. Herein, the prophylactic and therapeutic impact of a cellular vaccine on tumor growth and tumor-immune microenvironment was studied in a murine MLH1−/− knockout mouse model. Prophylactic application of the lysate (+/− CpG ODN 1826) delayed tumor development, accompanied by increased levels of circulating T cell numbers. Therapeutic application of the vaccine prolonged overall survival (median time: 11.5 (lysate) and 12 weeks (lysate + CpG ODN) vs. 3 weeks (control group), respectively) along with reduced tumor burden, as confirmed by PET/CT imaging and immune stimulation (increased CD3+CD8+ T – and NK cell numbers, reduced levels of TIM-3+ cells in both treatment groups). Coding microsatellite analysis of MMR-D-related target genes revealed increased mutational load upon vaccination (total mutation frequency within 28 genes: 28.6% vaccine groups vs. 14.9% control group, respectively). Reactive immune cells recognized autologous tumor cells, but also NK cells target YAC-1 in IFNγ ELISpot and, even more importantly, in functional kill assays. Assessment of tumor microenvironment revealed infiltration of CD8+ T-cells and granulocytes, but also upregulation of immune checkpoint molecules (LAG-3, PD-L1).
The present study is the first reporting in vivo results on a therapeutic cellular MMR-D vaccine. Vaccination-induced prolonged survival was achieved in a clinically-relevant mouse model for MMR-D-related diseases by long-term impairment of tumor growth and this could be attributed to re-activated immune responses.
KEYWORDS: cellular vaccine, MMR deficiency, in vivo imaging, tumor microenvironment, target gene identification
Introduction
Somatic hypermutation constitutes a molecular tumor make-up for which immunotherapy might be most effective.1 This phenomenon is attributable to alterations in the DNA polymerases encoded by the POLE/D1 genes, exposure to external (cigarette smoking, UV radiation) and endogenous mutagens.1,2 Besides, individuals who are inherited deficient in DNA replication and repair processes develop tumors with high mutational load mainly consisting of insertions/deletions at repetitive DNA sequences ( = microsatellites). MSI-induced neoantigens arise mostly from coding microsatellite frameshift mutations in specific target genes.3,4 The high number of mutational events in coding microsatellites leads to the microsatellite instability (MSI) phenotype. Due to their high immunogenicity, these frameshift mutations are perfect targets for immunological approaches.
Germline mutations in one of the mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS1, and PMS2) represent the underlying molecular mechanism causing malignancies in affected patients. Clinically, MMR-D related tumor syndromes include the autosomal dominant Lynch syndrome (LS) as well as their biallelic counterpart referred to as constitutional or compound MMR-D (CMMR-D).5 LS-affected patients develop tumors upon inactivation of the second allele. Tumors predominantly manifest in the gastrointestinal system as well as in the endometrium and with an average age of 45 years.6 By contrast, CMMR-D patients are prone to develop a more complex spectrum of ultra-mutated cancers during their (early childhood) life-time. CMMR-D-associated tumors include hematological, Lynch-like gastrointestinal tumors (GIT), and brain tumors.7 Despite biological and clinico-pathological differences, many features are common to both diseases, based on the initial driving MMR-mutations.5,8,9
However, when comparing with LS patients, the prognosis for CMMR-D patients is extremely bad. CMMR-D-associated tumors show intrinsic resistance mechanisms against several known standard chemotherapeutic drugs, including O6-methylating agents.7,10 This finding, together with the increasing evidence of a good response to immunotherapeutic drugs inhibiting the PD-1/PD-L1 pathway even in the metastatic setting,2,11,12 provides a rationale for investigation of immune-based strategies in general. Recent studies revealed that LS patients' own immune system is capable of recognizing a variety of neoantigens on the tumor cells' surface; thus contributing to a better prognosis.13–15 Additionally to immune-checkpoint inhibition, more “classical” strategies like application of autologous tumor lysate, containing a large repertoire of (shared) MSI-specific tumor antigens, might be an effective treatment regimen for hypermutated tumors. Given the extremely high mutational load, these cancers are under selective pressure for obliterating antigen presentation.16–18 Hence, simultaneous vaccination against a variety/multitude of tumor-specific antigens may prevent (or delay) the phenomenon of antigen-escape tumor cell variants.19
We here analyzed the impact of a cellular-based vaccine on tumor incidence, growth and immune response as a strategy to vaccinate mice with spontaneously developing MLH1−/− tumors, in both prophylactic and therapeutic settings. These mice combine characteristics of LS and CMMR-D, with the latter being represented by an early lymphomagenesis, while GIT development is seen at later age and thereby closely resembles LS-associated neoplasia.
Results
Whole exome sequencing analysis for the identification of vaccination antigens – a comparative analysis
Prior to vaccination, comparative whole exome sequencing was done on an MLH1−/−-derived GIT allograft (namely MLH1−/− A7450, which was used for vaccination) and the corresponding parental tumor to analyze the mutanome of these tumors (Fig. 1). Firstly, marked increases in total mutational events in the allograft as compared to the primary were evident with regard to missense, nonsense and silent mutations as well as the number of mutations per type (Fig. 1A). As expected, the number of silent mutations is high whereas the nonsense mutation rate is small in both tumor samples. Focusing on the mutation type “nonsense”, unexpectedly no SNP exclusive for the primary tumor and only one mutation of the MSH3 gene, at position 93110760, has been reported.
Figure 1.

Comparative mutational analysis of MLH1−/− GIT and allograft. (A) The statistics of each mutation type (missense, nonsense and silent) are shown for the MLH1−/− GIT allograft and the primary tumor as well as the exclusive distribution for each. Silent mutations are dominating both samples and missense mutations are equally distributed. (B) Ideogram plots showing the genomic distributions of the missense mutations occurring in the annotated/known genes for both samples. All missense SNVs for MLH1−/− GIT allograft and the primary tumor are shown in blue and yellow respectively with their corresponding coordinate on the mouse reference genome mm9 cytoband. The exclusive missense SNVs for the MLH1−/− GIT allograft and the primary tumor are shown in red and annotated with a corresponding ID containing the SNV position and the affected gene name. (C) and (D) Missense mutations mapped on human protein sequences for the FHIT and the NF1 genes, respectively. These two specific genes are frequently mutated in CMMR-D related diseases.
In the genomic genetic fingerprint (Fig. 1B) the focus is on visualizing missense mutations in a pairwise fashion for the allograft and for their parental counterpart. The primary tumor shows three genes being mutated: ERBB2, IDH2 and ARID1A, where ARID1A has eight mutations at different positions. These positions are exclusively mutated in the primary tumor (shown in red with the corresponding annotation containing the position and the gene, all SNPs are in yellow). For ERBB2 only some positions are shown for better visualization. Interestingly, the allograft has ARID1A SNPs as well, but at different positions (for comparisons please see supplementary Tables 1 and 2 summarizing the exclusive SNPs). Besides ARID1A, the other mutated genes are: CTNNBL1, FHIT, MET, MSH3, MYO1B, NF1, POLE, POLD1, and SMAD2. POLE and POLD1 frequently mutated in MMR-D related diseases20,21 and [Fig. 1C and D] show two or three mutated positions, respectively. An additional analysis on insertions/deletions– known to be interesting target structures for peptide-based vaccination – identified 16 candidates with potential relevance (supplementary Table 3). The resulting proteins are involved in different biological processes (such as signaling and growth control).
Table 1.
Tumor development after prophylactic vaccination and tumor spectrum.
| median age of onset [weeks] |
tumor type [%] |
mice [%] |
|||||
|---|---|---|---|---|---|---|---|
| Intervention | Lymphoma | GIT | Lymphoma | GIT | other | unexpected death | tumor free |
| CpG ODN 1826 | 26 | 32.5 | 60 | 20 | 20 | 0 | 0 |
| ODN 1826 ctrl | 26 | 38.5 | 60 | 20 | 10 | 10 | 0 |
| Lysate | 30 | 48 | 53.8 | 15.4 | 7.7 | 8.0 | 15.4 |
| Lysate + CpG ODN 1826 | 32.5 | 46.5 | 41.2 | 23.5 | 11.8 | 5.8 | 17.7 |
| Lysate + ODN 1826 ctrl | 31.5 | 45 | 40.0 | 20.0 | 20.0 | 6.7 | 13.3 |
ctrl – control; GIT – gastrointestinal tumor; unexpected death – died under isoflurane anesthesia, found dead
Table 2.
Analysis of coding microsatellite mutations in GIT from vaccinated and control tumors in comparing to the allograft, which was used for vaccination.
| cMS marker |
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | APC | Tmem60 | Senp6 | Phactr4 | Casc3 | SDCCAG1 | Fasl | Kit | Rasal2 | Tcf7l2 | Casc5 | Mbd6 | Lig4 | Bend5 | NKtr1 | Grb14 | Kcnma1 | Rfc3 | ERCC5 | Asnsd1 |
| control | 26.1 | 26.1 | 43.5 | 47.8 | 13.0 | 8.7 | 0.0 | 0.0 | 43.5 | 13.0 | 21.7 | 0.0 | 4.3 | 13.0 | 8.7 | 0.0 | 4.3 | 91.3 | 30.4 | 26.1 |
| lysate | 0.0 | 33.3 | 100.0 | 0.0 | 66.7 | 66.7 | 0.0 | 0.0 | 100.0 | 33.3 | 33.3 | 0.0 | 33.3 | 20.0 | 100.0 | 100.0 | 33.3 | 100.0 | 66.7 | 20.0 |
| lysate + CpG ODN 1826 | 0.0 | 40.0 | 20.0 | 20.0 | 0.0 | 80.0 | 20.0 | 20.0 | 20.0 | 20.0 | 20.0 | 40.0 | 20.0 | 60.0 | 20.0 | 20.0 | 0.0 | 80.0 | 40.0 | 20.0 |
| allograft* | wt/−1 | wt | wt/−1 | wt | wt | wt | wt | wt | wt/−1 | wt | n.d. | wt | wt/−1 | wt/−1/−2 | wt | wt | wt | wt/−1 | wt | wt |
Cumulative mutation frequency in %; differences between individual tumors (control, vaccine, allograft) are highlighted in green, confirming vaccination-induced increased mutational load;
used for vaccination; n.d. – not determined.
Delayed tumorigenesis after prophylactic vaccination due to immune stimulation
Next, the impact of the cellular lysate prepared from the MLH1−/− GIT allograft was tested on tumor development in syngeneic animals. A vaccination schedule is provided as supplementary Fig. 1. Mice without any clinical signs of tumor development (i.e. normal behavior, no ruffling of fur, normal blood cell numbers) were included in this study and assigned to the different treatment arms (lysate, lysate + CpG ODN 1826, lysate + control ODN 1826) and control groups (CpG ODN 1826, control ODN 1826). Mice repeatedly received applications of the vaccine to boost immune responses. CpG was chosen based on a preliminary in vitro assay to study the immune stimulatory effect of different adjuvants (Complete Freund's Adjuvant, a bacterial lysate from Streptococcus pyogenes,22 and CpG ODN 1826) (data not shown). In these experiments, lymphocytes from both heterozygous and homozygous mice (each n = 4) responded best towards CpG ODN 1826 in terms of activation (CD25+, CD69+) and viability.
Repeated application of the lysate was well-tolerated and without any signs of vaccination-induced side effects. To analyze the impact of the vaccine on the immune system, blood samples were taken routinely from mice of all groups. Mice of the vaccine groups (lysate +/− CpG ODN 1826 and control ODN 1826) had increased numbers of circulating T cells (Fig. 2A). Control mice did not show any altered immune response. As assessed on day 84, all vaccinated mice showed higher levels of cytotoxic T cells than control mice (lysate + CpG ODN 1826 up to 22% vs. control: 8 %). NK cell numbers were elevated as well (lysate up to 40 % vs. control: 23 %).
Figure 2.
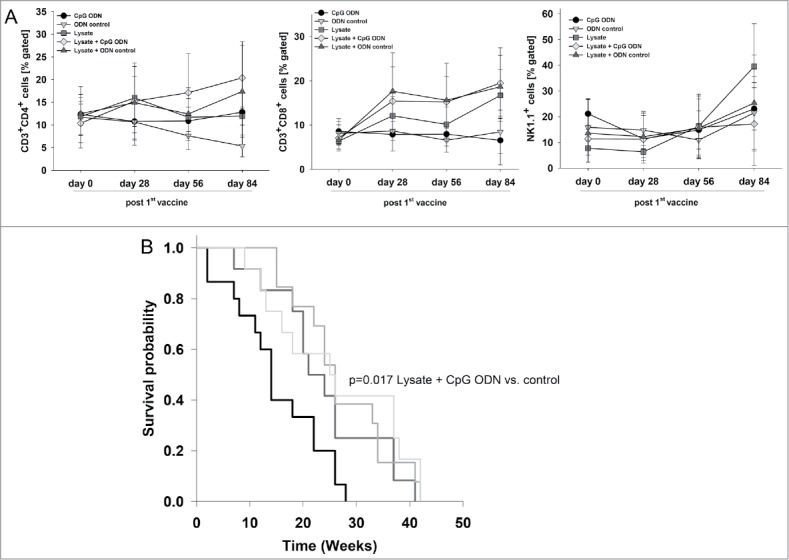
Prophylactic vaccination of MLH1−/− mice with a cellular lysate. (A) Flow cytometric phenotyping of peripheral blood leukocytes. Blood samples were taken at start of treatment ( = day 0) and regularly during the experiment. Given are the percentage numbers of positively stained cells as determined by gating on viable cells from vaccinated (lysate (n = 12) +/− CpG ODN 1826 (n = 15) and ODN control (n = 12)) and control mice (CpG ODN 1826or control ODN 1826; n = 9 and n = 7 mice per group, respectively). Values are given as mean ± SD; *p < 0.05 vs. control; t-test. (B) Kaplan Meier survival curve of vaccinated and control MLH1−/− mice. Log rank survival analysis, p = 0.017 Lysate + CpG ODN vs. control.
In addition to the observed immune stimulation, vaccinated mice lived longer than controls and developed tumors at later time points (Fig. 2B). Two and three mice of each of the two vaccination groups, respectively, even remained tumor free until the experimental end point ( = 42 weeks), while all control mice displayed tumorigenesis within the expected time frame, i.e. lymphomas at around 26 weeks and GIT at around 32.5 (CpG ODN 1826) or 38.5 weeks (control ODN 1826), respectively (Table 1). Vaccination slightly delayed lymphoma formation and GI tumorigenesis, the latter being detectable around nine weeks later (lysate group) than in control mice (Table 1). Unexpectedly, adding the adjuvant to the lysate played a minor role and did not decelerate tumor development when compared to the lysate.
Tumor microenvironment and target cell recognition by lymphocytes of prophylactically vaccinated mice
To further investigate the observed immune stimulation, the tumor microenvironment was studied in detail. Immunofluorescence staining of GIT from vaccinated and control mice revealed increased numbers of infiltrating CD4+ and CD8+ T cells, both being located within the tumors (representative images are shown in Fig. 3A). Besides, the immune-checkpoint molecule PD-L1 was upregulated, mainly on macrophages, upon vaccination. Numbers of LAG-3- and NK1.1-positive cells remained largely unchanged (Fig. 3A and data not shown).
Figure 3.
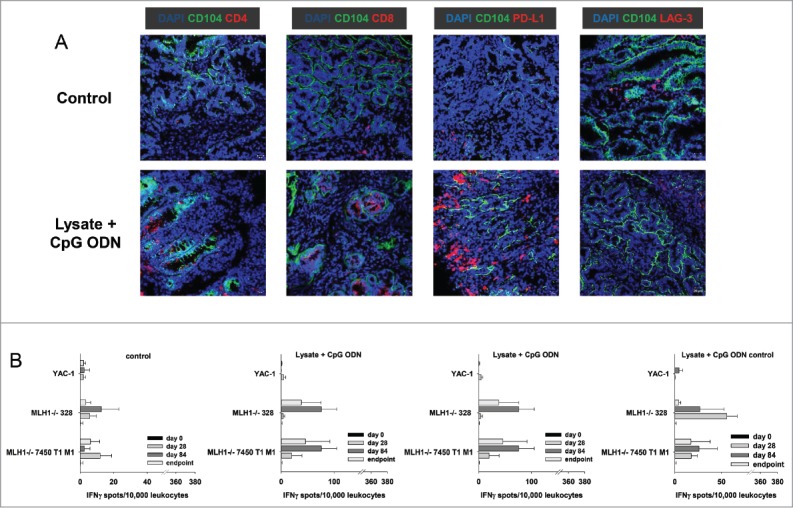
Immunofluorescence of MLH1−/− tumors and IFNγ–ELISpot after prophylactic vaccination. (A) Tumor microenvironment was studied from GIT cryostat sections of 4µm. Analyses were done on a laser scanning microscope (Zeiss) using 20x objectives. (B) Reactivity of PBL (during vaccination) or splenocytes (endpoint) against target cells (MLH1−/− 7450 T1 M1, MLH1−/− 328, and YAC-1) was examined after overnight co-incubation. Lymphocytes were isolated from vaccinated and control mice at different time points. Experiments revealed increased reactivity upon vaccination. Values are given as mean ± SD.
We next examined whether immune cells respond to stimulation from tumor cells. Therefore, the reaction of peripheral blood lymphocytes ( = during vaccination) and splenocytes ( = expe-rimental endpoint) co-incubated overnight with different target cell populations was measured in a classical IFNγ-ELISpot assay. The two tested MLH1−/− tumor target cell lines triggered IFNγ secretion of lymphocytes from vaccinated mice. Of note, there was a trend towards higher recognition at later time points, i.e. day 84. Strongest responses were seen in the lysate-treated group, followed by lymphocytes from lysate + adjuvant-treated mice (Fig. 3B). Control mice (CpG ODN 1826 and control ODN 1826) did not show any response towards the target cells (Fig. 3B).
Prolonged survival after therapeutic vaccination
Next, the impact of our vaccine was investigated in a therapeutic situation (supplementary Fig. 1). Mice with suspected GIT underwent in vivo PET/CT imaging, which was paralleled by weight monitoring and basic immune assays to determine the immune status before vaccination. When comparing with tumor-free young mice, T cell levels (CD4+, CD8+, γ/δ TCR+) were slightly reduced in tumor-bearing hosts. Vice versa, immune-suppressive molecules (i.e. LAG-3 and IDO) as well as ALCAM (CD166) increased in numbers (Fig. 4A). Granulocyte and NK cell numbers were not altered. As assessed by PET/CT imaging, mice developed 3.2 ± 1.3 tumors on average with a mean volume of 74.1 ± 51.8 mm3 (Fig. 4B, representative pictures showing PET/CT scan of a healthy mouse (left) in comparison to tumor-bearing mice (middle: GIT, right: lymphoma) are depicted in Fig. 4C). Tumorigenesis was accompanied by continuous weight loss (∼ 5 % during the last three weeks of observation) and impairment of general behavior (ruffling of fur, reduced mobility, and socio-physiological segregation).
Figure 4.
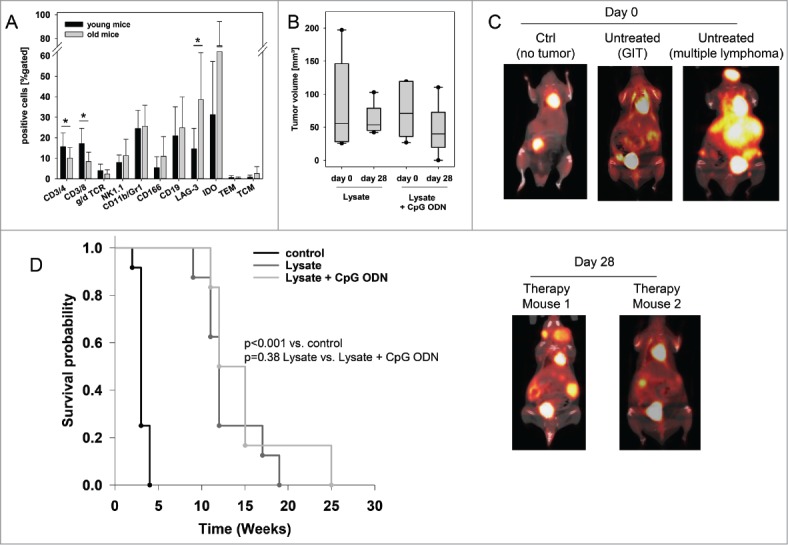
Therapeutic vaccination of MLH1−/− mice with a cellular lysate. (A) Comparative flow cytometric analyses from young and old mice for determining immune status prior to vaccination. * p<0.05 vs. young mice; t-test. (B) Tumor volume as determined by PET/CT in vivo imaging using radiopharmacon 18F-FDG. Values are given as mean ± SD. Whiskers (error bars) above and below the box indicate the 90th and 10th percentiles. (C) and (D) PET/CT in vivo imaging. Tumor nodules were visualized using radiopharmacom 18F-FDG. Given are representative summed images in axial (left) and coronal view (right) (C) For comparison, exemplary images of a tumor-free mouse as well as mice with a GIT and lymphoma are shown. Arrows indicate detected tumors. The color scale indicates SUV. (D) Repeated imaging at day 28 after vaccine revealed disappearance of single tumor nodules. Images at day 28 were taken from the same mouse as on day 0. (E) Kaplan Meier survival curve of vaccinated and control MLH1−/− mice. Log rank survival analysis. p<0.001 vs. control. p = 0.38 lysate vs. lysate + CpG ODN.
Upon vaccination, general state of health was improved. Additionally to stabilizing weight, mice showed improved mobility and parametric signs of disease vanished. Repeated PET/CT imaging on day 28 post vaccination revealed disease control, with tumor volumes being smaller than before vaccination (day 28: lysate: 60.2 ± 24.3mm3 and lysate + CpG ODN: 46.0 ± 37.3mm3). In some cases, single tumor nodules even disappeared (Fig. 4D). Disease control contributed to a better outcome and improved overall survival. Median survival time after vaccination was 11.5 ± 3.9 (lysate) and 12.0 ± 5.8 (lysate + CpG ODN 1826) weeks, compared to three weeks in the control group (median: 3.0 ± 0.6 weeks) (Fig. 4E). Therapeutic response was accompanied by immune stimulation in a way of reconstituting a “physiological” immune response. Of note, numbers of cytotoxic T and NK cells as well as granulocytes gradually increased during vaccination (Fig. 5). Amounts of TIM-3 positive T cells decreased, while CTLA-4, PD-1 and PD-L1 were only transiently down-regulated on immune cells (data not shown). Numbers of helper T cells did not significantly change during vaccination.
Figure 5.
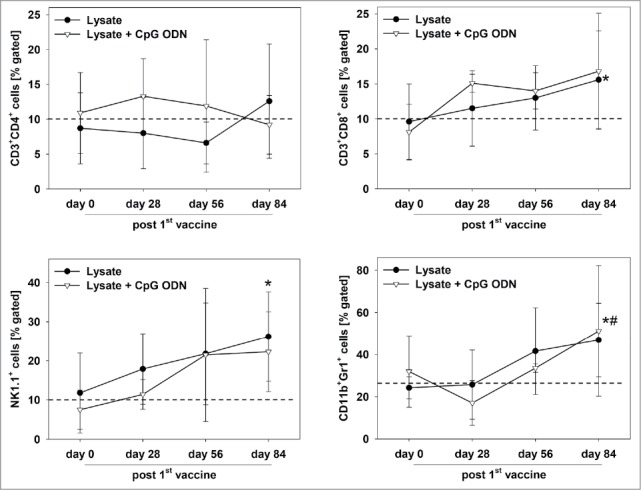
Immune status of therapeutically vaccinated mice. Flow cytometric phenotyping of peripheral blood leukocytes. Blood samples were taken at start of treatment ( = day 0) and regularly during the experiment. Given are the percentage numbers of positively stained cells as determined by gating on viable cells from vaccinated (lysate (n = 9) +/− CpG ODN 1826 (n = 7). Values are given as mean ± SD; *p < 0.05 lysate vs. day 0; t-test; # p < 0.05 vs. lysate + CpG ODN vs. day 0; t-test.
Considering treatment of highly aggressive lymphomas, median survival time upon vaccination with the GIT-derived lysate was 6.0 ± 2.5 weeks and thereby slightly prolonged compared to untreated control mice (2.0 ± 1.1 weeks). This finding suggests some overlapping but also different biological and immunological mechanisms among the two tumor types arising in MLH1−/− mice. Another interesting “single case” finding was successful eradication of a skin cancer by subcutaneous lysate application. The initial tumor (9 × 12 mm size) completely disappeared after three repetitive injections and the mouse remained tumor free for four months, before relapse. Re-administration of the vaccine mediated measurable tumor shrinkage, but not complete remission, and the mouse died from progressive disease four weeks later (supplementary Fig. 2).
Tumor microenvironment reveals increased T cell infiltration but also vaccine-induced up-regulation of immune-checkpoint molecules
The in vivo results obtained hint towards involvement of anti-tumoral immunological mechanisms. Consequently, tumor resection specimens were examined for infiltrating immune cells. Here, marked differences were seen between lysate and lysate + CpG ODN 1826 treated mice. In the latter group, infiltrating cytotoxic and helper T cell numbers markedly increased (Fig. 6). Although numbers of infiltrating granulocytes remained similar upon vaccination, their phenotype in lysate + CpG ODN 1826 treated tumors changed. Double positive CD11b+Gr1+ myeloid-derived suppressor cells were exclusively seen in this group; possibly documenting a compensatory immune-escape mechanism. In support of these findings, PD-L1 and LAG-3 were highly upregulated on infiltrating cells in the MLH1−/− tumor microenvironment. However, there was no expression of PD-L1 on vaccine-induced infiltrating CD11b+ granulocytes, a mechanism reported to counteract CD8+ T cell mediated tumor cell lysis,23 (Fig. 6). NK cell numbers were not altered at all (Fig. 6).
Figure 6.
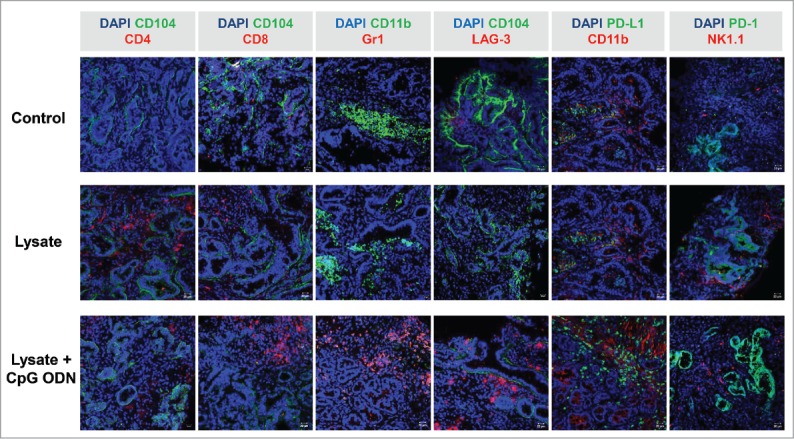
Immunofluorescence of MLH1−/− tumors after therapeutic vaccination. Tumor microenvironment was studied from GIT cryostat sections of 4µm. Pictures were done on a laser scanning microscope (Zeiss) using 20x objectives.
Increased mutational load in MSI-target genes upon vaccination
Prolonged survival of MLH1−/− mice was at least partially attributable to immune-stimulation. Since this might be associated with selective pressure and an accordingly increased mutational load, tumor resection specimens from control and vaccinated mice were subjected to molecular pathological analysis. Examination of coding microsatellites in selected MSI-target genes was based on our previous study to identify murine coding MSI target genes in MLH1−/− tumors.9
With this analysis, mutations were detectable in 31 % (lysate) and 25 % (lysate + CpG ODN 1826) of analyzed markers from vaccinated tumors, respectively (vs. 15 % controls; Table 2). Increased mutation frequencies were found in genes belonging to DNA repair (Lig4), signa-ling pathways that regulate growth and metabolism (Grb14), and transcriptional repression (Bend5). The latter gene shares functional and structural similarity with its human ortholog and is mutated in 10 % of cultured MSI-H cell lines [Seltarbase.org]. Besides, NKtr1, originally described to be present on the surface of natural killer cells to facilitate target cell binding, was more frequently mutated in vaccinated than in control tumors (100% (lysate) and 20% (lysate + CpG ODN 1826), respectively, vs. 9% in controls). In contrast, APC gene mutations, originally found in every fourth MLH1−/−−associated GIT as well as in the allograft that was used for vaccination, were no longer detectable (Table 2).
Additional direct comparison of such MSI-specific alterations between vaccinated tumors and the allograft confirmed an increased vaccination-induced mutational load. Here again, some mutations were exclusively found upon vaccination, forced by MSI+ tumor cells to escape immune attack (Table 2).
Immune responses against MLH1−/− targets upon vaccination
Finally, lymphocytes from vaccinated mice were analyzed for their tumor-recognizing and killing ability in immunological standard assays. By determining the number of IFNγ secreting cells, recognition of MLH1−/− target cell lines 328 and 7450 T1 M1 was confirmed (Fig. 7A). Numbers of reactive lymphocytes increased during vaccination. How-ever, recognition was not confined to tumor cells, since YAC-1 cells gave also rise to IFNγ secretion, especially at later times of vaccination (day 56 and endpoint).
Figure 7.
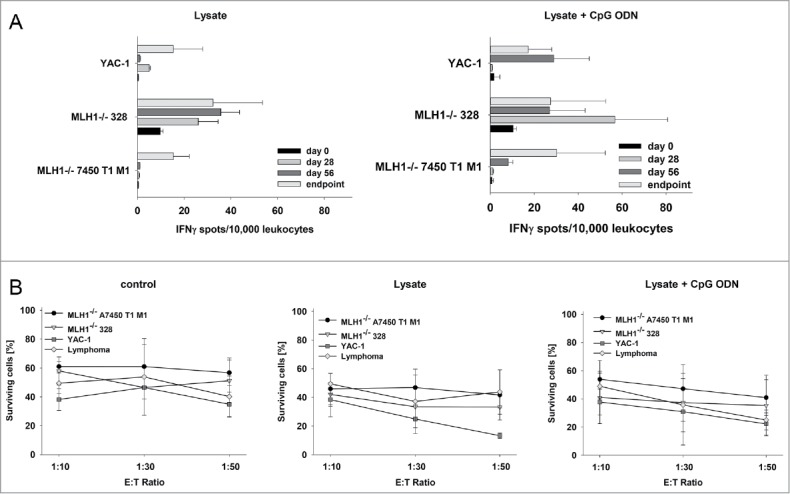
Functional immunological analysis. (A) IFNγ−ELISpot and (B) Flow cytometric cytotoxicity assay. (A) Reactivity of PBL (during vaccination) or splenocytes (endpoint) against target cells (MLH1−/− 7450 T1 M1, MLH1−/− 328, and YAC-1) was examined after overnight co-incubation. Lymphocytes were isolated from lysate-treated and control mice at different time points. These experiments identified increased reactivity post treatment. (B) Lytic activity of splenocytes against target cells (MLH1−/− 7450 T1 M1, MLH1−/− 328, and YAC-1) was examined after 12h co-incubation. Lymphocytes were isolated from spleens of lysate-treated and control mice. Again, an increased lytic activity upon vaccination was seen. Values are given as mean ± SD.
Results were confirmed on a functional level, in which splenocytes from vaccinated mice were capable of lysing MLH1−/− target cells. Of note, GIT and lymphoma target cells were both efficiently killed. Nonetheless, lytic activity was not exclusively antigen-restricted, since YAC-1 cells were again also recognized and killed (Fig. 7B). This is particularly interesting since T and NK cell proportions gradually increased upon vaccination. Hence, repeated application of the lysate seems to favor the induction of highly activated T as well as NK cells.
Discussion
In this study, we used a clinically relevant mouse model to answer the question whether a cellular vaccine, prepared from a MSI-H GIT, constitutes a successful immunotherapeutic strategy for MMR-D related diseases. In addition, we analyzed if this vaccine protects mice from tumors with known biological and molecular pathological heterogeneity (GIT vs. lymphomas and skin tumors), since MMR-D tumors (especially CMMR-D associated cases24) arise in virtually all cancer-prone organs and may share immunogenic target structures.25 We could show that: (I) repeated application of the vaccine stimulates both, the innate and the adaptive immune system, (II) it delays tumorigenesis of different origin in naïve mice, (III) it significantly prolongs survival of GI tumor-bearing mice by long-term impairment of tumor growth, but (IV) it has only marginal entity-overlapping antitumoral capacity in the therapeutic situation.
Application of tumor vaccines has a long-standing tradition in tumor immunology with proven efficacy in pre−(clinical) trials.19,26 Whole tumor lysates constitute an undefined mixture of “altered self” antigens; hence, identifying vaccination antigens is crucial for subsequent development of poly-epitope vaccines – made of stably presented T cell epitopes for activating cytotoxic T and helper lymphocytes simultaneously. In this study, we first analyzed the cancer mutanome of a GIT MLH1−/− allograft which was subsequently used for vaccination ( = lysate) and performed comparative analysis with the parental tumor counterpart. Firstly, by whole exome sequencing, an increased number of mutational events was confirmed in the allograft, a finding quite common to MMR-D-related tumors. These tumors acquire novel mutations after each cell cycle,21 putting them into the subtype of “ultra-mutated” cancers. Secondly, mutations identified in the allograft were defined as true target antigens based on their physiological functional role, i.e. involvement in different biological processes (signaling, growth control; supplementary Table 1).
A high number of frameshift-derived neoantigens is described in the literature for human MSI+ cancers [Seltarbase.org] and some were even identified in the murine system.3,9 Frameshift peptides (FSP) trigger T cell-mediated immune responses in LS patients as well as in healthy mutation carriers.15 Consequently, clinical trials aiming to decelerate or even prevent tumorigenesis in affected patients after repetitive FSP-based vaccination have been initiated [trial numbers: NCT01461148 & NCT01885702]. Preliminary results from single cancer patients describe FSP-specific immune responses in a Phase I clinical trial.27 By contrast, much less is known about the immunogenicity of CMMR-D associated cancers in terms of tumor microenvironment and antigenic profile. In the only prior published immunotherapeutic approach, two patients with recurrent multifocal brain tumor received complete remission after treatment with Nivolumab.28 Besides, no experimental or clinical trials on CMMR-D immunotherapy have been reported so far.
The MMR-D-related MLH1−/− tumor model is ideally suited to develop and test immunotherapeutic strategies in order to not only substantially improve the clinical situation for patients suffering from MMR-D-related tumors but moreover also for the development of prophylactic strategies delaying or on the long run even preventing tumor occurrence in CMMR-D individuals.
To test the prophylactic capacity of the GIT allograft lysate, we vaccinated mice repetitively prior to tumor formation. Vaccination was accompanied by increased levels of circulating (cytotoxic and helper) T as well as NK cells. T cells were additionally found elevated in situ in tumor resection specimens. Noticeably, numbers of both helper and cytotoxic T cells increased upon vaccination, most likely explaining delayed tumorigenesis. This approach did, however, not completely inhibit tumor formation; with only 15 % of mice being tumor-free at the experimental end-point.
The functional in vitro analysis confirmed initial immune stimulation with immune cells from lysate (+/− CpG ODN 1826) treated mice recognizing to autologous tumor target cells, especially at early time points. However, development of immunological memory was obviously counteracted by developing tumor cells through immunoediting. Tumor cells are not characterized by a conserved set of immunogenic antigens. Accordingly, the tumor is sculpted with a preponderance of tumor cells to which the immune system appears to be tolerant. Mechanistically, this has additionally been attributed to (I) the production of immunosuppressive cytokines (e.g. TGF-β, IL-10) or soluble factors (e.g. ROS, NO)23,26; (II) involving inhibitory receptors (upregulation of CTLA-4 and IDO) and (III) preventing co-stimulation (low amount of antigen-presenting cells). Adding other adjuvants or applying tumor lysate-loaded dendritic cells to ensure appropriate migration to T lymphocyte areas in draining lymphoid tissues may provide a strategy to improve therapy. Finally, conquering the plethora of adaptations malignant cells develop along with restoration of an intact CD4/CD8 interplay (“cancer immune cycle”) remains the main challenge to increase tumor rejection rates in the preclinical MLH1−/− model.
Therapeutic lysate application in GIT-bearing MLH1−/− mice significantly prolonged survival. PET/CT imaging with 18F-FDG, a sensitive parameter to monitor treatment response,29 confirmed vaccination-induced disease control with some tumor nodules even completely disappearing within four weeks. Immune responses, characterized by elevated numbers of cytotoxic T and NK cells as well as specific recognition of MLH1−/− GIT target cells by lymphocytes from vaccinated mice accompanied the therapeutic response. Marked immune cell infiltration was evident in residual tumors dominated by T cells and granulocytes; but also immunosuppressive checkpoint molecules expressed by the majority of cellular infiltrates.30 In cytotoxicity assays, reactivity was further emphasized by the lymphocytes' ability to kill MLH1−/− tumor target cells. Interestingly, we also observed a cross-reactivity of lymphocytes isolated from lymphomas, hinting to the recognition of shared tumor antigens. This is particularly surprising since in vivo treatment of lymphomas did not significantly affect tumor growth and overall survival. In support of these findings, a distinct mutational susceptibility of target genes among human hematological and gastrointestinal (sporadic) MSI tumors was reported.31 Besides, immune responses observed in our study were not exclusively antigen-restricted since NK cells target YAC-1 were also lysed. Confirmed by flow cytometric phenotyping, a mixture of highly activated, efficiently target cell killing NK cells and cytotoxic T cells attack MLH1−/− tumor targets; the latter in an MHC-I restricted manner.32,33
Another interesting finding of this study was the increased mutational load in selected MSI-target genes from vaccinated tumors, which likely constitutes some kind of escape mechanism by triggering antigenic drift. Together with the missing activation of T helper cells and upregulation of immune-checkpoint molecules this best explains final disease progression and treatment failure. Consequently, combining checkpoint-inhibition with vaccination represents a very promising strategy as has only recently been proposed for experimental glioblastomas (DC vaccine + anti-PD-1 antibody + colony stimulating factor 1 receptor inhibitor) and prostate cancer (anti-CTLA-4 antibody + GM-CSF-secreting cellular vaccine).23,34
Finally, we would like to bring forward the argument that the MLH1−/− mice that we used mirror – at least from an immunological point of view – many features of clinical CMMR-D. These include a biallelic MMR-D as the driver of tumorigenesis and the high incidence and early manifestation of lymphomas. Although not associated with clinically noticeable immune defects, impairment of Ig class switch recombination has been frequently found in CMMR-D patients.24,35 Even though not analyzed in detail in this study, we observed a slightly impaired response towards antigenic stimuli of MLH1−/− lymphocytes when compared to lymphocytes from heterozygous and wildtype mice. This fact in mind, we want to emphasize that the overall success of the cellular lysate vaccination strategy used is unlikely to be over-estimating the underlying potential. Due to the hypermutative nature of (C)MMR-D-associated cancers, which allows such tumor cells to easily gain novel (escape) mutations rapidly, multifaceted treatment strategies will most likely be key to their successful long-term disease management.
Materials and methods
Whole exome sequencing (WES) analysis. Comparative WES analysis was done on MLH1−/− GIT allograft 7450 and their matched primary9 using the Agilent SureSelectXT exome capture and sequencing via Genome Sequencer Illumina HiSeq in paired-end mode (GATC, Konstanz, Germany; coverage: 90x (primary) and 60x (allograft), respectively). Data preprocessing: For each sequence read the base quality is inspected for low quality calls and subsequently removed before proceeding with further processing using a sliding window approach. This means that bases with low quality are removed from the 3′ and 5′ ends. Additionally, bases are removed if the average quality is below 15. The whole exome reads were mapped to the published mouse genome build ENSEMBL mm9.64 reference using Burroughs Wheeler Aligner (BWA)36 with the default parameters. PCR duplicate reads were removed by using Picard (http://picard.sourceforge.net) in order to prevent artificial coverage brought on by the PCR amplification step during library preparation. SNP and InDel calling was performed using the GATK Unified Genotyper37,38 and the detected variants were annotated based on their gene context using snpEff.39
Data visualization
The summarized mutational profiles with the corresponding mutation type of the MLH1−/− GIT allograft and the primary tumor were generated as stacked bar plot with ggplot2.40 To better compare the molecular profiles between the allograft and primary tumor, whole genome ideograms were generated using ggplot2 package40 as well as circlize package41 in R. The mutation data were filtered for the exclusive SNVs for each condition (allograft or primary). Additional mutation filters such as the mutation type (missense and nonsense) as well as those mutations occurring in known annotated genes, were applied. Next, the filtered mutations were presented as ideogram based on the mm9 mouse assembly provided by the circlize package.41 The first track is the cytoband of the mm9 assembly. The following tracks include SNVs belonging to same condition (MLH1−/− GIT allograft or primary tumor). Further details about the overlaps in the mutation type profiles have been generated customized with the VennDiagram42 package in R. The mutation mapper tool was utilized for mapping the mutations and its statistics on a linear gene product (proteins of interest).43,44 For analysis, genes were chosen with high probability of mutation based on knowledge from the human MMR-D counterpart and general involvement in tumorigenesis.
MLH1−/− mouse model and vaccine preparation
Homozygous mice were generated by breeding heterozygous males and females of the ≥F5 generation. All animals received standard laboratory chow and free access to water. Mice breeding took place in the animal facilities (University of Rostock) under specified pathogen-free conditions. Trials were performed in accordance with the German legislation on protection of animals and the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council; NIH Guide, vol.25, no.28, 1996; approval number: LALLF M-V/TSD/7221.3-1.1-053/12). Mlh1 genotyping was done according to.45 In each generation, offspring of all three classes in the expected ratios were obtained (i.e. about 20–25% homozygous mice).
The vaccine originated from a molecularly and immunologically well-characterized MLH1−/− GIT,9 grown as allograft in NMRI Foxn1nu mice and serially passaged. The primary tumor grew in the small intestine of a female MLH1−/− mouse. Histopathologically, the tumor presented as well-differentiated adenocarcinoma with abundant extracellular mucin. Outgrowing allografts were resected, homogenized and washed. The vaccine was obtained after repetitive freeze/thaw cycles (n = 4), prior to heat-shock (42°C, 5 min). Protein lysates were gamma irradiated (60 Gy) and frozen immediately in aliquots at -80°C according to the procedure by.46
In vivo vaccination schedule
Mice were either vaccinated prophylactically or therapeutically. In case of prophylactic application, 8–10 week-old homozygous mice received four weekly subcutaneous injections of the vaccine (10 mg/kg bw, s.c., n = 12 mice) with or without CpG ODN 1826 (Invivogen, tlrl-1826) as adjuvant or its irrelevant control ODN 1826 (tlrl-1826c; 2.5 mg/kg bw, s.c., n = 15 and 12 mice per group, respectively). Control mice were given equivalent concentrations of CpG ODN or control ODN 1826 (n = 9 and n = 7 mice per group, respectively). Vaccination was continued until tumor development (monthly injections: 2.5mg/kg bw, without adjuvant). Therapeutic vaccinations started after in vivo confirmation of GI tumor growth, as determined by small animal PET/CT imaging.29 Mice received four weekly injections of the vaccine (10 mg/kg bw, s.c.) again with or without CpG ODN 1826 (2.5 mg/kg bw, s.c., n = 9 and n = 7, respectively), followed by biweekly applications of the lysate only (2.5 mg/kg bw) until tumor progression. PET/CT imaging was repeated on day 28 of therapy. Control mice were given no treatment. Blood samples were taken at start of treatment (prophylactic and therapeutic) and regularly during the experiment. Mice were sacrificed followed by collecting blood samples, tumors and spleens from all animals for further analysis.
PET/CT imaging
Mice with suspected GIT or healthy control mice (n = 30 in total) were anaesthetized by isoflurane (1.5–2.5 %, Baxter) supplemented with oxygen. Mice received a mean dose of 16,91 ± 1,73 MBq Radiopharmacon 18F-FDG intravenously via a microcatheter placed in a tail vein (1 h uptake). Afterwards, mice were imaged in prone position in the Inveon PET/CT scanner (Siemens Preclinical Solutions, Knoxville, TN, USA) for 15 min. During measurement animals were kept at constant temperature of 38 °C by an electrical heating pad and respiration was monitored.29 CT images were reconstructed with a Feldkamp algorithm. PET data were first Fourier rebinned into a 2D dataset from which real-space images were reconstructed with an ordered subset expectation maximization (OSEM) algorithm with 16 subsets and four iterations. Attenuation correction was carried out using the CT data. Metabolic volumes and SUVs were determined using Inveon Research Workplace 4.2 software.
Flow cytometry on blood samples
Blood samples were taken routinely from the retrobulbar venous plexus of vaccinated and control MLH1−/− mice, as well as naïve young homozygous mice (<14 weeks, n = 15). Blood samples were stained with a panel of conjugated monoclonal antibodies (mAb, 1 µg each) followed by lysis of erythrocytes (155 mM NH4Cl (MERCK Millipore, 101145), 10 mM KHCO3 (MERCK Millipore, 104854), 0.1 mM EDTA (Applichem, A5097)). Negative controls consisted of lymphocytes stained with the appropriate isotypes (BD Pharmingen). Cells were washed, resuspended in PBS and analyzed by flow cytometry on a FACS Verse Cytometer (BD Pharmingen). Data analysis was performed using BD FACSuite software (BD Pharmingen).
Microsatellite and coding microsatellite (cMS) frameshift mutation analysis
The mutational profile of MLH1−/− tumors from vaccinated mice was compared with those of control mice analyzing a panel of n = 26 mononucleotide repeats in MSI target genes. Genes included in this study were described before.9 PCR conditions were: 94 °C, 4 min (1 cycle); 94 °C, 30 s, 58 °C, 45 s and 72 °C, 30 s (35 cycles); and 72 °C, 6 min (1 cycle). Fluorescently labeled DNA fragments were analyzed on a 3500 Genetic Analyzer. In each reaction, normal tail DNA served as microsatellite stable (MSS) controls. Tumor samples were scored as instable if novel peaks were obtained compared to MSS controls or if the ratio of peak areas in MLH1−/− samples and stable controls revealed values ≤0.5or ≥2.
Immunofluorescence
Cryostat sections of 4 µm were air-dried and fixed in cold pure methanol for 8 min. Unspecific binding sites were 2 hours blocked in 2 % BSA (Roth, T844.4) followed by incubation with 1 µg of the following FITC- and PE-labeled mAbs: CD4, CD8α, CD11b, Gr1 (Immunotools), CD104, LAG-3, PD-1, NK1.1 and PD-L1 (Biolegend). Sections were washed and nuclei were stained with DAPI (0.5 µg/ml, Thermo Fisher Scientific, D1306). Target protein visualization was done on a laser scanning microscope (Zeiss, Jena, Germany) using 20x objectives.
IFNγ–ELISpot assay
2.5 × 103 target cells/well (MLH1−/− A7450, MLH1−/− 328, MLH1−/− lymphoma cells, YAC-1) were seeded in IFNγ–specific mAb (Mabtech, 3321–3)–coated, 96-well microtiter plates and incubated for 2 hours. Peripheral blood leukocytes or splenocytes (1 × 104/Well) were added to targets in triplicates and co-cultured overnight. Finally, bound antibody (Mabtech, 3321–6) was visualized by BCIP/NBT (KPL, Gaithersburg, Maryland, USA); spots were counted using an ELISpot reader. Presented are the numbers of IFNγ–secreting cells per 10,000 effector cells corrected for background levels counted in the absence of target cells, which was always ≤5 spots/well. Target cells without effector cells showed no background level.
Flow cytometric cytotoxicity assay
In a more functional cytotoxicity assay, lytic activity of effector cells against tumor target cells was determined by flow cytometry. Prior to co-culture, target cells (MLH1−/− A7450, MLH1−/− 328, MLH1−/− lymphoma cells, YAC-1) were labeled with CFDA-SE (carboxyfluorescein diacetate succinimidyl ester, Thermo Fisher Scientific, C1157; final concentration: 2 µmol/l). Target cells without effector cells were used as negative controls. Following co-incubation for twelve hours at an effector to target cell ratio of 10:1, 30:1, and 50:1, propidium iodide (PI, Sigma Aldrich, 11348639001; final concentration: 50µg/ml) was added to measure target cell death based on CFDA/PI double positive cells. Percentage (%) numbers of surviving cells were calculated from differences between normal and co-cultured cells in relation to the absolute number of measured events.
Statistics
All values are expressed as mean ± SD. After proving the assumption of normality (Kolmogorov-Smirnov test), differences between vaccinated and control mice were determined using the unpaired Student's t-test. Kaplan-Meier survival analysis was done by applying log rank test. The tests were performed by using Sigma-Stat 3.0 (Jandel Corp, San Rafael, CA). The criterion for significance was set to p < 0.05.
Supplementary Material
Funding Statement
German research fundation (MA5799/2-1)
Abbreviations
- CMMR-D
constitutional mismatch repair deficiency
- CRC
colorectal cancer
- FSM
frameshift mutation
- GIT
gastrointestinal tumor
- LS
Lynch Syndrome
- MMR-D
mismatch repair deficiency
- MSI
microsatellite instability
Disclosure of interest
The authors report no conflict of interest.
Acknowledgments
We gratefully thank Brigitte Vollmar and Bernd Krause for their continuous support in their efforts of chairing the Core Facility of Multimodal Small Animal Imaging. We also gratefully acknowledge the excellent technical assistance of Mrs. Anne Möller and Mrs. Joanna Förster. Furthermore, we thank Alexander Hohn, radiopharmacy team of the Department of Nuclear Medicine of the University Medical Centre Rostock, for providing 18F-FDG for the small animal PET/CT experiments.
References
- 1.Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin Cancer Res. 2016;22(4):813–20. doi: 10.1158/1078-0432.CCR-15-1678. PMID:26880610. [DOI] [PubMed] [Google Scholar]
- 2.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al.. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woerner SM, Tosti E, Yuan YP, Kloor M, Bork P, Edelmann W, Gebert J. Detection of coding microsatellite frameshift mutations in DNA mismatch repair-deficient mouse intestinal tumors. Mol Carcinog. 2015;54(11):1376–86. doi: 10.1002/mc.22213. PMID:25213383. [DOI] [PubMed] [Google Scholar]
- 4.Speetjens FM, Lauwen MM, Franken KL, Janssen-van Rhijn CM, van Duikeren S, Bres SA, van de Velde CJ, Melief CJ, Kuppen PJ, van der Burg SH, et al.. Prediction of the immunogenic potential of frameshift-mutated antigens in microsatellite instable cancer. Int J Cancer. 2008;123(4):838–45. doi: 10.1002/ijc.23570. PMID:18506693. [DOI] [PubMed] [Google Scholar]
- 5.Westdorp H, Kolders S, Hoogerbrugge N, de Vries IJM, Jongmans MCJ, Schreibelt G. Immunotherapy holds the key to cancer treatment and prevention in constitutional mismatch repair deficiency (CMMRD) syndrome. Cancer Lett. 2017;403:159–64. doi: 10.1016/j.canlet.2017.06.018. PMID:28645564. [DOI] [PubMed] [Google Scholar]
- 6.Lynch HT, Drescher K, Knezetic J, Lanspa S. Genetics, biomarkers, hereditary cancer syndrome diagnosis, heterogeneity and treatment: a review. Curr Treat Options Oncol. 2014;15(3):429–42. doi: 10.1007/s11864-014-0293-5. PMID:24827900. [DOI] [PubMed] [Google Scholar]
- 7.Vasen HF, Ghorbanoghli Z, Bourdeaut F, Cabaret O, Caron O, Duval A, Entz-Werle N, Goldberg Y, Ilencikova D, Kratz CP, et al.. EU-Consortium Care for CMMR-D (C4CMMR-D). Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium “Care for CMMR-D” (C4CMMR-D). J Med Genet. 2014;51(5):283–93. doi: 10.1136/jmedgenet-2013-102238. PMID:24556086. [DOI] [PubMed] [Google Scholar]
- 8.Maletzki C, Huehns M, Bauer I, Ripperger T, Mork MM, Vilar E, Klöcking S, Zettl H, Prall F, Linnebacher M. Frameshift mutational target gene analysis identifies similarities and differences in constitutional mismatch repair-deficiency and Lynch syndrome. Mol Carcinog. 2017;56(7):1753–64. doi: 10.1002/mc.22632. PMID:28218421. [DOI] [PubMed] [Google Scholar]
- 9.Maletzki C, Beyrich F, Hühns M, Klar E, Linnebacher M. The mutational profile and infiltration pattern of murine MLH1−/− tumors: concurrences, disparities and cell line establishment for functional analysis. Oncotarget. 2016;7(33):53583–98. doi: 10.18632/oncotarget.10677. PMID:27447752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodo S, Colas C, Buhard O, Collura A, Tinat J, Lavoine N, Guilloux A, Chalastanis A, Lafitte P, Coulet F, et al.. European Consortium “Care for CMMRD”. Diagnosis of Constitutional Mismatch Repair-Deficiency Syndrome Based on Microsatellite Instability and Lymphocyte Tolerance to Methylating Agents. Gastroenterology. 2015;149(4):1017–29.e3. doi: 10.1053/j.gastro.2015.06.013. PMID:26116798. [DOI] [PubMed] [Google Scholar]
- 11.Ryan E, Sheahan K, Creavin B, Mohan HM, Winter DC. The current value of determining the mismatch repair status of colorectal cancer: A rationale for routine testing. Crit Rev Oncol Hematol. 2017;116:38–57. doi: 10.1016/j.critrevonc.2017.05.006. PMID:28693799. [DOI] [PubMed] [Google Scholar]
- 12.Czink E, Kloor M, Goeppert B, Froehling S, Uhrig S, Weber TF, Meinel J, Sutter C, Weiss KH, Schirmacher P, et al.. Successful immune checkpoint blockade in a patient with advanced stage microsatellite unstable biliary tract cancer. Cold Spring Harb Mol Case Stud. 2017;3:a001974. doi: 10.1101/mcs.a001974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bauer K, Nelius N, Reuschenbach M, Koch M, Weitz J, Steinert G, Kopitz J, Beckhove P, Tariverdian M, von Knebel Doeberitz M, et al.. T cell responses against microsatellite instability-induced frameshift peptides and influence of regulatory T cells in colorectal cancer. Cancer Immunol Immunother. 2013;62(1):27–37. doi: 10.1007/s00262-012-1303-8. PMID:22729559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reuschenbach M, Kloor M, Morak M, Wentzensen N, Germann A, Garbe Y, Tariverdian M, Findeisen P, Neumaier M, Holinski-Feder E, et al.. Serum antibodies against frameshift peptides in microsatellite unstable colorectal cancer patients with Lynch syndrome. Fam Cancer. 2010;9(2):173–9. doi: 10.1007/s10689-009-9307-z. PMID:19957108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, Tariverdian M, Benner A, von Knebel Doeberitz M. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134(4):988–97. doi: 10.1053/j.gastro.2008.01.015. PMID:18395080. [DOI] [PubMed] [Google Scholar]
- 16.Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology. 2017;150(1):16–24. doi: 10.1111/imm.12675 Epub 2016 Oct 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernal M, Ruiz-Cabello F, Concha A, Paschen A, Garrido F. Implication of the β2-microglobulin gene in the generation of tumor escape phenotypes. Cancer Immunol Immunother. 2012;61(9):1359–71. doi: 10.1007/s00262-012-1321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirata T, Yamamoto H, Taniguchi H, Horiuchi S, Oki M, Adachi Y, Imai K, Shinomura Y. Characterization of the immune escape phenotype of human gastric cancers with and without high-frequency microsatellite instability. J Pathol. 2007;211(5):516–23. doi: 10.1002/path.2142. [DOI] [PubMed] [Google Scholar]
- 19.Chiang CL, Coukos G, Kandalaft LE. Whole Tumor Antigen Vaccines: Where Are We? Vaccines (Basel). 2015;3(2):344–72. doi: 10.3390/vaccines3020344. PMID:26343191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bourdais R, Rousseau B, Pujals A, Boussion H, Joly C, Guillemin A, Baumgaertner I, Neuzillet C, Tournigand C. Polymerase proofreading domain mutations: New opportunities for immunotherapy in hypermutated colorectal cancer beyond MMR deficiency. Crit Rev Oncol Hematol. 2017 May;113:242–8. doi: 10.1016/j.critrevonc.2017.03.027. [DOI] [PubMed] [Google Scholar]
- 21.Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D, Van Loo P, Tarpey PS, Coupland P, Behjati S, et al.. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet. 2015;47(3):257–62. doi: 10.1038/ng.3202. PMID:25642631. [DOI] [PubMed] [Google Scholar]
- 22.Linnebacher M, Maletzki C, Emmrich J, Kreikemeyer B. Lysates of S. pyogenes serotype M49 induce pancreatic tumor growth delay by specific and unspecific antitumor immune responses. J Immunother. 2008;31(8):704–13. doi: 10.1097/CJI.0b013e3181829f62. PMID:18779749. [DOI] [PubMed] [Google Scholar]
- 23.Antonios JP, Soto H, Everson RG, Moughon D, Orpilla JR, Shin NP, Sedighim S, Treger J, Odesa S, Tucker A, et al.. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro Oncol. 2017;19(6):796–807. doi: 10.1093/neuonc/now287. PMID:28115578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wimmer K, Rosenbaum T, Messiaen L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin Genet. 2017;91(4):507–19. doi: 10.1111/cge.12904. PMID:27779754. [DOI] [PubMed] [Google Scholar]
- 25.Maletzki C, Schmidt F, Dirks WG, Schmitt M, Linnebacher M. Frameshift-derived neoantigens constitute immunotherapeutic targets for patients with microsatellite-instable haematological malignancies: frameshift peptides for treating MSI+ blood cancers. Eur J Cancer. 2013 Jul;49(11):2587–95. doi: 10.1016/j.ejca.2013.02.035. [DOI] [PubMed] [Google Scholar]
- 26.Thomas S, Prendergast GC. Cancer Vaccines: A Brief Overview. Methods Mol Biol. 2016;1403:755–61. doi: 10.1007/978-1-4939-3387-7_43. PMID:27076165. [DOI] [PubMed] [Google Scholar]
- 27.Inderberg EM, Wälchli S, Myhre MR, Trachsel S, Almåsbak H, Kvalheim G, Gaudernack G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology. 2017;6(4):e1302631. doi: 10.1080/2162402X.2017.1302631. PMID:28507809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, et al.. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol. 2016;34(19):2206–11. doi: 10.1200/JCO.2016.66.6552. PMID:27001570. [DOI] [PubMed] [Google Scholar]
- 29.Rohde S, Lindner T, Polei S, Stenzel J, Borufka L, Achilles S, Hartmann E, Lange F, Maletzki C, Linnebacher M, et al.. Application of in vivo imaging techniques to monitor therapeutic efficiency of vemurafenib in an experimental model of microsatellite instable colorectal cancer. Oncotarget. 2017;8:69756–67. doi: 10.18632/oncotarget.19263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishikawa E, Yamamoto T, Matsumura A. Prospect of Immunotherapy for Glioblastoma: Tumor Vaccine, Immune Checkpoint Inhibitors and Combination Therapy. Neurol Med Chir (Tokyo). 2017;57(7):321–30. doi: 10.2176/nmc.nmc.ra.2016-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borie C, Colas C, Dartigues P, Lazure T, Rince P, Buhard O, Folliot P, Chalastanis A, Muleris M, Hamelin R, et al.. The mechanisms underlying MMR deficiency in immunodeficiency-related non-Hodgkin lymphomas are different from those in other sporadic microsatellite instable neoplasms. Int J Cancer. 2009;125(10):2360–6. doi: 10.1002/ijc.24681. PMID:19551857. [DOI] [PubMed] [Google Scholar]
- 32.Tian Y, Li M, Yu C, Zhang R, Zhang X, Huang R, Lu L, Yuan F, Fan Y, Zhou B, et al.. The novel complex combination of alum, CpG ODN and HH2 as adjuvant in cancer vaccine effectively suppresses tumor growth in vivo. Oncotarget. 2017;8(28):45951–64. doi: 10.18632/oncotarget.17504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talmadge JE, Adams J, Phillips H, Collins M, Lenz B, Schneider M, Schlick E, Ruffmann R, Wiltrout RH, Chirigos MA. Immunomodulatory effects in mice of polyinosinic-polycytidylic acid complexed with poly-L-lysine and carboxymethylcellulose. Cancer Res. 1985;45(3):1058–65. [PubMed] [Google Scholar]
- 34.Wada S, Jackson CM, Yoshimura K, Yen HR, Getnet D, Harris TJ, Goldberg MV, Bruno TC, Grosso JF, Durham N, et al.. Sequencing CTLA-4 blockade with cell-based immunotherapy for prostate cancer. J Transl Med. 2013;11:89. doi: 10.1186/1479-5876-11-89. PMID:23557194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Péron S, Metin A, Gardès P, Alyanakian MA, Sheridan E, Kratz CP, Fischer A, Durandy A. Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination. J Exp Med. 2008;205(11):2465–72. doi: 10.1084/jem.20080789. PMID:18824584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. PMID:19451168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al.. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. PMID:20644199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al.. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. doi: 10.1038/ng.806. PMID:21478889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cingolani P. snpEff: Variant effect prediction". http://snpeff.sourceforge.net; 2012.
- 40.Wickham H. ggplot2. Elegant Graphics for Data Analysis Book Use R; 2009. [Google Scholar]
- 41.Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2. doi: 10.1093/bioinformatics/btu393. PMID:24930139. [DOI] [PubMed] [Google Scholar]
- 42.Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. doi: 10.1186/1471-2105-12-35. PMID:21269502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. PMID:23550210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. PMID:22588877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edelmann W, Yang K, Kuraguchi M, Heyer J, Lia M, Kneitz B, Fan K, Brown AM, Lipkin M, Kucherlapati R. Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res. 1999;59(6):1301–7. PMID:10096563. [PubMed] [Google Scholar]
- 46.Vandenberk L, Belmans J, Van Woensel M, Riva M, Van Gool SW. Exploiting the Immunogenic Potential of Cancer Cells for Improved Dendritic Cell Vaccines. Front Immunol. 2016;6:663. doi: 10.3389/fimmu.2015.00663. PMID:26834740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.