

Abstract
Disruption of post-transcriptional gene regulation is a critical step in oncogenesis that can be difficult to observe using traditional molecular techniques. To overcome this limitation, a modified polyadenylation site sequencing (PAS-seq) protocol was used to generate a genome-wide map of alternative polyadenylation (APA) events in human primary breast tumor specimens and matched normal tissue. This approach identified an APA event in the PRELID1 mRNA that enhances its steady state level and translational efficiency, and is a strong breast cancer subtype-dependent predictor of patient clinical outcomes. Furthermore, it is demonstrated that PRELID1 regulates stress response and mitochondrial reactive oxygen species (ROS) production in a cell-type specific manner. Modulation of PRELID1 expression, including its post-transcriptional control, appears to be a common stress response across different cancer types. These data reveal that PRELID1 mRNA processing is an important regulator of cell-type specific responses to stress used by multiple cancers and is associated with patient outcomes.
Implications
This study suggests that the regulation of PRELID1 expression, by APA and other mechanisms, plays a role in mitochondrial ROS signaling and represents a novel prognostic factor and therapeutic target in cancer.
Keywords: alternative polyadenylation, PRELID1, cancer, mitochondria, ROS signaling
INTRODUCTION
Aberrant gene regulation plays a central role in the initiation and progression of cancer. In healthy cells, redundant pathways guard against unchecked proliferation and maintain genomic integrity. Perturbation of normal regulatory mechanisms can cause overexpression of tumor-promoting oncogenes or repression of tumor suppressor genes. In many cases, these changes in expression are the result of genomic mutation or altered transcriptional regulation. Disruption of post-transcriptional regulatory mechanisms, including miRNA targeting, is another important mode of oncogene activation. Oncogenes may avoid 3′ UTR-mediated repression by removing regulatory sites using alternative splicing or alternative polyadenylation (APA). Evidence suggests that APA may play a significant role in cancer (1,2) (for a review, see ref. 3). Mayr and Bartel reported that several oncogenes, including IMP-1 and Cyclin D1 (CCND1), have shortened 3′ UTRs in cancer cell lines relative to normal tissue controls. This follows similar reports that show global and coordinated changes in 3′ UTR length in T cell activation (shortening), neuronal activation (shortening) and embryonic development (lengthening) (4–6). More recent evidence suggests that an APA event in the PD-L1 gene (CD274) plays a direct role in PD-1/PD-L1-mediated immune escape in cancer development (7). While the physiological relevance of genome wide APA may not always be obvious (8), it is clear that individual APA events have a profound impact on cell biology.
Here, we provide evidence for a functional APA event in the PRELID1 mRNA. The PRELID1 protein, which localizes to the mitochondrial intermembrane space (IMS), exerts a cytoprotective effect in many cell types (9). Consistent with its well-defined, evolutionarily conserved cytoprotective role, PRELID1 is also linked to cellular metabolism and survival through its interaction with TRIAP1 (TP53 Regulated Inhibitor of Apoptosis 1) (10). The PRELID1/TRIAP1 complex transfers phosphatidic acid across the intermembrane space to the inner membrane where it is used to synthesize cardiolipin, an evolutionally conserved diphosphatidylglycerol lipid that is nearly exclusive to the mitochondria in eukaryotes (10–14). Cardiolipin plays a key role in virtually all mitochondrial functions, including the accumulation, anchoring and stabilization of the oxidative phosphorylation and respiratory chain proteins (15–19), mitochondrial fission (20), formation of mitochondrial membrane pores and the release of cytochrome c during apoptosis (21), as well as mitochondrial signaling (through its translocation to the outer membrane) (22–24). Furthermore, there is evidence that loss of cardiolipin increases production of reactive oxygen species (ROS) in the mitochondria (25). Here, we present a genome-wide map of APA events in primary human breast tumors, including a biologically and clinically relevant APA event at the PRELID1 locus that influences mitochondrial ROS production and response to cellular stress, with clinical impact reaching across cancer types.
MATERIALS AND METHODS
Oligonucleotide Sequences
Sequences for all shRNAs and oligonucleotide primers used in this study are shown in Supp. Table 1.
PAS-seq
We performed PAS-seq (26,27) with extensive modifications. Total RNA was fragmented and DNase treated by incubating with 1.5X First Strand Buffer (ThermoFisher Scientific) and 50U/mL RQ1 RNase-free DNase (Promega) for 30 minutes at 37C, followed by 3 minutes at 95C. The RNA was then concentrated using the RNeasy minElute kit (Qiagen) and reverse transcribed using SuperScript II (ThermoFisher Scientific) as follows: First, 1μg of RNA was mixed with 1μL each of primers 1320 UMI-DT2 (TVN primer with UMI) and 1290 SMART-TRUSEQ (template switching primer) in 12.5μL total volume and incubated at 65C for 5 minutes, followed by a hold at 42C. Next, 4 μL 5X First-strand buffer, 2 μL 100 mM DTT, 0.5 μL RNasin Plus (Promega), and 1 μL SuperScript II were added to the RNA+primers (all pre-warmed to 42C). The reaction was held at 42C for 50 minutes, followed by 15 minutes at 70C. The resulting cDNA was then amplified using Phusion Hot Start II (ThermoFisher Scientific) as follows: 4μL of cDNA and 1μL of TRU-SEQ barcoded primer mix (Illumina) were used in a standard 25μL reaction with the following program:
| 98° | 2:00 | |
| 98° | 0:10 | 3x |
| 60° | 0:30 | |
| 72° | 0:30 | |
| 98° | 0:10 | 25x |
| 72° | 1:00 | |
| 72° | 10:00 |
Resulting PCR products were then run on a 2% SeaPlaque agarose (Lonza) gel. The region between 250 and 380bp was excised and the DNA purified using the minElute Gel Kit (QIagen). The resulting libraries were then sequenced on an Illumina HiSeq 2000 (50bp, single end).
After sequencing, bases were called on raw CIF image files using AYB (http://www.ebi.ac.uk/goldman-srv/AYB/; (28)), which simultaneously removed the 14 nt poly(A) tail in bases 7–20, leaving the 6 bp UMI followed by the remainder of the read. Reads were then demultiplexed with fastq-multx (https://github.com/brwnj/fastq-multx) and the demultiplexed reads were processed with cutadapt (https://code.google.com/p/cutadapt/; (29)) to remove 3′ adaptor sequences and 3′ bases with QUAL < 10. Trimmed reads were aligned to the genome using NovoAlign (http://www.novocraft.com/products/novoalign/), and UMIs were collapsed using umitools (https://github.com/brwnj/umitools). Strand-specific peaks were called on alignments with using MACS2 (https://github.com/taoliu/MACS, (30)). Called peaks from all samples were then merged to create a master list of breast poly(A) sites. All sites with more than 6 adenosine residues in the 10 bases immediately downstream of the called poly(A) site were excluded from further analysis due to likelihood of priming on genomically encoded poly(A) tracts, and all sites within 5 nt of each other were merged into one site. Count tables were then generated for all samples with the merged, filtered poly(A) site list. Significant shifts were detected between pairs of samples by performing Fisher’s exact test on all pairwise sets of sites in each transcript. Raw sequence data and peak calls available at GSE87914.
Ribosome Profiling
Ribosome profiling libraries were prepared using the TruSeq Ribo Profile kit (RPHMR12126; Epicentre) with some modifications. Specifically, after ribosome protected RNA was purified with an S400 column (GE Healthcare), the RNA was extracted by phenol:chloroform and precipitated with isopropanol instead of using an RNA clean and concentrator-5 column (Zymo Research). Additionally, all RNA purification steps using Zymo columns were performed according to manufacturer protocols. Total RNA libraries were also prepared using TruSeq Ribo Profile kit. All libraries were sequenced on the Illumina HiSeq 2000 platform (50bp, single end).
Ribosomal profiling reads were processed with cutadapt to remove 3′ adaptor sequences and 3′ bases with QUAL < 10. Reads originating from human rRNA sequences (nuclear and mitochondrial) were filtered using TagDust2 (http://tagdust.sourceforge.net/, (31)), and the remaining reads were aligned to the human genome (grch37 build) using hisat2 (https://ccb.jhu.edu/software/hisat2/index.shtml, (32)). Aligned reads were assigned to features using the featureCounts component of the RSubread (https://bioconductor.org/packages/release/bioc/html/Rsubread.html, (33)), and translational efficiency was calculated using DESeq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html; (34)), both in R (35).
Analysis of TCGA RNA-seq data
All available RNA-seq V2 data and clinical annotations were downloaded from the Genomic Data Commons Data portal (https://gdc-portal.nci.nih.gov/). Exon and transcript level counts were used for differential expression testing with DESeq2, and survival curves were generates using the rms (https://cran.r-project.org/web/packages/rms/index.html, (36)), survival (https://cran.r-project.org/web/packages/survival/index.html, (37)) and dplyr (https://cran.r-project.org/web/packages/dplyr/index.html, (38)) packages in R. Gene ontology term enrichment was performed using g:Profiler (http://biit.cs.ut.ee/gprofiler/; (39)). To account for the disproportionate influence of the more numerous ER+ (IHC) tumors, we focused on genes that were differentially expressed in both ER− (IHC) and ER+ (IHC) tumors classified as PRELID1-associated/independent for GO term enrichment analysis.
Cell culture
The human cell lines MCF-10A and MDA-MB-231 were originally obtained from ATCC. The HEK293-FT cell line was obtained from Invitrogen. The MCF-7 cell line was obtained from Sam Brooks, the Michigan Cancer Foundation. Cells were maintained in DMEM with 10% FBS (MDA-MB-231), DMEM with 10% FBS and 6 mM L-glutamine (HEK-293FT), MEM with 5% FBS (MCF-7), or MEGM (Lonza). Cell line authenticity was confirmed by short tandem repeat analyses in the University of Colorado DNA Sequencing Core Laboratory. Cell lines were tested for mycoplasma contamination every 6 months using the Universal Mycoplasma Detection Kit (ATCC). No cell lines used in this manuscript have ever tested positive for mycoplasma contamination in this lab (most recent test: July, 2017).
Plasmids and transient transfections
PRELID1 expression constructs were created by reverse transcribing total RNA from the MCF-7 and MCF-10A cell lines with the TVN-PCRv2 RT primer using SuperScript III (ThermoFisher Scientific). The resulting cDNA was then amplified by PCR using the PRELID1-clone-F and TVN-REVv2 primers with Q5 polymerase (NEB). The products were run on a 1% agarose gel and excised bands at ~800 bp (proximal site) in MCF-7 lane and ~1100 bp (distal site) in MCF-10A lane. Purified excised bands using minElute Gel Kit (Qiagen) and cloned each into the pSC-B blunt cloning vector (Agilent Technologies). The inserts were excised from each pSC-B construct using BamHI and XhoI (NEB) and ligated into similarly cut pCMV-Cyclin D1 (gift from Yue Xiong; Addgene plasmid #19927) to create pCMV-PRELID1 and pCMV-PRELID1-Proximal. pCMV-PRELID1 and pCMV-PRELID1-Proximal were transfected into MCF-7 cells in Lipofectamine LTX (ThermoFisher Scientific) according to the manufacturer’s recommendations.
Drug treatments and cell growth assays
MCF-7 cells were treated with Docextaxel or Thasigargin for 48 hours or with for Thapsigargin for 4.5 hours, followed by a wash in 1X PBS and 43.5h recovery. Growth was estimated at 0, 24 and 48 hours by total protein sulforhodamine assay (SRB). Cells were processed for the sulforhodamine B colorimetric (SRB) assay 48 hours after treatment. Briefly, culture media was removed and cells were gently washed 1X with PBS followed by a 30-minute incubation at 4C in 10% trichloroacidic acid. Cells were washed 5 times with deionized water and plates were dried for 2 hours. Sulforhodamine B (0.2%) dye was added for 20 minutes at room temperature, followed by 5 washes with 1% acetic acid and a 2-hour drying period. Each well of a 96-well plate received 0.2 mL of 10mM unbuffered tris base and was agitated for 10 minutes. Total protein was assessed using a BioTek (Winooski, VT) Synergy 2 microplate reader (565 nm – 690 nm background). Key SRB assay results were validated by counting viable cells with a hemocytometer following staining with trypan blue.
shRNA viral packaging, transduction and clonal selection
PRELID1, TRIAP1 and controls shRNA constructs were obtained from the University of Colorado Cancer Center Functional Genomics Core. The shRNAs were packaged in HEK-293FT and viral supernatant was collected according to standard protocols available from the Functional Genomics Core. MCF-7 and MDA-MB-231 were transduced with viral supernatant according to standard protocols, and 1 mg/mL Puromycin (ThermoFisher Scientific) selection was added 24h post-transduction. After 96h of selection, the cells were trypsinized and plated in 96-well plates at serial dilutions of 16 cells/well to 0.5 cells/well to obtain single clones. Wells that received exactly one cell were identified by microscopy, and these clonal lines were expanded and characterized. Clonal lines arising from at least two different shRNAs were used for each target.
RT-qPCR
Total RNA for qRT-PCR was isolated using TRIzol (ThermoFisher Scientific). PRELID1 expression was assayed by SYBR green qPCR with the PRELID1-qRT-F and PRELID1-qRT-R primers. 18S rRNA was used as an endogenous normalization control (18S-F and 18S-R primers). The transient PRELID1 overexpression experiment was also normalized to PRELID1 DNA content by qPCR with the PRELID1-Dq-F and PRELID1-Dq-R primers. All qPCRs were performed in duplicate on at least two separate occasions.
Western Blotting
Whole cell lysates were lysed in RIPA buffer (150mM NaCl, 150mM NaCl, 50mM Tris-HCl, pH 7.5, 0.1% SDS, 0.5% Sodium Deoxycholate, 1% Triton X-100), separated by SDS-PAGE on 4–15% gradient gels and transferred onto Immobilon-FL PVDF membranes (IPFL00010; EMD Millipore) using the NuPage system (ThermoFisher Scientific). After transfer, membranes were blocked and probed with antibodies against anti-PRELID1 (ab174149, 1:200 dilution; Abcam), anti-GAPDH (NB300-221SS, 1:2000; Novus Biologicals) or anti-SDHA (A2594, 1:1000; ABclonal Biotechnology) followed by washing and incubating with secondary antibodies (Goat anti-Mouse IRDye 680LT, 926-68020 1:20,000 and Goat anti-Rabbit IRDye 800CW, 926-32211, 1:20,000; LI-COR Biosciences). Membranes were visualized by Oddysey fluorescence imagining scanner (LI-COR Biosciences).
Flow Cytometry
Superoxide production was measured by staining cells with MitoSOXTM Superoxide Indicator (ThermoFisher Scientific) according to the manufacturer’s recommendations. Apoptosis was measured by staining cells with Annexin V-FITC and Propidium Iodide (Miltenyi Biotec) according to the manufacturer’s recommendations. Flow cytometric analysis was performed at the University of Colorado Cancer Center on a Gallios Flow Cytometer (Beckman Coulter).
RESULTS
PAS-seq reveals alternative polyadenylation events in primary breast tumors
To determine the extent of APA in human breast cancer, we mapped polyadenylation sites transcriptome-wide using a modified version of PAS-Seq (26,27) in 12 pairs of primary human breast tumor/adjacent normal samples: six estrogen receptor (ER) positive (ER+) and six ER negative (ER-) tumors, each with adjacent normal tissue. Participants were selected by consecutive sampling of consenting patients, with lack of HER2 amplification as the only selection criteria. In order to minimize individual variation, during analysis we created three pooled samples containing normalized reads from all six ER+ tumors (TS-ERP), all six ER− tumors (TS-ERN) and all 12 adjacent normal samples (NBT), respectively. To validate our primary sample results, we also used our modified PAS-seq protocol in the ER+ breast cancer cell line MCF-7 and the non-tumor derived, non-tumorigenic breast cell line MCF-10A. We identified significant APA events by comparing data among different primary sample pools (NBT, TS-ERN, TS-ERP) using Fisher’s exact test for pairwise combinations of poly(A) sites. The results of these comparisons are summarized in Table 1. The relatively small number of APA events between normal tissue (NBT) and tumor (11 for ER-, 46 for ER+ out of 45,770 total tests) demonstrate that APA events between normal breast tissue and breast cancer are rare. In fact, none of the events described in breast cell lines by Mayr and Bartel (1), such as CCND1 (Fig. 1A), are detectable by PAS-seq in our primary samples. A small number of significant APA events, summarized in Supp. Table 2, are also detectable between ER+ and ER− tumors. Most notable among these events was a high amplitude proximal shift in PRELID1 (Fig. 1B) that removes 77% (290 nt) of the 3′ UTR in ER+ tumors relative to ER− tumors (q=0.0068) and adjacent normal tissue (q=0.012). In addition to validation with PAS-seq in cell lines (Fig. 1B), we also designed a multiplex RT-PCR assay (diagrammed at the bottom of Fig. 1B) to determine poly(A) site utilization. This assay confirmed that, like the ER+ tumors, the ER+ MCF-7 cell line utilizes the proximal poly(A) site, justifying its use as a model for the short form of PRELID1. Interestingly, all other cell lines tested, including the ER− MDA-MB-231 and the non-tumor derived, non-tumorigenic (“normal”) MCF-10A, utilize the distal poly(A) site, akin to ER− tumors and adjacent normal tissue (Fig. 1C). The ER+ ZR-75-1 cell line uses the proximal site at a higher rate than any cell line tested except MCF-7, but the distal site still predominates in this line as well.
Table 1.
Statistically significant APA events in breast cancer (q <= 0.01, fold-change >= 2).
| Comparison | Proximal Shifts | Distal Shifts |
|---|---|---|
| NBT to TS-ERN | 7 | 4 |
| NBT to TS-ERP | 26 | 20 |
| TS-ERN to TS-ERP | 16 | 9 |
Figure 1. Alternative polyadenlylation in breast cancer measured by PAS-seq.

A–B. PAS-seq coverage at the (A) CCND1 and (B) PRELID1 loci for a pool of 12 normal breast tissue samples (NBT), the MCF-10A cell line, a pool of 6 matched ER− breast tumor samples (TS-ERN), a pool of 6 matched ER+ breast tumor samples (TS-ERP) and the MCF-7 cell line. C. RT-PCR validation of PRELID1 APA in breast cell lines. Utilization of the proximal site product produces a 355 bp band, while utilization of the distal site produces a 312 bp band. Figure based on output from http://genome.ucsc.edu (hg19 assembly) (56,57).
APA in the PRELID1 gene results in transcript stabilization and increased translational efficiency
To assess the functional consequences of PRELID1 APA, we measured PRELID1 expression by RT-qPCR following transient transfection of an ER+ breast cancer cell line with equimolar amounts of pCMV-derived plasmids containing the PRELID1 cDNA with either the full 3′ UTR or the short 3′ UTR (Fig. 2A). The full 3′ UTR construct increased PRELID1 expression by 28-fold relative to endogenous expression (pCMV-Empty control), while the short 3′ UTR construct increased expression by 134-fold relative to endogenous expression. The 4.7-fold difference reflects substantial stabilization of the short from relative to the long form in breast cancer cells. In contrast, transient overexpression of PRELID1 had no effect on expression of the MXD3 gene (Fig. 2A, black bars), which overlaps the 3′ end of the PRELID1 locus and is transcribed in the opposite direction. Next, to assess the role of PRELID1 APA in a larger cohort of primary tumor samples, we obtained all available RNA-Seq V2 data from the cancer genome atlas (TCGA; https://gdc-portal.nci.nih.gov/). To measure APA, we calculated the ratio of reads mapping to PRELID1 exon 4, which has a consistent length in all isoforms, to the reads mapping to exon 5, which contains the 3′ UTR and varies in length due to APA. We then plotted the PRELID1 exon 4/exon 5 ratio vs. transcript abundance (exon 4 only, to account for the 3′ bias of oligo-d(T) RNA-seq) (Fig. 2B). Consistent with our transient transfection data, utilization of the short form of PRELID1 (high Exon 4/Exon 5 ratio) in all primary tumor samples found in TCGA is positively correlated with increased PRELID1 transcript abundance (R2 = 0.37, p < 0.0001).
Figure 2. PRELID1 APA correlates with transcript abundance and translational efficiency.

A. Relative expression of PRELID1 (white) or the adjacent MXD3 (black) measured by RT-qPCR following overexpression of PRELID1-Dist/Prox in MCF-7. Expression is relative to transfection with pCMV-Empty (“Control”). RT-qPCRs were performed in duplicate on two separate biological replicates, and an unpaired t-test was used to assess significance. B. PRELID1 Exon 4 expression (y-axis) vs. PRELID1 Exon 4/Exon 5 ratio (APA; x-axis) for all tumors in TCGA. n=10033; Pearson coefficient (r) from least-squares fit. C. PRELID1 ORF translational efficiency (y-axis) vs. PRELID1 Exon 4/Exon 5 ratio (APA; x-axis) for MCF-7 (filled circles) and UCD-12 (empty circles). n=10; Pearson coefficient (r) from linear regression. D. Representative Western blot showing PRELID1, SHDA and GAPDH in MCF-7 and MDA-MB-231 whole cell lysates. Western blot was performed twice on separate biological replicates. An unpaired t-test was used to assess significance of western blot densitometry. E. PRELID1 protein expression (western blot) relative to PRELID1 mRNA (measured by RT-qPCR) in MCF-7 and MDA-MB-231 clonal cell lines, normalized to MDA-MB-231. F. Quantitation of PRELID1 relative to GAPDH in (D). G. Quantitation of SDHA relative to GAPDH in (D). H. Quantitation of PRELID1 relative to SDHA in (D). A Two-way ANOVA (cell line x shRNA) followed by Tukey’s post hoc test was used to determine significance. *p<0.05, ***p<0.001
To determine if PRELID1 APA influences translatability of the PRELID1 mRNA as well as steady state transcript level, we utilized ribosome profiling data generated in our laboratory from two ER+ cell lines (MCF-7 and UCD12). Using the ratio of PRELID1 exon 4/exon 5 in the total RNA fraction as a measure of APA (as in Fig. 2B), we plotted APA vs. translational efficiency (Figure 2C). As with steady state transcript levels, translational efficiency is positively correlated with utilization of the short form of PRELID1 (R2 = 0.56, p = 0.012). These data suggest that the variable region of the PRELID1 3′ UTR contains repressive cis-elements that reduce transcript abundance and decrease translation of the PRELID1 mRNA, reducing the amount of PRELID1 protein produced. To further study this relationship, we generated shRNA knockdowns of PRELID1 in cell line models using the proximal (MCF-7) and distal (MDA-MB-231) PRELID1 poly(A) sites, and isolated individual cellular clones. We characterized triplicate clones from each group (shRNA control and PRELID1 knockdown) by RT-qPCR and by western blot with antibodies against PRELID1, SHDA (mitochondrial complex II subunit; surrogate for functional mitochondrial protein) and GAPDH (Fig. 2D). We obtained significant (>60%) knockdown of PRELID1 protein in both MCF-7 and MDA-MB-231 relative to both total cellular protein (GAPDH; Fig. 2F) and functional mitochondrial protein content (SDHA; Fig. 2H). Consistent with our ribosome profiling results, we found that PRELID1 translational efficiency was higher in MCF-7 (proximal poly(A) site) relative to MDA-MB-231 (distal poly(A) site) when comparing PRELID1 mRNA and protein levels in all 12 shRNA clones (Fig. 2E). We also noted that PRELID1 expression was higher per-cell in MDA-MB-231 relative to MCF-7 (Fig. 2F, “NC” groups), but SHDA expression was proportionally higher as well (Fig. 2G), resulting in similar PRELID1 expression relative to functional mitochondrial protein content (PRELID1/SDHA ratios) in both cell lines (Fig. 2H). Further, while knockdown of PRELID1 had no effect on SHDA levels in MCF-7, which are known to rely heavily on oxidative phosphorylation (typical for ER+ tumors (40)), knockdown of PRELID1 in MDA-MB-231 caused a significant reduction in SHDA expression (Fig. 2G).
PRELID1 expression modulates response to cellular stress in breast cancer cells
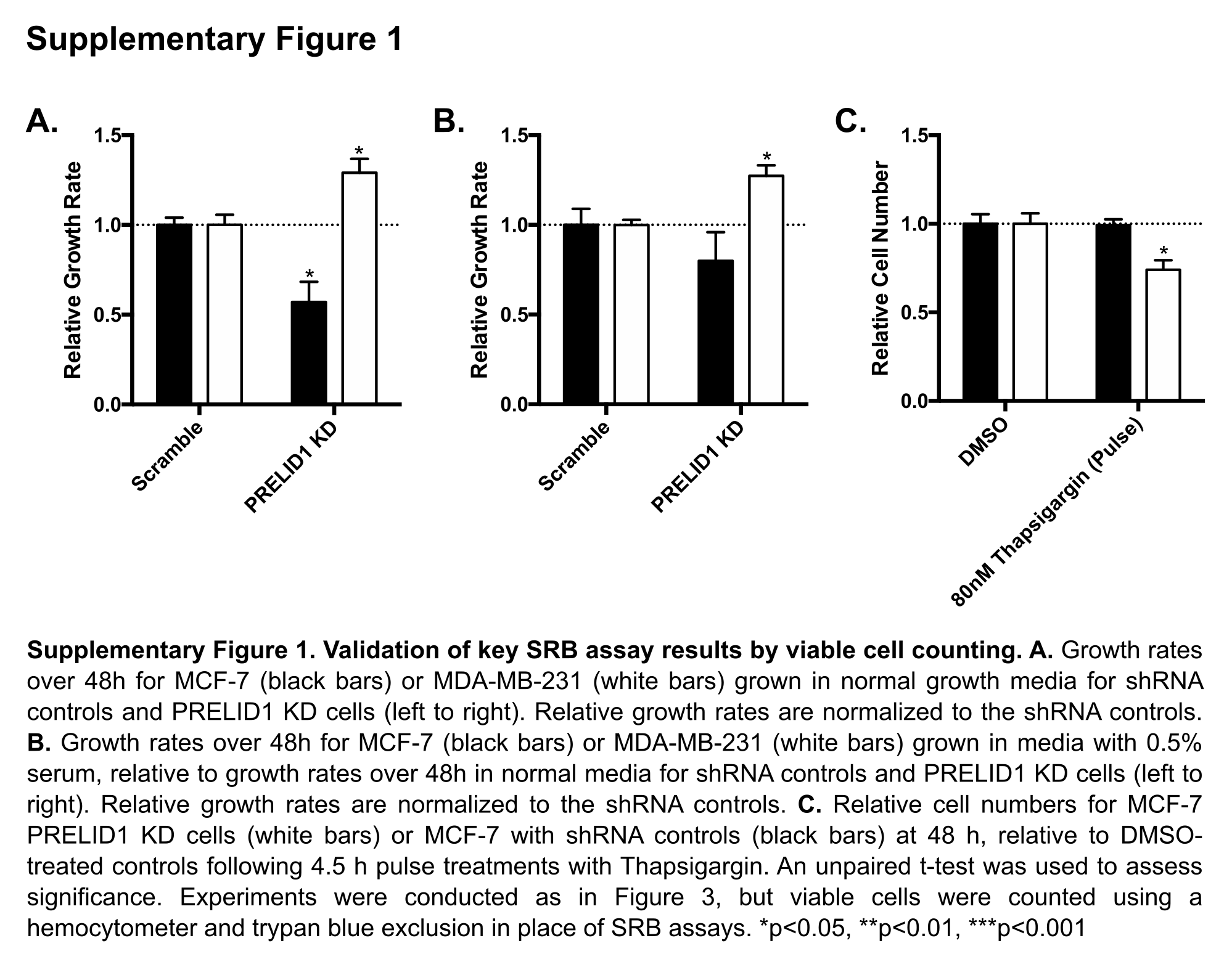
Given the known functions of PRELID1 (9,10,41,42), we hypothesized that modulation of its expression may play a role in breast cancer cell growth. Using our clonal MCF-7 and MDA-MB-231 shRNA cell lines, we determined the effect of PRELID1 loss on growth rates under both normal and nutrient deprived conditions by performing SRB assays to measure cellular protein content over 48h. Under normal growth conditions, MCF-7 grew significantly slower with reduced PRELID1, while MDA-MB-231 surprisingly grew significantly faster (Fig. 3A; validated by viable cell counts in Supp. Fig. 1A). Importantly, this effect was not driven by changes in the proportion of apoptotic cells in either cell line (Supp. Fig. 2A–D). Knockdown of PRELID1’s partner, TRIAP1, had no significant effect in either cell line under normal growth conditions (Fig. 3A). To determine the contribution of PRELID1 expression in the cellular response to stress (nutrient deprivation), we grew MCF-7 PRELID1 KD and MDA-MB-231 PRELID1 KD cells and controls in media containing either 0.5% serum or normal serum concentrations for 48 hours and again measured cellular protein content by SRB assay. Both cell lines grew more slowly in reduced serum conditions than normal growth conditions (55% of normal for MCF-7, 43% for MDA-MB-231). Consistent with the results in normal growth media, PRELID1 KD again had the opposite effect in the ER+ and ER− cell lines: in low serum conditions, MCF-7 grew more slowly with reduced PRELID1 than controls, while MDA-MB-231 grew significantly faster with reduced PRELID1 compared to controls (Fig. 3B; validated by viable cell counts in Supp. Fig. 1B). In this case, knockdown of TRIAP1 mirrored knockdown of PRELID1, suggesting that the shared role of phosphatidic acid transport across the mitochondrial intermembrane space becomes more important under stress conditions.
Figure 3. PRELID1 modulates cell growth response to cellular stress in breast cancer.

A. Growth rates over 48h for MCF-7 (black bars) or MDA-MB-231 (white bars) grown in normal growth media for (left to right) shRNA controls, PRELID1 KD cells, and TRIAP1 KD cells. Relative growth rates are normalized to the shRNA controls. B. Growth rates over 48h for MCF-7 (black bars) or MDA-MB-231 (white bars) grown in media with 0.5% serum, relative to growth rates over 48h in normal media for (left to right) shRNA controls, PRELID1 KD cells, and TRIAP1 KD cells. Relative growth rates are normalized to the shRNA controls. C. Relative cell numbers for MCF-7 PRELID1 KD cells (white bars) or MCF-7 with shRNA controls (black bars) at 48 h, relative to DMSO-treated controls following 4.5 h pulse treatments with Thapsigargin. D. Relative cell numbers for MCF-7 PRELID1 KD cells (white bars) or MCF-7 with shRNA controls (black bars), relative to DMSO-treated controls following 48 h treatments with Docetaxel or Thapsigargin. SRB assays were performed with 8 wells/condition for three separate biological replicates. An unpaired t-test was used to assess significance. *p<0.05, **p<0.01, ***p<0.001
Considering the impact on growth rates, especially under stress, we hypothesized that PRELID1 may also modulate response to other stressors. To test this, we induced endoplasmic reticulum stress, a common result of nutrient deprivation in cancer (43,44). We treated MCF-7 PRELID1 KD cells and controls with 10nM or 80nM Thapsigargin, which induces endoplasmic reticulum stress via inhibition of sarcoplasmic/endoplasmic reticulum Ca2+ ATPases [SERCAs] (45), for 4.5 h and allowed them to recover for 43.5 h. We performed SRB assays at 0h and 48h to measure cellular protein content, and found a significant difference between controls and PRELID1 KD cells at 80nM (Fig. 3C; validated by viable cell counts in Supp. Fig. 1C). This difference was not due to apoptosis, which was negligible under all conditions (Supp. Fig. 2E–H). We next treated the MCF-7 PRELID1 KD cells and controls with 10nM Thapsigargin or 20nM Docetaxel for 48 h and again measured cellular protein content by SRB assay (Fig. 3D). No significant difference was observed with either compound in PRELID1 KD cells relative to controls, suggesting that main function of the PRELID1/TRIAP complex is to buffer stress and not as a chemotherapy protective mechanism.
PRELID1 knockdown increases mitochondrial ROS production in ER− breast cancer cells
As PRELID1 regulates cardiolipin content in the mitochondrial membranes and cardiolipin content is correlated with mitochondrial ROS production, we hypothesized that PRELID1 may influence cell growth and stress response through regulation of mitochondrial ROS production. To test this, we measured mitochondrial ROS production in our PRELID1 shRNA clones. Breast cancer cell clones were treated with MitoSOX Red and analyzed by flow cytometry (Fig. 4). MDA-MB-231 shRNA controls had higher steady-state ROS levels than MCF-7 shRNA controls (Fig. 4A & 4D), consistent with increased utilization of mitochondrial ROS signaling in these cells. While MCF-7 did not experience a significant increases in mitochondrial ROS production with PRELID1 knockdown, but the increase was significant in MDA-MB-231 (Fig. 4B–D).
Figure 4. PRELID1 knockdown increases mitochondrial ROS production in ER− breast cancer cells.

A. MitoSOX Red fluorescence (x-axis) vs. proportion of gated events (y-axis) for MCF-7 shRNA control (dark grey) vs. MDA-MB-231 shRNA control (light grey; representative clones). B. MitoSOX Red fluorescence (x-axis) vs. proportion of gated events (y-axis) for MCF-7 shRNA control (dark grey) vs. MCF-7 PRELID1 shRNA knockdown (light grey; representative clones). C. MitoSOX Red fluorescence (x-axis) vs. proportion of gated events (y-axis) for MDA-MB-231 shRNA control (dark grey) vs. MDA-MB-231 PRELID1 shRNA knockdown (light grey; representative clones). D. Relative MitoSOX fluorescence (per-clone median) by shRNA group. Flow was performed on three separate clonal cell lines per group on two separate occasions. Histograms are from representative runs. A Two-way ANOVA (cell line x shRNA) followed by Tukey’s post hoc test was used to determine significance. *p<0.05, **p<0.01, ***p<0.001
PRELID1 APA, but not expression, is correlated with a subtype-dependent effect on breast cancer patient survival
Based on our experimental results, we wanted to determine the role of PRELID1 APA event on survival among breast cancer patients. Using the TCGA RNA-seq data for breast cancer, we split patients by ER status and PRELID1 APA (exon 4/exon 5 ratio) and compared their clinical outcomes (Fig. 5). Consistent with our experimental results, in patients with ER− disease (Fig. 5A), increased use of the proximal site conferred a significant survival advantage (p = 0.041), while the opposite was true in patients with ER+ disease (Fig. 5B; p = 0.036). In contrast to the strong and differential effect of APA on outcomes, PRELID1 transcript expression (measured as exon 4 expression) had no significant effect on outcomes in ER− or ER+ disease (Fig. 5C–D).
Figure 5. PRELID1 APA, but not transcript abundance, influences breast cancer outcomes in a subtype-dependent manner.

Overall survival split by median PRELID1 Exon 4/Exon 5 ratio (A–B) or median PRELID1 Exon 4 expression (C–D) in ER− (A,C) and ER+ (B,D) patients. *p<0.05, **p<0.01, ***p<0.001
We hypothesized that ER status was not the only factor that determined the effect of PRELID1 APA on tumor response to stress and ultimately on patient survival. However, other clinically relevant markers, including HER2 (ERBB2), progesterone receptor (PGR), p53 (TP53) and cancer stage failed to correlate significantly with PRELID1-associated outcomes in breast cancer (data not shown). To further investigate the potential mechanism of differential response to PRELID1 APA, we split all TCGA breast cancer patients who had died within five years of diagnosis into two groups by PRELID1 APA (median Exon 4/Exon 5 ratio). The first group, PRELID1-associated cancers (ER+ like), consisted of deceased patients with predominantly proximal PRELID1 poly(A) site usage. The second group, PRELID1-independent cancers (ER− like), consisted of deceased patients with largely distal PRELID1 poly(A) site. We then performed differential gene expression analysis between the two groups separately in ER+ and ER− tumors to avoid confounding influence from the more numerous ER+ tumors, and analyzed consensus differentially expressed genes using gProfiler (http://biit.cs.ut.ee/gprofiler/; (39); results in Supp. Table 3). Consistent with the results shown in Fig. 4A and 4C, genes overexpressed in PRELID1-independent tumors relative to PRELID1-associated tumors were enriched for terms related to ROS signaling, including transcriptional targets of HIF1a and NF-kappaB and biogenesis/assembly of complex I (a known source of mitochondrial ROS for cellular signaling) (46–51). These results support the critical role of ROS signaling for PRELID1-independent breast cancer cell growth in the absence of mitogenic estrogen signaling (52). PRELID1 appears to play a central role in the regulation of ROS signaling in breast cancer.
PRELID1 APA and expression are correlated with outcomes in multiple cancers
Given the role of PRELID1 in ROS production and signaling, we hypothesized that modulation of PRELID1 expression is a universal mechanism of stress response control, common to most cancers. To test if PRELID1 APA or expression is correlated with patient outcomes in other tumor types, we assessed all cancers in TCGA for dependence on PRELID1 APA and/or transcript expression for survival. For each cancer, we split patients by PRELID1 APA (exon 4/exon 5) and expression (exon 4). The results are summarized in Supp. Table 4 and representative survival curves are shown in Fig. 6. In total, patient outcomes in fourteen of the cancers contained in TCGA are significantly associated with either PRELID1 expression or APA. Of these, nine are PRELID1-associated, with lower transcript expression, increased distal poly(A) site utilization, or both conferring a survival advantage, including Cervical Squamous Cell Carcinoma (APA), Pancreatic Adenocarcinoma (APA), and Acute Myeloid Leukemia (mRNA Expression) (Fig. 6A,C). Three were PRELID1-independent, with higher expression conferring a survival advantage, including Bladder Urothelial Carcinoma (Fig. 6B), and two were confounding, appearing PRELID1-independent by APA but PRELID1-associated by expression: Uveal Melanoma and Liver Hepatocellular Carcinoma.
Figure 6. PRELID1 APA and expression influences outcomes in multiple cancers.

A–B. PRELID1 APA-associated outcomes: overall survival, split by median PRELID1 Exon 4/Exon 5 ratio in PRELID1-associated (A) and PRELID1-independent (B) tumors. C. PRELID1 expression-associated outcomes: overall survival, split by median PRELID1 Exon 4 expression. Dashed grey line is “Short PRELID1” (APA; A–B) or “High PRELID1” (Expression; C), black line is “Long PRELID1” (APA; A–B) or “Low PRELID1” (Expression; C).
DISCUSSION
Here, we report the first application of PAS-seq in primary human tumor samples. Based on our analysis, APA appears to be a rare event in breast carcinogenesis. While this result may be limited by the small size of our study (12 patients), we nevertheless demonstrate that individual APA events can be of high impact and capable of producing physiologically relevant changes in steady state transcript level and translational efficiency. These rare events, exemplified by PRELID1, cannot be simply detected in gene expression analyses, but can have dramatic impact on tumor cell proliferation and clinical outcomes. Our data demonstrate that PRELID1 is regulated both transcriptionally and post-transcriptionally. In some cancers, PRELID1 expression is primarily regulated at the transcript level, as mRNA expression directly correlates with outcome. In many other cancers, including breast cancer, PRELID1 APA appears to have a greater effect on protein expression and, as a result, clinical outcomes. Furthermore, PRELID1 APA and mRNA expression appear to have opposing effects in some cancers, suggesting that regulatory elements in the PRELID1 3′ UTR may not always be repressive.
Remarkably, PRELID1 expression appears to have a highly context-dependent role in regulating cancer cell growth and response to cellular stress. In breast cancer, the PRELID1 APA event has diametrically opposed effects on clinical outcomes in ER+ and ER− disease, as well as on the growth rates and stress responses of ER+ and ER− breast cancer cell lines. Many tumor cells, including ER− breast cancer cells, rely on supermetabolic levels of ROS to drive proliferation through activation of hypoxia inducible factors (HIF) and NF-kappaB, among others (46–49,52–54). As ROS produced by the mitochondria is known to have effects on cellular signaling, there may be negative pressure on PRELID1 expression in some contexts, despite its general pro-survival, pro-proliferation roles (51). This is especially likely in cells that rely upon aerobic glycolysis, as they are not dependent on oxidative phosphorylation for energy production and actively inhibit cytochrome c-mediated apoptosis through redox inactivation of cytochrome-c (55). Consistent with this, our data suggest that the variable impact of PRELID1 expression may be due to the context-dependent utilization of growth factor signaling and mitochondrial ROS signaling. PRELID1-associated tumors, typified by ER+ breast cancers, utilize a mitogen-driven proliferation system that requires PRELID1 expression for efficient mitochondrial respiration. In contrast, PRELID1-independent tumors such as ER− breast cancers, which rely on ROS-mediated HIF and NF-kappaB activity to drive proliferation, require low PRELID1 expression to induce mitochondrial ROS signaling. These tumors accommodate reduced mitochondrial function and increased cytochrome c release through the use of aerobic glycolysis to generate ATP and inhibit apoptosis. In light of this, it is likely that cancers in which PRELID1 APA or expression has no apparent impact on outcome are heterogeneous mixtures of these metabolic subtypes and that this event becomes clinically relevant upon clonal selection during disease progression. Importantly, our TCGA analysis shows that differential response to PRELID1 APA seen in breast cancers is not solely a result of hormone receptor status, but underlies a more fundamental survival mechanism of cancer cells. While our data are consistent with known functions of ROS in breast cancer, further studies are needed to determine the exact role of PRELID1 in breast cancer subtypes.
In summary, we describe PRELID1 as an important switch utilized by cancer cells to regulate mitochondrial ROS production and cell growth in a cell context-dependent manner. Further research is necessary to clarify the specific mechanisms, including metabolic activity and signaling, that define these cellular contexts. Better understanding of these mechanisms, including the variable effect of PRELID1 expression, may be further exploited for therapeutic purposes.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
Financial support: This work was supported by NIH K08CA164048 (PK), NIH R01CA205044 (PK), and Cancer League of Colorado (PK).
The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell [Internet] 2009;138:673–84. doi: 10.1016/j.cell.2009.06.016. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19703394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xia Z, Donehower LA, Cooper TA, Neilson JR, Wheeler DA, Wagner EJ, et al. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat Commun [Internet] 2014;5:5274. doi: 10.1038/ncomms6274. [cited 2017 Jun 6] Available from: http://www.ncbi.nlm.nih.gov/pubmed/25409906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erson-Bensan AE, Can T. Alternative Polyadenylation: Another Foe in Cancer. Mol Cancer Res [Internet] 2016:14. doi: 10.1158/1541-7786.MCR-15-0489. [cited 2017 Sep 6] Available from: http://mcr.aacrjournals.org/content/14/6/507. [DOI] [PubMed]
- 4.Ji Z, Lee JY, Pan Z, Jiang B, Tian B. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci U S A [Internet] 2009;106:7028–33. doi: 10.1073/pnas.0900028106. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19372383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, et al. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron [Internet] 2008;60:1022–38. doi: 10.1016/j.neuron.2008.11.029. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19109909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science (80- ) [Internet] 2008;320:1643–7. doi: 10.1126/science.1155390. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18566288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature [Internet] 2016;534:402–6. doi: 10.1038/nature18294. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/27281199. [DOI] [PubMed] [Google Scholar]
- 8.Spies N, Burge CB, Bartel DP. 3′ UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts. Genome Res [Internet] 2013;23:2078–90. doi: 10.1101/gr.156919.113. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24072873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKeller MR, Herrera-Rodriguez S, Ma W, Ortiz-Quintero B, Rangel R, Cande C, et al. Vital function of PRELI and essential requirement of its LEA motif. Cell Death Dis [Internet] 2010;1:e21. doi: 10.1038/cddis.2009.19. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21364629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Potting C, Tatsuta T, König T, Haag M, Wai T, Aaltonen MJ, et al. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab [Internet] 2013;18:287–95. doi: 10.1016/j.cmet.2013.07.008. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/23931759. [DOI] [PubMed] [Google Scholar]
- 11.Daum G, Vance JE. Import of lipids into mitochondria. Prog Lipid Res [Internet] 1997;36:103–30. doi: 10.1016/s0163-7827(97)00006-4. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/9624424. [DOI] [PubMed] [Google Scholar]
- 12.Hatch GM. Cardiolipin: biosynthesis, remodeling and trafficking in the heart and mammalian cells (Review) Int J Mol Med [Internet] 1998;1:33–41. doi: 10.3892/ijmm.1.1.33. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/9852196. [DOI] [PubMed] [Google Scholar]
- 13.Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res [Internet] 2000;39:257–88. doi: 10.1016/s0163-7827(00)00005-9. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/10799718. [DOI] [PubMed] [Google Scholar]
- 14.Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res [Internet] 2008;49:1607–20. doi: 10.1194/jlr.R700018-JLR200. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/18077827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haines TH, Dencher NA. Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett [Internet] 2002;528:35–9. doi: 10.1016/s0014-5793(02)03292-1. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/12297275. [DOI] [PubMed] [Google Scholar]
- 16.Camberg JL, Johnson TL, Patrick M, Abendroth J, Hol WGJ, Sandkvist M. Synergistic stimulation of EpsE ATP hydrolysis by EpsL and acidic phospholipids. EMBO J [Internet] 2007;26:19–27. doi: 10.1038/sj.emboj.7601481. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/17159897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res [Internet] 2004;94:53–9. doi: 10.1161/01.RES.0000109416.56608.64. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/14656928. [DOI] [PubMed] [Google Scholar]
- 18.Petrosillo G, De Benedictis V, Ruggiero FM, Paradies G. Decline in cytochrome c oxidase activity in rat-brain mitochondria with aging. Role of peroxidized cardiolipin and beneficial effect of melatonin. J Bioenerg Biomembr [Internet] 2013;45:431–40. doi: 10.1007/s10863-013-9505-0. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/23494666. [DOI] [PubMed] [Google Scholar]
- 19.Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J [Internet] 2003;17:714–6. doi: 10.1096/fj.02-0729fje. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/12586737. [DOI] [PubMed] [Google Scholar]
- 20.Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell [Internet] 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/20850011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell [Internet] 2002;111:331–42. doi: 10.1016/s0092-8674(02)01036-x. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/12419244. [DOI] [PubMed] [Google Scholar]
- 22.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol [Internet] 2013;15:1197–205. doi: 10.1038/ncb2837. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3806088&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Arriba G, Calvino M, Benito S, Parra T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol Lett [Internet] 2013;218:30–8. doi: 10.1016/j.toxlet.2013.01.007. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/23347876. [DOI] [PubMed] [Google Scholar]
- 24.Garcia Fernandez M, Troiano L, Moretti L, Nasi M, Pinti M, Salvioli S, et al. Cell Growth Differ [Internet] Vol. 13. AACR; 2002. Early changes in intramitochondrial cardiolipin distribution during apoptosis; pp. 449–55. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/12354754. [PubMed] [Google Scholar]
- 25.Schwall CT, Greenwood VL, Alder NN. The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim Biophys Acta [Internet] 2012;1817:1588–96. doi: 10.1016/j.bbabio.2012.04.015. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22575443. [DOI] [PubMed] [Google Scholar]
- 26.Shepard PJ, Choi E-A, Lu J, Flanagan LA, Hertel KJ, Shi Y. RNA [Internet] Vol. 17. Cold Spring Harbor Laboratory Press; 2011. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq; pp. 761–72. [cited 2016 Sep 14] Available from: http://www.ncbi.nlm.nih.gov/pubmed/21343387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao C, Shi Y. Methods Mol Biol [Internet] Vol. 1125. NIH Public Access; 2014. Global and quantitative profiling of polyadenylated RNAs using PAS-seq; pp. 179–85. [cited 2016 Sep 14] Available from: http://www.ncbi.nlm.nih.gov/pubmed/24590790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Massingham T, Goldman N, Varela I, Klijn C, Stephens P, Mudie L, et al. All your base: a fast and accurate probabilistic approach to base calling. Genome Biol [Internet] BioMed Central. 2012;13:R13. doi: 10.1186/gb-2012-13-2-r13. [cited 2016 Sep 13] Available from: http://genomebiology.biomedcentral.com/articles/10.1186/gb-2012-13-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011 May 17;17:10–2. [Google Scholar]
- 30.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol [Internet] BioMed Central. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/18798982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lassmann T. TagDust2: a generic method to extract reads from sequencing data. BMC Bioinformatics [Internet] 2015;16:24. doi: 10.1186/s12859-015-0454-y. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/25627334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods [Internet] 2015;12:357–60. doi: 10.1038/nmeth.3317. Available from: http://www.nature.com/nmeth/journal/v12/n4/full/nmeth.3317.html?WT.ec_id=NMETH-201504%5Cnhttp://www.nature.com/nmeth/journal/v12/n4/extref/nmeth.3317-S1.pdf%5Cnhttp://www.nature.com/nmeth/journal/v12/n4/pdf/nmeth.3317.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao Y, Smyth GK, Shi W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013:41. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol [Internet] 2014;15:550. doi: 10.1186/s13059-014-0550-8. Available from: http://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2016. [Google Scholar]
- 36.Harrell FE. R package version 4.5-0. 2016. rms: Regression Modeling Strategies. [Google Scholar]
- 37.Therneau T. A Package for Survival Analysis in R. version 2.38. [Google Scholar]
- 38.Wickham H, Francois R. R package version 0.5.0. 2016. dplyr: A Grammar of Data Manipulation. [Google Scholar]
- 39.Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, et al. g:Profiler—a web server for functional interpretation of gene lists (2016 update) Nucleic Acids Res [Internet] 2016 doi: 10.1093/nar/gkw199. Available from: http://nar.oxfordjournals.org/content/early/2016/04/29/nar.gkw199.abstract. [DOI] [PMC free article] [PubMed]
- 40.Rodríguez-Enríquez S, Carreño-Fuentes L, Gallardo-Pérez JC, Saavedra E, Quezada H, Vega A, et al. Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int J Biochem Cell Biol. 2010;42:1744–51. doi: 10.1016/j.biocel.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 41.Kawakami K, Enokida H, Chiyomaru T, Tatarano S, Yoshino H, Kagara I, et al. The functional significance of miR-1 and miR-133a in renal cell carcinoma. Eur J Cancer [Internet] 2012;48:827–36. doi: 10.1016/j.ejca.2011.06.030. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21745735. [DOI] [PubMed] [Google Scholar]
- 42.Uchida Y, Chiyomaru T, Enokida H, Kawakami K, Tatarano S, Kawahara K, et al. MiR-133a induces apoptosis through direct regulation of GSTP1 in bladder cancer cell lines. Urol Oncol [Internet] 2013;31:115–23. doi: 10.1016/j.urolonc.2010.09.017. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21396852. [DOI] [PubMed] [Google Scholar]
- 43.Tsai YC, Weissman AM. The Unfolded Protein Response, Degradation from the Endoplasmic Reticulum, and Cancer. Genes Cancer [Internet] 2010;1:764–78. doi: 10.1177/1947601910383011. [cited 2017 May 27] Available from: http://www.ncbi.nlm.nih.gov/pubmed/21331300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell [Internet] 2000;101:249–58. doi: 10.1016/s0092-8674(00)80835-1. [cited 2017 May 27] Available from: http://www.ncbi.nlm.nih.gov/pubmed/10847680. [DOI] [PubMed] [Google Scholar]
- 45.Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem [Internet] 1991;266:17067–71. [cited 2017 May 27] Available from: http://www.ncbi.nlm.nih.gov/pubmed/1832668. [PubMed] [Google Scholar]
- 46.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A [Internet] 1998;95:11715–20. doi: 10.1073/pnas.95.20.11715. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/9751731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem [Internet] 2000;275:25130–8. doi: 10.1074/jbc.M001914200. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/10833514. [DOI] [PubMed] [Google Scholar]
- 48.Manna SK, Zhang HJ, Yan T, Oberley LW, Aggarwal BB. Overexpression of manganese superoxide dismutase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappaB and activated protein-1. J Biol Chem [Internet] 1998;273:13245–54. doi: 10.1074/jbc.273.21.13245. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/9582369. [DOI] [PubMed] [Google Scholar]
- 49.Li Q, Engelhardt JF. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J Biol Chem [Internet] 2006;281:1495–505. doi: 10.1074/jbc.M511153200. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/16286467. [DOI] [PubMed] [Google Scholar]
- 50.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–72. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J [Internet] 2009;417:1–13. doi: 10.1042/BJ20081386. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/19061483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi J-A, Lee J-W, Kim H, Kim E-Y, Seo J-M, Ko J, et al. Pro-survival of estrogen receptor-negative breast cancer cells is regulated by a BLT2-reactive oxygen species-linked signaling pathway. Carcinogenesis [Internet] 2010;31:543–51. doi: 10.1093/carcin/bgp203. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/19748928. [DOI] [PubMed] [Google Scholar]
- 53.Ruiz-Ramos R, Lopez-Carrillo L, Rios-Perez AD, De Vizcaya-Ruíz A, Cebrian ME. Sodium arsenite induces ROS generation, DNA oxidative damage, HO-1 and c-Myc proteins, NF-kappaB activation and cell proliferation in human breast cancer MCF-7 cells. Mutat Res [Internet] 2009;674:109–15. doi: 10.1016/j.mrgentox.2008.09.021. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/18996220. [DOI] [PubMed] [Google Scholar]
- 54.Chandel NS, Schumacker PT. Cells depleted of mitochondrial DNA (rho0) yield insight into physiological mechanisms. FEBS Lett [Internet] 1999;454:173–6. doi: 10.1016/s0014-5793(99)00783-8. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/10431801. [DOI] [PubMed] [Google Scholar]
- 55.Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol [Internet] 2008;10:1477–83. doi: 10.1038/ncb1807. [cited 2016 Sep 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/19029908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res [Internet] 2002;12:996–1006. doi: 10.1101/gr.229102. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res [Internet] 2013;41:D64–9. doi: 10.1093/nar/gks1048. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23155063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.