

Summary
Cellular signaling networks coordinate physiological processes in all multicellular organisms. Within networks, modules switch their function to control signaling activity in response to the cellular context. However, systematic approaches to map the interplay of such modules have been lacking. Here, we generated a context-dependent genetic interaction network of a metazoan's signaling pathway. Using Wnt signaling in Drosophila as a model, we measured >290,000 double perturbations of the pathway in a baseline state, after activation by Wnt ligand or after loss of the tumor suppressor APC. We found that genetic interactions within the Wnt network globally rewired after pathway activation. We derived between-state networks that showed how genes changed their function between state-specific networks. This related pathway inhibitors across states and identified genes required for pathway activation. For instance, we predicted and confirmed the ER-resident protein Catsup to be required for ligand-mediated Wnt signaling activation. Together, state-dependent and between-state genetic interaction networks identify responsive functional modules that control cellular pathways.
Keywords: genetic interactions, signaling, epistatic mapping, Wnt, APC
Graphical Abstract
Highlights
-
•
Genetic interaction networks of Wnt signaling in three cellular states
-
•
Networks rewire upon activation of Wnt pathway by ligand or by loss of APC
-
•
Interaction profiles identify known and novel state-dependent pathway modules
-
•
State-specific and between-state profile similarity identify signaling regulators
Systematic measurement of genetic interactions between components of the Wnt pathway before and after pathway activation reveals how the pathway is rewired upon perturbation.
Introduction
Genetic interactions can reconstruct the wiring diagram of biological processes in health and disease. Such interactions are identified by simultaneously perturbing multiple gene products and identifying instances when the expected and experimentally measured phenotypes significantly deviate (Baryshnikova et al., 2013, Bateson, 1909). Systematic experimental approaches have been deployed to build maps of cellular processes in unicellular (Costanzo et al., 2010, Costanzo et al., 2016, Typas et al., 2008) and multicellular organisms (Fischer et al., 2015, Horn et al., 2011, Lehner et al., 2006), to place novel components into known pathways, and to delineate hierarchical relationships between components (Fischer et al., 2015, Horn et al., 2011).
Wnt signaling is a key signaling pathway conserved from hydra to man and has been implicated in cell differentiation, cell proliferation, and stem cell maintenance (Clevers and Nusse, 2012). Aberrant activation of Wnt pathways has been found in many cancers. For example, loss-of-function mutations in the adenomatous polyposis coli (APC) gene are found in more than 80% of all colorectal cancers (Korinek et al., 1997). Under physiological conditions, the tumor suppressor APC is required for the function of the destruction complex, which prevents β-catenin/Arm from triggering Wnt signaling target gene expression (Behrens et al., 1998). Loss of APC function leads to pathway activation in a largely ligand-independent fashion. Upstream and downstream of the destruction complex, pathway activity is additionally regulated by feedback loops acting at the ligand and receptor level, respectively (Kakugawa et al., 2015, Zeng et al., 2000), and context-dependent modulators of target gene transcription (Cavallo et al., 1998, Li et al., 2007). Moreover, target genes at the transcriptional level (Ji et al., 2013) and the presentation of receptors at the membrane determine pathway activity (Niehrs, 2012). Together, these factors control pathway activity in the absence of ligands. Understanding how these modules are linked in different pathway states and how components may “switch” their role has remained a question not fully understood.
In this study, we used genetic interaction analysis before and after activation of the Wnt pathway by a ligand or by loss of function of APC to investigate stable and “switchable” functional relationships. We found that state-specific interaction profiles describe stable functional relationships. In contrast, between-state correlations identified instances when gene function adapted after induction of a specific state. This categorized genes required for ligand-mediated pathway activation or restriction of pathway activity after specific modes of induction.
Results
State-Specific Genetic Interactions of Wnt Signaling
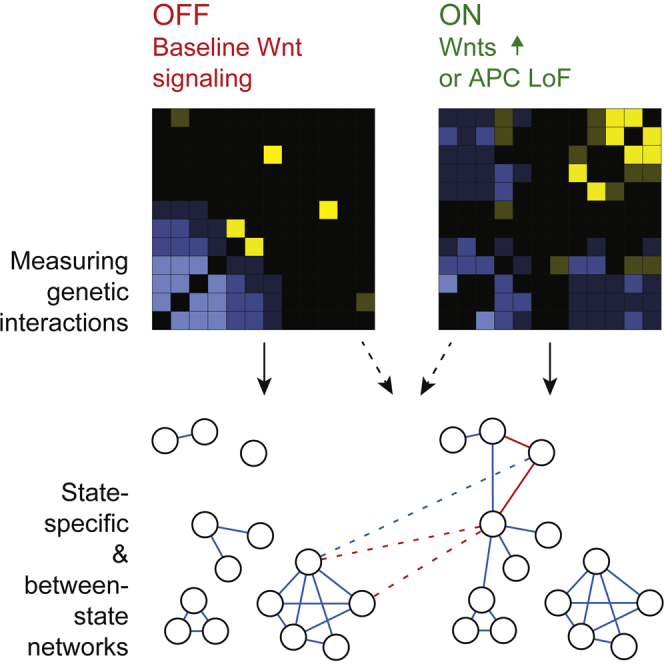
To dissect functional relationships in Wnt signaling, we chose a two-step approach (Figure 1A). We first screened genome-wide for Wnt signaling modulators in Wnt signaling competent Drosophila S2R+ and 1182-4H cell lines using at least two sequence-independent RNAi reagents per gene (Figures 1B, S1A, and S1B). We selected genes for genetic network analysis, prioritizing hits that were consistently identified in both cell lines (Figure S1B). This resulted in 336 genes, including genes involved in transcriptional regulation, chromatin remodeling, cell communication or vesicle trafficking, and other biological processes (Figure S1C), covering a wide range of Wnt signaling phenotypes in the genome-wide screen (Figure S1D). We then generated a state-dependent genetic interaction network in the baseline state and when the pathway was induced by the canonical Drosophila Wnt ligand Wingless (Wg) (Figures S2A and S2B) or simultaneous knockdown of the functionally redundant Drosophila Apc and Apc2 (Figures S2B and S2C), which mimicked the APC loss-of-function pathway state. We co-depleted 336 selected genes with a functionally representative subset of 72 genes in quadruplicates (all combinations of two independent RNAi reagents) across three pathway states. In total, 290,304 co-RNAi experiments (plus 13,824 experiments including one non-targeting control RNAi reagent) were performed in S2R+ cells using viability-normalized dTCF-luciferase reporter gene activity as a phenotypic readout. Genetic interactions were inferred from these co-RNAi Wnt signaling activity phenotypes by computing the deviation from the expected phenotype (π-score; Figures 1C, 1D, and S3A–S3C) (Horn et al., 2011).
Figure 1.

State-Dependent Genetic Interactions of Wnt Signaling Enable the Estimation of Signed Functional Relations
(A) Schematic workflow for state-dependent genetic interaction analysis.
(B) Wnt pathway activity z-scores in S2R+ cells of 28,950 independent double-stranded RNAs (dsRNAs) covering 14,331 genes of the Drosophila genome. The scores of the two independent dsRNA designs against core Wnt pathway components known to affect the activity positively (green) or negatively (red) are labeled.
(C and D) Combinatorial RNAi to quantify genetic interactions within the Wnt pathway. Single knockdown phenotypes were estimated from phenotypes of two independent dsRNAs in 144 different genetic backgrounds each (error bars show SE of median determined by bootstrapping). The white dashed bar indicates the expected combinatorial knockdown effect for each gene pair using a multiplicative neutrality function. The measured combinatorial phenotype illustrates the median of the four possible combinations of two independent dsRNA designs against each gene. The difference between the expected and measured combinatorial phenotype is quantified as a π-score for each genetic interaction (yellow if positive, blue if negative at false discovery rate <1%). The data are presented at log2 scale. Examples show measurements in the Wnt-active state, for all three states see Figures S3A, S3B, S3D, and S3E.
(E) Reproducibility of π-scores with rescreened (four replicates each) query genes evi/wls and Axn, which cover the secreting cell and the destruction complex. Further rescreened query genes are illustrated in Figure S2E. Correlations are highly significant (p < 2.2e−16).
(F) Genetic interaction (π) scores between 14 core pathway components in Drosophila S2R+ and 1182-4H cells (PCC, 0.78; p < 2.2e−16). Gene pairs from (C) and (D) and Figure S3C are highlighted.
(G–I) Genetic interaction scores in the three Wnt pathway states: baseline state, and after induction by Wg expression or Apc and Apc2 knockdown (low Wg levels). Candidate and query genes have the same order in each panel.
Genetic interactions of Wnt signaling in the same state were highly reproducible between biological replicates (Figures 1E, S2D, and S2E) and in different cell lines (Figure 1F), while they showed global pathway state dependency (Figures 1G–1I, S2D, S3D, and S3E). For example, co-depletion of the Wnt secretion machinery and the destruction complex by knockdown of the Wnt cargo receptor Evi/Wls and the negative regulator Axn was equivalent to the single knockdown effect of Axn alone (Figure 1C). This positive genetic interaction was only present in the Wnt-active state (BH-adjusted p-values: 0.95 [baseline], 0.002 [Wnt-active], 0.76 [Apc-loss]; Figure S3A). In contrast, a negative genetic interaction between Axn and the downstream target gene transcription activator β-catenin/Arm was measured in all pathway states (BH-adjusted p-value: <0.0005 [all states]; Figures 1D and S3B). Genetic interactions distinguished different negative regulators of Wnt signaling. Destruction complex components (e.g., Axn) and feedback regulators such as nkd act downstream of the ligand/receptor complex. While their individual depletion increases pathway activity, in contrast to Axn, nkd showed a negative genetic interaction with Evi/Wls (Figures S3A and S3C). At the level of functional complexes, it is well established that members of such a complex have the same interaction sign with each other (within complex) as well as with members of other complexes (between complexes) (Segre et al., 2005). We observed that this so-called monochromaticity within and between functional modules of the Wnt signaling route was highly dependent on its state (Figures S3D and S3E). Overall, genetic interactions both among and between functional modules of the Wnt pathway depended on the mode of activation.
State-Specific Relations Connect Functionally Similar or Opposing Genes
Signaling routes are controlled by modules, which activate or inhibit the signal in a context-dependent fashion (Housden and Perrimon, 2014). To understand the interplay of such modules after pathway activation, we reconstructed functional relationships at different levels. First, we inferred functional relations between genes by calculating the Pearson correlation coefficient (PCC) between the genetic interaction profiles of all gene pairs and each state (Baryshnikova et al., 2010, Horn et al., 2011). This reconstructed functional similarities of genes, such as the high correlation between profiles of Axn and CkIα (positive correlation, p < 2.2e−16, Pearson product-moment correlation) (Figure 2A). We also identified inhibitory relations, such as the negative regulation of the Wnt receptor complex component DVL1-3/Dsh by Nkd in the Wnt-active state (negative correlation, p = 8.5e−14) (Figure 2B). Overall, genetic interaction profile correlation was higher for gene pairs for which protein-protein interactions were measured in S2R+ cells (Figures S3F–S3I) (Guruharsha et al., 2011). Together, this enabled systematic identification of relations between components that act in the same functional module such as the ligand secretion machinery, the receptor or destruction complex or target gene transcription, and relations between modules that affect Wnt signaling in a similar (positive) or antagonizing (negative) fashion.
Figure 2.

Between-State and State-Specific Functional Relations Visualize Change of Gene Function and Rewiring of the Core Wnt Signaling Network
(A–D) State-specific correlation between genetic interaction profiles of the destruction complex components Axn and CkIα (both states: p < 2.2e−16, Pearson product-moment correlation) (A), dsh and nkd (baseline: p = 0.88; Wnt-active: p = 8.5e−14) (B) or receptor complex components LRP5/6/arr and dsh (baseline: p = 0.02; Wnt-active: p < 2.2e−16) (D) in the baseline and Wnt-active state. Each profile contains 72 quantitative genetic interaction (π) scores. (C) Correlation coefficients from (A), (B), and (D) in relation to the 56,280 measured coefficients in each respective state.
(E and F) State-specific correlation-based networks connecting core Wnt pathway components involved in ligand secretion and receptor complex binding (green), negative feedback (orange), the destruction complex (red), or target gene transcription (dark green) in baseline (E) or Wnt-active (F) states. The similarity or dissimilarity of each gene pair was estimated by computing Pearson correlation coefficient (PCC) between the genetic interaction profiles. Genes with an absolute PCC > 0.65 were connected. The edge width represents the absolute value of the coefficient; the color indicates positive (blue) or negative (red) correlation.
(G–I) Between-state correlation between genetic interaction profiles of Axn and dsh (p = 1.08e−9) (G), or Axn and nkd (p = 2.13e−6) (H). (I) Correlation coefficients from (G) and (H) in relation to the 112,896 measured coefficients between baseline and Wnt-active state.
(J) Between-state correlation-based network connecting core Wnt pathway components involved in ligand secretion and receptor complex binding (green), negative feedback (orange), the destruction complex (red), or target gene transcription (dark green) between baseline and Wnt-active state. Genes with an absolute PCC > 0.5 were connected.
Next, we investigated the dependency of positive and negative functional relations on the state of the Wnt pathway. The components of the destruction complex Axn and CkIα showed high genetic interaction profile correlation both in baseline and Wnt-active state (p < 2.2e−16 in both states; Figures 2A and 2C). Similarly, additional proteins of the destruction complex and the nuclear factors arm, pan, pygo, and BCL9/lgs were connected in each state (Figures 2E and 2F). In contrast, functional relations between receptor complex components Dsh and Arr (baseline: p = 0.02; Wnt-active: p < 2.2e−16), or Dsh and the feedback regulators Nkd (p = 0.88, p = 8.5e−14) occurred in the Wnt-active state only (Figures 2B–2D). In the derived network, components required for Wnt signaling were connected by positive edges, whereas inducible negative regulators of the pathway showed negative associations with those components (Figures 2E and 2F). Together, the data demonstrated state-dependent rewiring of functional relations between genes.
Between-State Relations Identify Functional Adaptation to Control Pathway Activation
The destruction complex is the central regulatory module of Wnt signaling. Surprisingly, the state-specific network did not connect the destruction complex to other modules of the Wnt pathway (Figures 2E and 2F). We hypothesized that state-specific interaction analysis missed functional relationships of genes that act mutually exclusively in either state. For instance, the destruction complex inhibits pathway activity in the absence of ligands whereas the ligand-bound receptor complex prevents the destruction complex from doing so. Therefore, only one of those functional modules can be active at a time, and visualizing their relationship requires the comparison of genetic interaction profiles between the states. Our data showed that the profile of Dsh in the ligand-induced state—where it is active—anticorrelated with the profile of Axn in the baseline state, where it inhibits Wnt signaling (p = 1.08e−9; Figures 2G and 2I). Moreover, Axn in the baseline state showed high similarity with Nkd in the induced state (p = 2.13e−6; Figures 2H and 2I), which takes over as a negative regulator of Wnt signaling once the pathway is induced and the destruction complex allows pathway activity to increase. In the core Wnt signaling pathway, we observed a similar pattern for all the destruction complex components with the receptors Dsh, Arr, or Fz2 or the negative regulators of the ligand-induced pathway Nkd and Notum (Figure 2J). Globally, such between-state relations were rare, with 1.8% (Wnt-active) and 0.3% (Apc-loss), or 0.9% (Wnt-active) and 0.3% (Apc-loss) of all gene pairs showing a coefficient larger than 0.5 or smaller than −0.5, respectively. Our data suggest that between-state genetic interaction profile correlation identifies how functional relationships change after pathway activation. This involved cases in which both partners switch their function in Wnt signaling after activation. Together, positive and negative state-specific and between-state relations detect the process-level structure of regulatory modes to control pathway activity.
Functional Adaptation of Genes only Partially Responsible for Network Rewiring
To assess how genes changed in function between states, we identified two distinct scenarios of between-state relations. Here, a gene's reference profile in a given state can be compared with profiles in a different state of the same gene (which we term self-correlation or self-similarity) or another gene (gene-gene correlation). To investigate how gene function adapts to pathway activation, we first estimated between-state self-correlations after ligand- or Apc-loss-mediated pathway activation. We observed that, globally, the complete set of genes tested exhibited higher self-similarity than expected by chance (both modes of activation: p < 2.2e−16, one-sided Welch’s t-test). However, many individual genes exhibited relatively low self-similarity, including some that exhibited non-significant between-state profile correlation (Figure 3A). To account for low self-correlation due to a lack of genetic interaction variation in the profiles, we defined genes that changed their profile to develop high state-specific similarity or dissimilarity with other genes in the network (Figure 3B). We found that components facilitating ligand secretion and receptor binding, or mediating feedback regulation such as nkd and Notum, showed low self-correlation after Wnt-mediated pathway activation, suggesting that this metric identifies functional adaptation. Several factors of other signaling routes exhibited large changes in their interaction profiles (Figure 3B). This involved yki (Hippo signaling), Pi3K59F, c-Fos/kay, and c-Jun/Jra (phosphatidylinositol 3-kinase and JNK signaling), Mek1/2/Dsor1 and Ets1/2/pnt (Ras signaling), or smo (Hedgehog [Hh] signaling). Dsor1 and pnt were connected to yki via the chromatin remodeler trx, ash1, and Br140 in the state-specific Wnt-active network (Figure 3C). Another state-specific subnetwork connected the Hh signaling receptor smo to the non-canonical Wnt ligand Wnt5 and Golgi trafficking components Gmap, Rab6, and CG6761, which had low self-similarity (Figure 3C). Interestingly, low self-correlation predicted rewiring of the state-dependent network for some modules only: While profiles of Wnt receptor complex components arr and dsh showed low self-correlation and their functional similarity only emerged after pathway activation (Figures S4A and S4B), JNK pathway components Jra and kay showed high similarity in each pathway state despite their low self-correlation (Figures 3D and 3E). Other state-specific subnetworks connecting genes with low self-correlation showed an emerging similarity between pnt and trx after pathway activation (Figures S4C and S4D), whereas trx and ash1 shared high similarity in both states (Figures S4D–S4F). Together, this illustrated that self-similarity of genes between states can indicate that their functions change even if their state-specific relationship to other genes does not rewire.
Figure 3.

Rewiring after Pathway Activation Partially Caused by Change in Gene Function
(A) Distribution of self-correlation of all tested genes between baseline and Wnt-active (left) or Apc-loss (right) state.
(B) Self-correlation (x axis) and maximal association (y axis) after pathway induction. The maximal association represents the highest absolute PCC of each gene in the Wnt-active state. The color illustrates positive (red) or negative (green) single knockdown Wnt signaling phenotypes in the Wnt-active state.
(C) Zoomed-in networks illustrate state-specific similarity (PCC > 0.65) between selected genes with a low autocorrelation.
(D) Change of genetic interaction profiles of JNK signaling transcription factors Jra and kay from baseline to Wnt-active state.
(E) Self-correlation of Jra and kay between baseline and Wnt-active state.
Functional Plasticity and Compensation within Modules
Gene-gene correlations after pathway activation were significantly lower than self-correlations (Figure S5A). Therefore, we assessed how well between-state relations predicted intra-module functional plasticity of genes after pathway activation. We used the destruction complex as an example and computed the cumulative rank distance of the correlation coefficients of its components in and between the baseline and Wnt-active states. In both states, all components of this complex were clearly distinguishable from the remaining genes (Figure 4A). In contrast, between-state relations showed that after ligand induction, those genes diverged from destruction complex baseline state function (Figures 4B, 4C, S5B, and S5C). In particular, sgg/GSK3 showed strongly decreased similarity to all destruction complex components in the baseline state (Figures S5B–S5D). Moreover, its depletion alone unexpectedly decreased pathway activity in the Wnt-active state (Figure S5E), possibly by supporting ligand-mediated pathway activation by triggering endocytosis of the ligand-bound receptor complex (Piao et al., 2008). Importantly, the between-state relations predicted similarity between baseline destruction complex function and feedback regulators in the Wnt-active state (Figures 4B and 4C). Many of those factors had not previously been described as Wnt pathway regulators and also showed an inhibitory effect on Wnt pathway activity after ligand-mediated pathway induction (Figure S5F, red lines). Together, between-state relations, inferred from between-state interaction profile correlation, can be used to visualize how gene function adapts in the activated pathway and pinpoint genes that promote or restrict pathway activity after induction.
Figure 4.

Plasticity of Functional Relations in Modules after Pathway Activation
(A) State-specific cumulative rank-based distance to the six destruction complex components Axn, CkIα, slmb, sgg, skpA, and lin19.
(B) Between-state cumulative rank-based distance from the destruction complex. The rank of the PCC of each gene with a destruction complex component was computed and ranks were summed per gene.
(C) Comparison of baseline-specific similarity (PCC between genetic interaction profiles) with baseline and Wnt-active state similarity of baseline state Axn with other destruction complex components (red) or negative feedback regulators (nkd, Notum) (orange).
(D) Between-state relations among components of stable functional modules. Stable functional relations exceeded a connection specificity index (CSI) of 0.9 in the baseline and respective activated state. Self-correlation of all tested genes is illustrated in black; the dark-blue line represents the self-correlation of genes in stable modules.
(E) Between-state relations among components state-specific functional modules. State-specific relations exceeded a CSI of 0.9 in either the baseline or activated state but not in the respective other state (CSI < 0.7). Self-correlation of all tested genes is illustrated in black. Curves were compared using a one-sided Wilcoxon rank-sum test.
Finally, we compared between-state gene-gene similarity in modules that were either functionally stable or state-specific after pathway activation. First, we identified such modules by computing the connection specificity index (CSI), which corrects the genetic interaction-based PCC for unspecific high coefficients for each gene (Billmann et al., 2016, Fuxman Bass et al., 2013, Green et al., 2011) (Figures S5G and S5H). Using a CSI larger than 0.9, which was observed for 1.4% (baseline), 1.8% (Wnt-active), or 1.3% (Apc-loss) of all gene pairs, we identified two types of modules: stable modules associated genes in the baseline and the respective activated state, whereas state-specific modules showed associations in either the baseline or the activated state. We observed that between-state gene-gene relations in the stable modules were significantly higher than self-similarity of the genes regardless of the mode of pathway activation (Figure 4D). In contrast, gene-gene relations in modules specific to one state were lower than self-similarity of genes in those modules (Figure 4E). In conclusion, gene-gene relations in functional modules can strongly contribute to plasticity or functional stability.
A Regulatory Network at and above the Wnt Receptor Level
To place potentially novel regulators identified by genome-wide screening in the architecture of the Wnt pathway at and above the receptor level, we selected the genes evi/wls, opm, and CHOp24, which are required for ligand secretion, and receptor complex components fz, fz2, arr, and dsh (Figure 5A, green nodes). To this seed network, we connected genes with a functional similarity (CSI > 0.9) with at least one of the selected genes in the Wnt-active state. Those potentially novel pathway factors comprised components of the ER-resident N-glycosylation machinery including the acetyl-coenzyme A transporter CG9706, galactose-binding motif containing CG31678, the putative mannosyltransferase CG11999 and CG14476, as well as genes regulating ion levels in the secretory pathway such as CG32495 and Catsup (Figure 5A, light-blue nodes). Interestingly, the human CG14476 ortholog and glucosidase IIα subunit GANAB has recently been identified as a positive regulator of canonical Wnt signaling in human HAP1 cells (Lebensohn et al., 2016). Next, to identify genes that control ligand-mediated pathway activity, we selected genes if their Wnt-active interaction profile was similar to that of the baseline state destruction complex (between-state PCC > 0.5), indicating an inhibitory role in the ligand-activated pathway. We furthermore required those genes to also antagonize the ligand secretion and receptor binding apparatus as suggested by their high dissimilarity (state-specific PCC < −0.65) to the seed network (Figure 5A, orange nodes). For example, this subnetwork visualized high similarity between the feedback regulator and inhibitor of the Wnt receptor complex Nkd with factors regulating AP-2-mediated clathrin-dependent endocytosis, which had been reported to be required for ligand secretion and receptor binding (Pan et al., 2008). Finally, we extended this network by genes sharing high similarity (CSI > 0.9) with the novel positive and negative regulators of the pathway (gray nodes). For instance, this showed a conserved role for Arf102F in Wg-activated signaling, whose human orthologs ARF4/5 have previously been found to be required for proper secretion of Wnt ligands (Yu et al., 2014). Together, this stepwise approach identified a network of known and potentially novel genes required for ligand secretion and receptor binding, and connected them to negative regulators of ligand-induced Wnt signaling.
Figure 5.

A Ligand Induction-Specific Network of Wnt Signaling
(A) Correlation-based network connecting components of Wnt secretion and receptor binding with potential co-regulatory modules. “Bait” genes required for ligand secretion and receptor binding (green nodes) were selected and potentially novel components (light-blue nodes) were called if they shared a CSI > 0.9 in the Wnt-active state. Potential negative regulators (orange nodes) were added if they shared a between-state similarity (PCC > 0.5) with the destruction complex (red nodes) and a Wnt-active state-specific dissimilarity (PCC < −0.65) with at least one “bait” gene. The network was further extended for genes sharing a CSI > 0.9 with potentially novel positive or negative regulators (gray nodes). Genes were connected if their state-specific profiles shared an absolute PCC > 0.65 or if their between-state shared an absolute PCC > 0.5 (dashed lines). Positive and negative correlations are shown as blue and red lines, respectively. Per state, the modules of positive regulators (green, light blue), negative regulators (orange), and the destruction complex (red) were separately arranged using a force-directed layout algorithm. The edge width represents the absolute PCC.
(B) State-specific (if |PCC| > 0.65) and between-state (if |PCC| > 0.5) relations of Catsup with the ligand secretion machinery and negative regulators.
(C) Catsup single depletion effect on Wnt signaling activity upon increasing Wg concentrations.
(D and E) Knockdown of Catsup in third instar wing imaginal disc leads to Wg accumulation in the Wg-producing cells. Total Wg staining intensity (red) was quantified using FIJI (n > 6 wing discs). enGal4, UAS-GFP/UAS-Catsup RNAi, posterior to the right (GFP positive). Scale bars, 20 μm.
(F and G) Knockdown of Catsup leads to depletion of extracellular Wg. Extracellular Wg (Wg ex) was stained on living third instar imaginal disc without cell permeabilization. Staining intensity (red) was quantified using FIJI (n > 6 wing discs). enGal4, UAS-GFP/UAS-Catsup RNAi, posterior to the right (GFP positive). Scale bars, 20 μm.
The Golgi-resident zinc transporter Catsup (Groth et al., 2013) showed high similarity with the ligand secretion machinery in the Wnt-active state and, similar to those factors, a strong dissimilarity with destruction complex components in the baseline state (Figure 5B), suggesting its requirement in Wg secretion. To confirm the predicted role for Catsup, we first demonstrated that it is required for Wg ligand-dependent Wnt pathway activation in S2R+ cells (Figure 5C). We further observed that Catsup depletion in vivo in the developing wing imaginal disc led to accumulation of Wg in Wg-secreting cells and a strong reduction of extracellular Wg (Figures 5D–5G), resulting in the reduction of the high-threshold Wg/Wnt target gene sens (Figures S6A and S6B). The accumulation of Wg in the secreting cells was confirmed using clonal mosaic analysis of a loss-of-function Catsup47 allele (Figures S6C and S6D), indicating that Catsup is required for proper Wg secretion.
Regulation of Wnt Target Gene Transcription upon Ligand- versus Apc-Loss-Mediated Pathway Activation
Loss-of-function mutations in the destruction complex component APC lead to constitutively active Wnt signaling (Korinek et al., 1997, Morin et al., 1997). To reconstruct a regulatory network downstream of the destruction complex, we identified genes with a negative regulatory role in both active pathway states (Figure 6A), and genes with roles specific for the Wnt-active or Apc-loss states (Figures 6B and 6C). We assessed genetic interaction profile similarity of its components in the baseline state with genes in the active states. We further filtered for genes that also showed high dissimilarity (state-specific PCC < −0.65) with the Wnt target gene transcription apparatus represented by arm, pan, lgs, and pygo, and all genes with a CSI > 0.9 with at least one of those genes. In the Wnt-active state, our data predicted a role for the transcriptional activators Ada2b, Ada3, and the zinc-finger binding protein Zn72D. The importin msk was positively associated with the predicted negative regulators, but could only be positively associated with baseline destruction complex function in the Apc-loss state (Figures 6B and 6C). In the Apc-loss state, we also found the nuclear effector of the Hippo signaling pathway yki, whose human homologs YAP1/TAZ had been described in regulating β-catenin stability (Azzolin et al., 2012), or the methyltransferase Lpt (Figure 6C). Interestingly, Lpt depletion in the non-induced pathway did not affect activity levels, but co-depletion with destruction complex components showed a positive genetic interaction (Figure 6D), indicating that Lpt restricts Wnt pathway activity downstream of the destruction complex. Notably, mutations in its human homologs KMT2C and KMT2D co-occurred with mutations in the destruction complex components APC (odds ratio [OR] 2.6, p = 0.029 and OR 2.6, p = 0.033; Fisher's exact test) or AXIN1 (OR 6.8, p = 0.003 and OR 19.3, p = 9.7e−6; Fisher's exact test) in melanoma. In conclusion, our approach systematically reconstructs rewiring of functional similarities after signaling pathway activation and assesses how genes functionally adapt with regard to the non-induced condition.
Figure 6.

Genes Resuming a Destruction Complex-like Role in Wnt Signaling in Different Pathway Activation Modes
(A) Putative inhibitors of the Wnt target genes transcription machinery after ligand and Apc-loss-mediated pathway activation.
(B and C) Putative inhibitors of the Wnt target gene transcription machinery exclusively after ligand or Apc-loss-mediated pathway activation. “Bait” genes required for target gene transcription (green nodes) were selected and potentially novel components (light-blue nodes) were called if they shared a CSI > 0.9 in both active pathway states (A) or one but not the other active state (B and C). Potential negative regulators (orange nodes) were added if they shared a between-state similarity (PCC > 0.5) with the destruction complex (red nodes) and a state-specific dissimilarity (PCC < −0.65) with at least one “bait” gene. Genes were connected if their state-specific profiles shared an absolute PCC > 0.65 or if their between-state shared an absolute PCC > 0.5 (dashed lines). Positive and negative correlations are shown as blue and red lines, respectively. Per state, the modules of positive regulators (green, light blue), negative regulators (orange), and the destruction complex (red) were separately arranged using a force-directed layout algorithm. The edge width represents the absolute PCC.
(D) Lpt knockdown effect on Wnt signaling activity (upper panel) and the genetic interaction of Lpt with destruction complex components (lower panel) in the baseline state. The single knockdown phenotype was estimated from phenotypes of two independent dsRNAs in 144 different genetic backgrounds each (error bars show SE of median determined by bootstrapping). Error bars of the genetic interactions show the median absolute deviation of four independent genetic interaction scores for each gene pair.
Discussion
Signaling networks consist of modules, which regulate their activity in a context-dependent manner. For modules to control signal processing, genes take on or abandon certain roles together or individually. Here, we demonstrate a general approach to systematically reconstruct positive and negative functional relations between genes and assess how individual gene function and gene-gene relations change after pathway activation. We illustrate how functional similarity derived from genetic interaction profiles rewires and uses this information to define Wnt pathway state-specific modules. We further refined the state-dependent wiring diagram by inferring negative relations. In accordance with a previous study of protein-protein interactions (Vinayagam et al., 2014), our network of positive and negative functional similarities described similarities within and between modules as well as inhibitory relations between modules.
Interestingly, we observed that the regulatory hub of the Wnt signaling pathway, the β-catenin destruction complex, was not connected to other modules of the pathway by state-specific relations. This suggested that genetic interaction-based networks that focus on one or multiple distinct conditions incompletely reconstruct responsive processes such as cell signaling routes. We further hypothesized that components that are only active when the destruction complex is inactive can be functionally associated by correlating genetic interaction profiles of genes in their respective active state. We developed an approach that assesses how a given gene function changes after pathway activation with regard to any gene in the baseline state—for instance, ligand-induced for a Wnt receptor complex component. These between-state gene-gene relations connected the destruction complex to various modules of the pathway, illustrating, for example, the inhibition through the Wnt receptor complex in the presence of ligands. In conclusion, this approach can identify connections in networks that (1) rewire context-specifically to (2) adopt a role relative to a “baseline”’ condition. Thus, in agreement with previous studies in yeast (Bandyopadhyay et al., 2010, Guénolé et al., 2013), we found that conditional genetic interaction maps increase their resolution. We further found that systematically visualizing inhibitory relations as they occur in various signaling cascades requires the investigation of between-state gene-gene relations. In contrast to previous genetic interaction network approaches, between-state relations enable the use of a given profile as a query to screen for profiles in any condition to identify compensatory mechanisms.
In perspective, our approach can be applied to conditional network data from other cell models such as yeast and human cells to investigate how biological processes respond to defined stimuli. This could be instrumental in identifying both compensatory mechanisms and vulnerabilities that occur in different genetic backgrounds such as those induced by cancer-associated mutations. With the advent of genome-editing technologies exploiting CRISPR/Cas9 (Cong et al., 2013, Hart et al., 2015, Steinhart et al., 2016), a first “static” genetic interaction map in β-catenin-active cancer cell lines has successfully applied a co-single-guide RNA (sgRNA) approach to group Wnt pathway components (Rosenbluh et al., 2016). As such efforts continue and new co-sgRNA systems are being developed (Han et al., 2017), our approach can guide a more complete analysis of context-dependent networks to understand gene function across various model organisms. Moreover, expression profile correlation of patient sequencing data currently emerges as a promising tool to systematically assess gene function (Alvarez et al., 2016). Using such data, our approach could be used to test how gene function evolves in the presence of oncogenic mutations.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Wg | Developmental System Hybridoma Bank | N/A |
| Guinea Pig anti-Sens | Gross et al., 2012 | N/A |
| anti-guinea pig-Alexa594 | Invitrogen | Cat #: A11076; RRID: AB_2534073 |
| anti-mouse-Alexa594 | Invitrogen | Cat #: A11005; RRID: AB_2534120 |
| Recombinant DNA | ||
| Plasmid: pAc-wg | Bartscherer et al., 2006 | N/A |
| Plasmid: dTCF-luc | Bartscherer et al., 2006 | N/A |
| Plasmid: Rp128-Rluc | Bartscherer et al., 2006 | N/A |
| Deposited Data | ||
| Primer for dsRNAs targeting all query and template genes | This paper | Tables S1 and S2 |
| Documented R code vignette | This paper | Data S1 |
| Raw data | https://github.com/boutroslab/Supplemental-Material | WntSGI |
| Processed data | https://github.com/boutroslab/Supplemental-Material | WntSGI |
| Experimental Models: Cell Lines | ||
| D. melanogaster: Cell line S2R+ | Drosophila Genomics Resource Center | FBtc0000150 |
| D. melanogaster: 1182-4H | Drosophila Genomics Resource Center | FBtc0000177 |
| Experimental Models: Organisms/Strains | ||
| Drosophila: en-GAL4, UAS-GFP 2nd chr. | Thompson and Cohen, 2006 | N/A |
| Drosophila: Catsup mutant alleles Catsup47 | Groth et al., 2013 | N/A |
| Drosophila: RNAi line targeting Catsup | Vienna Drosophila RNAi Center | transformant ID: 103630 |
| Drosophila: fz3-GFP | Bloomington Drosophila Stock Center | Bloomington stock number 43831 |
| Software and Algorithms | ||
| R | www.r-project.org | N/A |
| Custom R code | https://github.com/boutroslab/Supplemental-Material | WntSGI |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author (m.boutros@dkfz.de).
Experimental Model and Subject Details
Cultured Drosophila cells were used to perform high-throughput screening and genetically modified flies were used for validation studies.
Tissue Culture
Wnt signaling competent Drosophila S2R+ cells (Cherbas et al., 2011, Yanagawa et al., 1998) were cultured in Schneider’s medium (Invitrogen by ThermoFischer, Waltham, MA) supplemented with 10% fetal calve serum (FCS, Biochrom, Cambourne, UK) and 1% Penicillin/Streptomycin (Gibco by ThermoFischer). They were grown to confluence, detached by scraping and passaged using a 1:12 dilution every three days. Wnt competent 1182-4H cells (Cherbas et al., 2011) were cultured in M3 insect medium (Gibco by ThermoFischer) supplemented with 10% FCS (Biochrom) and 1% Penicillin/Streptomycin (Gibco by ThermoFischer). They were grown to confluence, detached by scraping and passaged using a 1:3 dilution every three days.
In Vivo RNAi in the Drosophila Wing Disc
The following Drosophila stocks were used: en-GAL4, UAS-GFP 2nd chr. (Thompson and Cohen, 2006), Catsup mutant alleles used were Catsup47 (Groth et al., 2013), fz3-GFP (Bloomington stock number 43831), hh-Gal4 (Tanimoto et al., 2000). RNAi lines targeting Catsup (transformant ID: 103630) were obtained from the Vienna Drosophila RNAi Center. Knockdown were performed by using the UAS-Gal4 system (Brand and Perrimon, 1993). RNAi fly lines were crossed to Hh-Gal4 driver lines and kept at 25°C.
Method Details
Synthesis of RNAi Reagents
For each gene, two ∼200 bp long sequence-independent double-stranded (ds) RNA reagents were designed using NEXT-RNAi software (Horn et al., 2010). The reagents were designed to avoid mismatches of 19 nt or more, more than 6 tandem tri-nucleotides of the CAN type, low complexity region or UTRs. In a two-step approach (Billmann and Boutros, 2016), the fragments were first generated by PCR on genomic Drosophila DNA in a 96-well format, and amplified using a primer combination preventing cross-contamination of the reagents. The DNA was transcribed into dsRNA via in vitro transcription (IVT). IVT products were purified via gel filtration (Bio-Gel P-30, BIO-RAD, Hercules, CA). Concentrations were measured using a NanoDrop 8000 spectrophotometer (Thermo scientific) and adjusted to 50 ng/μl.
Genome-wide RNAi Screening
To target each gene in the Drosophila genome, the HD3 library was designed, which contained 28,950 sequence-independent dsRNAs covering 14,331 genes, 13,369 genes with at least two designs (see Figure S1A for Wnt signaling phenotype correlation between the two designs). For S2R+ and 1182-4H cells, two replicates each comprising 88 384-well assay plates were prepared (see Figure S1A for replicate correlation). To control the dynamic range of the data, each plate was supplied with dsRNA targeting the Wnt signaling regulators evi, dsh, Apc or Axn, as well as GFP as non-targeting controls and the Renilla luciferase (Rluc) and Firefly luciferase (Fluc) to assess the reporter signals. Each dsRNA was present at 250 ng (5 μl) per well. 11,000 S2R+ or 12,000 1182-4H cells were seeded in 20 μl serum-free medium per well and the starvation was allowed to proceed for 45 min. Subsequently, 25 μl serum and P/S-containing medium were added. The dual Wnt reporter system was used in the Wnt active state (see below).
Combinatorial RNAi
The 336 selected candidate (template) genes were co-depleted with 72 query genes in a template × query gene design (Horn et al., 2011), and covering each candidate and query gene by two sequence-independent dsRNA designs. For primer sequences and query genes please see Tables S1 and S2. 125 ng (2.5 μl) of each dsRNA against the 336 template genes were spotted on a 384-well assay plate. Non-targeting reagents designed against GFP were spotted in additional 16 wells. 125 ng (2.5 μl) dsRNA against a query gene was added to each of the 352 wells in a contact-independent manner using the NanodropII dispenser (GC Biotech, Netherlands). To control the dynamic range of the data, the remaining 32 wells (without query dsRNA) of each assay plate were equipped with dsRNA targeting known Wnt regulators (see above) and GFP, Rluc and Fluc. The 336 template × 72 query gene set-up resulted in 288 assay plates per pathway state. 11,000 S2R+ cells were seeded per well following the serum starvation protocol (see above). To measure combinatorial RNAi phenotypes in the baseline, Wnt active and Apc loss state, the dual Wnt reporter system was adjusted as described below.
To validate genetic interactions in 1182-4H cells, 14 well-described Wnt regulators were co-depleted in a template-query gene design and tested all against all. 125 ng (2.5 μl) of two sequence-independent dsRNAs targeting each gene were spotted, and 125 ng (2.5 μl) of the query dsRNA was added. Per well, 12,000 cells were seeded using the serum starvation protocol. The dual Wnt reporter system was used in the Wnt active state (see below).
Dual Wnt Reporter Activity Assay in Defined Pathway States
To quantify Wnt signaling activity, reporter plasmids were transfected 24 h past cell seeding using 0.1 μl FuGENE transfection reagent (Promega, Madison, WI) in a total volume of 10 μl. Per well 3 ng of the Rp128-Rluc (expressed the Renilla luciferase) and 0.5 ng of the dTCF-luc (expresses the Firefly luciferase behind a Wnt signaling-specific promoter) plasmid were used. For the Apc loss state, dsRNA against Apc and Apc2 were added to the cell suspension before seeding in a concentration equivalent to 100 ng per well. For the baseline and Apc loss state, 0.02 ng, and for the Wnt active state 2 ng of the pAc-wg plasmid were used. Cells were grown at standard conditions (25°C) for up to 108 h past seeding, and reporter levels were quantified using a Mithras LB Multimode Mircoplate Reader (Berthold, Bad Wildbad, Germany) without filter and 0.05 s exposure time for Fluc, and using 490 nm filter settings and 0.1 s exposure time for Rluc.
Quantification of mRNA Levels
Quantification of mRNA levels of the Wnt target gene nkd was performed in S2R+ cells. 5×105 cells were seeded in 1 ml Schneider’s medium (Gibco by Life Technologies) onto 10 μg dsRNA in a 6-well plate and starvation was allowed to proceed for 45 min. Total RNA was extracted after 72 h using the RNeasy Mini Kit protocol (Qiagen) followed by cDNA synthesis using the RevertAid First Strand cDNA Synthesis Kit and oligo(dT)18 primer (Fermentas by Life Technologies), including on-column DNaseI digest of residual genomic DNA. The quantitative RT-PCR was performed using the TaqMan Universal ProbeLibrary (UPL) system (Roche, Mannheim, Germany). Specific Cp values were normalized. The Cp values of the RpL32 (also rp49) house-keeping gene, and the fold-change was estimated via the 2-ddCt method (Livak and Schmittgen, 2001).
Immunostainings, Microscopy and Image Analysis of the Drosophila Wing Disc
Wing discs were dissected form 3rd instar larvae in Schneider’s medium (Gibco by Life Technologies) and fixed for 20 min in 4% PFA in PBS. Permeabilization was allowed to proceed for 30 min in 0.2% Triton X-100 in PBS (PTx), followed by 60 min blocking in 0.2% BSA and overnight incubation with the primary antibody at 4°C. After washing, the secondary antibody incubation was performed for 60 min at RT, and washed again in PTx and finally with PBS.
Extracellular Wg staining was performed as described previously (Strigini and Cohen, 2000). Wing discs were mounted in Vectashield Mounting Medium (Vector Laboratories). Images were acquired at a Leica TCS Sp5 confocal microscope. Signal intensities were assessed using FIJI. Separate channel images were assembled using Adobe Photoshop CS6.
The following antibodies were used: Mouse Anti-Wg (4D4s, obtained from Developmental System Hybridoma Bank) 1:5 for extracellular and 1:50 for total staining, Rabbit-anti-Dll 1:200 (a gift from S. Carroll), Guinea Pig anti-Sens 1:300 (Gross et al., 2012). Secondary antibodies used were anti-guinea pig-Alexa594 (1:500 (A11076), Invitrogen), anti-mouse-Alexa594 (1:500, (A11005), Invitrogen).
Quantification and Statistical Analysis
Screening Data Normalization
To account for plate effects in the genome-wide or combinatorial RNAi screening data, the Wnt signaling-specific (Firefly luciferase behind dTCF binding sites) and viability (Renilla luciferase) signals were separately normalized to a set of controls per 384-well plate by division by the median thereof. In the genome-wide screen the controls comprised 18 wells, and in the combinatorial RNAi screens 32 wells per assay plate. Each control well contained one dsRNA reagent. Controls covered biological (known regulators) and non-targeting controls. The data were log2-transformed. In each well, the viability effect in the Firefly luciferase signal was corrected by subtracting the (log2-transformed) Renilla luciferase signal.
For the genome-wide screening data, the global dependency of the Wnt signaling-specific signal on the viability was accounted for, using LOESS (LOcally WEighted Scatter-plot Smoother) regression. The residuals were bin-wise variance-corrected, generating Wnt signaling activity z-scores. For details and documented code, see Supplemental Information.
Modeling of Genetic Interactions
Genetic interaction scores (π-scores) were estimated for each pair of dsRNA reagents as previously described (Horn et al., 2011). This multiplicative model was applied on the normalized Wnt activity signal. This model uses a multiplicative neutrality function, assuming that if two genes act independently the product of their individual phenotypes equals the combined phenotype. The single gene effects were estimated by Tukey’s median polish procedure (using the R function medpolish). The function was applied to the matrix of log2-transformed values where rows represent template RNAi reagents (plus 32 non-targeting reagents) and columns represent query RNAi reagents. Row and column medians are iteratively subtracted until the sum of absolute residuals is close to ‘0’ (Tukey, 1977). Fitted row effects illustrate template and fitted column effects illustrate query RNAi effects, the residual represents the genetic interaction score. For each gene pair, four measurements, which comprise two sequence-independent dsRNAs against the template and query gene, respectively, were taken (see Figure S2D for comparison of genetic interaction scores of the four dsRNA design combinations of all tested gene pairs). Averaging the four interaction scores generated the π-score for each gene pair. To test the significance of all pair-wise genetic interactions, p-values were calculated by the moderated t-test (limma), which estimates the mean and standard errors of the four interaction scores for each gene pair, followed by empirical Bayes shrinkage of the SEM (Smyth, 2004). p-values were adjusting for multiple testing by controlling the false discovery rate (FDR) applying the method of Benjamini-Hochberg (Benjamini and Hochberg, 1995). Figure S2F illustrates how the FDR relates to the difference in π-scores in a replication study using five of the 72 query genes. For details and documented code, see Supplemental Information.
Estimating Functional Similarity
The state-specific similarity of gene function was estimated by calculating the pairwise Pearson correlation coefficients (PCC) between the genetic interaction profiles separately for each Wnt pathway state (state-specific PCC). The state-specific profiles included the π-scores between a given gene and the 72 query genes, which covered known regulators of the Wnt signaling pathway as well as representative components of ‘other’ signaling pathways, and cellular processes including vesicle trafficking, transcriptional, protein turnover or cell cycle regulation.
Between-state functional similarity was estimated by calculating the pairwise PCC between the genetic interaction profile (along the 72 query genes) for a given gene (A) in the baseline state and another gene (B) in an active state. Note that this provided two scores for each gene pair since both genes A and B can be in the baseline and a given active state. Moreover, the PCC of Abaseline and Aactive does not equal 1.
Connection Specificity Index (CSI)
The connectivity specificity index (CSI) is based on a correlation matrix. For each pair of target genes, the Pearson correlation coefficient (PCC) of the two genetic interaction profiles along all query genes was computed. The CSI of a gene pair A-B was then defined as the fraction of genes connected to A and B that have a PCC smaller than the PCC of A and B. A constant of 0.1 was applied in the CSI definition of (Green et al., 2011).
Signed Similarity Networks
To visualize potential functional connections between genes, they were placed in a network graph and connected if the absolute PCC of their genetic interaction profiles exceeded a given threshold. A force-directed layout using the absolute PCC over the given threshold as weighted edges was applied to determine the position of nodes (genes), unless indicated otherwise in the figure legend. Therefore, positive and negative coefficients were given the same attraction force based on their absolute value. For details and documented code, see Supplemental Information.
Data and Software Availability
All computational analyses are available through the WntSGI R package containing the raw data and the code to process and illustrate the data. The WntSGI package is available through Github (https://github.com/boutroslab/Supplemental-Material). The documented R code vignette is also available with this article online (see Data S1).
Additional Resources
Author Contributions
Conceptualization, M. Billmann and M. Boutros; Methodology, M. Billmann; Software, M. Billmann and B.F.; Formal Analysis, M. Billmann and B.F.; Investigation, M. Billmann, V.C., and M.F.E.; Resources, M. Boutros; Writing – Original Draft, M. Billmann, V.C., B.F., and M. Boutros; Writing – Review & Editing, M. Billmann and M. Boutros; Visualization, M. Billmann; Supervision, M. Boutros and B.F.; Funding Acquisition, M. Boutros.
Acknowledgments
We would like to dedicate this paper to Bernd Fischer (deceased February 22, 2017). We thank Thomas Horn, Thomas Sandmann, Marija Buljan, Marco Breinig, and Chad Myers for helpful comments on the manuscript, and members of the Boutros lab for critical discussions. M. Billmann was in part supported by the Helmholtz International School of Cancer Research and an EMBO Short-term Fellowship (ASTF 489-2014). B.F. was supported by the Helmholtz Association (VH-NG-1010). Work in the lab of M. Boutros was funded by an ERC Advanced grant (“Syngene”) of the European Research Council.
Published: November 29, 2017
Footnotes
Supplemental Information includes six figures, two tables, and one data file and can be found with this article online at https://doi.org/10.1016/j.cels.2017.10.015.
Supplemental Information
References
- Alvarez M.J., Shen Y., Giorgi F.M., Lachmann A., Ding B.B., Ye B.H., Califano A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016;48:838–847. doi: 10.1038/ng.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzolin L., Zanconato F., Bresolin S., Forcato M., Basso G., Bicciato S., Cordenonsi M., Piccolo S. Role of TAZ as mediator of Wnt signaling. Cell. 2012;151:1443–1456. doi: 10.1016/j.cell.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S., Mehta M., Kuo D., Sung M.-K., Chuang R., Jaehnig E.J., Bodenmiller B., Licon K., Copeland W., Shales M. Rewiring of genetic networks in response to DNA damage. Science. 2010;330:1385–1389. doi: 10.1126/science.1195618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartscherer K., Pelte N., Ingelfinger D., Boutros M. Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell. 2006;125:523–533. doi: 10.1016/j.cell.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Baryshnikova A., Costanzo M., Kim Y., Ding H., Koh J., Toufighi K., Youn J., Ou J., San Luis B.-J., Bandyopadhyay S. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat. Methods. 2010;7:1017–1024. doi: 10.1038/nmeth.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A., Costanzo M., Myers C.L., Andrews B., Boone C. Genetic interaction networks: toward an understanding of heritability. Annu. Rev. Genomics Hum. Genet. 2013;14:111–133. doi: 10.1146/annurev-genom-082509-141730. [DOI] [PubMed] [Google Scholar]
- Bateson W. Cambridge Univ. Press; 1909. Mendel’s Principles of Heredity. [Google Scholar]
- Behrens J., Jerchow B.A., Würtele M., Grimm J., Asbrand C., Wirtz R., Kühl M., Wedlich D., Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. 1995;57:289–300. [Google Scholar]
- Billmann M., Boutros M. Methods for high-throughput RNAi screening in Drosophila cells. Methods Mol. Biol. 2016;1478:95–116. doi: 10.1007/978-1-4939-6371-3_5. [DOI] [PubMed] [Google Scholar]
- Billmann M., Horn T., Fischer B., Sandmann T., Huber W., Boutros M. A genetic interaction map of cell cycle regulators. Mol. Biol. Cell. 2016;27:1397–1407. doi: 10.1091/mbc.E15-07-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Cavallo R.A., Cox R.T., Moline M.M., Roose J., Polevoy G.A., Clevers H., Pfeifer M., Bejsovec A. Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature. 1998;395:604–608. doi: 10.1038/26982. [DOI] [PubMed] [Google Scholar]
- Cherbas L., Willingham A., Zhang D., Yang L., Zou Y., Eads B.D., Carlson J.W., Landolin J.M., Kapranov P., Dumais J. The transcriptional diversity of 25 Drosophila cell lines. Genome Res. 2011;21:301–314. doi: 10.1101/gr.112961.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H., Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E.D., Sevier C.S., Ding H., Koh J.L.Y., Toufighi K., Mostafavi S. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S.D. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353 doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B., Sandmann T., Horn T., Billmann M., Chaudhary V., Huber W., Boutros M. A map of directional genetic interactions in a metazoan cell. Elife. 2015 doi: 10.7554/eLife.05464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxman Bass J.I., Diallo A., Nelson J., Soto J.M., Myers C.L., Walhout A.J.M. Using networks to measure similarity between genes: association index selection. Nat. Methods. 2013;10:1169–1176. doi: 10.1038/nmeth.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R., Kao H., Audhya A., Arur S., Mayers J.R., Fridolfsson H., Schulman M., Schloissnig S., Niessen S., Wang S. A high-resolution C. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell. 2011;145:470–482. doi: 10.1016/j.cell.2011.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross J.C., Chaudhary V., Bartscherer K., Boutros M. Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 2012;14:1036–1045. doi: 10.1038/ncb2574. [DOI] [PubMed] [Google Scholar]
- Groth C., Sasamura T., Khanna M.R., Whitley M., Fortini M.E. Protein trafficking abnormalities in Drosophila tissues with impaired activity of the ZIP7 zinc transporter catsup. Development. 2013;140:3018–3027. doi: 10.1242/dev.088336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guénolé A., Srivas R., Vreeken K., Wang Z.Z., Wang S., Krogan N.J., Ideker T., van Attikum H. Dissection of DNA damage responses using multiconditional genetic interaction maps. Mol. Cell. 2013;49:346–358. doi: 10.1016/j.molcel.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha K.G., Rual J.-F., Zhai B., Mintseris J., Vaidya P., Vaidya N., Beekman C., Wong C., Rhee D.Y., Cenaj O. A protein complex network of Drosophila melanogaster. Cell. 2011;147:690–703. doi: 10.1016/j.cell.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K., Jeng E.E., Hess G.T., Morgens D.W., Li A., Bassik M.C. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 2017;35:463–474. doi: 10.1038/nbt.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Horn T., Sandmann T., Boutros M. Design and evaluation of genome-wide libraries for RNA interference screens. Genome Biol. 2010;11:R61. doi: 10.1186/gb-2010-11-6-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn T., Sandmann T., Fischer B., Axelsson E., Huber W., Boutros M. Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat. Methods. 2011;8:341–346. doi: 10.1038/nmeth.1581. [DOI] [PubMed] [Google Scholar]
- Housden B.E., Perrimon N. Spatial and temporal organization of signaling pathways. Trends Biochem. Sci. 2014;39:457–464. doi: 10.1016/j.tibs.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji N., Middelkoop T.C., Mentink R.A., Betist M.C., Tonegawa S., Mooijman D., Korswagen H.C., van Oudenaarden A. Feedback control of gene expression variability in the Caenorhabditis elegans Wnt pathway. Cell. 2013;155:869–880. doi: 10.1016/j.cell.2013.09.060. [DOI] [PubMed] [Google Scholar]
- Kakugawa S., Langton P.F., Zebisch M., Howell S.A., Chang T.-H., Liu Y., Feizi T., Bineva G., O’Reilly N., Snijders A.P. Notum deacylates Wnt proteins to suppress signalling activity. Nature. 2015;519:187–192. doi: 10.1038/nature14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V., Barker N., Morin P.J., van Wichen D., de Weger R., Kinzler K.W., Vogelstein B., Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Lebensohn A.M., Dubey R., Neitzel L.R., Tacchelly-Benites O., Yang E., Marceau C.D., Davis E.M., Patel B.B., Bahrami-Nejad Z., Travaglini K.J. Comparative genetic screens in human cells reveal new regulatory mechanisms in WNT signaling. Elife. 2016;5 doi: 10.7554/eLife.21459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B., Crombie C., Tischler J., Fortunato A., Fraser A.G. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat. Genet. 2006;38:896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- Li J., Sutter C., Parker D.S., Blauwkamp T., Fang M., Cadigan K.M. CBP/p300 are bimodal regulators of Wnt signaling. EMBO J. 2007;26:2284–2294. doi: 10.1038/sj.emboj.7601667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Morin P.J., Sparks A.B., Korinek V., Barker N., Clevers H., Vogelstein B., Kinzler K.W. Activation of beta-catenin—Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Niehrs C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- Pan C.-L., Baum P.D., Gu M., Jorgensen E.M., Clark S.G., Garriga G. C. elegans AP-2 and retromer control Wnt signaling by regulating mig-14/Wntless. Dev. Cell. 2008;14:132–139. doi: 10.1016/j.devcel.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao S., Lee S.-H., Kim H., Yum S., Stamos J.L., Xu Y., Lee S.-J., Lee J., Oh S., Han J.-K. Direct inhibition of GSK3β by the phosphorylated cytoplasmic domain of LRP6 in Wnt/β-catenin signaling. PLoS One. 2008;3:e4046. doi: 10.1371/journal.pone.0004046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh J., Mercer J., Shrestha Y., Oliver R., Tamayo P., Doench J.G., Tirosh I., Piccioni F., Hartenian E., Horn H. Genetic and proteomic interrogation of lower confidence candidate genes reveals signaling networks in β-catenin-active cancers. Cell Syst. 2016;3:302–316.e4. doi: 10.1016/j.cels.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segre D., DeLuna A., Church G.M., Kishony R. Modular epistasis in yeast metabolism. Nat. Genet. 2005;37:77–83. doi: 10.1038/ng1489. [DOI] [PubMed] [Google Scholar]
- Smyth G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- Steinhart Z., Pavlovic Z., Chandrashekhar M., Hart T., Wang X., Zhang X., Robitaille M., Brown K.R., Jaksani S., Overmeer R. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2016;23:60–68. doi: 10.1038/nm.4219. [DOI] [PubMed] [Google Scholar]
- Strigini M., Cohen S.M. Wingless gradient formation in the Drosophila wing. Curr. Biol. 2000;10:293–300. doi: 10.1016/s0960-9822(00)00378-x. [DOI] [PubMed] [Google Scholar]
- Tanimoto H., Itoh S., ten Dijke P., Tabata T. Hedgehog creates a gradient of DPP activity in Drosophila wing imaginal discs. Mol. Cell. 2000;5:59–71. doi: 10.1016/s1097-2765(00)80403-7. [DOI] [PubMed] [Google Scholar]
- Thompson B.J., Cohen S.M. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell. 2006;126:767–774. doi: 10.1016/j.cell.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Tukey J.W. Addison-Wesley; 1977. Exploratory Data Analysis. [Google Scholar]
- Typas A., Nichols R.J., Siegele D.A., Shales M., Collins S.R., Lim B., Braberg H., Yamamoto N., Takeuchi R., Wanner B.L. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat. Methods. 2008;5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayagam A., Zirin J., Roesel C., Hu Y., Yilmazel B., Samsonova A.A., Neumüller R.A., Mohr S.E., Perrimon N. Integrating protein-protein interaction networks with phenotypes reveals signs of interactions. Nat. Methods. 2014;11:94–99. doi: 10.1038/nmeth.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa S., Lee J.S., Ishimoto A. Identification and characterization of a novel line of Drosophila Schneider S2 cells that respond to wingless signaling. J. Biol. Chem. 1998;273:32353–32359. doi: 10.1074/jbc.273.48.32353. [DOI] [PubMed] [Google Scholar]
- Yu J., Chia J., Canning C.A., Jones C.M., Bard F.A., Virshup D.M. WLS retrograde transport to the endoplasmic reticulum during Wnt secretion. Dev. Cell. 2014;29:277–291. doi: 10.1016/j.devcel.2014.03.016. [DOI] [PubMed] [Google Scholar]
- Zeng W., Wharton K.A., Jr., Mack J.A., Wang K., Gadbaw M., Suyama K., Klein P.S., Scott M.P. naked cuticle encodes an inducible antagonist of Wnt signalling. Nature. 2000;403:789–795. doi: 10.1038/35001615. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.