

Abstract
Alignment is the first step in most RNA-seq analysis pipelines, and the accuracy of downstream analyses depends heavily on it. Unlike most steps in the pipeline, alignment is particularly amenable to benchmarking with simulated data. We performed a comprehensive benchmarking of 14 common splice-aware aligners for base, read, and exon junction-level accuracy and compared default with optimized parameters. We found that performance varied by genome complexity, and accuracy and popularity were poorly correlated. The most widely cited tool underperforms for most metrics, particularly when using default settings.
The majority of RNA-seq studies start with alignment to a reference genome or transcriptome. Analysis is also possible without a reference genome but generally underperforms alignment-guided analysis1. Many algorithms have been developed for this critical alignment step (Supplementary Fig. 1). Most of these are specific to RNA-seq, but BWA and Bowtie are designed for DNA alignment and do not properly handle intron-sized gaps; therefore we strongly advise against using these tools for genome alignment.
Polymorphisms, sequencing error, low-complexity sequences, intron-sized gaps, intron signal, incomplete annotation, alternative splicing, and pathological splicing can all complicate alignment. For an aligner to be viable for RNA-seq it must (i) align reads across splice junctions, (ii) handle paired-end reads, (iii) handle strand-specific data, and (iv) run efficiently. Since annotation is never perfect, the ability to align reads across unannotated splice junctions is also a plus. We identified 14 algorithms which satisfy these four basic requirements: CLC Genomics Workbench v8.5 (http://www.qiagenbioinformatics.com/products/clc-genomics-workbench/), ContextMap2 v2.6.0 (ref. 2), CRAC v2.4.0 (ref. 3), GSNAP v2015-9-29 (ref. 4), HISAT v0.1.6beta5, HISAT2 v2.0.0beta5, MapSplice2 v2.2.0 (ref. 6), Novoalign v3.02.13 (http://www.novocraft.com/products/novoalign/), OLego v1.1.6 (ref. 7), RUM v2.0.5_06 (ref. 8), SOAPsplice v1.10 (ref. 9), STAR v2.5.0a (ref. 10), Subread v1.5.0 (ref. 11), and TopHat v2.1.0 (ref. 12).
Simulating data for benchmarking alignment algorithms is straightforward on account of the discrete nature of the data. Simulated data were used for comprehensive RNA-seq alignment benchmarking studies in 2011 (ref. 8) and 2013 (ref. 13), but alignment methods have undergone considerable development since then. Here we analyze performance at the base, read, and junction levels using default and optimized parameters. We also examine execution time and memory usage; differential behavior at canonical versus noncanonical junctions; the effect of untrimmed adapters; performance on indels, reads that map to multiple sites (multimappers); and other factors.
Even aligning human reads to a human reference presents difficulties for some genes, and aligning across different strains or species can be globally difficult. It is therefore necessary to use aligners that handle both low- and high-complexity regions effectively. Thus it is important to simulate different levels of complexity and preferentially use aligners that generally perform well in all scenarios.
RESULTS
We simulated data from human and the malaria parasite Plasmodium falciparum at three complexity levels (T1, T2 and T3) for each of the two organisms. Each data type was simulated three times, giving a total of 18 data sets, that are used throughout (Online Methods). P. falciparum was chosen because it is a commonly studied organism with a very different genome from that of the human; its genes are 80% AT rich on average14. For each data set, 10 million 100-base read pairs (2 × 109 bases) were generated.
The least complex data sets, denoted T1, were generated with low polymorphism rates (0.001 substitution, 0.0001 indel) and error rates (0.005, a typical Illumina error rate15), similar to aligning most RNA-seq reads to the reference human genome. T2-level complexity data has moderate polymorphism and error rates (0.005 substitution, 0.002 indel, 0.01 error), similar to data from model organisms. T3 has high polymorphism and error rates (0.03 substitution, 0.005 indel, 0.02 error).
Base and read level
Of our base-, read- and junction-level metrics, base-level metrics are the strictest and require the highest degree of accuracy (Fig. 1). Each base of each read constitutes an ‘event’ which can be either right or wrong. Roughly speaking, recall measures the fraction of all bases that were aligned correctly, and precision measures the fraction of all aligned bases that were aligned correctly. Precision is high for most aligners, even at T3-level complexity. In other words, what the aligners do align, they tend to align well, at least at the base level. The greatest variance in performance is seen in recall.
Figure 1.
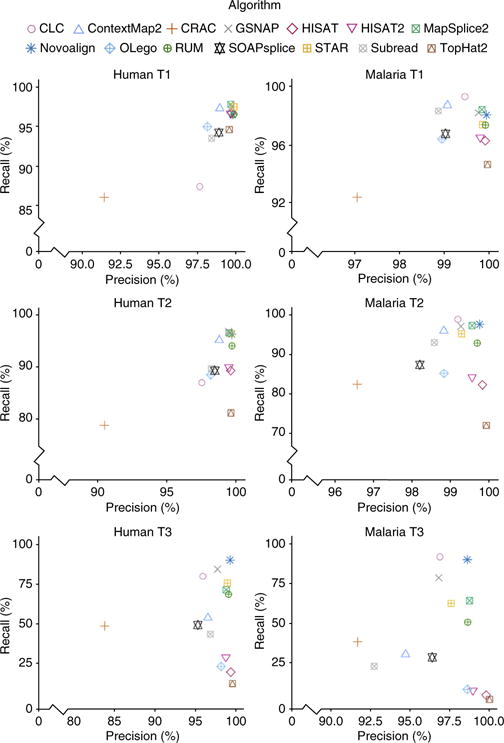
Base-level precision and recall for human and malaria data sets.
On T1 libraries, base-level recall is high for most tools, ranging from 86.1% (CRAC) to 97.8% (MapSplice2) on human data and from 92.4% (CRAC) to 99.3% (CLC) on malaria data. For results organized by class of misalignment (misaligned, aligned ambiguously, and unaligned) see Supplementary Figure 2.
In contrast, T2 libraries reveal significant differences in performance, ranging from 78.8% (CRAC) to 98.9% (GSNAP) on human data. Five tools maintain a recall greater than 95% (Contextmap2, GSNAP, Mapsplice2, Novoalign, STAR). On malaria data, performance ranges from 72.1% (TopHat) to 98.9% (CLC).
The T3 libraries show a vast difference in recall, ranging from 12.5% (TopHat2) to 90.3% (Novoalign) for human and 2.1% (TopHat2) to 91.2% (CLC) for malaria data. Novoalign, GSNAP, CLC, STAR, Mapsplice2, and RUM exceed 50% on both organisms.
For malaria data at the base level, CLC consistently has the best recall, while Novoalign and GSNAP also do well. For human data, Novoalign, GSNAP, Mapsplice2, and STAR are the best. Despite its popularity, TopHat2 is consistently among the worst performers on both human and malaria T2 and T3 libraries.
Reads are considered properly aligned if they are not multimappers and at least one base is aligned correctly. Read-level analysis is most relevant for gene-level quantification, because for a read that has at least one base aligned correctly, the correct gene which produced that read will usually be identified. Read-level results are similar to base-level results (Supplementary Figs. 3 and 4). On human and malaria T1 libraries, all the tools except CLC map more than 96% of the reads. On human data, Contextmap2, GSNAP, Mapsplice2, Novoalign, and STAR have recall ≥97%. On malaria, all the tools except HISAT, HISAT2, OLego, and TopHat2 have recall ≥97%. CRAC shows the greatest percentage of reads mapped incorrectly.
Junction level
A junction is where a read is spliced across an intron-sized gap that is typically thousands of bases long. When a read aligns across an intron gap, the shorter aligned segment is referred to as the anchor. Aligning across intron-sized gaps is particularly challenging, as the anchor can be as short as one base. For shorter anchors, more accurate annotation should help with the alignment.
Junction information is used extensively in downstream analysis programs; particularly in reconstructing alternative splicing events. Ambiguous cases, such as when the first base of an intron matches the first base of the next exon, can often be resolved by prioritizing canonical splice signals, but there is considerable latitude in how aligners accomplish this. Therefore, we assessed junction accuracy as a function of anchor length and splice signal.
An event is defined as a single read crossing a single splice junction. Not all reads involve events, and some reads involve multiple events. Postalignment analysis, which combines information across reads, is provided by some aligners and was not included in our tests; instead we assessed the quality of the individual read alignments. A correct event is scored when an algorithm aligns the read uniquely and properly identifies intron boundaries. The most consistently accurate performers are CLC, STAR, and NOVOALIGN (Fig. 2). As before, a much greater separation is seen with regards to the recall. CLC is the top performer in all data sets except human T1 and T2, two of the least complex data sets.
Figure 2.
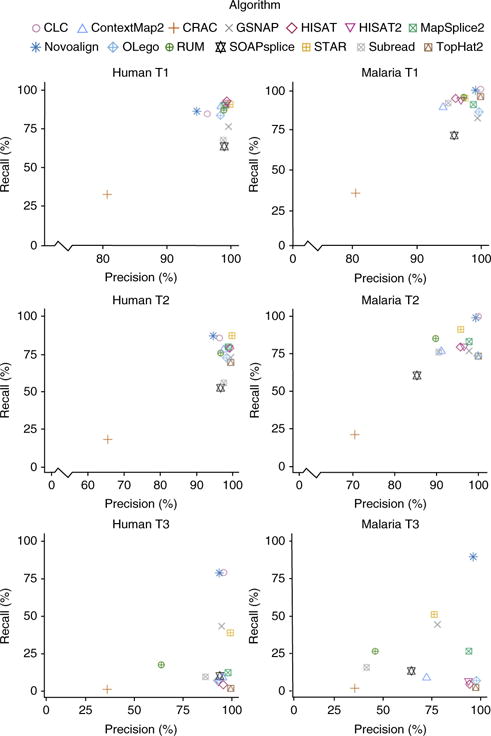
Junction-level precision and recall for human and malaria data sets.
This is somewhat surprising, as CLC only detects alternative splice sites at annotated junctions. If, for example, an exon ends at position N1, and the adjacent exon starts at position N2 according to the annotation, then CLC will align reads even if they splice from N1 to N2 + 3, for example. However, CLC does not connect exons that are never connected in the annotation; if, for example, a gene has only one annotated transcript consisting of exons E1, E2, and E3, then CLC will not properly align a read connecting E1 and E3. In contrast, Novoalign can identify unannotated connections, but it does not recognize changes from the annotated start or end position of an exon. In spite of these limitations, both CLC and Novoalign are among the top performers.
We further investigated the differential effect of anchor length on performance, both with and without annotation, and we found large variation (Supplementary Note 1 and Supplementary Fig. 5). HISAT, HISAT2, and ContextMap2 are remarkably accurate even on the shortest anchors and without annotation. CRAC, GSNAP, and SOAPsplice have the most trouble with short anchors; while OLego, STAR, and MapSplice2 have trouble with anchors of one or two bases but perform well on longer anchors. As long as annotation is provided, CLC, ContextMap2, HISAT, HISAT2, Novoalign, STAR, and TopHat2 perform well. CLC and Novoalign require annotation.
As the vast majority of splice junctions are canonical, we analyzed canonical and noncanonical junctions separately (Supplementary Fig. 6). All algorithms have significantly lower accuracy on noncanonical junctions as compared to canonical junctions, and no algorithm’s performance on noncanonical junctions improves much with annotation. As with short anchors, STAR, HISAT, HISAT2, and ContextMap2 perform best.
Annotation
At the base and read levels, the use of annotation does not provide significant improvement—most tools map just a few more reads (Supplementary Fig. 7) with annotation than without. This should not be surprising, since annotation is mainly expected to affect accurate placement of reads across exon–exon junctions; and the further a base is from a junction, the less likely it is to benefit. Only a small percent of bases are at exon–exon junctions, limiting the advantage of annotation at the base level, and even more so at the read level. The greatest effect of annotation is seen at the junction level (Supplementary Fig. 8).
Some algorithms require annotation (CLC and Novoalign), while others cannot utilize annotation (CRAC, SOAPsplice, Subread). Among those that can be run in both cases, annotation often helps to increase the junction-level recall, while it does not tend to increase the precision. Not surprisingly, the improvement of recall increases from T1 to T3 (Supplementary Note 2).
Overall the greatest improvements from annotation are seen in TopHat2, RUM, GSNAP, and STAR (Supplementary Table 1). Generally the two-pass aligners (such as HISAT or STAR-2-pass) have similar performance with and without annotations, while one-pass aligners (RUM, GSNAP, STAR-1-pass) benefit the most. It is worth nothing that one-pass performance with annotations is comparable to two-pass performance, which may reduce the required computing time two-fold, as the second pass is not necessary.
General improvement from annotation is perhaps more modest than one might expect. This could be because algorithms do not use annotation effectively, or because they achieve nearly optimal performance without the use of annotation. When no annotation is available, as in the case that sequencing of new organismal genomes outpaces their annotation, one should in fact favor algorithms that perform well without the need for annotation.
There may be other advantages of annotation which are not captured here. For example, several methods perform a postalignment analysis to produce a set of quality splice-junction calls. In this case annotation is likely to increase the accuracy of such calls, particularly on genes which are expressed at low levels.
Parameter optimization
It is important to explore the effect of parameters on performance and to identify algorithms which perform well with default settings. For each algorithm, the parameter space is enormous; we thus used a heuristic strategy to search the parameter space, which may not necessarily produce a global optimum (see Supplementary Note 3). Parameter optimization was performed on the T3-complexity data sets, which have the greatest room for improvement. It is generally not possible to optimize both precision and recall simultaneously or to optimize at the base, read, and junction level simultaneously. Furthermore, we found that optimizing the precision generally results in low recall. Since precision is already high in most cases, our focus was on optimizing the recall, which was done independently for base, read, and junction levels (Fig. 3 and Supplementary Figs. 9 and 10). The algorithm that benefits most dramatically from parameter tuning is TopHat2, while CLC, Novoalign, GSNAP, MapSplice2, and STAR perform the best with defaults. Unfortunately there is no clear way to optimize parameters on real data. Therefore, an algorithm that is robust to parameter settings and exhibits good performance using defaults is desirable (see Supplementary Note 3 and Supplementary Tables 2–43 for the most impactful parameters for each algorithm).
Figure 3.
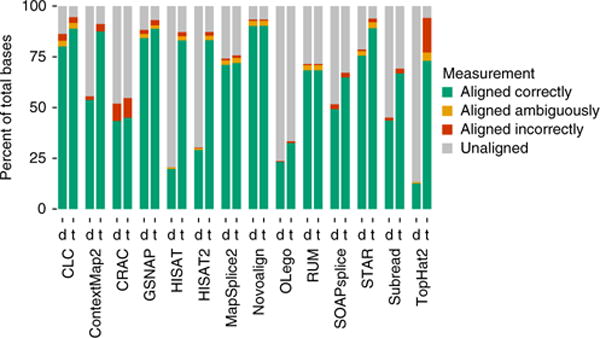
The effect of tuning parameters on the human-T3-data base-level statistics. For each tool, the figure shows the alignment statistics for the ‘default’ (d) and the ‘tuned’ (t) alignments.
Other analyses
In additional analyses, we addressed issues relating multimappers (Supplementary Note 4 and Supplementary Fig. 11), adapters (Supplementary Note 5 and Supplementary Fig. 12), indels (Supplementary Note 6 and Supplementary Figs. 13 and 14), two-pass modes (Supplementary Note 7), and computational performance (Supplementary Note 8 and Supplementary Fig. 15).
DISCUSSION
RNA-seq alignment has not undergone comprehensive benchmarking studies, making it hard to know how well individual algorithms work. Our results identify some effective aligners that are robust to parameter settings and others that display startling differences between default and optimized settings. TopHat2, for example, exhibits an alignment recall on malaria T3 that varies from under 3% using defaults to over 70% using optimized parameters. This is important since many TopHat2 users only use the defaults. The most important TopHat2 parameter is the number of mismatches. For a random set of 20 publications which used TopHat, the authors were contacted to determine the parameters they used. 10 of 13 authors who responded used the default mismatch parameter, and 5 used the defaults for all parameters. Since parameter optimization is not straightforward in practice, good default performance is an advantage. Based on this analysis the most reliable general-purpose aligners appear to be CLC, Novoalign, GSNAP, and STAR.
Three extensive RNA-seq alignment benchmarking studies that we are aware of considered both the accuracy and performance of spliced aligners8,13,16. They compared between four and seven spliced aligners plus some unspliced aligners. Other comparisons have considered only real data17,18 and are limited by the inability to know the ground truth. Fonseca et al.19 used simulated data to assess performance at the count level; however, the analysis focused on quantification output and not individual steps in the pipeline. Our results are consistent with prior studies of full RNA-seq aligners8,13 in spite of new versions of almost all applications; the notable difference being runtime (Fig. 4 and Supplementary Fig. 15). STAR was released in 2013 with a RAM-intensive approach that dramatically increased speed. Since then the STAR approach influenced other developers; for example, GSNAP, whose runtime has decreased dramatically. The new HISAT and HISAT2 also incorporate a fast search algorithm, yet their accuracy is comparable to that of TopHat2. Novoalign was available in the public domain when the previous studies were performed, while CLC has never been included in a benchmarking study, as far as we are aware.
Figure 4.
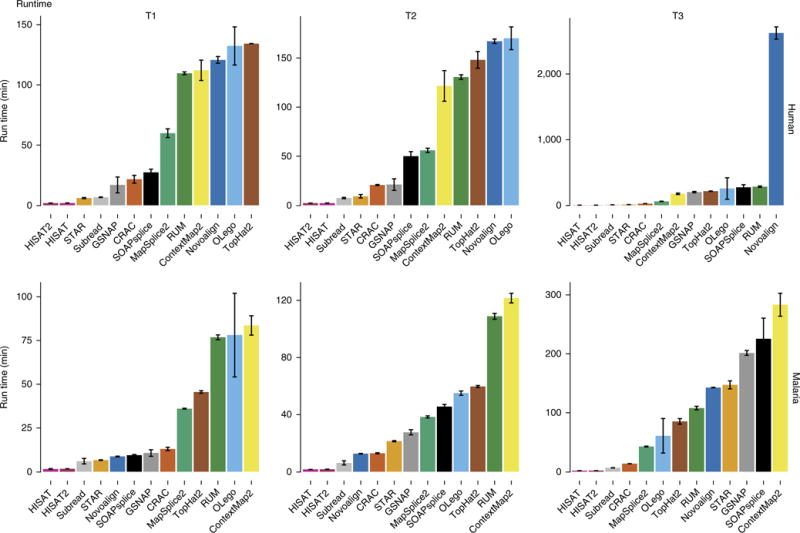
Runtime performance on human and malaria data. Bars show average runtime in minutes from three replicates. Error bars, s.d. Note that Novoalign has no multithreading in its free-license versions. To obtain comparable results, we divided the Novoalign runtime by the number of threads used (16). However, the real scalability could be different from the ideal one used here, resulting in a longer execution time.
One categorical difference between aligners that can help to explain the differences in performances is that several aligners are built on top of Bowtie or Bowtie2, which were designed to align DNA without intron-sized gaps.
Although the new results are largely consistent with those of past studies, this analysis should be updated regularly, as it is a fast-developing field. Standardizing methods for benchmarking will help to facilitate this in the future.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Simulated data
Simulating data for benchmark analyses of alignment algorithms is straightforward because of the discrete nature of the data. Since all algorithms align reads one at a time without combining information across reads or across samples, it is not necessary to model sample-to-sample variance or the dependence structure within or between samples—reads can therefore be generated from a reference genome and a set of accurate gene models, introducing polymorphisms (in the form of substitutions and indels), intron signal, and sequence errors, to varying degrees. Ultimately, it is introns and indels that give algorithms the greatest difficulty. All algorithms perform well when there are few indels or substitutions, as is the case for most regions of the human genome. By introducing an increasing number of polymorphisms, a separation of performance is observed, indicating which methods handle the complex regions better. Even aligning human to human presents a difficult challenge for some genes, while aligning across different strains or species, which is often necessary, can be difficult for all genes. Since most alignment tasks will involve some problematic regions, even when aligning human to human, it is always necessary to use an aligner than handles both low and high complexity most effectively.
The simulation engine BEERS8 (https://github.com/itmat/beers_simulator) was used to generate simulated data. Data of three different qualities were generated for each of two species, in triplicate, resulting in 18 data sets. Each data set consists of 10 million 100-base paired-end strand-specific reads. The genomes used were Homo sapiens hg19 and Plasmodium falciparum. Human data were limited to chromosomes 1–22, X and Y. For human data, 30,000 transcript models were chosen at random from a conglomeration of 858,063 gene models obtained by taking the union of ten annotation tracks: RefSeq, GeneID, Aceview, Augustus, ENSEMBL, UCSC, Vega, GenCode, GenScan, and lincRNA. This was done so as not to give unfair advantage to any algorithm that utilizes or was optimized on any particular set of annotation. For each gene an alternate splice form was generated by randomly including or excluding exons. Thus, a total of 60,000 transcript models were used. Expression levels were taken from an exponential distribution with P = 0.01 applied to a random 2/3 of the transcripts; the rest were left unexpressed. Intron signal was introduced at levels representative of real data, resulting in approximately 40% of reads coming from introns. Intron signal is introduced by inserting one intron back into the edited transcript before fragmentation. Two genomes, H. sapiens and P. falciparum, were simulated at each of three levels of complexity. Complexity level T1 had a substitution rate of 0.001, indel rate of 0.0005, and error rate of 0.005. Complexity level T2 had a substitution rate of 0.005, indel rate of 0.002, and error rate of 0.01. Complexity level T3 had a substitution rate of 0.03, indel rate of 0.005, and error rate of 0.02. In addition, in T3 there is a higher error rate equal to 0.5 in the last ten bases. The fragment length distribution has minimum length equal to 100 bases, mean equal to 200 bases, and maximum length equal to 500 bases. The P. falciparum genome was used because it is notorious for being difficult, mainly because it is approximately 80% AT rich in exons and 90% in introns and intergenic regions20. All simulated data are available at http://bioinf.itmat.upenn.edu/BEERS/bp1. Public repositories do not accept simulated data.
The T3 parameters were chosen to create a data set with uniformly high polymorphism rates. Data sets with uniform polymorphism rates are preferable to data sets with variable rates for benchmarking in order to isolate the performance in complex regions. T3 also represents polymorphism rates which can be observed when aligning across different (but similar) species–for example aligning Deer to Cow produces similar polymorphism rates. In practice, RNA-seq data is often generated for species for which the genome is not available or is of low quality. Aligning Deer to Cow, for example, enabled us to reconstruct the Deer clock pathway before any deer genomes were available (data not shown). In this way meaningful RNA-seq analysis of all mammals will be enabled if the genome is available from a sufficient number of mammalian organisms, even though it is unlikely there will be genomes of all mammals anytime soon; sequencing may be cheap, but genome assembly of new organisms is still very expensive. Therefore, RNA-seq aligners will continue to be applied to high-polymorphism data.
Data sets were generated in triplicate; however, as virtually no variance between replicates was observed, for the sake of efficiency the results shown are based on one replicate, except for the performance analyses (runtime and memory usage), where all three replicates were used.
Alignment metrics and statistics
Accuracy and performance metrics were compiled. The accuracy metrics consider accuracy on several levels: bases, reads, junctions, insertions, and deletions. Both the precision and recall were computed for each of these metrics.
An extensive set of metrics were defined in order to measure the most important aspects of the mapping process. First, the metrics already employed in previous studies were included8,13,16,17,19,21,22. Then additional metrics were defined with the goal of finding the smallest set of indices able to describe the most important characteristics of the RNA-seq data alignment. The resulting set of metrics can be organized into three levels, one for each basic concept of the RNA-Seq data alignment. As such, the metrics are based on events defined as follows: a single base of a single read aligning to the right location (base level), a single read having at least one base aligning to the right location (read level), and a single read crossing a single intron (junction level). Note that a single read may cross none, one, or multiple introns, in which case one read may involve none, one, or multiple junction-level events.
Metrics are then based on standard measures of accuracy for each type of event. In particular, we computed the standard accuracy metrics ‘precision’ and ‘recall’ for each level. Alternatively, the results can be presented as the ‘false negative rate’ (FNR) and ‘false discovery rate’ (FDR) using the relations FNR = 1 – recall and FDR = 1 – precision. Moreover, we collected summary statistics based on these basic concepts. As ground truth, we used the .cig file provided by the simulator engine. The .cig file describes the true position of the simulated reads in similar format to that of a SAM file23. The scripts developed to collect the alignment metrics and statistics are available at https://github.com/khayer/aligner_benchmark.
In the main body of this paper a few of the results were focused on and the rest can be found in Supplementary Notes 1–8. The base-wise accuracy involves the individual bases of the reads that aligned uniquely and to the correct location. There are three ways to be wrong at the base level: a base can either be not aligned at all, aligned to the wrong place, or aligned ambiguously to several places. The base-level ‘recall’ is defined as the ratio between the number of bases aligned correctly and uniquely to the total number of bases in the data set. The base-level ‘precision’ is the ratio of the number of bases that were aligned correctly and uniquely to the total number of bases that were aligned uniquely. There has to be some flexibility in this metric, in that some cases are ambiguous. For example, if GG in the reference is replaced by G in the read, then the aligner will typically choose one of the two G’s to call the aligned base and the other to call the deleted base. Ultimately the simulator did delete one of the two G’s specifically, but in reality evolution has replaced two G’s with one, so it does not make sense to indicate which one of the two G’s was retained and which was lost. Therefore, the aligner is credited for specifying either of the two possibilities. If one is interested only in gene-level quantification, then it may be sufficient to get the general location of the read correct without having to get every base correct. Thus accuracy is also measured at the read level, and accuracy in this case is determined by counting the percentage of reads for which at least one base is in the right location.
The SAM CIGAR string specifies whether indels are insertions, deletions, or introns. Junctions are differentiated from deletions in the SAM file, the former being indicated in the CIGAR string by an ‘N’ and the latter by a ‘D’. So the accuracy of each of these specifications can be measured. Furthermore, the left and right junctions were considered separately to determine whether any algorithm exhibits differential performance between left and right. Basic alignment statistics were collected on all algorithms. These consist of summary statistics such as the number of reads aligned and the number of reads aligned ambiguously. Supplementary Software developed to collect alignment metrics and statistics is available at http://bioinf.itmat.upenn.edu/BEERS/bp1.
For the performance metrics, the execution time, CPU time, and the maximum amount of RAM used by each tool were collected using the LSF tools provided by our HPC system. More details about the computational performance metrics are given in Supplementary Note 8.
Alignment of RNA-seq data
The goal of the alignment process is finding the right position of the input reads in the reference genome. Each read would be declared as ‘aligned’ or ‘unaligned’, depending on the ability of the aligner to find any putative position in the reference sequence. Obviously, where the minimum amount of information for a correct mapping is not available, the aligner cannot provide an alignment as output. However, with current sequencing technology the percent of reads that are impossible to align due to sequencing issues should be very small.
Except for being low quality, there are two main reasons why a read would be declared ‘unaligned’: the aligner is not able to find the right position in the reference sequence, or there is no right position in the reference sequence. The first scenario depends on the ability of each tool to manage the common alignment issues: sequencing errors, splicing events, intron-sized gaps, low-complexity sequence, and polymorphisms. The second scenario happens when a portion of the read comes from an adaptor or a contaminant, for which there are no reference sequences. Reads declared ‘aligned’ can be summarized in three main groups: reads aligned correctly, reads aligned incorrectly, and reads aligned ambiguously. Hopefully, an effective tool will report the majority of reads aligned correctly, with a few reads aligned ambiguously and very few reads aligned incorrectly. Of course this depends on exactly how we define ‘correct’ at the read level. The details of base-level, read-level, and junction-level accuracy are given below.
Base-level analysis
The base-level metrics focus on the behavior of the aligner with single-base resolution. The base-wise accuracy is calculated by determining whether individual bases of the reads align uniquely and to the correct location. Some flexibility was introduced in this metric, since some cases are ambiguous. Other metrics involve insertions and deletions.
The basic terms used in the base-level analysis are:
Aligned base: a base is defined as aligned if its read is aligned and its CIGAR character is different from ‘S’ and ‘H’ (clipping).
Unaligned base: a base is defined as unaligned if its read is unaligned or its read is aligned and its CIGAR character is ‘S’ or ‘H’ (clipping).
Ambiguously aligned base: a base is defined as ambiguously aligned if its read is ambiguously aligned.
Correctly aligned base: a base is defined as correctly aligned if it is aligned (uniquely, not ambiguously) and the CIGAR character in the SAM file is the same as the corresponding one in the .cig file (as provided by the simulator).
Incorrectly aligned base: a base is defined as incorrectly aligned if it is aligned (uniquely, not ambiguously) and the CIGAR character in the SAM file is different from the corresponding one in the .cig file (as provided by the simulator).
Insertion: a base is called insertion if its CIGAR character in the SAM file is an ‘I’.
Deletion: a base is called deletion if its CIGAR character in the SAM file is a ‘D’.
Skip: a base is called a skip if its CIGAR character in the SAM file is an ‘N’ (these are introns).
The base-level metrics are defined as follows:
Base-level precision: (no. correctly aligned bases) / (no. uniquely aligned bases)
Base-level recall: (no. correctly aligned bases) / (total no. bases)
Insertion precision: (no. insertions called correctly by the tool) / (no. insertions called by the tool)
Insertion recall: (no. insertions called correctly by the tool) / (total no. of real insertions)
Deletion precision: (no. deletions called correctly by the tool) / (no. deletions called by the tool)
Deletion recall: (no. deletions called correctly by the tool) / (total no. of real deletions)
Skip precision: (no. skips called correctly by the tool) / (no. skips called by the tool)
Skip recall: (no. skips called correctly by the tool) / (total no. of real skips)
The base-level statistics are defined as follows:
Percent of bases aligned correctly: (no. correctly aligned bases) / (total no. bases)
Percent of bases aligned incorrectly: (no. incorrectly aligned bases) / (total no. bases)
Percent of bases aligned ambiguously: (no. ambiguously aligned bases) / (total no. bases)
Percent of bases unaligned: (no. unaligned bases) / (total no. bases)
Percent of bases aligned: (no. aligned bases) / (total no. bases)
Read-level analysis
The read-level metrics focus on the read as a unit and are appropriate for gene-level quantification. Indeed, in gene-level quantification it is generally sufficient to get the location of the read correct without the constraint of having every single base correctly aligned. Thus we measure accuracy at the read level in terms of percentage of reads for which at least one base is in the right location.
The basic terms used in the read-level analysis are:
Aligned read: a read is defined as aligned if the SAM bit flag 0×4 is unset.
Unaligned read: a read is defined as unaligned if the SAM bit flag 0×4 is set.
Ambiguously aligned read: a read is defined as aligned ambiguously if either read in the read pair (fragment) was aligned but has multiple entries in the SAM file.
Correctly aligned read: a read is defined as aligned correctly if it is aligned (uniquely, not ambiguously) and at least one base of the read is mapped to the right position.
Incorrectly aligned read: a read is defined as aligned incorrectly if it is aligned (uniquely, not ambiguously) and no base of the read is mapped to the right position.
The read-level statistics are defined as follows:
Read-level precision: (no. correctly aligned reads) / (no. uniquely aligned reads)
Read-level recall: (no. correctly aligned reads) / (total no. reads)
The read-level statistics are defined as follows:
Percent of reads aligned correctly: (no. correctly aligned reads) / (total no. reads)
Percent of reads aligned incorrectly: (no. incorrectly aligned reads) / (total no. reads)
Percent of reads aligned ambiguously: (no. ambiguously aligned reads) / (total no. reads)
Percent of reads unaligned: (no. unaligned reads) / (total no. reads)
Percent of reads aligned: (no. aligned reads) / (total no. reads)
Junction-level analysis
Aligning over a junction is one of the most important features of RNA-seq aligners. This feature is so important that it defines one of the most relevant ways to classify an NGS aligner: ‘splice aware’ versus ‘splice unaware’. All the tools involved in our benchmark are splice-aware algorithms, since RNA-seq data require the ability to map reads across such junctions.
The basic terms used in the junction level analysis:
Correctly called junction: a junction is defined as being called correctly if both the junction start and the junction end sites were identified correctly.
Incorrectly called junction: if either (or both) junction sites were called incorrectly, the whole junction is classified as an incorrectly called junction.
Junction sides none: called junctions where neither side was identified correctly.
Junction sides left: called junctions where only the upstream junction was called correctly.
Junction sides right: called junctions where only the downstream junction was called correctly.
Junction sides both: correctly called junctions.
The junction-level metrics are defined as follows:
Junction-level precision: (no. junctions called correctly by the tool) / (no. junctions called by the tool)
Junction-level recall: (no. junctions called correctly by the tool) / (total no. of real junctions)
The junction-level statistics are defined as follows:
Percent of junction sides none: (no. junctions sides none) / (no. junctions called by the tool)
Percent of junction sides left: (no. junctions sides left) / (no. junctions called by the tool)
Percent of junction sides right: (no. junctions sides right) / (no. junctions called by the tool)
Percent of junction sides both: (no. junctions sides both) / (no. junctions called by the tool)
Multimapper analysis
To identify the recall and precision in the case of multimapping fragments, the alignment with the most correct bases aligned was chosen, and any further calculations were based on this best alignment. Here the same statistics were calculated as introduced in the read- and base-level analysis section.
Read alignment
In the alignment process only the information available in a typical real data set was used. This information consists of annotation, read length, fragment length distribution, and raw data. RefSeq was used as generic base annotation for all algorithms. In order to perform a fair comparison, an index was created for each aligner even though some indexes were already available. In this way all the aligners use the same version of the genome and the same annotation.
For each tool an alignment was performed starting from the default parameters. When the tool provides specific parameter presets or precise suggestions to increase the quality of the alignment, these suggestions were followed. In particular, parameters related to the read were set (i.e., read length, fragment length, inner mate distance) or related to the genome, for example, suggested seed size, k-mer size, etc. This set of alignments are referred to as ‘default’; they are the typical alignments obtained by following the tools′ documentation as the typical user would do.
Moreover, many alignments were performed with each tool in search of optimal parameter settings for our particular data sets. The documentation was followed to determine which parameters are most important. Usually the suggestions from the specific documentation of each tool are more qualitative than quantitative, for this reason they were not included in the default. Where these suggestions were not provided, the most exhaustive and reasonable sets of parameters we could identify were searched. Additionally all authors were contacted and given the opportunity to make further suggestions. This required performing thousands of alignments involving a large amount of computation. In order to search as large a space as possible, each set of parameters was run on 1 million reads in the T3 complexity level data sets. T3 data sets were used because the greatest improvement from the defaults can be made in these sets. This set of alignments is referred to as ‘tweaked’ or ‘tuned’. The goal of these alignments is to determine how far the default performance is from the real potential of the tool. Moreover, this information provides some general suggestion as to what are the most important parameters for each tool. Most tools will use annotation as a guide. In order to determine the effect of using annotation, alignment was performed both with and without providing this information. Parameter optimization was performed with annotation, as the goal was to try to get the best possible performance from each tool.
Both the default and the tweaking alignments were performed using 16 threads. When available, the performance parameters that guaranteed the shortest execution time were used (without any loss of precision). These options sometimes use more RAM than the default option. However, in practice the available amount of RAM is usually a smaller problem than the required execution time.
Details about each aligner are given in Supplementary Notes 9 and 10. More details about the default and the optimized (tweaked) alignments can be found in Supplementary Note 3.
Supplementary Material
Acknowledgments
We thank A. Srinivasan for his help administrating the PMACS cluster. We thank N. Lahens, T. Grosser, D. Sarantopoulou, F. Coldren, E. Scarci, and E. Ricciotti for support and helpful discussions. This work was funded in part by the National Heart Lung and Blood Institute (U54HL117798, G.A.F.) and The National Center for Advancing Translational Sciences (UL1-TR-001878, G.A.F.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
G.B. contributed research, analysis, and writing. K.E.H. contributed analysis, figures, and benchmarking scripts. E.J.K. contributed analysis. B.D.C. contributed analysis and formulation of ideas. G.A.F. contributed formulation of ideas and direction. G.R.G. contributed the simulated data, direction, ideas, and writing.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Data availability
No accession codes are associated with this study since public repositories do not maintain simulated data. All data used in this study, including data used for the figures and all scripts used in the analysis are available in the Supplementary Data 1–15 and at http://www.bioinf.itmat.upenn.edu/BEERS/bp1/.
Source data for Figures 1–4 and Supplementary Figures 1–15 are available online.
References
- 1.Hayer KE, Pizarro A, Lahens NF, Hogenesch JB, Grant GR. Benchmark analysis of algorithms for determining and quantifying full-length mRNA splice forms from RNA-seq data. Bioinformatics. 2015;31:3938–3945. doi: 10.1093/bioinformatics/btv488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonfert T, Kirner E, Csaba G, Zimmer R, Friedel CC. ContextMap 2: fast and accurate context-based RNA-seq mapping. BMC Bioinformatics. 2015;16:122. doi: 10.1186/s12859-015-0557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Philippe N, Salson M, Commes T, Rivals E. CRAC: an integrated approach to the analysis of RNA-seq reads. Genome Biol. 2013;14:R30. doi: 10.1186/gb-2013-14-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang K, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu J, Anczuków O, Krainer AR, Zhang MQ, Zhang C. OLego: fast and sensitive mapping of spliced mRNA-Seq reads using small seeds. Nucleic Acids Res. 2013;41:5149–5163. doi: 10.1093/nar/gkt216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grant GR, et al. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang S, et al. SOAPsplice: Genome-wide ab initio detection of splice junctions from RNA-Seq data. Front Genet. 2011;2:46. doi: 10.3389/fgene.2011.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engström PG, et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods. 2013;10:1185–1191. doi: 10.1038/nmeth.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aurrecoechea C, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37:D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glenn TC. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 2011;11:759–769. doi: 10.1111/j.1755-0998.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 16.Wang WA, et al. 2014 International Conference on Electrical Engineering and Computer Science 215–218. ICEECS; 2014. Comparisons and performance evaluations of RNA-seq alignment tools. [Google Scholar]
- 17.Benjamin AM, Nichols M, Burke TW, Ginsburg GS, Lucas JE. Comparing reference-based RNA-Seq mapping methods for non-human primate data. BMC Genomics. 2014;15:570. doi: 10.1186/1471-2164-15-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fonseca NA, Rung J, Brazma A, Marioni JC. Tools for mapping high-throughput sequencing data. Bioinformatics. 2012;28:3169–3177. doi: 10.1093/bioinformatics/bts605. [DOI] [PubMed] [Google Scholar]
- 19.Fonseca NA, Marioni J, Brazma A. RNA-Seq gene profiling—a systematic empirical comparison. PLoS One. 2014;9:e107026. doi: 10.1371/journal.pone.0107026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner MJ, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindner R, Friedel CC. A comprehensive evaluation of alignment algorithms in the context of RNA-seq. PLoS One. 2012;7:e52403. doi: 10.1371/journal.pone.0052403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatem A, Bozdağ D, Toland AE, Çatalyürek UV. Benchmarking short sequence mapping tools. BMC Bioinformatics. 2013;14:184. doi: 10.1186/1471-2105-14-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.