

Abstract
Suzuki, Negishi, and Kumada couplings are some of the most important named reactions for the formation of skeletal C–C linkages. Their widespread use to forge bonds between two different aromatic rings has enabled every branch of chemical science. The analogous union between alkyl halides and metallated aryl systems has not been as widely employed due to the lack of commercially available halide building blocks. Redox-active esters (RAEs) have recently emerged as useful surrogates for alkyl halides in cross-coupling chemistry. Such esters are easily accessible through reactions between ubiquitous carboxylic acids and coupling agents widely used in amide-bond formation. This article features an amalgamation of in-house experience bolstered by ca. 200 systematically designed experiments to accelerate the selection of ideal reaction conditions and activating agents for the cross-coupling of primary, secondary, and tertiary alkyl carboxylic acids with both aryl and heteroaryl organometallic species.
Graphical abstract
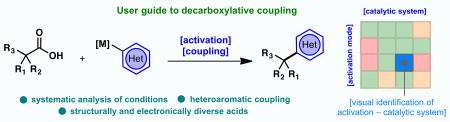
Cross-coupling meets amide-bond formation. Decarboxylative cross-coupling via redox-active esters forms C–C bonds using boronic acids, organozinc, and organomagnesium species with comparable simplicity to amide-bond formation. Here, an extensive study is performed, enabling the selection of the right activating agent and coupling conditions across a range of substrates including notoriously challenging aromatic heterocycles.
The formation of C–C bonds between alkyl fragments and aromatic systems is among the most useful transformations available to practitioners of chemical synthesis.[1] Over the past year our lab has reported Negishi,[2] Kumada,[2c] and Suzuki[3] couplings that employ simple alkyl carboxylic acid starting materials in lieu of alkyl halides to achieve such constructions. The success of these new transformations hinges on the use of redox-active esters (RAEs), repurposed from the realm of amide-bond formation (Figure 1A). From a practical standpoint, these reactions benefit from high operational simplicity: (1) an inexpensive Ni (ca. $10/mole) or Fe (ca. 1 cent/mole) precatalyst is used in combination with simple and widely available ligands, (2) common activators that are routinely used in peptide synthesis can be employed,[4] (3) the RAEs do not need to be isolated, (4) the reactions usually tolerate air and moisture, and (5) a simple, thermal batch process makes scale-up straightforward. Conceptually, the ability to substitute RAEs for alkyl halides in common cross-coupling reactions can expand retrosynthetic options for both specific targets and diversity-oriented platforms since alkyl carboxylic acids are both ubiquitous and stable.[5–8] By "piggybacking" on the ease and success of amide-bond formation, the barrier to performing a decarboxylative cross-coupling using RAEs is extremely low. Guides for the most popular C–C[9] and C–N[10] bond forming cross-couplings, and amide bond formation itself,[4] have been developed to aid the practitioner on the correct choice of catalyst and coupling partner. This study presents a comprehensive user-guide for coupling with RAEs across a range of representative substrate classes including notoriously challenging heteroarenes.
Figure 1.

(A) A user guide to decarboxylative couplings using simple and available carboxylic acid activators. (B) A systematic study of activating agents, catalytic systems and coupling agents is reported.
The goal of this study is to identify trends across substrate classes that would accelerate the selection of the best coupling partners and conditions for a given target of interest. Figure 1B presents the variables explored for each substrate and represents an amalgamation of internal knowledge gained from performing hundreds of such couplings. We have found that a broad spectrum of aryl coupling partners could be accommodated in RAE couplings, and its influence on the reaction outcome was only of secondary importance (with the exception of functionalized heterocycles). Thus, for the three major classes of cross-coupling (Negishi, Suzuki, and Kumada), three different RAEs were examined (both isolated and in situ derived), with particular emphasis on covering a broad chemical space for such coupling partners. For Negishi couplings, Ni/bipy and Fe/dppBz catalytic systems were employed. For Suzuki couplings, a Ni/phenanthroline was used and for Kumada couplings the simple Fe/DMPU catalytic system was evaluated.
Table 1 summarizes the cross-coupling of nine different alkyl carboxylic acids (three primary (1, 3 and 5), four secondary (7, 9, 11 and 13), and two tertiary (15 and 17), each under 16 different conditions, in a total of 144 reactions. These carboxylic acids were chosen to serve as a representative cross-section of substrates and functional groups one might encounter in the synthesis of modern medicinal or agrochemical substances. Several general trends can be distilled from Table 1 (independent of the type of carboxylic acid): (1) in situ activation with HATU in Fe-catalyzed Negishi cross-coupling proved to be a reliable approach across all compound classes analyzed. Primary, secondary and tertiary acids were amenable to coupling under these conditions, independent of electronic or structural features, or functional groups of the alkyl acid. (2) If in situ activation is desired, the activation mode has to be chosen according to the reaction type. For example, TCNHPI with DCC is the best choice for activation for Suzuki couplings, whereas HATU proved to be superior for Ni and Fe-based Negishi couplings.
Table 1.
A systematic study of Ni and Fe-based catalyst systems in the decarboxylative cross-coupling of structurally diverse alkylcarboxylic acids. All yields were determined using 1,4-difluorobenzene or 1,3,5-mesitylene. Yield in brackets indicates isolated pure material. For specific conditions, see SI.

|
Within the primary acids tested (1, 3 and 5) best yields were obtained with in situ activation with HATU and Fe as catalyst in the Negishi coupling with Ph2Zn (vide supra). Additionally, TCNHPI esters of 1 and 3 smoothly reacted under Ni-Suzuki and Ni-Negishi conditions. However, acid 5 bearing a basic nitrogen did not afford 6 under Suzuki conditions, not because of the nitrogen per se but rather due to RAE instability. Indeed, TCNHPI ester of 5 was found to hydrolyze prematurely under the reaction conditions. In such cases the coupling can be achieved using Fe-Negishi (HATU activation, 72%) or Fe-Kumada coupling (HATU activation, 45%). Importantly, ketone (1) and ester (3) functional groups are well tolerated even in the presence of Grignard reagents in Fe-Kumada reactions. This observation is consistent with the chemoselectivity observed in Fürstner's Fe-based cross-couplings.[11]
For secondary carboxylic acids, 7 and 9 were selected as fragments due to their high frequency of use in industry. This substance exhibited greater flexibility to different reaction conditions. While Ni-Negishi of the isolated TCNHPI gave the highest yield (79%), excellent results were also observed using isolated TCNHPI esters with Ni-Suzuki (60%) and isolated NHPI with Ni-Negishi (62%). The best choice for in situ activation was once again HATU in conjunction with Fe-Negishi (63%). These types of RAE cross-couplings are both orthogonal to other cross-couplings and function well across a variety of aryl-substituents. For example, with substrate 7, p-chlorophenyl was used instead of a simple phenyl species with no impact in isolated yield. Even when employing aryl groups having ortho-substitution, such as the use of 1-naphthyl to produce 10 from 9, RAE-coupling works under the right conditions. In this case, Fe-catalyzed reactions proved superior over Ni couplings, with HATU Fe-Negishi performing best (76% isolated). The clear importance of amino acids (AAs) as readily available substrates was next investigated using cyclic proline 11 and linear leucine 13 as examples. Not surprisingly, cyclic AA 11 and acyclic AA 13 show stark differences in reactivity under coupling conditions: Fe-Negishi under all activation modes investigated was best suited for coupling of 11, giving 12 in excellent yields (57–63%). Fe-Kumada using both HATU in situ or isolated NHPI also afforded workable yields of 12. Subjecting the TCNHPI ester of 11 to the Ni-Suzuki and Ni-Negishi conditions resulted in extremely poor yields of 12. In contrast, these very conditions proved to be the most effective to couple leucine derivative 13 (64 and 87%). The low yields obtained with the Fe-Kumada couplings further highlight the differences in reactivity between 11 and 13, which might be attributable to differences in radical geometry or the protective group used. During these studies, it was found that an N-phthalimide protecting group was optimal for coupling in open-chain AAs.
The synthesis of quaternary centers from tertiary alkyl radicals still represents a major challenge in catalysis. Efforts to fill this gap by means of Fe-catalysis using tertiary carboxylic RAEs were recently described.[2c] As a general requirement, only non-planar radicals with π-character were amenable to coupling, leaving substrates such as pivalic acid outside the scope of this methodology at the present time (although workable in a conjunctive sense).[2b] As a prototypical example of carboxylic acids forming non-planar carbon-centered radicals, 1-adamantylcarboxylic acid (15) was selected. The isolated RAE of NHPI afforded excellent yields of cross-coupling using Fe-catalysis (55% Negishi, 68% Kumada) while standard Ni-Suzuki and Ni-Negishi conditions completely failed to furnish 16. These results are in line with what has typically been observed in the reactions of tertiary carboxylic acids. [2b]
In contrast, this reactivity pattern was not observed when evaluating the cyclopropyl tertiary acid 17 under optimized protocols for arylation. Beyond the expected reactivity for tertiary centers with Fe, the isolated TCNHPI ester of 17 reacted smoothly under Ni-Suzuki (50%) and Ni-Negishi conditions (69%). It is worth noting that structurally closely related acids such as gem-dimethyl phenylacetic acid failed to react (formation of planar radical, vide supra). The unique trend seen with 17 probably results from the peculiar reactivity imparted by the phenylcyclopropyl motif.
The examples, thus far, clearly delineate important features and guidelines for achieving RAE cross-coupling with aryl-donors. For real-world applications, however, heteroaryl-donors are both the most sought-after and challenging. Thus, a separate study was conducted across a range of heteroaromatic donors using an isolated piperidine-based RAE (Table 2). The choice of metallated species used was based on commercial availability, pragmatic considerations, and stability. Of the thirteen substrates evaluated, all delivered the desired product to some extent (23–76% yield). Substrates are roughly ordered based on the perceived electron-density at the metallated carbon atom. In general, it is known that the ease of coupling and/or metallation can be correlated to the electronic character of the carbon atom.[12] For example, 3-pyridyl-based organometallic reagents usually undergo Suzuki/Negishi/Kumada coupling without incident. In contrast, specialized procedures have been developed for the 2-pyridyl isomer as the metallated species required for cross-coupling are unstable.[13] As a testament to the difficulty of forging such bonds with these heterocycles, most approaches favor a stepwise path through cross-coupling of a vinyl halide (or triflate) followed by hydrogenation or addition to a carbonyl followed by deoxygenation. For furan (19), thiophene (20), 4-pyrazole (21), and 3-pyridyl nucleophiles, a Fe-Kumada protocol was employed due to the low cost and ease of generating these nucleophilic species. The low yields observed when performing the Ni-Suzuki with 2-furan and 2-thiophene boronic acids may be ascribed to their instability and resulting decomposition under the reaction conditions. It is worth noting that the Hu group has successfully achieved cross coupling with these four heterocyclic Grignard reagents with alkyl iodides using an expensive Ni-pincer complex.[14] In contrast, there are no reported uses of a metallated 2-fluoro-5-pyridyl species 23 in an alkyl-aryl cross coupling of any kind. RAE-based coupling proceeded in serviceable yields under all three protocols tested. The chlorinated analog of this heterocycle (24) underwent coupling smoothly using a Ni-Suzuki protocol. This metallated heterocycle has only been reported by Knochel to participate in Fe- or Co-based cross coupling with two alkyl iodides and one alkyl bromide.[15] In the case of brominated pyridine 25, to our knowledge there are no reports using such a species in any alkyl cross coupling and even C(sp2) couplings are sparse. The Ni-Suzuki protocol yield for this substrate, although modest, would be sufficient for practicing medicinal chemists in need of a key analog. Quinoline-based systems such as 26 have only rarely been employed in alkyl-aryl cross coupling with the vast majority of such products being prepared instead by ring synthesis.[15a,16] In our hands, the Ni-Suzuki protol was the only viable option for the RAE coupling. Pyrimidine 27 is a popular coupling partner to make C(sp2)-C(sp2) linkages but has not been reported in the context of alkyl-aryl cross coupling; the Ni-Suzuki protocol delivered 56% isolated yield of product. As mentioned above, 2-pyridyl systems (28 and 29) are notoriusly capricious in cross-coupling chemistry. Alkyl-aryl cross coupling of such systems is quite rare but could be accomplished using RAEs under the Fe-Negishi system in reasonable yield. Although 4-pyridyl systems 30 and 31 are not known specifically in alkyl aryl cross-couplings, related substrates have been utilitized by Molander in Suzuki couplings of alkyl halides.[17] Similarly, it was found that the Ni-Suzuki process functioned most efficiently when employing RAEs.
Table 2.
A systematic study of decarboxylative cross-coupling using a broad spectrum of medicinally relevant heterocycles with differing electronic character. All yields refer to isolated pure material. For specific conditions, see SI

|
The user-guide presented herein is geared for practicing organic chemists in industry that are looking for a rapid route to challenging alkyl-aryl bonds. It is worth noting that of all of the substrates reported in this guide furnished the desired product. For those trying the reaction for the first time or with limited time for experimentation or optimization, the Fe-Negishi protocol using HATU and in situ activation is recommended as it had the highest rate of success. In the case of heteroaromatic couplings at electron-deficient carbons (the most challenging type), the Ni-Suzuki protocol proved most versatile. The experimental simplicity, low-cost, and broad scope exhibited in RAE-based cross coupling bodes well for its widespread adoption since the starting materials, alkyl carboxylic acids, are ever-present.
Supplementary Material
Acknowledgments
Financial support for this work was provided by NIH (GM-118176), Alfried Krupp von Bohlen und Halbach Foundation (F. S.), Glynn Family Honors Program (M. J. O), postdoctoral fellowships from the Catalan Government (to J. C.) and Austrian Science Fund (to L. W.).
Footnotes
Detailed Supporting Information and the ORCID identification number(s) for the author(s) of this article can be found online
References
- 1.Diederich F, Stang PJ. Metal-catalyzed Cross-coupling Reactions. Wiley-VCH; New York: 1998. [Google Scholar]
- 2.a) Cornella J, Edwards JT, Kawamura S, Wang J, Pan C-M, Gianatassio R, Schmidt M, Eastgate MD, Baran PS. J. Am. Chem. Soc. 2016;138:2174. doi: 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qin T, Cornella J, Li C, Malins L, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS. Science. 2016;352:801. doi: 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Toriyama F, Cornella J, Wimmer L, Chen T-G, Dixon DD, Creech G, Baran PS. J. Am. Chem. Soc. 2016;138:11132. doi: 10.1021/jacs.6b07172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J, Qin T, Chen T-G, Wimmer L, Edwards JT, Cornella J, Vokits B, Shaw SA, Baran PS. Angew. Chem. Int. Ed. 2016;55:9676. doi: 10.1002/anie.201605463. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016;128:9828. [Google Scholar]
- 4.Ishihara Y, Moreno A, Baran PS. The Portable Chemist’s Consultant. Apple Publishing Group; 2013. [Google Scholar]
- 5.For the use of aromatic carboxy groups in decarboxylative strategies, see: Rodriguez N, Goossen LJ. Chem. Soc. Rev. 2011;40:5030. doi: 10.1039/c1cs15093f.
- 6.For the use of aromatic carboxy groups in decarbonylative C–C bond formation, see: Muto K, Yamaguchi J, Musaev DG, Itami K. Nat. Commun. 2015;6:7508. doi: 10.1038/ncomms8508.Amaike K, Muto K, Yamaguchi J, Itami K. J. Am. Chem. Soc. 2012;134:13573. doi: 10.1021/ja306062c.
- 7.For the use of alkylcarboxy groups in Ni-catalysis in conjunction with PET chemistry, see: Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437. doi: 10.1126/science.1255525.Chu L, Lipshultz JM, MacMillan DWC. Angew. Chem. Int. Ed. 2015;54:7929. doi: 10.1002/anie.201501908.Angew. Chem. 2015;127:8040.
- 8.For the use of redox-active esters in reductive cross-couplings, see: Huihui KMM, Caputo JA, Melchor Z, Olivares AM, Spiewak AM, Johnson KA, DiBenedetto TA, Kim S, S, Ackerman LKG, Weix DJ. J. Am. Chem. Soc. 2016;138:5016. doi: 10.1021/jacs.6b01533.
- 9.Nishihara Y. Applied Cross-Coupling Reactions. Springer-Verlag; Berlin, Heidelberg: 2013. [Google Scholar]
- 10.Surry DS, Buchwald SL. Chem. Sci. 2011;2:27. doi: 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Fürstner A, Leitner A, Méndez M, Krause H. J. Am. Chem. Soc. 2002;124:13856. doi: 10.1021/ja027190t. [DOI] [PubMed] [Google Scholar]; b) Martin R, Fürstner A. Angew. Chem. Int. Ed. 2004;43:3955. doi: 10.1002/anie.200460504. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004;116:4045. [Google Scholar]
- 12.a) Thomas AA, Denmark SE. Science. 2016;352:329. doi: 10.1126/science.aad6981. [DOI] [PubMed] [Google Scholar]; b) Lennox AJJ, Lloyd-Jones GC. Angew. Chem. Int. Ed. 2013;52:7362. doi: 10.1002/anie.201301737. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013;125:7506. [Google Scholar]; c) Carrow BP, Hartwig JF. J. Am. Chem. Soc. 2011;133:2116. doi: 10.1021/ja1108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dick GR, Woerly EM, Burke MD. Angew. Chem. Int. Ed. 2012;51:2667. doi: 10.1002/anie.201108608. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012;124:2721. [Google Scholar]
- 14.Vechorkin O, Proust V, Hu X. J. Am. Chem. Soc. 2009;131:9756. doi: 10.1021/ja9027378. [DOI] [PubMed] [Google Scholar]
- 15.a) Steib AK, Thaler T, Komeyama K, Mayer P, Knochel P. Angew. Chem. Int. Ed. 2011;50:3303. doi: 10.1002/anie.201007187. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011;123:3361. [Google Scholar]; b) Hammann JM, Haas D, Steib AK, Knochel P. Synthesis. 2015;47:1461. [Google Scholar]
- 16.Krasovskiy A, Thomé I, Graff J, Krasovskaya V, Konopelski P, Duplais C, Lipshutz BH. Tetrahedron Lett. 2011;52:2203. [Google Scholar]
- 17.Molander GA, Argintaru OA, Aron I, Dreher SD. Org. Lett. 2010;12:5783. doi: 10.1021/ol102717x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.