

Abstract
Left ventricular hypertrophy is frequently observed in hypertensive patients and is believed to be due to the pressure overload and cardiomyocyte stretch. Three recent reports on mice with genetically engineered Na+ pumps, however, have demonstrated that cardiac ouabain-sensitive α2-Na+ pumps play a key role in the pathogenesis of transaortic constriction-induced hypertrophy. Hypertrophy was delayed/attenuated in mice with mutant, ouabain-resistant α2-Na+ pumps and in mice with cardiac-selective knockout or transgenic overexpression of α2-Na+ pumps. The latter, seemingly paradoxical, findings can be explained by comparing the numbers of available (ouabain-free) high-affinity (α2) ouabain-binding sites in wild-type, knockout, and transgenic hearts. Conversely, hypertrophy was accelerated in α2-ouabain-resistant (R) mice in which the normally ouabain-resistant α1-Na+ pumps were mutated to an ouabain-sensitive (S) form (α1S/Sα2R/R or “SWAP” vs. wild-type or α1R/R α2S/S mice). Furthermore, transaortic constriction-induced hypertrophy in SWAP mice was prevented/reversed by immunoneutralizing circulating endogenous ouabain (EO). These findings show that EO and its receptor, ouabain-sensitive α2, are critical factors in pressure overload-induced cardiac hypertrophy. This complements reports linking elevated plasma EO to hypertension, cardiac hypertrophy, and failure in humans and elucidates the underappreciated role of the EO-Na+ pump pathway in cardiovascular disease.
Keywords: cardiac hypertrophy, genetic engineering, ouabain, sodium pump, transaortic constriction
left ventricular (LV) hypertrophy occurs in about 10-15% of the general adult population (87) and in ~40% of hypertensive patients (18) and is a predictor of adverse cardiovascular events, including heart failure and death, in hypertensive patients (1, 49, 65). It is therefore important to understand the mechanism(s) that cause hypertrophy. In poorly controlled hypertension, it is widely accepted that the “pressure overload” and consequent increase in cardiac workload required to expel blood from the heart initiates the almost inevitable cardiac hypertrophy and dysfunction (16, 45, 72, 113, 120). The myocardial stretch is believed to trigger the cardiac changes (35, 40, 110, 122). However, two landmark, but underappreciated, articles published in this journal, and a more recent report in Circulation Research, in which transaortic constriction (TAC), a common model, was used to induce pressure overload, have demonstrated that this view is not entirely correct. Notably, experiments on mice with genetically engineered cardiac α2-Na+ pumps can exhibit substantial TAC-induced elevation of LV systolic pressure (LVSP) but greatly attenuated cardiac hypertrophy and dysfunction (15, 82, 112). Those observations, which are summarized here, provide the basis for a new explanation for how pressure overload triggers the cardiac structural and functional changes that characterize hypertrophy (Figs. 1 and 2).
Fig. 1.
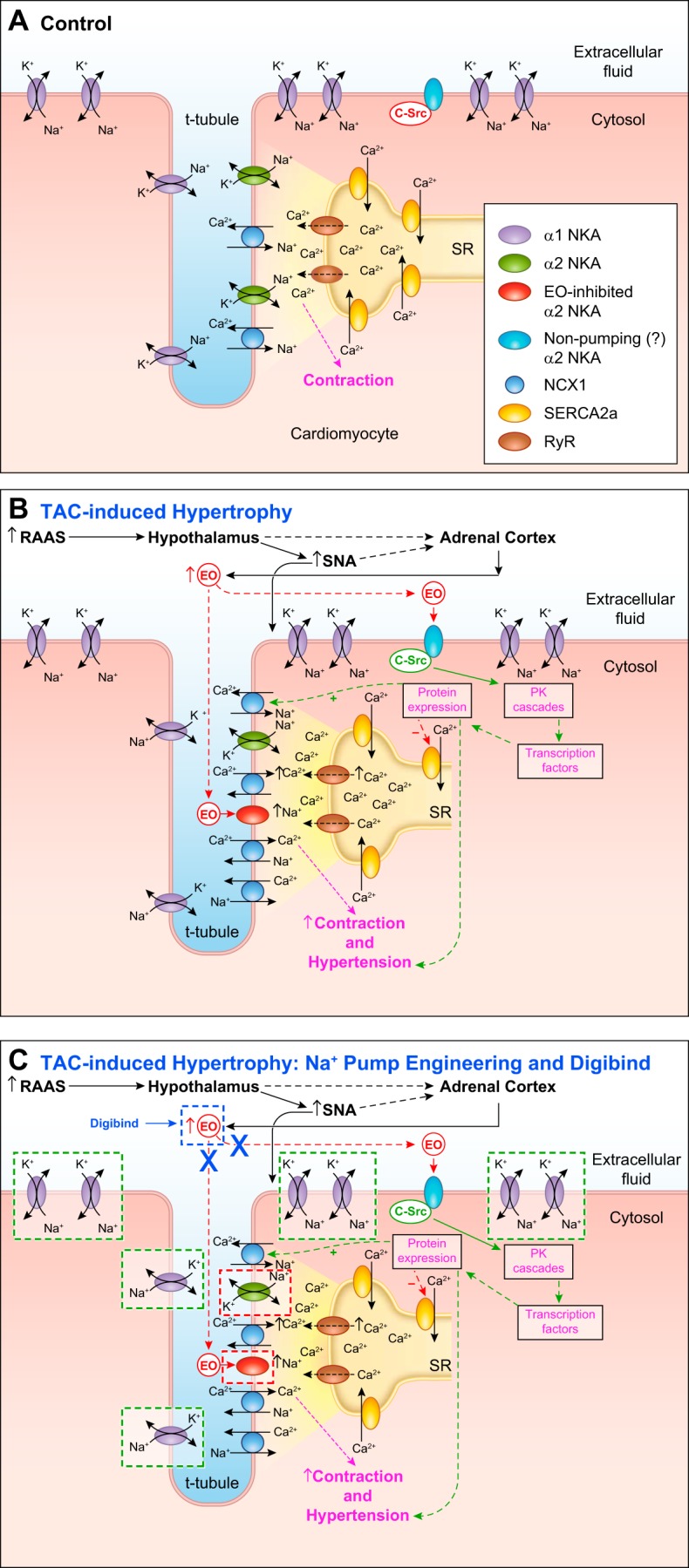
A: control conditions. Key transporters that link Na+ transport to Ca2+ signaling in cardiomyocytes; for simplicity, plasma membrane (PM) Ca2+ channels and Ca2+ pumps (PMCA), are omitted. Na+/Ca2+ exchanger 1 (NCX1), governed primarily by the α2-Na+ pump (NKA)-generated local Na+ electrochemical gradient at the PM-sarcoplasmic reticulum (SR) junction (the PLasmERosome; shaded area) helps regulate the local cytosolic Ca2+ concentration ([Ca2+]CYT) and SR Ca2+ stores ([Ca2+]SR). B: the hypothesized changes in these mechanisms during pressure overload [transaortic constriction (TAC)]-induced cardiac hypertrophy (see text and Ref. 10). Pressure overload (e.g., via TAC) activates the renin-angiotensin-aldosterone system (RAAS). This increases sympathetic nerve activity (SNA), which directly stimulates cardiomyocytes. RAAS activation also leads, via a hypothalamic-adrenocortical neurohumoral pathway, to elevation of plasma endogenous ouabain (EO), which selectively inhibits high-ouabain affinity α2-Na+ pumps, thereby enhancing NCX1-mediated net Ca2+ gain (see Fig. 2, left; acute effects of EO). This increases [Ca2+]SR, enhances Ca2+ signaling, and augments contraction (i.e., it increases cardiac workload). Sustained high plasma EO binding to (perhaps nontransporting) α2-Na+ pumps also activates protein kinase (PK) cascades that lead to cardiac hypertrophy (see Fig. 2, right; chronic effects of EO). C: as discussed in this review, the dashed-line boxes indicate where the mechanisms in B are influenced by genetic engineering of α1- and α2-Na+ pumps and by treatment with Digibind. In C, the two red-bordered boxes surrounding α2-Na+ pumps refer to the evidence that TAC-induced cardiac hypertrophy is attenuated or prevented by the following genetic alterations: mutation of α2 to an ouabain-resistant form (test 1), knockout of α2 (test 2), or overexpression of α2-Na+ pumps (test 3). The five green-bordered boxes surrounding α1-Na+ pumps refer to the evidence that hypertrophy is unaffected by overexpression of (normally) ouabain-resistant α1-Na+ pumps (test 4) but is augmented by mutation of α1 to an ouabain-sensitive form in α2-ouabain-resistant mice (i.e., α1S/Sα2R/R or “SWAP” mice) (test 5). The single blue-bordered “Digibind” box surrounding EO refers to the evidence that, in SWAP mice, hypertrophy is prevented or reversed by immunoneutralization of ouabain with Digibind (test 6). See text for further details. RyR, ryanodine receptor.
Fig. 2.
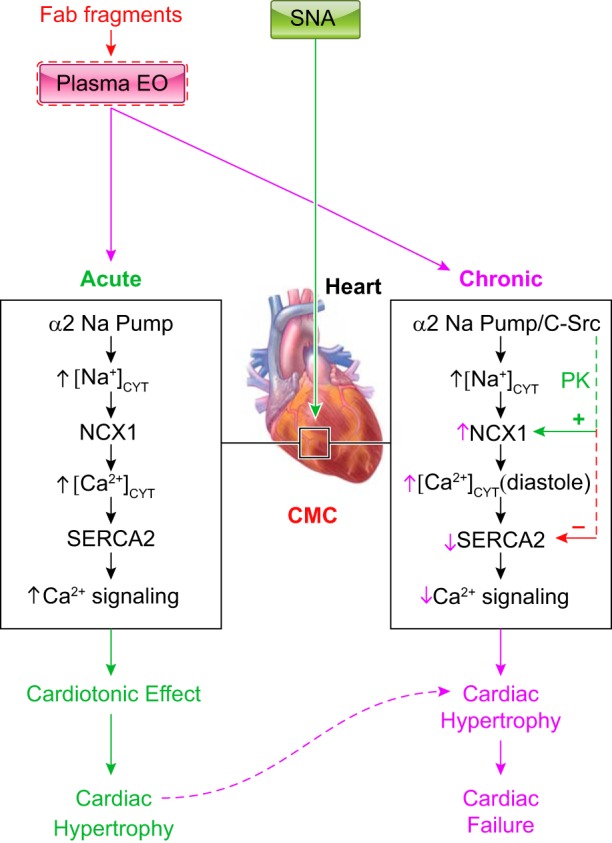
Cellular mechanisms that mediate the acute (left) and chronic (right) effects of ouabain/endogenous ouabain (EO) in the heart. Regulation of the heart by sympathetic nerve activity (SNA) is also shown. As described in the text, in rodents, low-dose EO binds exclusively to, and inhibits, α2-Na+ pumps, which are located in cardiomyocyte (CMC) PLasmERosomes (see Fig. 1A). The rise in local intracellular Na+ in the PLasmERosomes ([Na+]PL) rapidly induces Na+/Ca2+ exchanger (NCX)-mediated Ca2+ gain and the cardiotonic effect (left). If this is sustained, it leads to cardiac hypertrophy. EO can be immunoneutralized by commercial Fab fragments raised against digoxin (Digibind or DigiFab). Prolonged plasma EO elevation also activates a α2-Na+ pump-associated protein kinase (PK) cascade, e.g., c-Src mediated, that increases cardiac NCX expression (right; green dotted line). This promotes NCX-mediated Ca2+ extrusion during diastole. Prolonged α2-pump inhibition by EO, however, reduces the Na+ gradient driving force so that “global” cytosolic Na+ concentration ([Na+]CYT) ([Na+]CYT) [as well as [Na+]PL) and diastolic cytosolic Ca2+ concentration ([Ca2+]CYT)] are elevated; consequently, cardiac relaxation is slowed and/or incomplete. Also, cardiac SERCA2 expression is usually reduced in heart failure (perhaps due to the high EO; red dotted line), as are sarcoplasmic reticulum (SR) Ca2+ stores and Ca2+ transients, and systolic function is impaired. The diastolic dysfunction and attenuated cardiac contraction and stroke volume help explain the heart failure. [Modified from Ref. 10.]
Ouabain-Sensitive Na+ Pumps in the Heart
Evidence that cardiac α2-Na+ pumps (or Na+-K+-ATPase), their high-affinity ouabain-binding site, and its ligand endogenous ouabain (EO) are pivotal elements in the pathogenesis of cardiac hypertrophy and failure was recently reviewed (10, 36). The data demonstrate that hypothalamic mechanisms help regulate cardiovascular function not only via central modulation of sympathetic nerve activity but also via a novel neurohumoral pathway mediated by circulating EO and its cardiac and vascular receptors, the high-ouabain affinity Na+ pumps (10, 38).
Na+ pumps consist of an α-subunit and a β-subunit. The glycosylated β-subunit is required for catalytic activity, but the much larger α-subunit, of which there are four isoforms (α1−α4), contains the catalytic machinery and ouabain [cardiotonic steroid (CTS)]-binding site and mediates Na+ and K+ transport (8, 47, 104, 119). The Na+ pump subunit structure is shown in Fig. 3. Ouabain binds with its lactone ring deep in the cation transport pathway and its sugar moiety (rhamnose) in the wide external vestibule exposed to the extracellular fluid (47). Cardiac Na+ pumps are regulated by another small polypeptide, phospholemman (PLM), a member of the FXYD family, sometimes called the γ-subunit (7, 13, 55, 81). Cloning of the Na+ pump α- and β-subunit isoforms by Shull et al. (93–95) enabled subsequent mutation of the ouabain-binding site in the α1- and α2-isoforms and knockout (KO) or overexpression of individual isoforms in specific tissues. This provided an opportunity to examine the Na+ pump-endogenous CTS endocrine system and the different functions of these two α-isoforms.
Fig. 3.
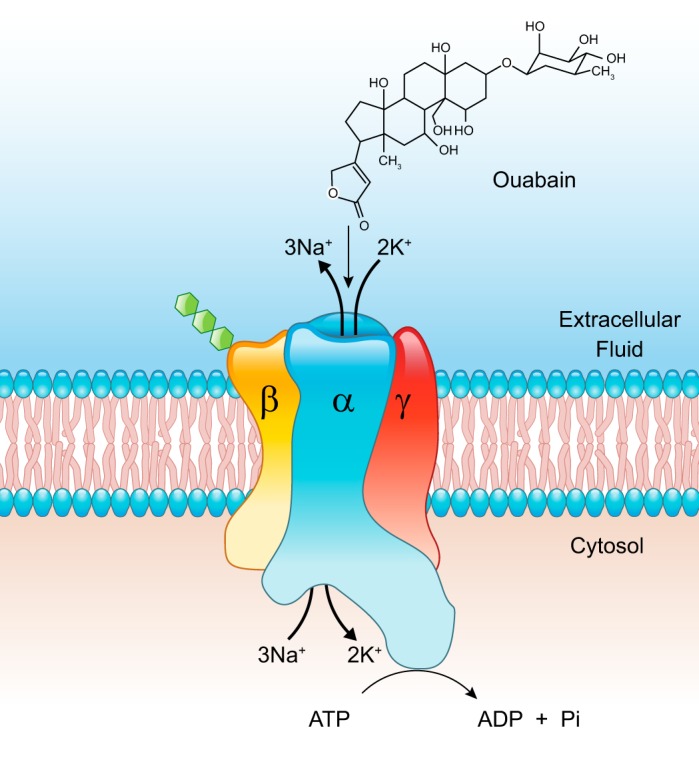
Model of a Na+ pump depicts the organization of the α- and-β subunits; as illustrated, the small β-subunit is glycosylated (hexoses shown in green). Also shown is the regulatory γ-subunit, phospholemman (PLM). Ouabain (at top) enters the cation transport pathway lactone ring first and binds close to one of the cation-binding sites in the pathway.
Adult rodent and human cardiomyocytes [and arterial myocytes (54)] express Na+ pumps with an α1-isoform (≈80–95% of pumps) and pumps with an α2-isoform (≈5–20% of pumps) (6, 23, 41, 115), but human hearts also express a very small number of α3-pumps (63, 91). Notably, α1 is widely distributed in the plasma membrane (PM) and is the “housekeeper” that maintains the low global cytosolic Na+ concentration ([Na+]CYT) (6, 62). The α1-to-α2 ratio (≈5–20:1) is similar in rodents and humans, but rodent α1-Na+ pumps are unusually resistant (R) to ouabain (8, 32, 54, 70), i.e., the wild-type (WT) mouse genotype is α1R/Rα2S/S (where S indicates ouabain sensitive).
In contrast to α1-pumps, cardiomyocyte α2-pumps are largely confined to PM microdomains at PM-sarcoplasmic reticulum (SR) junctions, often in t tubules (Fig. 1A); however, some α1-pumps also are located at PM-SR junctions (6, 66, 90). Because α2, but not α1, has a high affinity for ouabain in rodents, it is likely that low EO concentrations preferentially elevate the local Na+ concentration at these PM-SR junctions ([Na+]PL) (19, 107). Moreover, type 1 Na+/Ca2+ exchangers (NCX1) colocalize with α2-pumps, close to SR Ca2+ pumps (SERCA) at cardiac PM-SR junctions (66, 90, 109), in Ca2+ signaling units or “PLasmERosomes” (Fig. 1A) (12, 42). This enables “privileged communication” among the α2-Na+ pumps, NCX1 and SERCA. Thus, the elevated [Na+]PL in the tiny, diffusion-retarded volume of cytosol between the PM and “junctional SR” in the PLasmERosome (114)1 reduces NCX1-mediated Ca2+ extrusion and fosters net Ca2+ gain by the cardiomyocyte (Figs. 1B and 2). This underlies the well-documented “cardiotonic effect” of ouabain and other CTSs such as digoxin (12, 19).
EO and Other CTSs and Their Physiological Effects
In 1953, Schatzmann (86) reported that a variety of CTSs selectively inhibit the Na+ pump, and Szent-Gyorgi (102) postulated that the natural ligand for the Na+ pump CTS receptor (i.e., an endogenous CTS) modulates cardiac contraction. Nearly 25 yr later, but before Na+ pump α-subunit isoforms were recognized, two groups proposed that an EO-like substance, a Na+ pump inhibitor, behaved as both a hypertensinogenic and natriutretic agent (9, 33). That led to the search for a mammalian CTS and to the discovery of EO, an adrenocortical hormone, in human plasma (37). Mammalian EO has been analytically verified in several independent laboratories (37, 43, 44, 61, 76, 89, 103). A recent update and analysis (38) suggested that flawed chromatographic separation of the unusually polar steroid EO in plasma samples may explain why a few investigators failed to find EO in human plasma (50).
A related CTS, marinobufagenin (MBG), a bufadienolide first identified in amphibia (4), has also been linked to hypertension (24, 25). This linkage is based primarily on immunological (versus analytic) identification and on immunoneutralization by Digibind and DigiFab [commercial anti-digoxin Fab fragments with much higher affinity for digoxin and ouabain than for MBG (79, 80)] (24). Also, MBG binds preferentially to α1-Na+ pumps (25, 111). This article focuses on α2 and EO.
To determine the significance of the high-affinity α2-ouabain-binding site and its ligand, Lingrel et al. (21) generated mice in which the binding site was mutated to a low-affinity, “ouabain-resistant” form (α1R/Rα2R/R or “α2R/R” mice). This mutation of just two amino acids in the ouabain-binding site does not affect Na+-K+-ATPase activity (23), pump-mediated cation transport, or baseline cardiac function (21, 112). Nevertheless, pregnant dams from this line apparently have unusually low blood pressure (BP) during the third trimester of pregnancy (73), which may indicate that the ouabain-binding site and its ligand have a physiological role in the cardiovascular system when the body is stressed. Furthermore, hypertension induced by infused adrenocorticotropic hormone or ouabain or dietary or centrally infused NaCl is prevented or greatly attenuated in α1R/Rα2R/R mice (20, 22, 48, 58, 105). Interestingly, α1R/Rα2R/R mice are also learning-impaired and exhibit reduced dopamine-mediated locomotion (85), which implies that EO and its α2-Na+ pump receptor play a physiological role in these behaviors. This is likely related to the fact that all astroglia and some neurons also express α2-Na+ pumps (64).
Pressure Overload-Induced Hypertrophy Depends on α2-Na+ Pump Ouabain Sensitivity
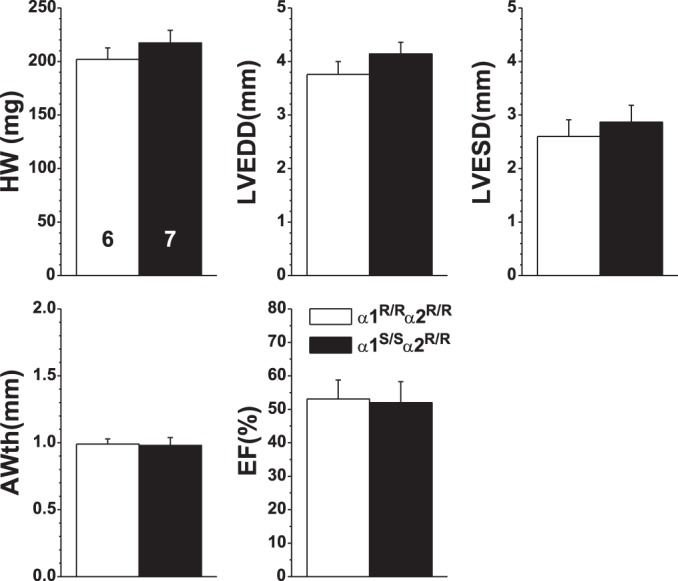
In a study of the role of the ouabain-binding site in the heart, Wansapura et al. (112) made the seminal observation that TAC-induced cardiac dysfunction, measured by echocardiography, was attenuated in α1R/Rα2R/R mice. WT (α1R/Rα2S/S) mice exhibited evidence of hypertrophy after 4 wk of TAC: LV end-diastolic and end-systolic diameters (LVEDD and LVESD, respectively) were decreased and anterior wall thickness (AWth) and ejection fraction (EF) were increased versus sham-operated mice (Fig. 4).2 In striking contrast, there was no alteration in any of these parameters, versus sham-operated mice, in ouabain-resistant α1R/Rα2R/R mice (Fig. 4 and see Fig. 1C, test 5), even though LVSP was significantly elevated by TAC in α1R/Rα2R/R mice (Table 1). This leads to the astonishing conclusion that counters current dogma: namely, that much of the cardiac dysfunction is not due directly to the pressure overload and cardiomyocyte stretch, per se, but to the interaction between α2-ouabain-binding sites and their endogenous ligand.
Fig. 4.

Ouabain resistance attenuates transaortic constriction (TAC)-induced cardiac hypertrophy. The effects of 4 wk of TAC (black bars) versus sham surgery (white bars) was compared in wild type (WT; α1R/Rα2S/S), ouabain-resistant (α1R/Rα2R/R), and SWAP (α1S/Sα2R/R) mice. Left ventricular (LV) structure and function were measured by echocardiography. Results (shown as means ± SE; n values on LVEDD bars apply to all graphs) were analyzed by two-way ANOVA; groups were compared post hoc using Tukey’s test. LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; AWth, anterior wall thickness; EF, ejection fraction. *P < 0.05 vs. the corresponding sham; †P <0.05 vs. TAC WT and TAC α1R/Rα2R/R. [Reproduced from Ref. 112 with permission.]
Table 1.
Left ventricular systolic pressure in 4 wk post-TAC and sham-operated wild-type (α1R/Rα2S/S), ouabain-resistant α2-Na+ pump (α1R/Rα2R/R), and SWAP (α1S/Sα2R/R) mice
| Wild Type | α1R/Rα2R/R | SWAP | |
|---|---|---|---|
| Sham operation | 96 ± 3 (5) | 104 ± 3 (6) | 101 ± 3 (5) |
| TAC | 162 ± 9*† (5) | 133 ± 7* (8) | 122 ± 12 (8) |
Data are means ± SE of left ventricular systolic pressure (in mmHg); values in parentheses are numbers of animals/group. TAC, transaortic constriction. Data were analyzed by two-way ANOVA, and groups were compared post hoc using Tukey’s test.
P < 0.05 vs. the corresponding sham group;
P < 0.05 vs. the TAC value in the other two groups. [Data from Ref. 112 are presented with permission.]
The effects of TAC on α2R/R mice, in which the α1-isoform was mutated to make it ouabain sensitive (α1S/Sα2R/R or “SWAP” mice), were also determined (112). In sham-operated mice, LVSP was low: only 96-104 mmHg in all three strains (Table 1), and there were no differences in baseline cardiac function. After 4 wk of TAC, LVSP was not as high in SWAP mice (122 mmHg) as in the other two strains (Table 1). Nevertheless, TAC increased LVEDD and LVESD much more in SWAP mice than in the other two strains, and EF was significantly reduced in TAC versus sham-operated mice (Fig. 4 and see Fig. 1C, test 6), i.e., SWAP mice with TAC exhibited evidence of heart failure (Fig. 2). Thus, SWAP mice were even more prone to TAC-induced cardiac dysfunction than were WT mice, but why?
The Number of High-Affinity Ouabain-Binding Sites Is Apparently Crucial
There are ~1 million α1-Na+ pumps and ~100,000 α2-pumps in a neonatal rat (and, we assume, mouse) cardiomyocyte (115). Let us suppose that, during TAC, a circulating endogenous ligand is elevated to a level sufficient to block two-thirds of ouabain-sensitive pumps. In WT (α1R/Rα2S/S) mice, 67,000 pumps will be inhibited, but the other 1,037,000 pumps (94%) will still function. The 67% reduction in functional α2-pumps in WT mice should elevate local [Na+]PL but not α1-controlled global [Na+]CYT (which may, however, partially buffer the rise in [Na+]PL) and thereby promote NCX1-mediated Ca2+ retention and enhanced Ca2+ signaling.
In contrast, in TAC-SWAP mice, 670,000 (α1) pumps will be blocked and only 430,000 (α1 + α2) pumps (39%) will be functional, and global [Na+]CYT would be expected to rise. Because PLasmERosomes are encircled by this cytosol and are, presumably, not completely impervious to Na+, Na+ will diffuse in and raise [Na+]PL. In other words, ouabain-induced, NCX1-mediated Ca2+ gain and augmented cardiac contraction in SWAP mice should be regulated by ouabain-sensitive α1-Na+ pumps when α2-pumps are ouabain resistant (19, 111). In addition, of course, no pumps will be inhibited in α1R/Rα2R/R mice, and there will be negligible “buffering” (see above) in the case of α1S/Sα2-pumps, as in humans. This raises the possibility that susceptibility to cardiac dysfunction during pressure overload is related to the number of cardiac ouabain-sensitive Na+ pumps that are inhibited by the endogenous ligand. [As discussed below, however, ouabain-induced activation of protein kinases may also be involved.]
Immunoneutralization of EO Attenuates Hypertrophy
Wansapura et al. (112) also performed another test of the role of the ouabain-binding site and its endogenous ligand: they administered Digibind to immunoneutralize ouabain (79, 80) in TAC and sham-operated SWAP mice 2 wk after the surgery. Two weeks later, Digibind-treated TAC-SWAP mice showed no alterations versus sham surgery (Fig. 4) or TAC-α1R/Rα2R/R mice in heart weight, LVEDD, LVESD, AWth, or EF (Fig. 5). In other words, the TAC-induced cardiac dysfunction was prevented or reversed by Digibind (see Figs. 1C, test 6, and 2).
Fig. 5.
Digibind, which immunoneutralizes ouabain, attenuates transaortic constriction (TAC)-induced cardiac hypertrophy in SWAP mice. Ouabain-resistant (α1R/Rα2R/R; n = 6) mice and SWAP (α1S/Sα2R/R; n = 7) mice were all infused with Digibind (50 μg/day) by minipump via a jugular vein catheter during the final 2 wk of TAC. The echocardiographic data (means ± SE) after 4 wk of TAC are shown. In contrast to results in the absence of Digibind (Fig. 2), there were negligible differences between the two genotypes. HW, heart weight; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; AWth, anterior wall thickness; EF, ejection fraction. [Reproduced from Ref. 112 with permission.]
Fig. 6.

A: cardio-specific knockout of α2-Na+ pumps (α2-KO) attenuates the increase in the heart weight-to-body weight ratio (HW/BW) after 9 wk of transaortic constriction (TAC). Sham-operated mice (0 wk) are indicated by the white bars and TAC mice by the black bars. The data, (reproduced from Ref. 82 with permission) are shown as means ± SE; numbers of mice are in parentheses. Note that the data from TAC weeks 3 and 6, presented in the original report, are omitted because each group contained only 2−3 mice. These data were included in the original statistical analysis by two-way ANOVA; they may have contributed to the failure to find a significant difference between wild-type (WT) and α2-KO mice after 9 wk of TAC (a type II error). B–D: cardio-specific α2-KO also delayed the development of TAC-induced cardiac hypertrophy [end-systolic volume (ESV; B); end-diastolic volume (EDV; C); and ejection fraction (EF; D)]. The echocardiographic results in WT and α2-KO mice after 3, 6, and 9 wk of TAC (means ± SE; n = 9 of each genotype) were analyzed by two-way ANOVA. Post hoc comparisons indicated that ESV was significantly lower (P < 0.05) in α2-KO than in WT mouse hearts at 3 and 6 wk and in ESV at 3 wk; EF was significantly greater in α2-KO than WT mice at 3 and 6 wk. [Reproduced from Ref. 82 with permission.]
These data of Wansapura et al. (112) are compelling evidence that the α2-ouabain-binding site and its endogenous ligand play a crucial role in the mechanism(s) that directly trigger the hypertrophic changes and the cardiac dysfunction during pressure overload. Indeed, shortly after the discovery of EO, elevated plasma levels were reported in patients in congestive heart failure (31). In those patients, the EO level was inversely correlated with cardiac index, which raised the possibility that the functional impairment may be related to EO. Several subsequent cardiac hypertrophy and heart failure studies indicated that the highest EO levels were associated with the most severe cardiac dysfunction and the worst clinical outcomes (76, 96, 100). Furthermore, LV hypertrophy was directly correlated with plasma EO (100).
KO of Cardiac α2-Na+ Pumps Delays TAC-Induced Cardiac Hypertrophy
The role of α2-Na+ pumps in TAC-induced cardiac dysfunction was also assessed in mice with cardiac-selective KO of α2 (“cardio-α2-KO”) by Rindler et al. (82). Heart weight (normalized to body weight) and basal cardiac performance were indistinguishable from normal in cardio-α2-KO mice (Fig. 6). The implication is that, in unstressed cardio-α2-KO mice, α1-Na+ pump function is sufficient to regulate NCX1-mediated Ca2+ transport and [Na+]PL as well as [Na+]CYT because, despite retarded diffusion, PLasmERosomes are likely not completely impervious to Na+.
TAC induced the same pressure gradient across the constriction in WT and KO mice. Nine weeks after TAC surgery, however, the heart weight-to-body weight ratio was ≈50% greater in WT-TAC mice than in KO-TAC mice (Fig. 6A). Also, at 3 and 6 wk post-TAC surgery, cardiac function was less impaired in KO mice than in WT mice, as indicated by significantly smaller LV end-systolic and end-diastolic volumes and significantly greater EF (Fig. 6, B–D). Although functional impairment in KO mouse hearts was similar to that in WT hearts by 9 wk postsurgery (Fig. 6, B–D) despite the heart weight/body weight difference, the data demonstrate that KO of cardiac α2-Na+ pumps delays the development of pressure overload-induced dysfunction (see Fig. 1C, test 2). This confirms the key role of cardiac ouabain-sensitive α2-Na+ pumps in the genesis of the structural and functional impairment.
TAC, the Renin-Angiotensin-Aldosterone System, and EO
If the endogenous α2-ligand EO plays a role in the pathogenesis of TAC-induced cardiac hypertrophy, as the α1R/Rα2R/R mouse and Digibind treatment data indicate, we need to explore the role of EO more carefully. As noted, human studies have indicated a direct correlation between plasma EO and LV hypertrophy (100). However, does elevated plasma EO actually contribute to cardiac hypertrophy? In addition, if so, how? Also, why should plasma EO be elevated in mice with TAC?
Much evidence indicates that the renin-angiotensin-aldosterone system is activated in suprarenal aortic coarctation, presumably because renal perfusion pressure and blood flow is reduced (28, 67, 74). In addition to the direct effects of ANG II and aldosterone on the heart (3, 14, 88, 92), these agents act on the brain to increase sympathetic drive to the cardiovascular system (116, 121, 123). Indeed, renin-angiotensin-aldosterone system antagonists and sympatholytic agents at least partially reduce BP elevation and cardiac damage in aortic coarctation (51, 69, 83). Recent observations have demonstrated that ANG II infusion, either into the cerebral ventricles at very low concentration (38) or subcutaneously (11), also activates a novel neurohumoral pathway in the hypothalamus that, likely via the adrenal cortex, elevates plasma EO. Specific blockade of this pathway prevents the ANG II-triggered elevation of plasma EO (38). Thus, although not yet determined (a critical shortcoming), we postulate that plasma EO is elevated during TAC. This view is supported by the observation that immunoneutralization of the endogenous ligand EO with Digibind (Fig. 5) attenuates the cardiac pathology (112).
EO Helps Trigger Cardiac Hypertrophy
The next question is: how does EO affect cardiac function? One way is as a classic, selective Na+ pump inhibitor: it raises [Na+]PL (10, 12). This increases the force of contraction [the “positive inotropic response” (10)] and thereby increases the cardiac workload (Figs. 1B and 2, left). A caveat is that some α1-Na+ pumps also colocalize with NCX1 (23, 66) and, especially in SWAP mice and α2-KO mice, can regulate cardiac contraction (23, 82). Nevertheless, under normal circumstances, α2, because of its localization and ouabain sensitivity, preferentially modulates Ca2+ signaling in rodents (19). In humans, the fact that α1, too, is ouabain sensitive suggests that α1-pumps might also mediate the effects of EO and modulate Ca2+ signaling, as observed in SWAP mice (with ouabain-resistant α2) (23). Comparison of WT (i.e., α2-sensitive) and SWAP (i.e., α1-sensitive) mouse cardiomyocyte responses to low-dose ouabain indicated, however, that α2-Na+ pumps preferentially regulates SR Ca2+ release, Ca2+ transients, and cardiac inotropy (19). Nevertheless, the accelerated hypertrophy (and failure) in TAC-SWAP mice (112) (Fig. 4) suggests that α1-pumps likely also contribute significantly to the pathology when plasma EO is elevated in humans.
A second possible mechanism by which EO affects cardiac structure and function relates to the fact that ouabain triggers cardiomyocyte Na+ pump-dependent, but cation transport-independent, protein kinase cascade-mediated hypertrophic signaling (Figs. 1B and 2, right) (26, 56, 57, 118). Furthermore, prolonged (48-72 h) exposure of cultured rat cardiomyocytes to 50 nM to 100 μM ouabain increases NCX1 protein expression (11, 68, 106), a cellular modification often associated with the progression to heart failure (71, 84, 101). Nanomolar ouabain exerts similar effects on protein kinase signaling and NCX1 expression in human (53) and rodent (78, 124) vascular myocytes. Some authors have suggested that low-ouabain affinity α1-Na+ pumps in rodents trigger the cardiomyocyte signaling cascade (17, 56, 57, 118). Rodent α1-pumps, however, do not respond to the low nanomolar ouabain/EO concentrations (21, 98) that are expected in the circulation of animals stimulated with ANG II (38). The implication is that this low concentration ouabain/EO-activated signaling is mediated by cardiac α2-Na+ pumps, although this needs to be tested directly (e.g., in WT vs. α2R/R-cardiomyocytes).
It seems likely that both of the EO-dependent, α2-mediated mechanisms, the cardiotonic effect and the protein kinase cascade activation, are involved in the generation of cardiac hypertrophy. The cardiotonic effect, as well as the stimulated protein kinase-mediated signaling and “reprogramming” of Ca2+ transporter expression, should, early on, enhance cardiac work and promote hypertrophy but, if sustained, should progress to heart failure (Fig. 2) (10).
The Paradox of Cardiac α2 KO Versus α2 Overexpression
One might anticipate that, if TAC-induced cardiac dysfunction is delayed and/or attenuated in cardio-α2-KO mice, cardiac-specific overexpression of α2-Na+ pumps in transgenic (TG) mice should have the opposite effect: it should sensitize the heart to pressure overload. This is not the case, however, as TAC-induced cardiac hypertrophy was also attenuated in cardio-TG(α2) but not cardio-TG(α1) mice, i.e., the attenuating effect is α2 specific (15) (see Fig. 1C, tests 3 and 4). As was true of cardio-α2-KO mice, baseline cardiac function was normal in cardio-TG(α2) mice. However, the expected, progressive TAC-induced increase in the ventricular weight-to-body weight ratio was attenuated in TG(α2) but not TG(α1) mice after 10−12 wk of TAC (Fig. 7, A and B). Several other measurements also indicate that TG(α2) but not TG(α1) mouse hearts were at least partially protected from TAC-induced hypertrophy. For example, the expected TAC-induced increases in lung weight (Fig. 7C), a measure of impending heart failure (52), and cardiomyocyte cross-sectional area (Fig. 7, D and E) were markedly diminished in TG(α2) mice. Also, the increased expression of hypertrophic markers (e.g., atrial natriuretic factor and brain natriuretic peptide) and decreased myocyte fractional shortening (Fig. 7F) were attenuated in TG(α2) hearts (15).
Fig. 7.

Cardio-specific overexpression of α2- but not α1-Na+ pumps attenuates transaortic constriction (TAC)-induced cardiac hypertrophy: echocardiographic and histological evidence. Transgenic α2 [TG(α2)] mice exhibited less cardiac hypertrophy than wild-type (Wt) mice or transgenic α1 [TG(α1)] mice after 10-12 wk (10w or 12w) of TAC. Sham-operated mice served as controls. This was exemplified by the left ventricular weight-to-body weight ratio (VB/BW; A and B), the lung weight-to-body weight ratio (LW/BW; C), cardiomyocyte cross-sectional area (D and E), and fractional cardiomyocyte shortening (FS; F). The bar graphs (reproduced from Ref. 15 with permission) are presented as means ± SE; n values are shown on the bars. Statistical analysis of the data was performed using unpaired two-tailed t-tests with Prism 5 (Graphpad Software). *P <0.05 vs. sham; #P <0.05 vs. WT TAC.
At baseline, the amplitudes of the Ca2+ transients and SR Ca2+ load were similarly decreased (vs. WT) in TG(α1) and TG(α2) mouse hearts (15). NCX1-mediated Ca2+ extrusion, however, was significantly more rapid in TG(α2) hearts than in hearts of WT or TG(α1) mice (Fig. 8A). This might indicate that NCX1 is functionally more closely coupled to α2 than α1 (16). Interestingly, cardiomyocyte [Na+]CYT was ~33% lower in α1-TG hearts than in WT or TG(α2) hearts both at rest and during 2-Hz stimulation (Fig. 8, B and C, respectively).3 This is consistent with other evidence showing that the more abundant α1-Na+ pumps are the “housekeepers” that normally maintain the low global [Na+]CYT (30, 62), even in cardio-TG(α2) mice, which overexpress α2 by approximately threefold (15), because α2 is still a minority (perhaps 25%) of total Na+ pumps (115). Thus, it is not surprising that overexpression of the low-abundance α2-Na+ pumps had minimal effect on global [Na+]CYT. In astrocytes, too, which have a similar α1-to-α2 ratio, reduced α2-expression had little effect on global [Na+]CYT but a large effect on Ca2+ signaling (30). Also, colocalization of cardiac α2-pumps with NCX1 at PM-SR junctions helps drive NCX1-mediated Ca2+ transport (10). Furthermore, phosphorylated and total PLM are markedly downregulated in TG(α2) but not TG(α1) mouse hearts both at baseline and after TAC (15). Phosphorylated PLM negatively regulates both α2 (39, 75) and NCX (34, 108). Therefore, downregulation of PLM expression and phosphorylated PLM should increase the ability of α2 to extrude Na+ at PM-SR junctions and increase the trans-PM Na+ electrochemical gradient at the junctions. Because NCX1 is functionally coupled to α2-Na+ pumps (10), the lower local [Na+]CYT and steeper Na+ gradient would explain the enhanced efficiency of NCX1-mediated Ca2+ extrusion observed in TG(α2) mouse hearts (15).
Fig. 8.
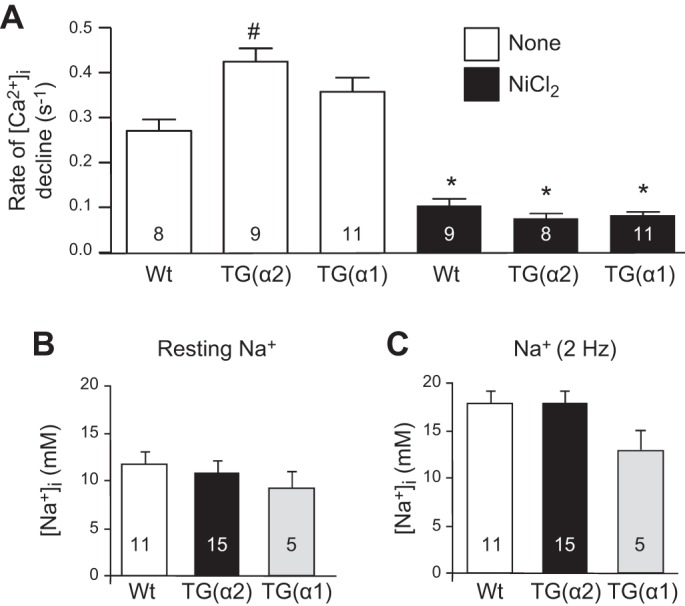
A: Na+/Ca2+ exchanger (NCX)-dependent rate of intracellular Ca2+ concentration ([Ca2+]i) decline after caffeine-induced unloading of the sarcoplasmic reticulum was faster in transgenic α2 [TG(α2)] than TG(α1) or wild-type (Wt) mouse cardiomyocytes. NCX was blocked by 10 mM NiCl2 (black bars), which lowered the rate of [Ca2+]i decline to about the same level in all three genotypes. *P < 0.05 vs. control (no nickel); #P < 0.05 vs. Wt mice with the same treatment. B and C: intracellular Na+ concentration ([Na+]i) was reduced by about 33% in TG(α1) but not TG(α2) versus sham cardiomyocytes, both at rest (B) and during 2-Hz stimulation (C). This reduction of [Na+]i did not reach statistical significance, likely because too few myocytes were studied (see footnote 3 in the text). [Data reproduced from Ref. 15 with permission.]
So what is the explanation for the paradox that both KO and overexpression of cardiac α2 result in a similar phenotype: delayed or attenuated development of pressure overload-induced cardiac hypertrophy? As summarized above, mutation of the (α2) high-affinity ouabain-binding site to an ouabain-resistant form or immunoneutralization of its ligand EO ameliorates hypertrophy (112). Therefore, it seems logical that, if hypertrophy depends on the number of ouabain-bound high-ouabain affinity pumps, KO of α2 (the EO receptors) should also ameliorate the hypertrophy (82) if NCX1-mediated Ca2+ transport is then regulated by ouabain-resistant α1-pumps. The α1-pumps can apparently prevent [Na+]PL from rising excessively, and they do not respond to elevated plasma EO. Conversely, making α1 ouabain sensitive (i.e., greatly increasing the number of high-ouabain affinity pumps: SWAP mice) should have an augmenting effect (112). However, to understand why α2 overexpression should also ameliorate the hypertrophy, consider how EO works: by binding to α2-Na+ pumps, EO blocks those transporters and should elevate [Na+]PL. With ≈100,000 ouabain-sensitive α2 and 1,000,000 ouabain-resistant α1 Na+ pumps per cardiomyocyte (115), and postulating, as above, that ≈67% of the α2 pumps are inhibited when plasma EO rises after TAC, ≈33,000 of the α2-pumps will still be functional. However, with a threefold overexpression of α2 (15) and thus a total of 300,000 α2-pumps, even if 67% are inhibited, ≈100,000 ouabain-sensitive α2-Na+ pumps will remain functional. This is the normal complement and is apparently sufficient to sustain ordinary functions including maintaining [Na+]PL at the normal level, thereby minimizing TAC-induced cardiac hypertrophy and remodeling in TG(α2) mice.
TG(α1) Mice Support the Concept of a Restricted Cytosolic Compartment at PM-SR Junctions
Much of the preceding discussion examines the evidence that the binding of ouabain and inhibition of cardiac α2-Na+ pumps play a key role in the development of cardiac hypertrophy. The SWAP mouse data suggest that when α1 is mutated to an ouabain-sensitive form, it, too, can mediate these pathophysiological processes. As noted above, two- to threefold or more (estimated from immunoblots; quantitation was not given) overexpression of ouabain-resistant cardiac α1 apparently lowers [Na+]CYT more than does α2 overexpression (Fig. 8, B and C) but does not ameliorate TAC-induced hypertrophy (15). This fits the view (10) that α2-Na+ pumps and NCX1 have “privileged access” to tiny, diffusion-restricted cytosolic compartments at PM-SR junctions, even though there is no direct evidence (but see Ref. 77) that Na+ diffusion is restricted. Additionally, possible differences between ouabain-triggered α1- and α2-mediated protein kinase cascade signaling may be a factor. If, as inferred above, however, EO is elevated by TAC, the overexpressed ouabain-resistant α1 should not be responsive to EO, in contrast to the situation when α1 is mutated to an ouabain-sensitive form (112).
Summary and Conclusions
The three key reports reviewed here (15, 82, 112) provide a total of six different experimental manipulations related to Na+ pumps and ouabain binding in cardiomyocytes (see Fig. 1C): 1) mutation of the α2-Na+ pump high-affinity ouabain-binding site to an ouabain-resistant form (Fig. 1C, test 1); 2) KO of cardiac high-ouabain affinity α2-Na+ pumps (Fig. 1C, test 2); 3) immunoneutralization of circulating EO (Fig. 1C, test 6); 4) overexpression of cardiac myocyte high-ouabain affinity α2-Na+ pumps (Fig. 1C, test 3); 5) overexpression of cardiac myocyte low-ouabain affinity α1-Na+ pumps (Fig. 1C, test 4); and 6) mutation of the normally low-affinity α1-Na+ pump ouabain-binding site to a high-affinity site in α2-ouabain-resistant mice (“ SWAP” mice; Fig. 1C, test 5). Tests 1–3 and 6 attenuate or delay TAC-induced structural and functional changes that constitute cardiac hypertrophy. While overexpression of ouabain-resistant α1-Na+ pumps (test 4) has no effect, simply making α1-ouabain-sensitive in α2R/R mice (test 5) accelerates the hypertrophy. Taken together, these findings provide compelling evidence that EO and cardiac high-ouabain affinity α2-Na+ pumps play a fundamental role in the pathogenesis of pressure overload-induced cardiac hypertrophy in rodents. Nevertheless, the SWAP mouse results raise the possibility that, in humans, ouabain-sensitive α1 may also be involved.
These data also suggest that blockade of the EO-binding site on α2-Na+ pumps may be a novel strategy to ameliorate the hypertrophy and eventual heart failure. Indeed, PST2238 (rostafuroxin), a digoxigenin derivative that preferentially blocks ouabain binding but not pump-mediated cation transport (27, 97), appears to be an effective antihypertensive agent in rodents (26) and selected humans (46) with high EO and elevated BP. The use of rostafuroxin in humans may be limited (99), however, because of its relatively low affinity for the Na+ pump ouabain-binding site (97). Thus, there is need for a more effective EO antagonist that might be useful for attenuating hypertension-induced cardiac hypertrophy. Recent evidence indicates that very low (quarter) doses of multiple antihypertensive agents may have therapeutic benefit with reduced side effects (5). In this context, it may be worth considering the use of very low-dose digoxin, an ouabain antagonist (60, 97), as adjunct therapy in hypertensives with cardiac hypertrophy.
In summary, the three studies that are the focus of this review (15, 82, 112) complement and extend the numerous reports that have linked elevated plasma EO to hypertension as well as cardiac hypertrophy and failure in humans (summarized in Refs. 10 and 36). They elucidate the underappreciated role of the EO-α2-Na+ pump endocrine pathway in cardiovascular physiology and pathophysiology.
GRANTS
This work was supported in part by American Heart Association Grant 15GRNT24940022 and National Heart, Lung, and Blood Institute Grant HL-107555 and funds from the University of Maryland Foundation (Hypertension Center Account).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
AUTHOR CONTRIBUTIONS
M.P.B. conceived and designed the work, prepared and revised the manuscript and figures, and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
I thank Donald M. Bers (University of California-Davis), Sanda Despa (University of Kentucky), and Jerry B. Lingrel, John N. Lorenz, and Jeffrey D. Molkentin (all University of Cincinnati) for very helpful discussions and for permission to reproduce some of their data.
Footnotes
A shortcoming is that such local Na+ concentration “hotspots” are still controversial. For example, two recent studies (29, 59) of electrogenic Na+ pump currents concluded that there are no restricted sub-PM Na+ spaces in cardiomyocytes. Those studies, however, almost certainly refer to the prevalent α1-Na+ pumps and “bulk” cytosol and not to the sparse α2-pumps and tiny spaces between the PM and junctional SR, where diffusion is largely limited to two directions between the closely apposed PM and SR membranes.
Despite the small standard errors, only LVESD was significantly different from sham-operated mice in WT mice (P < 0.05) because of the small sample size (sham: n = 3 and TAC: n = 5) and the rigorous statistical test (two-way ANOVA). The latter may lead to rejection of a valid hypothesis (i.e., a type II error). In larger studies (e.g., Refs. 2 and 117), significant cardiac hypertrophy has been reported after just 2−4 wk of TAC.
The study was underpowered because only five α1(TG) myocytes were tested (Fig. 8). Statistical analysis indicated that these large declines in [Na+]CYT in TG(α1) mouse cardiomyocytes were not significant, and the authors concluded that α1 “overexpression does not alter Na+ content” (15). This may, however, represent a type II error; a power analysis by one of the authors (personal communication, S. Despa) indicated that 20 myocytes would have been needed in each group to demonstrate significance.
REFERENCES
- 1.Antikainen RL, Peters R, Beckett NS, Fagard RH, Wang JG, Rajkumar C, Bulpitt CJ. Left ventricular hypertrophy is a predictor of cardiovascular events in elderly hypertensive patients: hypertension in the Very Elderly Trial. J Hypertens 34: 2280–2286, 2016. doi: 10.1097/HJH.0000000000001073. [DOI] [PubMed] [Google Scholar]
- 2.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1α. Proc Natl Acad Sci USA 103: 10086–10091, 2006. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Araujo CM, Hermidorff MM, Amancio GC, Lemos DS, Silva ME, de Assis LV, Isoldi MC. Rapid effects of aldosterone in primary cultures of cardiomyocytes–do they suggest the existence of a membrane-bound receptor? J Recept Signal Transduct Res 36: 435–444, 2016. doi: 10.3109/10799893.2015.1122042. [DOI] [PubMed] [Google Scholar]
- 4.Barbier M, Shroter H, Meyer K, Schindler O, Reichstein T. Die Bufogenine des Paratoidensekrets von Bufo marinus (L.) Schneider. Helv Chim Acta 42: 2486–2506, 1959. doi: 10.1002/hlca.19590420720. [DOI] [Google Scholar]
- 5.Bennett A, Chow CK, Chou M, Dehbi HM, Webster R, Salam A, Patel A, Neal B, Peiris D, Thakkar J, Chalmers J, Nelson M, Reid C, Hillis GS, Woodward M, Hilmer S, Usherwood T, Thom S, Rodgers A. Efficacy and safety of quarter-dose blood pressure-lowering agents: a systematic review and meta-analysis of randomized controlled trials. Hypertension 70: 85–93, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09202. [DOI] [PubMed] [Google Scholar]
- 6.Berry RG, Despa S, Fuller W, Bers DM, Shattock MJ. Differential distribution and regulation of mouse cardiac Na+/K+-ATPase α1 and α2 subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc Res 73: 92–100, 2007. doi: 10.1016/j.cardiores.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Bibert S, Liu CC, Figtree GA, Garcia A, Hamilton EJ, Marassi FM, Sweadner KJ, Cornelius F, Geering K, Rasmussen HH. FXYD proteins reverse inhibition of the Na+-K+ pump mediated by glutathionylation of its β1 subunit. J Biol Chem 286: 18562–18572, 2011. doi: 10.1074/jbc.M110.184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol Renal Physiol 275: F633–F650, 1998. [DOI] [PubMed] [Google Scholar]
- 9.Blaustein MP. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol Cell Physiol 232: C165–C173, 1977. [DOI] [PubMed] [Google Scholar]
- 10.Blaustein MP, Chen L, Hamlyn JM, Leenen FH, Lingrel JB, Wier WG, Zhang J. Pivotal role of α2 Na+ pumps and their high affinity ouabain binding site in cardiovascular health and disease. J Physiol 594: 6079–6103, 2016. doi: 10.1113/JP272419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaustein MP, Chen L, Song H, Leenen FH, Hamlyn JM. How does the brain talk to the arteries and heart? (Abstract). FASEB J 29: 984.983, 2015. [Google Scholar]
- 12.Blaustein MP, Juhaszova M, Golovina VA. The cellular mechanism of action of cardiotonic steroids: a new hypothesis. Clin Exp Hypertens 20: 691–703, 1998. doi: 10.3109/10641969809053247. [DOI] [PubMed] [Google Scholar]
- 13.Bossuyt J, Despa S, Han F, Hou Z, Robia SL, Lingrel JB, Bers DM. Isoform specificity of the Na/K-ATPase association and regulation by phospholemman. J Biol Chem 284: 26749–26757, 2009. doi: 10.1074/jbc.M109.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cannavo A, Liccardo D, Eguchi A, Elliott KJ, Traynham CJ, Ibetti J, Eguchi S, Leosco D, Ferrara N, Rengo G, Koch WJ. Myocardial pathology induced by aldosterone is dependent on non-canonical activities of G protein-coupled receptor kinases. Nat Commun 7: 10877, 2016. doi: 10.1038/ncomms10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Correll RN, Eder P, Burr AR, Despa S, Davis J, Bers DM, Molkentin JD. Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ Res 114: 249–256, 2014. doi: 10.1161/CIRCRESAHA.114.302293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crozatier B, Hittinger L. Mechanical adaptation to chronic pressure overload. Eur Heart J 9, Suppl E: 7−11, 1988. [DOI] [PubMed] [Google Scholar]
- 17.Cui X, Xie Z. Protein interaction and Na/K-ATPase-mediated signal transduction. Molecules 2017: E990, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuspidi C, Sala C, Negri F, Mancia G, Morganti A; Italian Society of Hypertension . Prevalence of left-ventricular hypertrophy in hypertension: an updated review of echocardiographic studies. J Hum Hypertens 26: 343–349, 2012. doi: 10.1038/jhh.2011.104. [DOI] [PubMed] [Google Scholar]
- 19.Despa S, Lingrel JB, Bers DM. Na+/K+-ATPase α2-isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc Res 95: 480–486, 2012. doi: 10.1093/cvr/cvs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dostanic-Larson I, Van Huysse JW, Lorenz JN, Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci USA 102: 15845–15850, 2005. doi: 10.1073/pnas.0507358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dostanic I, Lorenz JN, Schultz JJ, Grupp IL, Neumann JC, Wani MA, Lingrel JB. The α2 isoform of Na,K-ATPase mediates ouabain-induced cardiac inotropy in mice. J Biol Chem 278: 53026–53034, 2003. doi: 10.1074/jbc.M308547200. [DOI] [PubMed] [Google Scholar]
- 22.Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW, Lingrel JB. The α2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol 288: H477–H485, 2005. doi: 10.1152/ajpheart.00083.2004. [DOI] [PubMed] [Google Scholar]
- 23.Dostanic I, Schultz JJ, Lorenz JN, Lingrel JB. The α1 isoform of Na,K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J Biol Chem 279: 54053–54061, 2004. doi: 10.1074/jbc.M410737200. [DOI] [PubMed] [Google Scholar]
- 24.Fedorova OV, Kolodkin NI, Agalakova NI, Lakatta EG, Bagrov AY. Marinobufagenin, an endogenous α-1 sodium pump ligand, in hypertensive Dahl salt-sensitive rats. Hypertension 37: 462–466, 2001. doi: 10.1161/01.HYP.37.2.462. [DOI] [PubMed] [Google Scholar]
- 25.Fedorova OV, Shapiro JI, Bagrov AY. Endogenous cardiotonic steroids and salt-sensitive hypertension. Biochim Biophys Acta 1802: 1230–1236, 2010. doi: 10.1016/j.bbadis.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrandi M, Molinari I, Barassi P, Minotti E, Bianchi G, Ferrari P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J Biol Chem 279: 33306–33314, 2004. doi: 10.1074/jbc.M402187200. [DOI] [PubMed] [Google Scholar]
- 27.Ferrari P, Torielli L, Ferrandi M, Padoani G, Duzzi L, Florio M, Conti F, Melloni P, Vesci L, Corsico N, Bianchi G. PST2238: a new antihypertensive compound that antagonizes the long-term pressor effect of ouabain. J Pharmacol Exp Ther 285: 83–94, 1998. [PubMed] [Google Scholar]
- 28.Freeman RH, Davis JO, Spielman WS. Renin-angiotensin system and aldosterone secretion during aortic constriction in the rat. Am J Physiol Renal Physiol 232: F434–F437, 1977. [DOI] [PubMed] [Google Scholar]
- 29.Garcia A, Liu CC, Cornelius F, Clarke RJ, Rasmussen HH. Glutathionylation-dependence of Na+-K+-pump currents can mimic reduced subsarcolemmal na(+) diffusion. Biophys J 110: 1099–1109, 2016. doi: 10.1016/j.bpj.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golovina VA, Song H, James PF, Lingrel JB, Blaustein MP. Na+ pump α2-subunit expression modulates Ca2+ signaling. Am J Physiol Cell Physiol 284: C475–C486, 2003. doi: 10.1152/ajpcell.00383.2002. [DOI] [PubMed] [Google Scholar]
- 31.Gottlieb SS, Rogowski AC, Weinberg M, Krichten CM, Hamilton BP, Hamlyn JM. Elevated concentrations of endogenous ouabain in patients with congestive heart failure. Circulation 86: 420–425, 1992. doi: 10.1161/01.CIR.86.2.420. [DOI] [PubMed] [Google Scholar]
- 32.Habeck M, Tokhtaeva E, Nadav Y, Ben Zeev E, Ferris SP, Kaufman RJ, Bab-Dinitz E, Kaplan JH, Dada LA, Farfel Z, Tal DM, Katz A, Sachs G, Vagin O, Karlish SJ. Selective assembly of Na,K-ATPase α2β2 heterodimers in the heart: Distinct functional properties and isoform-elective inhibitors. J Biol Chem 291: 23159–23174, 2016. doi: 10.1074/jbc.M116.751735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haddy FJ, Overbeck HW. The role of humoral agents in volume expanded hypertension. Life Sci 19: 935–947, 1976. doi: 10.1016/0024-3205(76)90284-8. [DOI] [PubMed] [Google Scholar]
- 34.Hafver TL, Hodne K, Wanichawan P, Aronsen JM, Dalhus B, Lunde PK, Lunde M, Martinsen M, Enger UH, Fuller W, Sjaastad I, Louch WE, Sejersted OM, Carlson CR. Protein phosphatase 1c associated with the cardiac sodium calcium exchanger 1 regulates its activity by dephosphorylating serine 68-phosphorylated phospholemman. J Biol Chem 291: 4561–4579, 2016. doi: 10.1074/jbc.M115.677898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haggart CR, Ames EG, Lee JK, Holmes JW. Effects of stretch and shortening on gene expression in intact myocardium. Physiol Genomics 46: 57–65, 2014. doi: 10.1152/physiolgenomics.00103.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamlyn JM, Blaustein MP. Endogenous ouabain: recent advances and controversies. Hypertension 68: 526–532, 2016. doi: 10.1161/HYPERTENSIONAHA.116.06599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR, Ludens JH. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci USA 88: 6259–6263, 1991. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamlyn JM, Linde CI, Gao J, Huang BS, Golovina VA, Blaustein MP, Leenen FH. Neuroendocrine humoral and vascular components in the pressor pathway for brain angiotensin II: a new axis in long term blood pressure control. PLoS One 9: e108916, 2014. doi: 10.1371/journal.pone.0108916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han F, Tucker AL, Lingrel JB, Despa S, Bers DM. Extracellular potassium dependence of the Na+-K+-ATPase in cardiac myocytes: isoform specificity and effect of phospholemman. Am J Physiol Cell Physiol 297: C699–C705, 2009. doi: 10.1152/ajpcell.00063.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Honsho S, Nishikawa S, Amano K, Zen K, Adachi Y, Kishita E, Matsui A, Katsume A, Yamaguchi S, Nishikawa K, Isoda K, Riches DW, Matoba S, Okigaki M, Matsubara H. Pressure-mediated hypertrophy and mechanical stretch induces IL-1 release and subsequent IGF-1 generation to maintain compensative hypertrophy by affecting Akt and JNK pathways. Circ Res 105: 1149–1158, 2009. doi: 10.1161/CIRCRESAHA.109.208199. [DOI] [PubMed] [Google Scholar]
- 41.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA, Lingrel JB. Identification of a specific role for the Na,K-ATPase α 2 isoform as a regulator of calcium in the heart. Mol Cell 3: 555–563, 1999. doi: 10.1016/S1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- 42.Juhaszova M, Blaustein MP. Distinct distribution of different Na+ pump alpha subunit isoforms in plasmalemma. Physiological implications. Ann N Y Acad Sci 834: 524–536, 1997. doi: 10.1111/j.1749-6632.1997.tb52310.x. [DOI] [PubMed] [Google Scholar]
- 43.Kawamura A, Guo J, Itagaki Y, Bell C, Wang Y, Haupert GT Jr, Magil S, Gallagher RT, Berova N, Nakanishi K. On the structure of endogenous ouabain. Proc Natl Acad Sci USA 96: 6654–6659, 1999. doi: 10.1073/pnas.96.12.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komiyama Y, Nishimura N, Dong XH, Hirose S, Kosaka C, Masaki H, Masuda M, Takahashi H. Liquid chromatography mass spectrometric analysis of ouabainlike factor in biological fluid. Hypertens Res 23, Suppl: S21–S27, 2000. doi: 10.1291/hypres.23.Supplement_S21. [DOI] [PubMed] [Google Scholar]
- 45.Krayenbühl HP. [Is secondary myocardial hypertrophy a physiological or pathological adaptive mechanism?]. Z Kardiol 71: 489–496, 1982. [PubMed] [Google Scholar]
- 46.Lanzani C, Citterio L, Glorioso N, Manunta P, Tripodi G, Salvi E, Carpini SD, Ferrandi M, Messaggio E, Staessen JA, Cusi D, Macciardi F, Argiolas G, Valentini G, Ferrari P, Bianchi G. Adducin- and ouabain-related gene variants predict the antihypertensive activity of rostafuroxin, part 2: clinical studies. Sci Transl Med 2: 59ra87, 2010. doi: 10.1126/scitranslmed.3001814. [DOI] [PubMed] [Google Scholar]
- 47.Laursen M, Yatime L, Nissen P, Fedosova NU. Crystal structure of the high-affinity Na+K+-ATPase-ouabain complex with Mg2+ bound in the cation binding site. Proc Natl Acad Sci USA 110: 10958–10963, 2013. doi: 10.1073/pnas.1222308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leenen FH, Hou X, Wang HW, Ahmad M. Enhanced expression of epithelial sodium channels causes salt-induced hypertension in mice through inhibition of the α2-isoform of Na+, K+-ATPase. Physiol Rep 3: e12383, 2015. doi: 10.14814/phy2.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levy D. Left ventricular hypertrophy. Epidemiological insights from the Framingham Heart Study. Drugs 35, Suppl 5: 1–5, 1988. doi: 10.2165/00003495-198800355-00002. [DOI] [PubMed] [Google Scholar]
- 50.Lewis LK, Yandle TG, Hilton PJ, Jensen BP, Begg EJ, Nicholls MG. Endogenous ouabain is not ouabain. Hypertension 64: 680–683, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03919. [DOI] [PubMed] [Google Scholar]
- 51.Liao Y, Asakura M, Takashima S, Ogai A, Asano Y, Shintani Y, Minamino T, Asanuma H, Sanada S, Kim J, Kitamura S, Tomoike H, Hori M, Kitakaze M. Celiprolol, a vasodilatory β-blocker, inhibits pressure overload-induced cardiac hypertrophy and prevents the transition to heart failure via nitric oxide-dependent mechanisms in mice. Circulation 110: 692–699, 2004. doi: 10.1161/01.CIR.0000137831.08683.E1. [DOI] [PubMed] [Google Scholar]
- 52.Liao Y, Ishikura F, Beppu S, Asakura M, Takashima S, Asanuma H, Sanada S, Kim J, Ogita H, Kuzuya T, Node K, Kitakaze M, Hori M. Echocardiographic assessment of LV hypertrophy and function in aortic-banded mice: necropsy validation. Am J Physiol Heart Circ Physiol 282: H1703–H1708, 2002. doi: 10.1152/ajpheart.00238.2001. [DOI] [PubMed] [Google Scholar]
- 53.Linde CI, Antos LK, Golovina VA, Blaustein MP. Nanomolar ouabain increases NCX1 expression and enhances Ca2+ signaling in human arterial myocytes: a mechanism that links salt to increased vascular resistance? Am J Physiol Heart Circ Physiol 303: H784–H794, 2012. doi: 10.1152/ajpheart.00399.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lingrel JB. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu Rev Physiol 72: 395–412, 2010. doi: 10.1146/annurev-physiol-021909-135725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu CC, Karimi Galougahi K, Weisbrod RM, Hansen T, Ravaie R, Nunez A, Liu YB, Fry N, Garcia A, Hamilton EJ, Sweadner KJ, Cohen RA, Figtree GA. Oxidative inhibition of the vascular Na+-K+ pump via NADPH oxidase-dependent β1-subunit glutathionylation: implications for angiotensin II-induced vascular dysfunction. Free Radic Biol Med 65: 563–572, 2013. doi: 10.1016/j.freeradbiomed.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem 275: 27838–27844, 2000. [DOI] [PubMed] [Google Scholar]
- 57.Liu L, Wu J, Kennedy DJ. Regulation of cardiac remodeling by cardiac Na+/K+-ATPase isoforms. Front Physiol 7: 382, 2016. doi: 10.3389/fphys.2016.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lorenz JN, Loreaux EL, Dostanic-Larson I, Lasko V, Schnetzer JR, Paul RJ, Lingrel JB. ACTH-induced hypertension is dependent on the ouabain-binding site of the α2-Na+-K+-ATPase subunit. Am J Physiol Heart Circ Physiol 295: H273–H280, 2008. doi: 10.1152/ajpheart.00183.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu FM, Hilgemann DW. Na/K pump inactivation, subsarcolemmal Na measurements, and cytoplasmic ion turnover kinetics contradict restricted Na spaces in murine cardiac myocytes. J Gen Physiol 149: 727–749, 2017. doi: 10.1085/jgp.201711780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manunta P, Hamilton J, Rogowski AC, Hamilton BP, Hamlyn JM. Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res 23, Suppl: S77–S85, 2000. doi: 10.1291/hypres.23.Supplement_S77. [DOI] [PubMed] [Google Scholar]
- 61.Mathews MR, DuCharme DW, Hamlyn JM, Harris DW, Mandel F, Clark MA, Ludens JH. Mass spectral characterization of an endogenous digitalis-like factor from human plasma. Hypertension 17: 930–935, 1991. doi: 10.1161/01.HYP.17.6.930. [DOI] [PubMed] [Google Scholar]
- 62.McDonough AA, Azuma KK, Lescale-Matys L, Tang MJ, Nakhoul F, Hensley CB, Komatsu Y. Physiologic rationale for multiple sodium pump isoforms. Differential regulation of α1 vs α2 by ionic stimuli. Ann NY Acad Sci 671: 156−168, 1992. [DOI] [PubMed] [Google Scholar]
- 63.McDonough AA, Zhang Y, Shin V, Frank JS. Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am J Physiol 270: C1221–C1227, 1996. [DOI] [PubMed] [Google Scholar]
- 64.McGrail KM, Phillips JM, Sweadner KJ. Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J Neurosci 11: 381–391, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Messerli FH, Schmieder R. Left ventricular hypertrophy. A cardiovascular risk factor in essential hypertension. Drugs 31, Suppl 4: 192–201, 1986. doi: 10.2165/00003495-198600314-00023. [DOI] [PubMed] [Google Scholar]
- 66.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogné K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421: 634–639, 2003. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 67.Morris BJ, Davis JO, Zatzman ML, Williams GM. The renin-angiotensin-aldosterone system in rabbits with congestive heart failure produced by aortic constriction. Circ Res 40: 275–282, 1977. doi: 10.1161/01.RES.40.3.275. [DOI] [PubMed] [Google Scholar]
- 68.Müller-Ehmsen J, Nickel J, Zobel C, Hirsch I, Bölck B, Brixius K, Schwinger RH. Longer term effects of ouabain on the contractility of rat isolated cardiomyocytes and on the expression of Ca and Na regulating proteins. Basic Res Cardiol 98: 90–96, 2003. doi: 10.1007/s00395-003-0396-9. [DOI] [PubMed] [Google Scholar]
- 69.Nakamura K, Stefanescu Schmidt A. Treatment of hypertension in coarctation of the aorta. Curr Treat Options Cardiovasc Med 18: 40, 2016. doi: 10.1007/s11936-016-0462-x. [DOI] [PubMed] [Google Scholar]
- 70.O’Brien WJ, Lingrel JB, Wallick ET. Ouabain binding kinetics of the rat alpha two and alpha three isoforms of the sodium-potassium adenosine triphosphate. Arch Biochem Biophys 310: 32–39, 1994. doi: 10.1006/abbi.1994.1136. [DOI] [PubMed] [Google Scholar]
- 71.O’Rourke B, Kass DA, Tomaselli GF, Kääb S, Tunin R, Marbán E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. I: experimental studies. Circ Res 84: 562–570, 1999. doi: 10.1161/01.RES.84.5.562. [DOI] [PubMed] [Google Scholar]
- 72.Oparil S. Pathogenesis of ventricular hypertrophy. J Am Coll Cardiol 5, Suppl: 57B–65B, 1985. doi: 10.1016/S0735-1097(85)80528-3. [DOI] [PubMed] [Google Scholar]
- 73.Oshiro N, Dostanic-Larson I, Neumann JC, Lingrel JB. The ouabain-binding site of the α2 isoform of Na,K-ATPase plays a role in blood pressure regulation during pregnancy. Am J Hypertens 23: 1279–1285, 2010. doi: 10.1038/ajh.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parker FB Jr, Streeten DH, Farrell B, Blackman MS, Sondheimer HM, Anderson GH Jr. Preoperative and postoperative renin levels in coarctation of the aorta. Circulation 66: 513–514, 1982. doi: 10.1161/01.CIR.66.3.513. [DOI] [PubMed] [Google Scholar]
- 75.Pavlovic D, Fuller W, Shattock MJ. Novel regulation of cardiac Na pump via phospholemman. J Mol Cell Cardiol 61: 83–93, 2013. doi: 10.1016/j.yjmcc.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 76.Pitzalis MV, Hamlyn JM, Messaggio E, Iacoviello M, Forleo C, Romito R, de Tommasi E, Rizzon P, Bianchi G, Manunta P. Independent and incremental prognostic value of endogenous ouabain in idiopathic dilated cardiomyopathy. Eur J Heart Fail 8: 179–186, 2006. doi: 10.1016/j.ejheart.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 77.Poburko D, Fameli N, Kuo KH, van Breemen C. Ca2+ signaling in smooth muscle: TRPC6, NCX and LNats in nanodomains. Channels (Austin) 2: 10–12, 2008. doi: 10.4161/chan.2.1.6053. [DOI] [PubMed] [Google Scholar]
- 78.Pulina MV, Zulian A, Berra-Romani R, Beskina O, Mazzocco-Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP, Golovina VA. Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol 298: H263–H274, 2010. doi: 10.1152/ajpheart.00784.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pullen MA, Brooks DP, Edwards RM. Characterization of the neutralizing activity of digoxin-specific Fab toward ouabain-like steroids. J Pharmacol Exp Ther 310: 319–325, 2004. doi: 10.1124/jpet.104.065250. [DOI] [PubMed] [Google Scholar]
- 80.Pullen MA, Harpel MR, Danoff TM, Brooks DP. Comparison of non-digitalis binding properties of digoxin-specific Fabs using direct binding methods. J Immunol Methods 336: 235–241, 2008. doi: 10.1016/j.jim.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 81.Rembold CM, Ripley ML, Meeks MK, Geddis LM, Kutchai HC, Marassi FM, Cheung JY, Moorman JR. Serine 68 phospholemman phosphorylation during forskolin-induced swine carotid artery relaxation. J Vasc Res 42: 483–491, 2005. doi: 10.1159/000088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rindler TN, Lasko VM, Nieman ML, Okada M, Lorenz JN, Lingrel JB. Knockout of the Na,K-ATPase α2-isoform in cardiac myocytes delays pressure overload-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol 304: H1147–H1158, 2013. doi: 10.1152/ajpheart.00594.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rockman HA, Wachhorst SP, Mao L, Ross J Jr. ANG II receptor blockade prevents ventricular hypertrophy and ANF gene expression with pressure overload in mice. Am J Physiol Heart Circ Physiol 266: H2468–H2475, 1994. [DOI] [PubMed] [Google Scholar]
- 84.Rodriguez JS, Velez Rueda JO, Salas M, Becerra R, Di Carlo MN, Said M, Vittone L, Rinaldi G, Portiansky EL, Mundiña-Weilenmann C, Palomeque J, Mattiazzi A. Increased Na+/Ca2+ exchanger expression/activity constitutes a point of inflection in the progression to heart failure of hypertensive rats. PLoS One 9: e96400, 2014. doi: 10.1371/journal.pone.0096400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schaefer TL, Lingrel JB, Moseley AE, Vorhees CV, Williams MT. Targeted mutations in the Na,K-ATPase α 2 isoform confer ouabain resistance and result in abnormal behavior in mice. Synapse 65: 520–531, 2011. doi: 10.1002/syn.20870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schatzmann HJ. [Cardiac glycosides as inhibitors of active potassium and sodium transport by erythrocyte membrane]. Helv Physiol Pharmacol Acta 11: 346–354, 1953. [PubMed] [Google Scholar]
- 87.Schirmer H, Lunde P, Rasmussen K. Prevalence of left ventricular hypertrophy in a general population; the Tromsø Study. Eur Heart J 20: 429–438, 1999. doi: 10.1053/euhj.1998.1314. [DOI] [PubMed] [Google Scholar]
- 88.Schlüter KD, Wenzel S. Angiotensin II: a hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks. Pharmacol Ther 119: 311–325, 2008. doi: 10.1016/j.pharmthera.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 89.Schneider R, Wray V, Nimtz M, Lehmann WD, Kirch U, Antolovic R, Schoner W. Bovine adrenals contain, in addition to ouabain, a second inhibitor of the sodium pump. J Biol Chem 273: 784–792, 1998. doi: 10.1074/jbc.273.2.784. [DOI] [PubMed] [Google Scholar]
- 90.Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J 79: 2682–2691, 2000. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shamraj OI, Grupp IL, Grupp G, Melvin D, Gradoux N, Kremers W, Lingrel JB, De Pover A. Characterisation of Na/K-ATPase, its isoforms, and the inotropic response to ouabain in isolated failing human hearts. Cardiovasc Res 27: 2229–2237, 1993. doi: 10.1093/cvr/27.12.2229. [DOI] [PubMed] [Google Scholar]
- 92.Shanmugam P, Valente AJ, Prabhu SD, Venkatesan B, Yoshida T, Delafontaine P, Chandrasekar B. Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy. J Mol Cell Cardiol 50: 928–938, 2011. doi: 10.1016/j.yjmcc.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shull GE, Lane LK, Lingrel JB. Amino-acid sequence of the beta-subunit of the (Na+ + K+)ATPase deduced from a cDNA. Nature 321: 429–431, 1986. doi: 10.1038/321429a0. [DOI] [PubMed] [Google Scholar]
- 94.Shull GE, Schwartz A, Lingrel JB. Amino-acid sequence of the catalytic subunit of the (Na+ + K+)ATPase deduced from a complementary DNA. Nature 316: 691–695, 1985. doi: 10.1038/316691a0. [DOI] [PubMed] [Google Scholar]
- 95.Shull MM, Lingrel JB. Multiple genes encode the human Na+,K+-ATPase catalytic subunit. Proc Natl Acad Sci USA 84: 4039–4043, 1987. doi: 10.1073/pnas.84.12.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Simonini M, Pozzoli S, Bignami E, Casamassima N, Messaggio E, Lanzani C, Frati E, Botticelli IM, Rotatori F, Alfieri O, Zangrillo A, Manunta P. Endogenous ouabain: an old cardiotonic steroid as a new biomarker of heart failure and a predictor of mortality after cardiac surgery. BioMed Res Int 2015: 714793, 2015. doi: 10.1155/2015/714793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Song H, Karashima E, Hamlyn JM, Blaustein MP. Ouabain-digoxin antagonism in rat arteries and neurones. J Physiol 592: 941–969, 2014. doi: 10.1113/jphysiol.2013.266866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song H, Lee MY, Kinsey SP, Weber DJ, Blaustein MP. An N-terminal sequence targets and tethers Na+ pump α2 subunits to specialized plasma membrane microdomains. J Biol Chem 281: 12929–12940, 2006. doi: 10.1074/jbc.M507450200. [DOI] [PubMed] [Google Scholar]
- 99.Staessen JA, Thijs L, Stolarz-Skrzypek K, Bacchieri A, Barton J, Espositi ED, de Leeuw PW, Dłużniewski M, Glorioso N, Januszewicz A, Manunta P, Milyagin V, Nikitin Y, Souček M, Lanzani C, Citterio L, Timio M, Tykarski A, Ferrari P, Valentini G, Kawecka-Jaszcz K, Bianchi G. Main results of the ouabain and adducin for Specific Intervention on Sodium in Hypertension Trial (OASIS-HT): a randomized placebo-controlled phase-2 dose-finding study of rostafuroxin. Trials 12: 13, 2011. doi: 10.1186/1745-6215-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stella P, Manunta P, Mallamaci F, Melandri M, Spotti D, Tripepi G, Hamlyn JM, Malatino LS, Bianchi G, Zoccali C. Endogenous ouabain and cardiomyopathy in dialysis patients. J Intern Med 263: 274–280, 2008. doi: 10.1111/j.1365-2796.2007.01883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Studer R, Reinecke H, Bilger J, Eschenhagen T, Böhm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res 75: 443–453, 1994. doi: 10.1161/01.RES.75.3.443. [DOI] [PubMed] [Google Scholar]
- 102.Szent-Gyorgi A. Chemical Physiology of Contraction in Body and Heart Muscle. New York: Academic, 1953, p. 135. [Google Scholar]
- 103.Tamura M, Harris TM, Phillips D, Blair IA, Wang YF, Hellerqvist CG, Lam SK, Inagami T. Identification of two cardiac glycosides as Na+-pump inhibitors in rat urine and diet. J Biol Chem 269: 11972–11979, 1994. [PubMed] [Google Scholar]
- 104.Toyoshima C, Kanai R, Cornelius F. First crystal structures of Na+,K+-ATPase: new light on the oldest ion pump. Structure 19: 1732–1738, 2011. doi: 10.1016/j.str.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 105.Van Huysse JW, Dostanic I, Lingrel JB, Hou X, Wu H. Hypertension from chronic central sodium chloride in mice is mediated by the ouabain-binding site on the Na,K-ATPase α2-isoform. Am J Physiol Heart Circ Physiol 301: H2147–H2153, 2011. doi: 10.1152/ajpheart.01216.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vemuri R, Longoni S, Philipson KD. Ouabain treatment of cardiac cells induces enhanced Na+-Ca2+ exchange activity. Am J Physiol Cell Physiol 256: C1273–C1276, 1989. [DOI] [PubMed] [Google Scholar]
- 107.Verdonck F, Mubagwa K, Sipido KR. [Na+] in the subsarcolemmal “fuzzy’ space and modulation of [Ca2+]i and contraction in cardiac myocytes. Cell Calcium 35: 603–612, 2004. doi: 10.1016/j.ceca.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 108.Wang J, Gao E, Rabinowitz J, Song J, Zhang XQ, Koch WJ, Tucker AL, Chan TO, Feldman AM, Cheung JY. Regulation of in vivo cardiac contractility by phospholemman: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol 300: H859–H868, 2011. doi: 10.1152/ajpheart.00894.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang J, Schwinger RH, Frank K, Müller-Ehmsen J, Martin-Vasallo P, Pressley TA, Xiang A, Erdmann E, McDonough AA. Regional expression of sodium pump subunits isoforms and Na+-Ca++ exchanger in the human heart. J Clin Invest 98: 1650–1658, 1996. doi: 10.1172/JCI118960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang S, Gong H, Jiang G, Ye Y, Wu J, You J, Zhang G, Sun A, Komuro I, Ge J, Zou Y. Src is required for mechanical stretch-induced cardiomyocyte hypertrophy through angiotensin II type 1 receptor-dependent β-arrestin2 pathways. PLoS One 9: e92926, 2014. doi: 10.1371/journal.pone.0092926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wansapura AN, Lasko V, Xie Z, Fedorova OV, Bagrov AY, Lingrel JB, Lorenz JN. Marinobufagenin enhances cardiac contractility in mice with ouabain-sensitive α1-Na+-K+-ATPase. Am J Physiol Heart Circ Physiol 296: H1833–H1839, 2009. doi: 10.1152/ajpheart.00285.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wansapura AN, Lasko VM, Lingrel JB, Lorenz JN. Mice expressing ouabain-sensitive α1-Na-K-ATPase have increased susceptibility to pressure overload-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol 300: H347–H355, 2011. doi: 10.1152/ajpheart.00625.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weber KT, Clark WA, Janicki JS, Shroff SG. Physiologic versus pathologic hypertrophy and the pressure-overloaded myocardium. J Cardiovasc Pharmacol 10, Suppl 6: S37–S50, 1987. doi: 10.1097/00005344-198700106-00006. [DOI] [PubMed] [Google Scholar]
- 114.Wendt-Gallitelli MF, Voigt T, Isenberg G. Microheterogeneity of subsarcolemmal sodium gradients. Electron probe microanalysis in guinea-pig ventricular myocytes. J Physiol 472: 33–44, 1993. doi: 10.1113/jphysiol.1993.sp019934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Werdan K, Wagenknecht B, Zwissler B, Brown L, Krawietz W, Erdmann E. Cardiac glycoside receptors in cultured heart cells–II. Characterization of a high affinity and a low affinity binding site in heart muscle cells from neonatal rats. Biochem Pharmacol 33: 1873–1886, 1984. doi: 10.1016/0006-2952(84)90542-2. [DOI] [PubMed] [Google Scholar]
- 116.Westcott KV, Huang BS, Leenen FH. Brain renin-angiotensin-aldosterone system and ventricular remodeling after myocardial infarct: a review. Can J Physiol Pharmacol 87: 979–988, 2009. doi: 10.1139/Y09-067. [DOI] [PubMed] [Google Scholar]
- 117.Witt H, Schubert C, Jaekel J, Fliegner D, Penkalla A, Tiemann K, Stypmann J, Roepcke S, Brokat S, Mahmoodzadeh S, Brozova E, Davidson MM, Ruiz Noppinger P, Grohé C, Regitz-Zagrosek V. Sex-specific pathways in early cardiac response to pressure overload in mice. J Mol Med (Berl) 86: 1013–1024, 2008. doi: 10.1007/s00109-008-0385-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xie Z, Askari A. Na+/K+-ATPase as a signal transducer. Eur J Biochem 269: 2434–2439, 2002. doi: 10.1046/j.1432-1033.2002.02910.x. [DOI] [PubMed] [Google Scholar]
- 119.Yatime L, Laursen M, Morth JP, Esmann M, Nissen P, Fedosova NU. Structural insights into the high affinity binding of cardiotonic steroids to the Na+,K+-ATPase. J Struct Biol 174: 296–306, 2011. doi: 10.1016/j.jsb.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 120.Yazaki Y, Tsuchimochi H, Kurabayashi M, Komuro I. Molecular adaptation to pressure overload in human and rat hearts. J Mol Cell Cardiol 21, Suppl 5: 91–101, 1989. doi: 10.1016/0022-2828(89)90775-X. [DOI] [PubMed] [Google Scholar]
- 121.Yu Y, Wei SG, Zhang ZH, Gomez-Sanchez E, Weiss RM, Felder RB. Does aldosterone upregulate the brain renin-angiotensin system in rats with heart failure? Hypertension 51: 727–733, 2008. doi: 10.1161/HYPERTENSIONAHA.107.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zablocki D, Sadoshima J. Solving the cardiac hypertrophy riddle: The angiotensin II-mechanical stress connection. Circ Res 113: 1192–1195, 2013. doi: 10.1161/CIRCRESAHA.113.302501. [DOI] [PubMed] [Google Scholar]
- 123.Zucker IH, Xiao L, Haack KK. The central renin-angiotensin system and sympathetic nerve activity in chronic heart failure. Clin Sci (Lond) 126: 695–706, 2014. doi: 10.1042/CS20130294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zulian A, Linde CI, Pulina MV, Baryshnikov SG, Papparella I, Hamlyn JM, Golovina VA. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca(2+) signaling in arterial smooth muscle cells. Am J Physiol Cell Physiol 304: C324–C333, 2013. doi: 10.1152/ajpcell.00337.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]