

Abstract
Autophagy is one of the major degradative mechanisms that can eliminate excessive nutrients, toxic protein aggregates, damaged organelles and invading microorganisms. In response to obesity and obesity-associated lipotoxic, proteotoxic and oxidative stresses, autophagy plays an essential role in maintaining physiological homeostasis. However, obesity and its associated stress insults can often interfere with the autophagic process through various mechanisms, which result in further aggravation of obesity-related metabolic pathologies in multiple metabolic organs. Paradoxically, inhibition of autophagy, within specific contexts, indirectly produces beneficial effects that can alleviate several detrimental consequences of obesity. In this minireview, we will provide a brief discussion about our current understanding of the impact of obesity on autophagy and the role of autophagy dysregulation in modulating obesity-associated pathological outcomes.
Keywords: autophagy, diabetes, metabolism, obesity, stress
INTRODUCTION
As highlighted by the 2016 Nobel Prize in Physiology or Medicine, autophagy is now considered the major mechanism through which cells remove unnecessary or toxic cellular constituents (Tooze and Dikic, 2016). The process of autophagy and its critical role in physiological homeostasis have been extensively reviewed in recent literature (Choi et al., 2013; Feng et al., 2014; Kaur and Debnath, 2015), so will be only briefly summarized in this introduction.
Initial genetic screens in yeast identified autophagy-regulating genetic components, now catalogued as ATG genes (Feng et al., 2014; Klionsky et al., 2003). ATG genes constitute the pathways that lead to the formation of the isolation membrane, which sequesters target substrates from the cytoplasm through autophagosome formation (Feng et al., 2014). Mature autophagosomes then fuse with lysosomes, which contain digestive enzymes, to form autolysosomes, in which the substrates are degraded and the simple molecules are released for recycling through lysosomal channels. Although many key genetic components of autophagy have been characterized, further investigations are still necessary to understand how each step of autophagy is mechanistically mediated by its biochemical components (Feng et al., 2014).
The physiological importance of autophagy has been clearly demonstrated by genetic studies in model animals such as mice and Drosophila (Mizushima and Levine, 2010; Mulakkal et al., 2014). For instance, autophagy defects in neurons provoke neurodegenerative phenotypes associated with toxic protein aggregates (Kim et al., 2017; Menzies et al., 2017). These aggregates are often observed in human patients with neurodegenerative diseases (Ross and Poirier, 2004). In addition, autophagy defects also generate prominent phenotypes in metabolic organs, such as liver, adipose tissue, skeletal muscle and pancreas (Kim and Lee, 2014). These studies indicate that autophagy indeed plays an important catabolic role and that proper regulation of autophagy is important for metabolic homeostasis.
Obesity, which affects up to 2.1 billion people or ~30% of the population worldwide (Ng et al., 2014), is now a global health concern. Obesity is frequently associated with its diverse comorbidities such as diabetes, hypertension, dyslipidemia, cardiovascular disease and cancer (Collaborators et al., 2017; Kopelman, 2000). As autophagy may play a critical role in preventing or attenuating these diseases, recent studies have focused on how the autophagy process is altered during obesity and how autophagic catabolism plays a role in preventing the comorbidities of obesity.
OBESITY-ASSOCIATED STRESS INSULTS
Over-nutrition and lack of physical activity, prevalent in modern society, are among the major causes of the current obesity epidemic (Hill et al., 2003; 2012; Stubbs and Lee, 2004). These conditions lead to a chronic surplus of calories, which is stored as fat in adipose tissue. Accumulation of fat in non-adipose tissue, such as liver and skeletal muscle, can provoke tissue damage (Brookheart et al., 2009; Lelliott and Vidal-Puig, 2004). In addition, excessive fat accumulation can elevate serum free fatty acid level, resulting in systemic lipotoxicity (Browning and Horton, 2004; Brookheart et al., 2009; Goldberg et al., 2012; Lelliott and Vidal-Puig, 2004).
Lipid is the major constituent of the biological membrane, and excessive lipid influx and accumulation of byproducts from lipid metabolism can alter cellular processes by affecting membrane integrity and fluidity. For instance, sarco-ER calcium pump (SERCA) activity requires a fluidic ER membrane and is inhibited by saturated fatty acids and cholesterol that rigidify the membrane (Li et al., 2004) or even by obesity-associated moderate changes in ER membrane lipidomic profile (Fu et al., 2011). SERCA inhibition and subsequent reduction in ER calcium level can lead to a decreased activity of calcium-dependent ER chaperones, resulting in decreased protein folding capacity and subsequent accumulation of unfolded proteins (Arruda and Hotamisligil, 2015; Ji and Kaplowitz, 2006). Furthermore, unfolded protein accumulation induces proteotoxic stresses in the ER (ER stress), which can promote lipogenesis (Basseri and Austin, 2008; Zheng et al., 2010) and interfere with the normal lipoprotein secretion pathway (Ota et al., 2008; Zhang et al., 2011). These outcomes can further aggravate lipotoxic pathologies. Accumulation of cytotoxic lipids also provokes oxidative stress, as the reactive lipids damage critical biomolecules such as membrane-bound mitochondrial enzymes (Hauck and Bernlohr, 2016). The oxidative stress can be further propagated by mitochondrial damages that lead to production of reactive oxygen species (ROS) (Bournat and Brown, 2010). Excessive oxidative damage in DNA, in turn, can also produce genotoxic stresses (Cerda et al., 2014; Cooke et al., 2003). Membrane lipid alteration can also provoke stress signaling through modification of lipid subdomains known as lipid rafts, and induce stress-activated protein kinase (SAPK)/JNK signaling that aggravates obesity-associated metabolic pathologies (Holzer et al., 2011). In addition, extracellular fatty acids are directly sensed by toll-like receptors to elicit stress responses (Kennedy et al., 2009; Shi et al., 2006), and various metabolites of fatty acids indirectly provoke activation of stress signaling pathways (Schenk et al., 2008). These responses collectively lead to an aggravation of obesity-associated pathologies, such as chronic inflammation and insulin resistance (Kennedy et al., 2009; Schenk et al., 2008).
Autophagy plays an important protective role against these stress insults (Kroemer et al., 2010; Murrow and Debnath, 2013). For example, elimination of lipid droplets through the autophagic pathway (known as lipophagy) provides a mechanism for reducing fat content and thereby normalizing lipid metabolism in tissues of obese individuals (Singh and Cuervo, 2012; Singh et al., 2009a). Autophagy can also remove dysfunctional mitochondria (known as mitophagy), limiting production of ROS in obesity-associated pathological conditions (Sarparanta et al., 2017). During ER stress, autophagy can eliminate a portion of the ER (known as ER-phagy), which removes damaged or redundant parts of the ER restoring ER homeostasis (Bernales et al., 2007). Therefore, to resolve the obesity-associated stresses, autophagic responses should be properly coordinated; however, these stresses can often impair autophagic processes, as highlighted below.
IMPACT OF OBESITY ON AUTOPHAGY
ATG1 is one of the first genes isolated to mediate the autophagy response in yeast (Kamada et al., 2000; Matsuura et al., 1997). Unc-51-like kinase 1 (ULK1) and 2 (ULK2), ATG1’s mammalian homologs, are essential for initiation of autophagy (Alers et al., 2012a). Activity of ULK1 is controlled by two nutrient-regulated protein kinases, AMP-activated kinase (AMPK) and mTOR complex 1 (mTORC1) (Alers et al., 2012b). AMPK is activated upon cellular energy (ATP) depletion while mTORC1 is activated upon nutrient abundance (Jewell and Guan, 2013). AMPK is known to inhibit mTORC1 through several distinct mechanisms (Jewell and Guan, 2013). AMPK and mTORC1 produce diametrically opposing effects on ULK1; ULK1 is activated by AMPK-mediated phosphorylation but inhibited by mTORC1-mediated phosphorylation. Through these mechanisms, autophagy can be activated by starvation while suppressed during nutritional affluence (Galluzzi et al., 2014). Indeed, hepatic autophagy is strongly upregulated during starvation and coordinates liver metabolism to meet the metabolic needs of the organism during the nutritional stringency (Ezaki et al., 2011).
Autophagy was thought to be inactive during obesity because hypernutrition can inhibit AMPK and subsequently activate mTORC1. Indeed, mTORC1 activity is chronically upregulated during obesity, which is associated with increased anabolic metabolism in liver (Dann et al., 2007; Um et al., 2004). Consistent with this premise, initial studies on obese mice showed dramatic downregulation of autophagic activities, associated with reduced expression of ATG5 and ATG7 and the subsequent inhibition in autophagosome biogenesis (Yang et al., 2010). Insulin resistance and hyper-insulinemia were also suggested to contribute to autophagy inhibition during obesity (Liu et al., 2009). In addition, recent studies suggest that lipotoxic insults can downregulate AMPK signaling, thereby decreasing autophagosome production in macrophages (Wen et al., 2011) and liver cells (Cho et al., 2017; Li et al., 2017).
In contrast, other studies involving both human and mouse tissues demonstrated that autophagosomes can accumulate in response to obesity and lipotoxicity in multiple tissues, including liver and adipose tissues (Jansen et al., 2012; Kovsan et al., 2011; Mei et al., 2011; Nunez et al., 2013; Ost et al., 2010). These findings suggest that the relationship between obesity and autophagy is not as simple as originally speculated. For instance, ER stress, which can be provoked by obesity and lipotoxicity as reviewed above, can induce autophagy through multiple mechanisms (Ogata et al., 2006; Qin et al., 2010; Rashid et al., 2015; Senft and Ronai, 2015). Obesity per se is an established strong inducer of ER stress in liver (Hotamisligil, 2010; Ozcan et al., 2004) and obesity-associated ER stress aggravates fat accumulation, insulin resistance and liver damage (Ozcan et al., 2006; Park et al., 2014a). Therefore, it is plausible that, as a defensive mechanism against ER stress-induced damages, cells upregulate autophagy. In fibroblasts, lipotoxic activation of protein kinase C (PKC) can upregulate autophagic flux thereby protecting cells from apoptotic cell death (Tan et al., 2012). Other stresses associated with obesity, such as inflammation and oxidative stress, can also upregulate autophagy through various mechanisms (Filomeni et al., 2015; Qian et al., 2017). Autophagy induction in this context can be viewed as part of a cellular defense mechanism that is coordinated to maintain cellular homeostasis under obesity-associated stresses.
Efficient autophagy should result in decreased accumulation of autophagy substrates, such as lipid droplets and a ubiquitin adaptor protein p62/SQSTM1. However, in virtually all of the studies, these autophagy substrates were found to prominently accumulate during conditions of obesity and lipotoxicity (Gonzalez-Rodriguez et al., 2014; Park et al., 2014b; Yang et al., 2010). These findings suggest that obesity, in actuality, interferes with the autophagy process. Although early studies interpreted obesity-induced accumulation of autophagosomes as activated autophagy, this interpretation is currently being challenged, as many of these studies did not appropriately measure autophagic flux (Klionsky et al., 2016; Mizushima et al., 2010). For instance, pancreatic beta cells were originally reported to upregulate autophagosome formation in response to lipotoxic insults during obesity (Ebato et al., 2008; Lupi et al., 2002; Martino et al., 2012). However, it was later revealed that lipotoxicity inhibits the autophagic flux in these cells, and defective degradation is instead the main cause of autophagosome accumulation during obesity (Las et al., 2011; Mir et al., 2015). Also, in liver and kidney cells, excessive autophagosome accumulation during obesity was again revealed to be primarily due to decreased degradation (Park and Lee, 2014; Park et al., 2014b; Takabatake et al., 2017).
Interestingly, the mechanism of how lipotoxic insults interfere with autophagic flux is dependent on tissue type. In liver cells, the defect was observed at the autophagosomal-lysosomal fusion step; autophagosomes cannot fuse with lysosomes, primarily due to elevated levels of cytosolic calcium during lipotoxicity (Park et al., 2014b). Lipotoxic inhibition of SERCA is likely to be involved in this type of autophagy dysregulation (Czaja, 2015; Park and Lee, 2014). In addition to the calcium-dependent mechanism of inhibition, lipotoxicity and obesity can upregulate expression of Rubicon, a protein that can inhibit the fusion between autophagosomes and lysosomes (Tanaka et al., 2016). In contrast, kidney cells do not display defects in autophagosomal-lysosomal fusion in response to lipotoxicity or obesity (Yamamoto et al., 2017). Rather, they showed defects in lysosomal acidification, which is essential for the activity of lysosomal enzymes, suggesting that lysosomal degradative function was impaired during obesity (Yamamoto et al., 2017). Interestingly, lysosomal acidification was not found to become defective in liver cells after lipotoxicity (Park et al., 2014b). In pancreatic beta cells, defects in autophagosomal-lysosomal fusion and lysosomal acidification were both observed after lipotoxic insults (Las et al., 2011; Mir et al., 2015). Recent genetic study showed that autophagosomal-lysosomal fusion is not controlled by lysosomal acidification, indicating that these two processes are independently regulated by separate mechanisms (Mauvezin and Neufeld, 2015; Mauvezin et al., 2015). Therefore, the mechanism of how lipotoxicity alters autophagic flux seems to be different between tissues.
Most of the studies mentioned above focused on the effects of obesity and lipotoxicity on the conventional autophagy process, which involves bulk isolation of cytoplasm and membrane fusion with lysosomes (also known as macroautophagy). In addition to macroautophagy, obesity has also been demonstrated to interfere with chaperone-mediated autophagy (also known as CMA) (Kaushik and Cuervo, 2012), which directly targets specific proteins into the lysosome (Rodriguez-Navarro et al., 2012). In this context, obesity-associated changes in the lysosomal membrane lipid profile can alter lipid microdomains, which results in degradation of LAMP2A, the CMA receptor (Rodriguez-Navarro and Cuervo, 2012).
IMPACT OF AUTOPHAGY ALTERATION ON OBESITY-ASSOCIATED PATHOLOGIES
Considering the intricate relationship between obesity and the autophagy process, it can be easily presumed that autophagy plays a critical role in regulating the pathological outcomes of obesity. Obesity-associated pathologies are accompanied by prominent accumulation of lipid droplets, protein aggregates and damaged mitochondria, all of which are major substrates of autophagy. Therefore, it can be expected that autophagy abrogation would accelerate obesity-associated pathologies in multiple tissue systems.
Indeed, genetic autophagy ablation in liver resulted in several pathologies that resemble those observed in obesity-associated non-alcoholic steatohepatitis (NASH), such as protein inclusion formation, fat accumulation and liver injury (Komatsu et al., 2005; 2007; Singh et al., 2009a). Systemic reduction of autophagic activity through Atg7 haploinsufficiency accelerated progression of diabetic pathologies during obesity (Lim et al., 2014). Conversely, systemic overexpression of Atg5, which upregulates autophagy, protected mice from age-associated obesity and insulin resistance (Pyo et al., 2013). Liver-specific overexpression of Atg7 or TFEB was also shown to improve obesity-associated ER stress and insulin resistance, confirming the protective role of autophagy in the organ (Settembre et al., 2013; Yang et al., 2010). Pharmacological activation of autophagy, through rapamycin or carbamazepine, also alleviated fat accumulation and liver injury during alcoholic and non-alcoholic fatty liver conditions (Ding et al., 2010; Lin et al., 2013). Calcium channel blockers, which can restore autophagic flux inhibited by lipotoxicity and obesity, almost completely normalized liver fat level and insulin sensitivity during high fat diet (HFD)-induced obesity (Park et al., 2014b). Genetic deletion of Rubicon, which can alternatively restore autophagosomal-lysosomal fusion during obesity, also substantially ameliorated obesity-associated liver fat accumulation (Tanaka et al., 2016). Collectively, these results indicate that autophagy has a protective role against obesity-associated pathologies in liver.
However, mice with liver- or skeletal muscle-specific deletions of Atg7 paradoxically improved multiple obesity-associated pathologies, such as adipogenesis, insulin resistance and hepatic fat accumulation (Kim et al., 2013). This observation was explained by an increased production of FGF21 in autophagy-defective tissues, which improves metabolism through a tissue non-autonomous manner (Kim et al., 2013). Similarly, mice with liver-specific deletion of FIP200, another gene essential for ATG1-dependent autophagosome formation (Hara et al., 2008), showed decreased levels of hepatic steatosis upon HFD (Ma et al., 2013). These results indicate that, even though physiological autophagy is important for suppressing obesity-associated metabolic derangements, complete ablation of autophagy can induce compensatory mechanisms that can protect against the detrimental consequences of obesity and autophagy dysfunction.
Autophagy may also play a role in modifying the pathological outcome of obesity in non-parenchymal cells of liver, such as macrophages (Liu et al., 2015) and hepatic stellate cells (Hernandez-Gea et al., 2012). Loss of autophagy in macrophages can promote inflammatory macrophage polarization, provoking inflammation at both systemic and hepatic levels (Liu et al., 2015). This exacerbated liver injury upon HFD and lipopolysaccharide (LPS) challenges (Liu et al., 2015). Autophagy in macrophages is demonstrated to be important for suppressing atherosclerosis, another obesity-associated pathology in the cardiovascular system (Liao et al., 2012; Razani et al., 2012). These results, as well as other results indicating that autophagy in immune cells is important for suppressing inflammation (Netea-Maier et al., 2016), suggest that autophagy in macrophages has a role in limiting obesity-associated inflammation and pathologies. In contrast, autophagy in hepatic stellate cells has a function in promoting fibrotic liver pathologies (Hernandez-Gea et al., 2012). Upon liver injury, hepatic stellate cells upregulate autophagy to utilize lipid droplets as an energy source, and this process is required for full activation of the hepatic stellate cells that results in promotion of liver fibrosis (Hernandez-Gea et al., 2012). Autophagy-mediated degradation of p62/SQSTM1 can also contribute to fibrosis aggravation because p62/SQSTM1 suppresses fibrogenic processes through vitamin D receptor activation (Duran et al., 2016). Consequently, inhibition of autophagy in hepatic stellate cells decreased liver fibrosis after chemical injuries (Hernandez-Gea et al., 2012).
The homeostatic role of autophagy was also established in mice with the pancreatic beta cell-specific deletion of Atg7. Autophagy inhibition resulted in beta cell dysfunction and reduced mass, leading to diabetic phenotypes such as hyperglycemia and glucose intolerance due to decreased insulin production (Ebato et al., 2008; Jung et al., 2008). These phenotypes were aggravated when combined with dietary or genetic obesity induction (Ebato et al., 2008; Quan et al., 2012). The aggravation of beta cell pathologies was partially due to the damages caused by excessive ER stress (Bartolome et al., 2012; Quan et al., 2012).
Finally, autophagy ablation in adipose tissue produced some beneficial effects against obesity phenotypes. Because autophagy is required for adipogenic processes, autophagy ablation decreased adipogenesis thereby reduced body weight gain during obesity (Singh et al., 2009b; Zhang et al., 2009). In addition, autophagy is potentially involved in whitening of adipose tissue by reducing the amount of mitochondria through autophagic elimination. Therefore, suppression of autophagy led to retention of mitochondria in white adipose tissue, resulting in increased energy expenditure and subsequent reduction in body weight (Singh et al., 2009b). Therefore, similar to the cases observed in skeletal muscle and liver, autophagy suppression in adipose tissue paradoxically produced beneficial effects during the conditions of obesity (Singh et al., 2009b; Zhang et al., 2009).
CONCLUSION
The large amount of recent literature, among which some were discussed above, focused on understanding the intricate relationship between obesity, autophagy dysfunction and their pathological consequences (Fig. 1). Although nutritional abundance can generally suppress autophagy initiation, stress insults associated with obesity can stimulate autophagy as a stress defense mechanism. In addition, lipotoxic insults can interfere with autophagy by inhibiting autophagosome degradation through multiple independent mechanisms. Although defective autophagy generally leads to deterioration of metabolic homeostasis, autophagy inhibition in certain pathophysiological context can paradoxically upregulate compensatory pathways that can be beneficial for defending against the consequences of obesity.
Fig. 1. Relationship between Obesity and Autophagy.
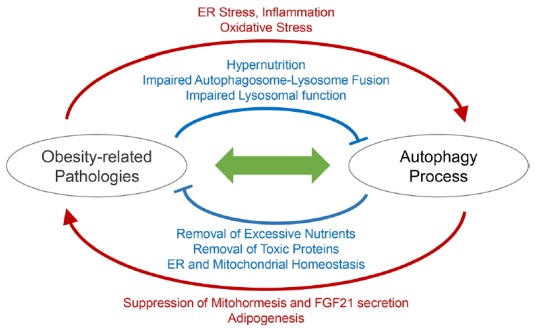
Sophisticated interaction between autophagy and obesity-associated pathologies is schematically illustrated.
Obesity and metabolic syndrome are not a homogeneous disorder in both humans and animal models. The composition of diets and the level of sedentariness are critical variables for determining the role of autophagy in homeostasis of different tissues. For instance, saturated and unsaturated fatty acids can produce distinct pathogenetic effects, while they have similar caloric values and both are obesogenic. Levels of physical activity can be also important for determining the metabolic contribution of different tissues as their rates of energy expenditure can be altered. Understanding the contribution of each of these factors have in autophagic regulation and pathological outcomes will be important for designing tailored therapeutic strategies for obese patients. Individual obese patients will have autophagy alterations through different molecular mechanisms, which may necessitate the development of personalized medicine that can precisely restore the specifically defective processes.
ACKNOWLEDGMENTS
This work was supported by the NIH (R01DK102850 and R21OD018265) to J.H.L.
REFERENCES
- Alers S, Loffler AS, Wesselborg S, Stork B. The incredible ULKs. Cell Commun Signal. 2012a;10:7. doi: 10.1186/1478-811X-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012b;32:2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda AP, Hotamisligil GS. Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab. 2015;22:381–397. doi: 10.1016/j.cmet.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolome A, Guillen C, Benito M. Autophagy plays a protective role in endoplasmic reticulum stress-mediated pancreatic beta cell death. Autophagy. 2012;8:1757–1768. doi: 10.4161/auto.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basseri S, Austin RC. ER stress and lipogenesis: a slippery slope toward hepatic steatosis. Dev Cell. 2008;15:795–796. doi: 10.1016/j.devcel.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy. 2007;3:285–287. doi: 10.4161/auto.3930. [DOI] [PubMed] [Google Scholar]
- Bournat JC, Brown CW. Mitochondrial dysfunction in obesity. Curr Opin Endocrinol Diabetes Obes. 2010;17:446–452. doi: 10.1097/MED.0b013e32833c3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookheart RT, Michel CI, Schaffer JE. As a matter of fat. Cell Metab. 2009;10:9–12. doi: 10.1016/j.cmet.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda C, Sanchez C, Climent B, Vazquez A, Iradi A, El Amrani F, Bediaga A, Saez GT. Oxidative stress and DNA damage in obesity-related tumorigenesis. Adv Exp Med Biol. 2014;824:5–17. doi: 10.1007/978-3-319-07320-0_2. [DOI] [PubMed] [Google Scholar]
- Cho CS, Lombard DB, Lee JH. SIRT3 as a regulator of hepatic autophagy. Hepatology. 2017;66:700–702. doi: 10.1002/hep.29271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- Collaborators GBDO, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, Marczak L, Mokdad AH, Moradi-Lakeh M, et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N Engl J Med. 2017;377:13–27. doi: 10.1056/NEJMoa1614362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- Czaja MJ. A new mechanism of lipotoxicity: Calcium channel blockers as a treatment for nonalcoholic steatohepatitis? Hepatology. 2015;62:312–314. doi: 10.1002/hep.27858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007;13:252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Hernandez ED, Reina-Campos M, Castilla EA, Subramaniam S, Raghunandan S, Roberts LR, Kisseleva T, Karin M, Diaz-Meco MT, et al. p62/SQSTM1 by binding to vitamin D receptor inhibits hepatic stellate cell activity, fibrosis, and liver cancer. Cancer Cell. 2016;30:595–609. doi: 10.1016/j.ccell.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8:325–332. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K, Yoshida M, et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7:727–736. doi: 10.4161/auto.7.7.15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22:377–388. doi: 10.1038/cdd.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–1276. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15:805–812. doi: 10.1016/j.cmet.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillon J, Lo Iacono O, Corazzari M, Fimia GM, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179. doi: 10.1038/cddis.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck AK, Bernlohr DA. Oxidative stress and lipotoxicity. J Lipid Res. 2016;57:1976–1986. doi: 10.1194/jlr.R066597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JO, Wyatt HR, Reed GW, Peters JC. Obesity and the environment: where do we go from here? Science. 2003;299:853–855. doi: 10.1126/science.1079857. [DOI] [PubMed] [Google Scholar]
- Hill JO, Wyatt HR, Peters JC. Energy balance and obesity. Circulation. 2012;126:126–132. doi: 10.1161/CIRCULATIONAHA.111.087213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, Karin M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–184. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen HJ, van Essen P, Koenen T, Joosten LA, Netea MG, Tack CJ, Stienstra R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology. 2012;153:5866–5874. doi: 10.1210/en.2012-1625. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Guan KL. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci. 2013;38:233–242. doi: 10.1016/j.tibs.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. ER stress: can the liver cope? J Hepatol. 2006;45:321–333. doi: 10.1016/j.jhep.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy A, Martinez K, Chuang CC, LaPoint K, McIntosh M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. J Nutr. 2009;139:1–4. doi: 10.3945/jn.108.098269. [DOI] [PubMed] [Google Scholar]
- Kim KH, Lee MS. Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10:322–337. doi: 10.1038/nrendo.2014.35. [DOI] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- Kim M, Ho A, Lee JH. Autophagy and human neurodegenerative diseases-A fly’s perspective. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18071596. pii: E1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Cregg JM, Dunn WA, Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Kopelman PG. Obesity as a medical problem. Nature. 2000;404:635–643. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- Kovsan J, Bluher M, Tarnovscki T, Kloting N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schon MR, et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96:E268–277. doi: 10.1210/jc.2010-1681. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in beta-cells. J Biol Chem. 2011;286:42534–42544. doi: 10.1074/jbc.M111.242412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelliott C, Vidal-Puig AJ. Lipotoxicity, an imbalance between lipogenesis de novo and fatty acid oxidation. Int J Obes Relat Metab Disord. 2004;28(Suppl 4):S22–28. doi: 10.1038/sj.ijo.0802854. [DOI] [PubMed] [Google Scholar]
- Li Y, Ge M, Ciani L, Kuriakose G, Westover EJ, Dura M, Covey DF, Freed JH, Maxfield FR, Lytton J, et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J Biol Chem. 2004;279:37030–37039. doi: 10.1074/jbc.M405195200. [DOI] [PubMed] [Google Scholar]
- Li S, Dou X, Ning H, Song Q, Wei W, Zhang X, Shen C, Li J, Sun C, Song Z. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology. 2017;66:936–952. doi: 10.1002/hep.29229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YM, Lim H, Hur KY, Quan W, Lee HY, Cheon H, Ryu D, Koo SH, Kim HL, Kim J, et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun. 2014;5:4934. doi: 10.1038/ncomms5934. [DOI] [PubMed] [Google Scholar]
- Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, Dong XC, Yin XM. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58:993–999. doi: 10.1016/j.jhep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284:31484–31492. doi: 10.1074/jbc.M109.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, Tanaka KE, Czaja MJ. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. doi: 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, Patane G, Boggi U, Piro S, Anello M, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51:1437–1442. doi: 10.2337/diabetes.51.5.1437. [DOI] [PubMed] [Google Scholar]
- Ma D, Molusky MM, Song J, Hu CR, Fang F, Rui C, Mathew AV, Pennathur S, Liu F, Cheng JX, et al. Autophagy deficiency by hepatic FIP200 deletion uncouples steatosis from liver injury in NAFLD. Mol Endocrinol. 2013;27:1643–1654. doi: 10.1210/me.2013-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino L, Masini M, Novelli M, Beffy P, Bugliani M, Marselli L, Masiello P, Marchetti P, De Tata V. Palmitate activates autophagy in INS-1E beta-cells and in isolated rat and human pancreatic islets. PLoS One. 2012;7:e36188. doi: 10.1371/journal.pone.0036188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene. 1997;192:245–250. doi: 10.1016/s0378-1119(97)00084-x. [DOI] [PubMed] [Google Scholar]
- Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–1438. doi: 10.1080/15548627.2015.1066957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C, Nagy P, Juhasz G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6:7007. doi: 10.1038/ncomms8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–498. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–1034. doi: 10.1016/j.neuron.2017.01.022. [DOI] [PubMed] [Google Scholar]
- Mir SU, George NM, Zahoor L, Harms R, Guinn Z, Sarvetnick NE. Inhibition of autophagic turnover in beta-cells by fatty acids and glucose leads to apoptotic cell death. J Biol Chem. 2015;290:6071–6085. doi: 10.1074/jbc.M114.605345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulakkal NC, Nagy P, Takats S, Tusco R, Juhasz G, Nezis IP. Autophagy in Drosophila: from historical studies to current knowledge. Biomed Res Int. 2014;2014:273473. doi: 10.1155/2014/273473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–137. doi: 10.1146/annurev-pathol-020712-163918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy. 2016;12:245–260. doi: 10.1080/15548627.2015.1071759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–781. doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez CE, Rodrigues VS, Gomes FS, Moura RF, Victorio SC, Bombassaro B, Chaim EA, Pareja JC, Geloneze B, Velloso LA, et al. Defective regulation of adipose tissue autophagy in obesity. Int J Obes (Lond) 2013;37:1473–1480. doi: 10.1038/ijo.2013.27. [DOI] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ost A, Svensson K, Ruishalme I, Brannmark C, Franck N, Krook H, Sandstrom P, Kjolhede P, Stralfors P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16:235–246. doi: 10.2119/molmed.2010.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest. 2008;118:316–332. doi: 10.1172/JCI32752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Lee JH. Calcium channel blockers as potential therapeutics for obesity-associated autophagy defects and fatty liver pathologies. Autophagy. 2014;10:2385–2386. doi: 10.4161/15548627.2014.984268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Park H, Ro SH, Jang I, Semple IA, Kim DN, Kim M, Nam M, Zhang D, Yin L, et al. Hepatoprotective role of Sestrin2 against chronic ER stress. Nat Commun. 2014a;5:4233. doi: 10.1038/ncomms5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Park H, Semple IA, Jang I, Ro SH, Kim M, Cazares VA, Stuenkel EL, Kim JJ, Kim JS, et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat Commun. 2014b;5:4834. doi: 10.1038/ncomms5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6:24. doi: 10.1186/s40169-017-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6:239–247. doi: 10.4161/auto.6.2.11062. [DOI] [PubMed] [Google Scholar]
- Quan W, Hur KY, Lim Y, Oh SH, Lee JC, Kim KH, Kim GH, Kim SW, Kim HL, Lee MK, et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia. 2012;55:392–403. doi: 10.1007/s00125-011-2350-y. [DOI] [PubMed] [Google Scholar]
- Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11:1956–1977. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro JA, Cuervo AM. Dietary lipids and aging compromise chaperone-mediated autophagy by similar mechanisms. Autophagy. 2012;8:1152–1154. doi: 10.4161/auto.20649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro JA, Kaushik S, Koga H, Dall’Armi C, Shui G, Wenk MR, Di Paolo G, Cuervo AM. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci USA. 2012;109:E705–714. doi: 10.1073/pnas.1113036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Sarparanta J, Garcia-Macia M, Singh R. Autophagy and mitochondria in obesity and type 2 diabetes. Curr Diabetes Rev. 2017;13:352–369. doi: 10.2174/1573399812666160217122530. [DOI] [PubMed] [Google Scholar]
- Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40:141–148. doi: 10.1016/j.tibs.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol. 2012;2012:282041. doi: 10.1155/2012/282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009a;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009b;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs CO, Lee AJ. The obesity epidemic: both energy intake and physical activity contribute. Med J Aust. 2004;181:489–491. doi: 10.5694/j.1326-5377.2004.tb06406.x. [DOI] [PubMed] [Google Scholar]
- Takabatake Y, Yamamoto T, Isaka Y. Stagnation of autophagy: A novel mechanism of renal lipotoxicity. Autophagy. 2017;13:775–776. doi: 10.1080/15548627.2017.1283084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SH, Shui G, Zhou J, Li JJ, Bay BH, Wenk MR, Shen HM. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin) J Biol Chem. 2012;287:14364–14376. doi: 10.1074/jbc.M111.294157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Hikita H, Tatsumi T, Sakamori R, Nozaki Y, Sakane S, Shiode Y, Nakabori T, Saito Y, Hiramatsu N, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016;64:1994–2014. doi: 10.1002/hep.28820. [DOI] [PubMed] [Google Scholar]
- Tooze SA, Dikic I. Autophagy Captures the Nobel Prize. Cell. 2016;167:1433–1435. doi: 10.1016/j.cell.2016.11.023. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Takabatake Y, Takahashi A, Kimura T, Namba T, Matsuda J, Minami S, Kaimori JY, Matsui I, Matsusaka T, et al. High-Fat Diet-Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Lipotoxicity in the Kidney. J Am Soc Nephrol. 2017;28:1534–1551. doi: 10.1681/ASN.2016070731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7). in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, Chang L, Xu W, Miao H, Leonardi R, et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011;30:1357–1375. doi: 10.1038/emboj.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Zhang C, Zhang K. Role of unfolded protein response in lipogenesis. World J Hepatol. 2010;2:203–207. doi: 10.4254/wjh.v2.i6.203. [DOI] [PMC free article] [PubMed] [Google Scholar]