

Abstract
Metastasis is the leading cause of death in patients with advanced melanoma, yet the somatic alterations that aid tumour cell dissemination and colonisation are poorly understood. Here, we deploy comparative genomics to identify and validate clinically relevant drivers of melanoma metastasis. To do this, we identified a set of 976 genes whose expression level was associated with a poor outcome in patients from two large melanoma cohorts. Next, we characterised the genomes and transcriptomes of mouse melanoma cell lines defined as weakly metastatic, and their highly metastatic derivatives. By comparing expression data between species, we identified lunatic fringe (LFNG), among 28 genes whose expression level is predictive of poor prognosis and whose altered expression is associated with a prometastatic phenotype in mouse melanoma cells. CRISPR/Cas9‐mediated knockout of Lfng dramatically enhanced the capability of weakly metastatic melanoma cells to metastasise in vivo, a phenotype that could be rescued with the Lfng cDNA. Notably, genomic alterations disrupting LFNG are found exclusively in human metastatic melanomas sequenced as part of The Cancer Genome Atlas. Using comparative genomics, we show that LFNG expression plays a functional role in regulating melanoma metastasis.
Keywords: comparative genomics, CRISPR, melanoma, RNA‐Seq
Abbreviations
- Bp
base pair
- CNV
copy number variants
- CRISPR
clustered regularly interspaced short palindromic repeats
- DASL
cDNA‐mediated Annealing, Selection, Ligation
- Dll3
Delta‐like ligand‐3
- DMEM
Dulbecco's modified Eagle's medium
- FDR
false discovery rate
- FPKM
fragments per kilobase per million
- HDC
histidine decarboxylase
- HR
hazard ratio
- Kb
kilobase
- LFNG
lunatic fringe
- MSS
melanoma‐specific survival
- NDRG2
N‐myc downstream‐regulated gene 2
- NICD
Notch intracellular domain
- OS
overall survival
- qPCR
quantitative polymerase chain reaction
- RT‐PCR
reverse transcription polymerase chain reaction
- SD
standard deviation
- SNV
single nucleotide variation
- TCGA
The Cancer Genome Atlas
- TPM
Transcripts per million
- UV
ultraviolet
1. Introduction
Melanoma is an aggressive cancer that develops from the pigment‐producing cells of the skin. In melanoma, as in other cancers, metastasis accounts for the majority of the mortality of patients with advanced disease (Chaffer and Weinberg, 2011; Damsky et al., 2010). This complex multistep process requires melanoma cells to invade adjacent tissues, intravasate into the lymphatics or blood vasculature, extravasate at distant sites and ultimately colonise an organ or tissue. For this to happen, melanoma cells must evade the immune system and sculpt the host microenvironment (Fidler, 2003).
Several models of metastasis have been proposed including those that describe monoclonal and polyclonal seeding. It is also clear that once a cell has left the primary tumour, it may undergo further evolution (Turajlic and Swanton, 2016). This complex pattern of tumour cell dissemination and ongoing evolution complicates the identification of the genetic events that drive the metastatic process. Importantly, transcriptome profiling of primary tumours has identified expression changes shown to be predictive of metastasis (Paik et al., 2004; van de Vijver et al., 2002), and alterations found in metastases have been shown to be present in subclones in early primary lesions (Wardwell‐Ozgo et al., 2013). These data support the idea that a proportion of cells within primary tumours may evolve, acquire or have intrinsic metastatic capabilities. Identifying those patients with tumours at high risk of metastasising could help identify individuals who may benefit from adjuvant therapies or more regular screening (Eggermont, 2016).
In this study, we set out to identify clinically relevant genes that confer enhanced metastatic capabilities upon melanoma cells. To do this, we used comparative functional genomics applied to gene expression predictors of patient survival, combined with expression data from murine cell line models that have different capabilities to colonise the lung, a major site of human melanoma metastasis. In this way, we identified a set of 28 genes associated with patient outcome that were also differentially expressed when weakly and highly metastatic mouse melanoma lines were compared. We focused on lunatic fringe (LFNG) that encodes for a glycosylating enzyme (O‐fucosylpeptide 3‐beta‐N‐acetylglucosaminyltransferase) that regulates NOTCH signalling (Moloney et al., 2000), and show an important role for this gene in controlling melanoma metastasis.
2. Materials and methods
2.1. Survival analysis
Gene expression data generated using whole‐genome cDNA‐mediated annealing, selection, ligation and extension (DASL) arrays (Illumina Inc., San Diego, CA, USA) from 217 (Leeds) (Nsengimana et al., 2015) and 222 (Lund) (Jonsson et al., 2010) primary melanomas (209 cutaneous, 13 mucosal) were obtained. The Leeds data set (Leeds melanoma cohort, N = 204, and chemotherapy study, N = 13) was profiled on the human HT12.4 array, while the Lund cohort was profiled on the earlier HT8.3 version. Quality control and normalisation of these data sets has been published elsewhere (Jonsson et al., 2010; Nsengimana et al., 2015). Briefly, the HT8.3 version had a lower performance with only 7752 genes passing QC filters. The overlap between this set and the Leeds data set using HT12.4 was 7584 genes. Survival benefit of each gene (log2 scale) was assessed in a Cox proportional hazards model using STATA v14.2 (STATACorp, Texas, USA) for melanoma‐specific survival (MSS) in the Leeds data and overall survival (OS) using the data from Lund. Analysis of the Leeds data set was adjusted for patient age and sex. P‐values were corrected for multiple testing (Benjamini–Hochberg false discovery rate, FDR). Kaplan–Meier curves were plotted comparing high to low gene expression relative to the median. Functional gene annotation and enrichment analyses of the genes that showed the same direction of association in both patient cohorts were performed using DAVID (Huang et al., 2009).
2.2. Cell lines
B16‐F0 and B16‐F10 cell lines were purchased from the American Type Culture Collection (ATCC), and the B16‐BL6, K1735‐P and K1735‐M2 lines were obtained from the University of Texas M.D. Anderson Cancer Centre. All cell lines were screened for the presence of mycoplasma and other mouse pathogens (Charles River Laboratories, Wilmington, MA, USA). Cells were cultured at 37°C in 5% CO2 in high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 29.2 mg·mL−1 L‐glutamine, 10 000 units·mL−1 penicillin and 10 000 μg·mL−1 streptomycin.
2.3. Nucleic acid extraction and sequencing
For whole‐genome sequencing, DNA was extracted from cell pellets using the QIAGEN Puregene Core Kit A. Paired‐end 75‐bp libraries were prepared and sequenced using the Illumina HiSeq platform. Data have been deposited in the European Nucleotide Archive (ERP001691). RNA was extracted from cell pellets using the QIAGEN RNeasy Mini Kit. Five different vials were cultured and extracted per cell line, to obtain five independent biological replicates for each line. 1 μg of total RNA per sample was submitted for sequencing. Unstranded 75‐bp paired‐end barcoded libraries were prepared with the standard Illumina library preparation kit. RNA libraries were sequenced on the Illumina Hiseq platform and the data deposited in public databases (European Nucleotide Archive (ERP001690) and ArrayExpress (E‐ERAD‐94)).
2.4. Whole‐genome data processing and somatic variant calling
Raw reads were mapped to the mouse reference genome (GRCm38p1) using bwa‐mem (Li, 2013) v0.7.5 and PCR duplicates marked using Picard tools MarkDuplicates v1.72 (http://broadinstitute.github.io/picard). Single nucleotide variants (SNVs) and short indels were called using Samtools mpileup (Li et al., 2009) v0.1.19‐58‐g3d123 cd, and the resulting variants were filtered using VCFTools (Danecek et al., 2011). Variants with variant quality QUAL < 20, or number of reads supporting the variant less than 5 (DP < 5) or SNPGAP < 10, were discarded. Due to the absence of a matched germline/normal sample from the exact mouse from which the cell lines were generated, we removed all variants reported by the mouse genomes project (Keane et al., 2011) for the genetic backgrounds of each cell line group. Similarly, variants located within ± 50 bp of structural variants reported by the mouse genomes project (Keane et al., 2011) were also discarded. Finally, functional consequences were predicted using ENSEMBL's variant effect predictor (v74) (McLaren et al., 2016).
2.5. Orthogonal validation of single nucleotide variants using Sequenom
A total of 262 SNVs (116 for the K1735 lines and 146 for the B16 lines) were selected for orthogonal validation using the Sequenom platform. These variants were randomly chosen using GATK's ValidationSiteSelector (v2.8‐1‐g932cd3a) from the set of variants that were identified to be present in all the cell lines from each group. All assays using the Sequenom platform were performed with three biological replicates of each line.
2.6. Somatic signature identification and comparison
Somatic mutational signatures were identified for each mouse cell line group using the filtered somatic single nucleotide variants (above). Signatures were identified using the non‐negative matrix factorisation method from the SomaticSignatures R package (Gehring et al., 2015) (v 2.6.1). To compare these signatures to those reported in COSMIC (http://cancer.sanger.ac.uk/cancergenome/assets/signatures_probabilities.txt), we calculated cosine similarities as previously reported (Alexandrov et al., 2015).
2.7. Copy number calling
Copy number alterations were identified using Control‐FREEC (Boeva et al., 2011) v6.7 with 50‐Kb windows. Due to the lack of a matched normal for each cell line, CNVs were called relative to parental cell lines (B16‐F0 and K1735‐P); somatic CNVs for the B16‐BL6 line were called using the BAM file for B16‐F10.
2.8. Identification of differentially expressed genes
Raw paired‐end reads were aligned to the mouse reference genome (GRCm38p1) using the splice‐aware aligner Tophat2 (Kim et al., 2013) guided by ENSEMBL mouse annotation (v73). Subsequently, the number of uniquely mapped read pairs that were aligned to each gene within the annotation with a mapping quality > 10 were counted using htseq‐count (Anders et al., 2015). Raw counts were normalised by calculating the fragments per kilobase per million (FPKM) values for each gene for each replicate. As a ‘fit for use’ quality control, blind pairwise comparisons across all the RNA‐Seq samples were performed by calculating the Pearson's correlation coefficient based on the FPKM values of all protein‐coding genes of the 25 sequenced samples. This information was used to group the samples using unsupervised hierarchical clustering using the package gplots in R (Gregory et al., 2013). To identify differentially expressed genes, all of the four possible paired comparisons between cell lines and their more metastatic derivatives were made (B16‐F10 vs B16‐F0, B16‐BL6 vs B16‐F10, B16‐BL6 vs B16‐F0 and K1735‐M2 vs K1735‐P) using DESeq2 (Love et al., 2014). Once dispersion estimates and normalised counts were calculated, genes with mean normalised counts < 10 were filtered out and P‐values were re‐adjusted using the Benjamini–Hochberg correction for multiple testing. All genes with P < 0.01 and a log2(foldchange) ≤−2 or ≥ 2 were considered as differentially expressed.
2.9. Mouse–human orthologue identification
To identify the human orthologues of mouse genes, the Compara module from the ENSEMBL Perl API was used (Herrero et al., 2016). In cases where a mouse gene had multiple orthologues in humans, the gene with the highest percentage of identity when comparing the human and mouse proteins was selected. Genes that had an ortholog classification of ‘many2many’ were not considered for further analysis.
2.10. Randomisation test to identify the expected number by chance of differentially expressed mouse genes overlapping and concordant with the list of genes associated with poor survival in melanoma patients
Two independent randomisation tests were performed using two different sample sizes 1290 or 388. A total of 1000 samples with randomly selected mouse genes (out of the 15 412 genes with normalised fragment counts > = 10 expressed by B16‐F0 or B16‐BL6) of each sample size were generated. Genes were selected without replacement. For each randomly selected gene, a random direction of expression was assigned with the same probability as the one observed in the mouse data: underexpressed (0.4621429) or overexpressed (0.5378571). Then, each random sample was compared to the list of human genes associated with poor outcome with an FDR < 0.1 in our combined patient survival analysis, to identify the number of overlapping and concordant genes. Finally using the distribution of the number of overlapping and concordant genes across the 1000 samples, we calculated the probability of obtaining a number of overlapping genes or more as the one observed in the mouse cell line/human data comparison.
2.11. Cas9 gRNA selection
To select suitable gRNAs, we identified sequences in the exons of candidate genes in the ENSEMBL v71 annotation of the GRCm38 mouse reference genome (′5‐NNNNNNNNNNNNNNNNNNNNNGG‐3′). For each sequence, possible off‐targets were identified using Cas‐Offinder (Bae et al., 2014). We then used biomaRt (Durinck et al., 2009) to identify all possible off‐targets with up to three mismatches whose expected cutting site overlapped an exon. Targeting sequences with zero exonic off‐targets with up to three mismatches were selected. See Table S7.
2.12. Lfng disruption using a single gRNA (g2d1 clone generation)
Oligos with the Lfng gRNA sequence (Sigma‐Aldrich Corp, St. Louis, MO, USA) were cloned into the vector PX459 (Addgene #48139) following the Zhang laboratory protocol (Ran et al., 2013). Plasmids were validated by Sanger sequencing using a U6 oligo (Table S7). To obtain stable transfectants, the region containing the U6 promoter, gRNA, gRNA scaffold and the CBh‐hSpCsn1‐PURO‐PolyA was excised and cloned into the PiggyBac plasmid PB713B‐1 to make PX459_Lfng_g2_gRNA‐PB713B‐1 (Fig. S12B). B16‐F0 cells (6 × 105) were cotransfected with 0.5 μg pCMV‐PiggyBac PBase (System Biosciences) and 5 μg of either PX459_Lfng_g2_gRNA‐PB713B‐1 plasmid (to generate Lfng‐targeted cells) or empty PB713B‐1 plasmid (to generate ‘control’ cells) using Fugene HD (Promega Corporation, Madison, WI, USA). Twenty‐four hours later, 5 μg puromycin was added to the medium and after 7 days individual colonies were isolated. Sequences amplified from the Lfng locus were analysed with TIDE (Brinkman et al., 2014) to identify clones with disruptive mutations. Clone ‘g2d1’, carrying a homozygous 1‐bp insertion in Lfng, and clone ‘ca4’ (from the control plate) were selected for further analysis.
2.13. Lfng disruption using two gRNAs (L1 clone generation)
Oligos with the Lfng targeting sequences (Sigma‐Aldrich Corp) were cloned into the PiggyBac gRNA expressing vector, Piggy_gRNAScaffold_BLASTO (S12B), following the Zhang's laboratory protocol (Ran et al., 2013). Plasmids carrying gRNA sequences were validated by Sanger sequencing using a U6 oligo (Table S7). To target Lfng using two different gRNA sequences (gRNAs Lfng_g2 and Lfng_g3; Table S7), we first generated a Cas9 stably expressing B16‐F0 cell line by cotransfecting 6 × 105 B16‐F0 cells with 5 μg of pPB‐LR5.1‐EF1a‐puro2ACas9 (Koike‐Yusa et al., 2014) and 0.5 μg pCMV‐piggyBac. From this experiment, we cloned a single cell line and cotransfected 6 × 105 cells with 2.5 μg of Piggy_gRNAscaffold_Lfng_g2 and 2.5 μg of Piggy_gRNAscaffold_Lfng_g3 (Fig. S12B), or LMDJ‐Piggy_gRNAscaffold to generate a control cell line. Twenty‐four hours later, 10 μg blasticidin was added to the medium, and after 7 days, individual colonies were isolated and assessed for targeting of Lfng by PCR. Clone ‘L1’, carrying a 4.8‐kb deletion encompassing exons 1–4 of Lfng, and clones ‘C1’ and ‘C2’ (from the control plate) were selected for further analysis.
2.14. Lfng cDNA rescue experiments
To confirm that the metastasis phenotypes we observed were due to the disruption of Lfng, we used plasmid rescue in the L1 cell line using the vector PB533A‐2 carrying a flag‐tagged full‐length Lfng cDNA (synthesised by GeneArt) to generate the cell line L1‐Lfng. L1‐PB cells carrying the empty vector were used as a control.
2.15. Assessment of Lfng expression in cell lines by quantitative RT‐PCR
For the comparison of Lfng expression levels between cell lines, RNA was extracted from 1 × 106 cells using the RNAeasy mini Kit (QIAgen, Manchester, UK) and cDNA was prepared using the SuperScript VILO Master Mix (Thermo) according to the manufacturers’ instructions. RT‐qPCR was performed using the TaqMan Fast Advanced Master Mix. Lfng (Mm01201988_m1) and B2m (Mm00437762_m1) assays were used for these studies. Reactions were performed in quadruplicate using the StepOnePlus system (Thermo Fischer Scientific, Waltham, MA, USA), and analysis was performed using the ΔΔCt method (Schmittgen and Livak, 2008).
2.16. Western blotting
Western blotting was performed using standard approaches. Anti‐vinculin (clone V284) and anti‐Flag (clone M2) antibodies were used (Sigma‐Aldrich Corp).
2.17. In vivo experimental metastasis assays
The experimental metastasis assay was performed as described previously (van der Weyden et al., 2017). For testing of the K1735‐P and K1735‐M2 cell lines, 1 × 105 cells were tail‐vein‐dosed into six‐ to eight‐week‐old wild‐type C3H/HeJ mice. After 10 days, mice were humanely sacrificed and their lungs were collected into 10% neutral buffered formalin and then processed for histopathological analysis. For testing of the B16‐F0, B16‐F10, and B16‐BL6 cell lines, 0.75 × 105 cells were tail‐vein‐dosed into six‐ to eight‐week‐old wild‐type C57BL6/NTac mice and their pulmonary metastatic burden was determined 7 days later by macroscopic counting. For testing the Lfng‐targeted g2d1 cells (and respective ca4 control cells), 4 × 105 cells were tail‐vein‐dosed, and for testing the Lfng‐targeted L1 cells (and respective C1/C2 control cells), 5 × 105 cells were tail‐vein‐dosed; both into six‐ to eight‐week‐old wild‐type C57BL/6NTac mice. cDNA rescue experiments, using the cell line L1‐Lfng and the control L1‐PB, were performed using 4 × 105 cells. The pulmonary metastatic burden was determined 10 days postdosing by macroscopic counting. In all cases, sex‐matched mice were used. The care and use of all mice in this study was in accordance with the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012, and all procedures were performed under a UK Home Office Project licence (PPL 80/2562). All mice were housed in individually ventilated cages (Techniplast GM500) receiving 60 air changes per hour, in a specific pathogen‐free environment with ad libitum access to autoclaved water and food (Mouse Breeders Diet, Laboratory Diets, 5021‐3). Cages were filled with aspen bedding substrate, with a nestlet and fun tunnel for environmental enrichment. There was a 12‐h light/dark cycle with no twilight period with a temperature of 21 °C ± 2 °C and a humidity of 55% ± 10%. Throughout the experiment, the welfare of the mice was monitored with daily visual checks.
2.18. Whole exome sequencing of the L1 cell line
DNA from L1 cells was exome‐sequenced using Agilent mouse whole exome baits. A 75‐bp paired‐end library was prepared and sequenced on the Illumina HiSeq2500 platform. Data were analysed as above and are available in the European Nucleotide Archive (ERP015062).
3. Results
3.1. mRNA expression predictors of prognosis in primary melanoma
Both tumour depth (Breslow thickness) and ulceration are established predictors of melanoma metastasis (Nsengimana et al., 2015), but the underlying mechanisms that drive metastasis are unknown. We first set out to identify genes whose expression levels were associated with poor outcome. To do this, we analysed the expression profiles of primary melanomas from two previously published studies from Leeds (the Leeds Melanoma Cohort and chemotherapy studies, n = 217) and from the Lund Melanoma Research Group (n = 222) (Jonsson et al., 2010; Nsengimana et al., 2015). Demographic information for these cohorts is provided in Table S1. Tumours from both cohorts have been analysed using Illumina DASL arrays such that the expression of 7584 genes may be assessed. For the Leeds cohort, melanoma‐specific survival (MSS) data were available, whereas overall survival (OS) was recorded for the Lund cohort. Survival analyses stratifying by gene expression were performed using the Cox proportional hazards model. In this way, we identified 976 genes whose expression levels were significantly associated with patient outcome in both cohorts (FDR < 0.1; Fig. 1A, Table S2). Of these genes, 78.17% (763/976) showed the same direction of association in both cohorts. These genes included SKP2 (Chen et al., 2011), TOP2A (Song et al., 2013), SOX4 (Jafarnejad et al., 2010), MAP2 (Soltani et al., 2005) and CTLA4 (Hannani et al., 2015), all of which have been associated with patient outcome in melanoma. Gene enrichment analysis found that biological processes including epidermis development, keratinocyte differentiation and immune response were overrepresented (FDR < 0.01; Table S3) – biological processes previously reported to be important in the development of melanoma metastasis (Bald et al., 2014; Golan et al., 2015).
Figure 1.
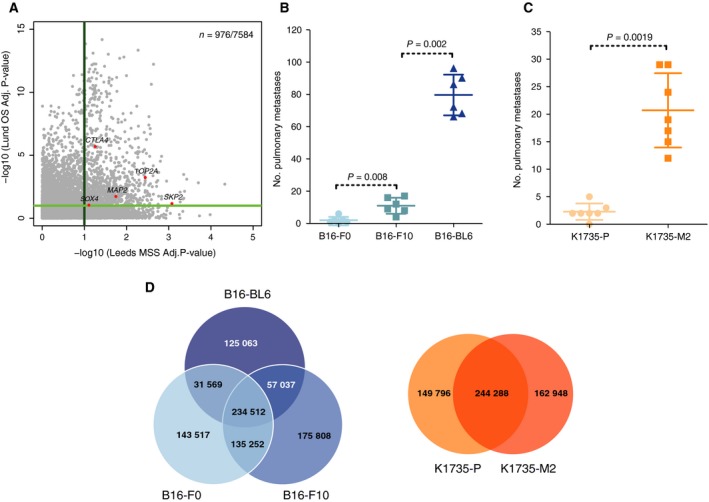
Patient sample and mouse cell line characteristics. (A) Scatter plot showing the −log10‐corrected P‐values for the 7584 genes analysed in both cohorts in association with melanoma‐specific survival in the Leeds cohort (x‐axis) and overall survival in the Lund cohort (y‐axis). (B‐C) Experimental metastasis assay using (B) B16 cell lines and (C) K1735 cell lines in wild‐type female mice (symbols representing individual mice with horizontal bar at the mean ± SD and statistics performed using a Mann–Whitney test; data shown are representative of two independent experiments). (D) Venn diagrams showing the number of variants shared between the mouse melanoma cell lines.
3.2. Genomic characterisation of murine melanoma cell lines with contrasting metastatic capabilities
To facilitate comparative analyses, we selected five mouse melanoma cell lines with different metastatic capabilities: B16‐F0, B16‐F10 and B16‐BL6 derived from C57BL/6 mice and K1735‐P and K1735‐M2, derived from the C3H strain (Fidler, 1970, 1973; Kripke, 1979; Kripke et al., 1978). Prior to genomic analysis, we validated the metastatic capabilities of these lines in vivo using an experimental metastasis assay (Fig. 1B–C). Consistent with previous reports (Poste et al., 1980; Talmadge and Fidler, 1982), B16‐BL6 cells were highly metastatic when compared to B16‐F10 or B16‐F0 cells and K1735‐M2 cells were highly metastatic when compared to K1735‐P cells. Spectral karyotyping of these cell lines showed high levels of polyploidy and multiple chromosomal aberrations (Figs. S1‐S2 and Table S4). We sequenced each of these lines to 30‐56x whole‐genome coverage, using the Illumina HiSeq platform. To identify somatic mutations (SNVs), we mapped these data to the reference C57BL/6J genome (GRCm38) and filtered the calls using variants described by the Mouse Genomes Project (Keane et al., 2011) and for quality (as detailed in Fig. S3). The number of variants shared among the lines within the B16 and K1735 groups is shown in Fig. 1D. The B16 lines showed higher numbers of somatic SNVs and short indels (~ < 50 bp) than the K1735 lines, with an average of 267 566 and 243 913 SNV, respectively (Fig. S4). A copy number analysis was also performed (Fig. S5 and Table S5). To assess our variant calling, we randomly selected 262 variants for validation by Sequenom genotyping (146 identified from the B16 cell lines and 116 from the K1735 lines), obtaining an overall validation rate of 90.86% for the B16 lines and 76.72% for the K1735 lines (Fig. S6). In B16 cell lines, the predominant mutation type was T > G (Fig. 2A) and the predominant mutational signature was Mmus‐S1 (Fig. 2B), which shows highest similarity to human mutational signature, signature 17 (Alexandrov et al., 2013) (cosine similarity 0.872) – a signature whose aetiology is currently unknown but has been observed in melanoma tumours (http://cancer.sanger.ac.uk/cosmic/signatures). In K1735 cell lines, the predominant mutational signature was Mmus‐S2 (Fig. 2B), with similarity to the UV light signature reported by COSMIC (signature 7; cosine similarity 0.597), which is in keeping with the genesis of these lines following the combined administration of UV light and croton oil (Kripke, 1979; Kripke et al., 1978; Talmadge and Fidler, 1982). Further characterisation revealed that both K1735 cell lines carried a homozygous activating mutation in Nras (p.G13D) and deletion of the first exon of Cdkn2a (p19 gene), as previously reported (Melnikova et al., 2004) (Fig. S7), as well as an unreported Trp53 mutation (p.T74P) in K1735‐P cells (Fig. 2C). B16 cell lines carried a deletion of the entire Cdkn2a locus, as previously reported (Melnikova et al., 2004) (Fig. S7), as well as an unreported heterozygous missense mutation in Braf (p.C263R; predicted to be deleterious by SIFT), a missense Trp53 mutation (p. N125D) and a mutation in Pten (p.T131P; Fig. 2C). Finally, we could observe that mutations in Rac1 and Nf1 were only present in the more invasive derivative lines. For instance, the Rac1 missense mutation (p.A59S) was present only in B16‐F10 cells. Similarly, the splice site variant (Chr11.79408779T>G) within Nf1, was observed in B16‐BL6 and K1735‐M2.
Figure 2.
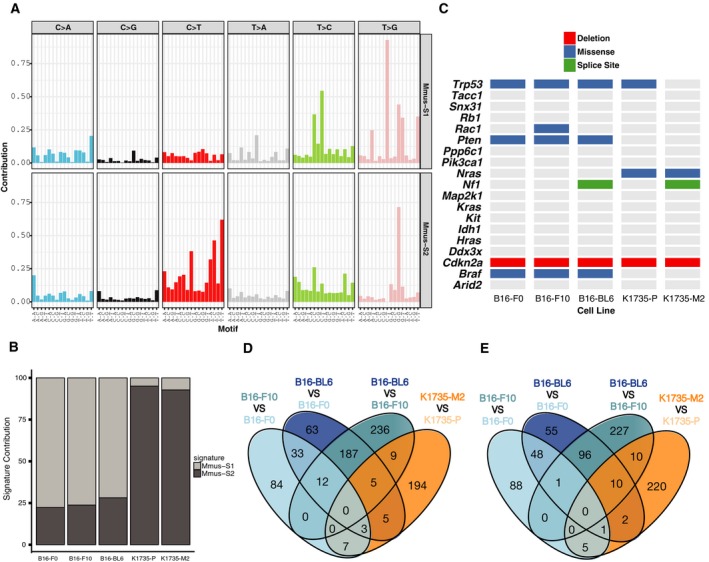
Characterisation of mouse melanoma cell line series. (A) Somatic mutational signatures operative in the genomes of mouse melanoma cell lines. (B) Signature contribution in the mouse melanoma cell line genomes for each process identified. (C) Matrix showing the mutations in known melanoma driver genes found in mouse melanoma cell lines. Venn diagrams showing the number of genes identified as differentially (D) overexpressed or (E) underexpressed across the multiple paired comparisons between a cell line with higher metastatic potential and its parental line.
3.3. Transcriptomic characterisation of murine melanoma cell lines
We generated RNA‐seq data from five biological replicates for each of the five melanoma cell lines. We mapped the reads against the GRCm38 mouse reference genome, counted the number of read pairs and verified the correlation among biological replicates (r > 0.95; Fig. S8). In an effort to identify changes in RNA levels that associate with higher metastatic capabilities, we identified all genes that were differentially expressed between the parental lines (B16‐F0, K1735‐P) and their more metastatic derivatives (B16‐F10, B16‐BL6 and K1735‐M2). To do this, we performed all possible paired comparisons within each group using DESeq2 (Love et al., 2014). Genes were classified as differentially expressed if their P‐value, after multiple testing correction, was P‐adj < 0.01, with an expression change of fourfold or more. In this way, we identified a total of 1430 genes that were differentially expressed (Table S6). qPCR was performed on selected genes for validation (Fig. S9). Notably, no genes were consistently differentially expressed across the comparisons of B16 and K1735 parental lines to their more metastatic derivatives (Fig. 2D–E), suggesting that different mechanisms confer metastatic potential in these cell lines series.
3.4. Identification of conserved putative regulators of metastatic colonisation in melanoma
To identify putative regulators of metastatic colonisation in melanoma, we next took a comparative genomics approach (Fig. 3A). We identified the human orthologues for all 1430 differentially expressed genes identified from the mouse melanoma cell line comparisons. For each mouse gene, we selected the orthologue with the highest protein sequence identity between mouse and humans. All paralogous genes were discarded. These criteria retained 1290 of the 1430 differentially expressed genes. We intersected these genes with the 7584 genes analysed in both human cohorts, which left 338 genes; 61 of which were significantly predictive of survival in both human cohorts (FDR < 0.1). Of these 61 genes, 28 genes showed the same direction of expression change (up‐ or downregulation) in relation to poor patient outcome and cell line metastasis phenotype (Table 1, Fig. 3B). A summary of the gene numbers obtained through each stage of our analysis is presented in Fig. S10. To assess the statistical significance of this result, we performed two independent randomisation tests revealing that the probability of obtaining 28 concordant genes by chance when intersecting a gene set with the human survival data was P (x ≥ 28) = 0.024 (when n = 1290) and P (x ≥ 28) = 0 (when n = 388) (Fig. S11).
Figure 3.
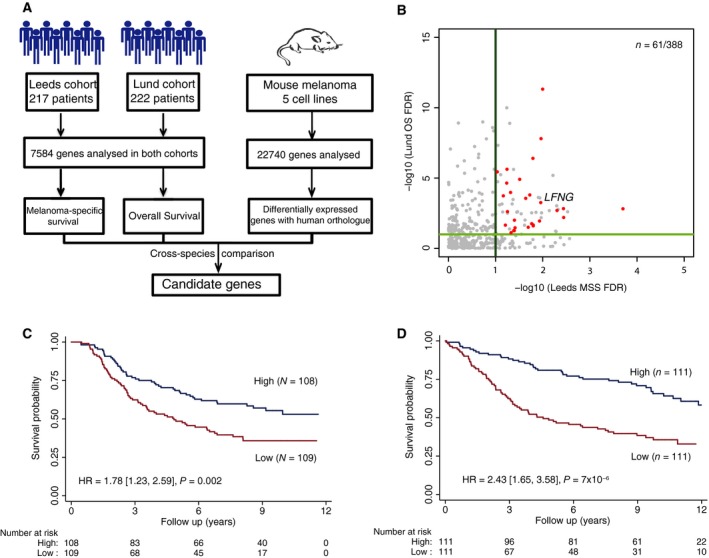
Cross‐species metastasis colonisation gene candidate identification. (A) Diagram showing the cross‐species approach used to identify gene candidates. (B) Scatter plot showing the corrected P‐values (−log10) obtained from the survival analysis for the 388 genes that can be analysed in both human patient cohorts and whose orthologue in mouse were identified as differentially expressed in metastatic cell lines. In red, genes with concordance between the expression changes in the mouse cell lines comparisons and the gene levels associated with poor outcome in both patient cohorts with an FDR < 0.1 are shown. (C) Kaplan–Meier curve showing the melanoma‐specific patient survival in the Leeds cohort when stratified by LFNG expression. (D) Kaplan–Meier curve showing the overall patient survival in the Lund cohort when stratified by LFNG expression. The plots show the results when the data are stratified by median expression into high and low LFNG expression groups. The hazard ratio shown is for the low expression group vs the high expression group.
Table 1.
Candidate genes identified in this study. Genes identified to be associated with metastasis and poor patient prognosis. This table shows gene expression on a continuous scale. The hazard ratios (HR) shown are per each additional unit of log2 gene expression
| Gene | Leeds cohort Melanoma‐Specific Survival | Lund cohort overall survival | ||||||
|---|---|---|---|---|---|---|---|---|
| Hazard Ratio | 95% C.I. | P‐val | FDR | Hazard Ratio | 95% C.I. | P‐val | FDR | |
| CD82 | 0.58415 | 0.48411, 0.70485 | 2.03E‐08 | 0.00020 | 0.52520 | 0.37432, 0.73692 | 0.00019 | 0.00153 |
| LFNG | 0.60248 | 0.45858, 0.79153 | 0.00027 | 0.01102 | 0.52328 | 0.38240, 0.71605 | 5.18E‐05 | 0.00055 |
| PTK2B | 0.64250 | 0.52325, 0.78893 | 2.41E‐05 | 0.00364 | 0.47279 | 0.31916, 0.70039 | 0.00019 | 0.00148 |
| CCL5 | 0.67761 | 0.56575, 0.81157 | 2.35E‐05 | 0.00360 | 0.57686 | 0.41392, 0.80393 | 0.00116 | 0.00629 |
| LSP1 | 0.68714 | 0.57354, 0.82325 | 4.71E‐05 | 0.00497 | 0.48760 | 0.33130, 0.71764 | 0.00027 | 0.00201 |
| DDX60 | 0.71537 | 0.59150, 0.86520 | 0.00056 | 0.01594 | 0.53727 | 0.34560, 0.83524 | 0.00578 | 0.02156 |
| PARP14 | 0.71561 | 0.58722, 0.87208 | 0.00091 | 0.02035 | 0.57359 | 0.37679, 0.87319 | 0.00953 | 0.03145 |
| TUBA4A | 0.71722 | 0.60117, 0.85567 | 0.00022 | 0.01002 | 0.31663 | 0.23623, 0.42440 | 1.42E‐14 | 4.79E‐12 |
| GPX3 | 0.71882 | 0.60071, 0.86016 | 0.00031 | 0.01170 | 0.26009 | 0.10878, 0.62186 | 0.00246 | 0.01130 |
| NDUFA4L2 | 0.72297 | 0.60147, 0.86901 | 0.00055 | 0.01588 | 0.67309 | 0.50482, 0.89746 | 0.00700 | 0.02488 |
| GABRE | 0.72349 | 0.59532, 0.87925 | 0.00114 | 0.02295 | 0.42004 | 0.28150, 0.62674 | 2.16E‐05 | 0.00028 |
| BTBD6 | 0.72365 | 0.58391, 0.89682 | 0.00313 | 0.04005 | 0.47218 | 0.25171, 0.88574 | 0.01938 | 0.05445 |
| ITM2A | 0.72406 | 0.60252, 0.87011 | 0.00057 | 0.01613 | 0.35698 | 0.25098, 0.50776 | 1.00E‐08 | 3.96E‐07 |
| HDC | 0.73176 | 0.61012, 0.87764 | 0.00076 | 0.01881 | 0.63449 | 0.51806, 0.77708 | 1.09E‐05 | 0.00016 |
| ELF4 | 0.73257 | 0.61273, 0.87584 | 0.00064 | 0.01716 | 0.56910 | 0.38619, 0.83865 | 0.00438 | 0.01738 |
| RIPK3 | 0.73743 | 0.62606, 0.86861 | 0.00027 | 0.01085 | 0.40256 | 0.30353, 0.53388 | 2.67E‐10 | 1.57E‐08 |
| CCBE1 | 0.73817 | 0.59969, 0.90862 | 0.00418 | 0.04704 | 0.35930 | 0.14251, 0.90584 | 0.03004 | 0.07619 |
| PON3 | 0.74237 | 0.61458, 0.89673 | 0.00200 | 0.03083 | 0.64187 | 0.53994, 0.76304 | 5.02E‐07 | 1.23E‐05 |
| BOC | 0.74269 | 0.61053, 0.90345 | 0.00292 | 0.03872 | 0.67546 | 0.50016, 0.91219 | 0.01048 | 0.03398 |
| RUNX1T1 | 0.74951 | 0.61916, 0.90730 | 0.00310 | 0.03987 | 0.57779 | 0.40754, 0.81917 | 0.00207 | 0.00983 |
| FILIP1L | 0.76881 | 0.64177, 0.92100 | 0.00433 | 0.04808 | 0.49836 | 0.36807, 0.67478 | 6.67E‐06 | 0.00011 |
| NDRG2 | 0.77243 | 0.64328, 0.92751 | 0.00567 | 0.05603 | 0.56030 | 0.40811, 0.76923 | 0.00034 | 0.00241 |
| TSPAN33 | 0.77331 | 0.65148, 0.91792 | 0.00329 | 0.04124 | 0.46715 | 0.24663, 0.88486 | 0.01953 | 0.05474 |
| FAM110C | 0.77589 | 0.64552, 0.93259 | 0.00686 | 0.06188 | 0.62169 | 0.44282, 0.87281 | 0.00603 | 0.02221 |
| JUP | 0.77882 | 0.65184, 0.93053 | 0.00591 | 0.05722 | 0.61515 | 0.51528, 0.73437 | 7.62E‐08 | 2.36E‐06 |
| RUNX3 | 0.78039 | 0.64937, 0.93784 | 0.00818 | 0.06857 | 0.34593 | 0.21468, 0.55742 | 1.29E‐05 | 0.00018 |
| EGLN3 | 0.78592 | 0.64904, 0.95166 | 0.01361 | 0.09212 | 0.42935 | 0.31385, 0.58735 | 1.23E‐07 | 3.61E‐06 |
| MID1 | 1.37339 | 1.09496, 1.72262 | 0.00606 | 0.05803 | 3.45380 | 2.09940, 5.68197 | 1.06E‐06 | 2.28E‐05 |
Of the above‐mentioned 28 genes, only one was upregulated in poor outcome patients, specifically MID1. Notably, 5 of 28 genes we identified have previously been reported to affect melanoma metastasis: CD82, NDRG2, RUNX3, CCL5 and HDC. For example, reports suggest that CD82 expression in melanoma cells inhibits tumour cell extravasation and lung metastasis formation in vivo (Khanna et al., 2014); upregulation of CD82 predicted better outcome in our analysis of two independent cohorts (Table 1). In addition to this, the tumour suppressor N‐myc downstream‐regulated gene 2 (NDRG2) is known to restrict melanomagenesis by regulating Mitf expression (Kim et al., 2008). Similarly, the tumour suppressor RUNX3 has been shown to be downregulated in metastatic melanoma lines when compared to primary melanoma or healthy skin (Kitago et al., 2009), while expression of the chemokine and leucocyte chemoattractant CCL5 in B16 cells strongly suppresses lung metastasis (Aravindaram et al., 2009). Finally, the histidine decarboxylase (HDC) gene encodes a protein whose function is to convert L‐histidine to histamine. Histamine has been shown to play an important role in immune cell function (Hansson et al., 1999), and a histamine/IL‐2 combination has been used to increase T‐cell responses in stage IV melanoma patients (Asemissen et al., 2005). In addition to the five genes mentioned above, an additional nine genes (CCBE1, GPX3, PTK2B, LSP1, DDX60, PARP14, GABRE, JUP and MID1) have been associated with metastasis in other types of epithelial or solid cancers (Aktary and Pasdar, 2013; Bachmann et al., 2014; Hao et al., 2010; Jeltsch et al., 2014; Koral et al., 2015; Yue et al., 2015; Zhang et al., 2014b).
Next, we ranked the 28 identified genes based on their hazard ratios and overall patient survival, as well as their P‐values after multiple test correction (Table 1). Based on these criteria, we selected the gene LFNG for further analysis.
Lunatic fringe (LFNG) is a glycosylating enzyme that post‐translationally modifies Notch receptor proteins (LeBon et al., 2014). LFNG‐mediated glycosylation of Notch receptors alters the binding affinity of Notch proteins with their ligands and activation of Notch receptors by delta‐like ligands (Kakuda and Haltiwanger, 2017). In our analyses, low RNA levels of LFNG were shown to be associated with poor outcome in both patient cohorts (Fig. 3C–D). Moreover, LFNG is prognostic for melanoma‐specific survival independent of Breslow thickness. Furthermore, B16‐BL6 cells had decreased Lfng expression when compared to its B16‐F0 parental line (log2(foldchange) = −2.185773, P = 1.069061 × 10−6, negative binomial Wald test with Benjamini–Hochberg correction), which was confirmed by qPCR (Fig. S12A).
3.5. Lfng disruption enhances the lung colonisation capabilities of CRISPR/Cas9‐targeted melanoma cells
To test the effect of Lfng disruption on the metastatic capabilities of melanoma cells, we used CRISPR/Cas9 to target Lfng in order to determine whether this may confer enhanced metastatic capabilities upon the weakly metastatic B16‐F0 cell line (Fig. S12B). We generated two independently targeted Lfng null B16‐F0 clones, termed g2d1 and L1 (Fig. 4A–B). G2d1 cells carried a single base insertion resulting in a frameshift loss‐of‐function mutation (Fig. S13) and L1 cells carried a 4.8‐kb deletion encompassing exons 1–4 which we verified by exome sequencing (Fig. S14). Additionally, exome sequencing identified that L1 cells carried 413 variants (334 SNVs and 79 Indels) not present in B16‐F0 cells. Of these variants, 120 were missense, two were nonsense (altering genes Aqp3 and Vmn2r115) and three were frameshift mutations (affecting Cyp7b1, Olfr657, and Vmn2r115).
Figure 4.
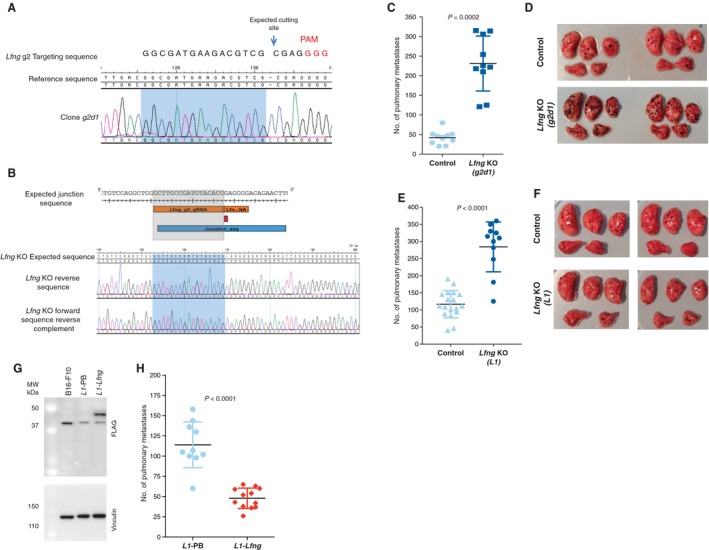
In vivo validation of the role of Lfng in metastasis. To validate the role of Lfng in metastasis, two independent Lfng targeting experiments were performed in B16‐F0 cells: one using a single gRNA to introduce a single base pair insertion (A, C and D) and another using two gRNAs to induce a 4.8‐kb deletion (B, E and F). (A) Sanger sequence trace of the targeted region in clone g2d1 carrying a homozygous 1‐bp insertion. (B) Image showing from top to bottom, the expected junction sequence after the deletion caused by the targeting of Lfng using two gRNAs, the expected reference sequence and the Sanger sequence traces observed and assembled with SeqMan Pro (Lasergene) against the expected reference sequence. The expected junction sequence separated by a single base insertion can be observed. Experimental metastasis assays using control and Lfng‐deficient cell lines (tail‐vein‐injected into wild‐type female mice (symbols representing individual mice with horizontal bar at the mean ± SD and statistics performed using a Mann–Whitney test; data shown are representative of two independent experiments)). Photographs are representative images of the lungs from mice injected with control and Lfng‐deficient cell lines. Plasmid rescue showing that introduction of the Lfng cDNA reverts the metastatic phenotype of L1 cells (L1‐Lfng; Lfng‐transfected cells, L1‐PB, vector‐only controls) (G and H). (G) A western blot with an anti‐Flag antibody shows restoration of Lfng expression (clone L1‐Lfng). An anti‐vinculin antibody was used as a loading control. These results are representative of three independent experiments. (H) Experimental metastasis assays using control L1‐PB cells and Lfng‐transfected cells. Please note experiments in e and h were performed with 5 × 105 and 4 × 105 cells, respectively, hence the different metastasis counts.
In an experimental metastasis assay, both g2d1 cells (Fig. 4C–D) and L1 cells (Fig. 4E–F) showed significantly increased numbers of pulmonary metastases when compared to control cells (transfected with an empty guide vector). These results directly demonstrate that Lfng loss enhances the metastatic capabilities of B16‐F0 cells. It was notable that compared to clone g2d1, clone L1 reproducibly produced numerous smaller lung foci. To further confirm the role of Lfng in metastasis, we used a full‐length Lfng cDNA to rescue the metastatic phenotype of L1 cells (Fig 4G–H).
3.6. Analysis of somatic LFNG mutations in human melanomas
Using two large patient cohorts (Jonsson et al., 2010; Nsengimana et al., 2015), we showed that reduced expression of LFNG is associated with poor patient outcome. We next evaluated the prevalence of inactivating somatic LFNG mutations in The Cancer Genome Atlas (TCGA) cutaneous melanoma collection (Cancer Genome Atlas Network, 2015) comparing primary vs metastatic melanoma. In this way, we identified five samples (5/481) with nonsilent mutations (four missense and one nonsense), all of which were in metastases. Due to short follow‐up times reported for melanoma primary tumours by TCGA, assessment of the association between LFNG expression and survival is not possible.
3.7. Analysis of NOTCH pathway gene expression in B16‐derived melanoma lines
To explore the role of LNFG in metastasis further, we next used our cell line transcriptome sequence data to examine the expression of Notch pathway components (Fig. 5). We observed significant changes in Notch pathway elements in B16‐BL6 such as upregulation of Notch2, Jag1, Hes1, Esr1 and Rbpj, as well as downregulation of Rfng and Dll3, when compared to the B16‐F0 line. This result is in keeping with a functional role for Lfng expression in the metastatic phenotypes observed.
Figure 5.
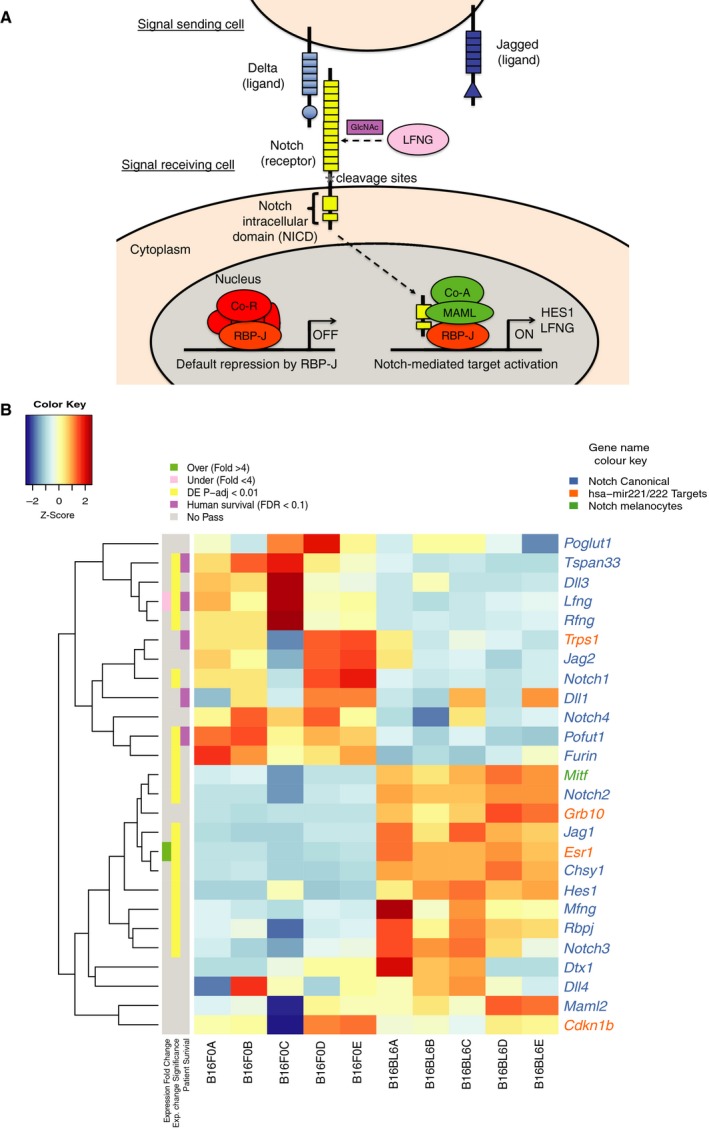
Notch pathway expression. (A) Diagram showing the canonical Notch pathway (LFNG‐mediated glycosylation occurs in the Golgi but is shown to depict the effect it has in mediating NOTCH–delta‐like ligand interactions). (B) Heatmap showing the z‐scores calculated using the normalised read counts obtained from DESeq2 for the multiple components of the Notch signalling pathway in the transcriptomes of B16‐F0 and B16‐BL6 mouse melanoma cell lines. Gene names are coloured according to their established relationship with the Notch pathway. On the left is indicated if a gene passed the thresholds of expression fold change (> 4), statistical significance of the differential expression, as calculated by DESeq2 (P‐adj < 0.01) and FDR threshold (FDR < 0.1) of the human orthologue in the human survival analysis.
4. Discussion
In this study, we combined expression data from two cohorts of melanoma patients with the analysis of a selection of mouse cell lines with different metastatic capabilities to identify LFNG as a regulator of metastasis. We validated these results using CRISPR genome editing to transform a weakly metastatic mouse cell line to a highly metastatic line by the disruption of Lfng. Although our focus here was LFNG, many of the other genes we discovered in our analysis may also regulate melanoma metastasis. For example, the DEXD/H box helicase 60 (DDX60) gene, a known regulator of the antiviral response and a DNA‐/RNA‐binding protein, has been reported to be associated with the development and prognosis of squamous cell carcinoma (Fu et al., 2016). Similarly, PTK2B has been reported to be involved in the CCR7‐mediated regulation of metastasis in squamous cell carcinoma (Yue et al., 2015) and was found to be downregulated in our study.
Notch signal transduction occurs when a Notch ligand (Jag or delta‐like) from a sender cell binds to a Notch receptor on an adjacent receiver cell (Bray, 2016). This event triggers the proteolytic cleavage of the Notch intracellular domain (NICD). Subsequently, NICD migrates to the nucleus and binds RBP‐JK to transcribe target genes. Notch receptor–delta‐like ligand binding affinity is regulated by the fringe glycosylating enzymes (LFNG, MFNG and RFNG) (Kakuda and Haltiwanger, 2017). In this study, we show that low RNA levels of LFNG predict worse outcome in patients with melanoma. Further, we find that Lfng is downregulated in B16‐BL6 cells when compared to parental B16‐F0 cells. Lfng ablation in B16‐F0 cells using CRISPR enhanced the number of pulmonary metastases in support of its role in regulating metastasis. Previous work in pancreatic cancer has showed that deletion of Lfng in a Kras LSL‐G12D mouse model upregulates Notch3 and Hes1, accelerating cell proliferation (Zhang et al., 2016). A role for LFNG in breast and prostate cancer has also been reported (Xu et al., 2012; Zhang et al., 2014a). In highly metastatic B16‐BL6 cells, we observed overexpression of the notch effectors, Hes1 and Notch2 (Fig. 5) as well as Rbpj, Jag1, Maml2 and Mitf, relative to B16‐F0 cells. Notably, delta‐like ligand (Dll3), as well as the fringe genes, Lfng and Rfng, were underexpressed (Fig. 5) in comparison with the Jag ligands, Jag1 and Jag2. This suggests that B16 cells are more suited to activate the Notch signalling pathway via Jag ligands.
In summary, this study shows how a cross‐species approach provides a useful framework for the identification of clinically relevant genes that play a role in metastasis. Identifying genetic markers such as LNFG is important as it helps to identify those patients at greatest risk of disease spread and may help guide their management.
Author contributions
MDCV‐H, LvdW and DJA devised the experiments. MDCV‐H performed the analysis of the mouse cell line data, the comparisons with the human survival data, designed the plasmids and gRNAs, carried out the double gRNA targeting experiments and validation of all the targeting experiments on the mouse cell lines. LvdW performed all the in vivo experimental metastasis assays and the single gRNA targeting of the mouse cells. JN performed the QC and survival analysis on the human patient cohorts. MKS and AOS performed the RT‐qPCRs on the targeted cell lines. JN‐B and DTB provided the data for the Leeds cohort. GJ provided the data for the Lund cohort. MDCV‐H, LvdW and DJA led the project. MDCV‐H, LvdW and DJA wrote the manuscript with contributions from all authors.
Supporting information
Fig. S1. Spectral Karyotyping of the B16 cell lines. Spectral karyotype analysis of ten different metaphases from (A) B16‐F0, (B) B16‐F10 and (C) B16‐BL6 cells. High levels of polyploidy, multiple chromosomal aberrations and at least one event of whole genome amplification can be observed.
Fig. S2. Spectral Karyotyping of the K1735 cell lines. Spectral karyotype analysis of ten different metaphases from (A) K1735‐P and (B) K1735‐M2. High levels of polyploidy, multiple chromosomal aberrations and at least one event of whole genome amplification can be observed.
Fig. S3. Cell line somatic variant calling and filtering strategy. Diagram showing the multiple steps followed to call single nucleotide variants and short indels from whole genome data of the murine lines in the absence of a matched normal sample from the same mouse.
Fig. S4. Somatic variants in murine melanoma cell lines (A) Total number of SNV and indel variants identified in each cell line. (B) Mean number SNVs identified in each mouse melanoma cell line genome. (C) Bar plot showing the mutational spectra of base substitutions identified in the lines according to the 96‐substitution type and genomic context classification.
Fig. S5. Variation in highly metastatic mouse cell lines. (A) Circos plot showing from the innermost track; somatic short indels and SNVs identified uniquely in the B16‐BL6 cell line genome, the CNVs identified in the B16‐BL6 cell line against the B16‐F0 genome, and the CNVs identified in the B16‐F0. (B) Circos plot showing from the innermost track somatic short indels, SNVs identified uniquely in the K1735‐M2 cell line genome, the CNVs identified in the K1735‐M2 cell line against the K1735‐P and the CNVs identified in the K1735‐P parental line against the C3H/HeN genome.
Fig. S6. Orthogonal validation of SNVs identified in the murine melanoma lines. A total of 262 variants were tested; 146 from the B16 cell line group and 116 from the K1735 lines; using three biological replicates per cell line. (A) Bar plot showing the proportion of SNVs that were validated using the Sequenom technology across three different replicates per cell line. (B) Box and whisker plot showing the proportion of validated SNVs per cell line across the three replicates, whiskers represent the upper and lower quartiles and solid thick line represents the mean.
Fig. S7. Cdkn2a genomic deletions. (A) Screenshot from the integrated genomics viewer showing the coverage of the Cdkn2a locus, from top to bottom, on the C57BL/6 genome data from (Keane et al., 2011), the B16‐F0, B16‐F10 and B16‐BL6 cell line genomes. (B) Screenshot from the integrated genomics viewer showing the coverage of the Cdkn2a locus, from top to bottom, on the C3H/HeJ genome data from (Keane et al., 2011), the K1735‐P and K1735‐M2 cell line genomes.
Fig. S8. Hierarchical clustering of the murine cell line RNA‐seq data. Heat map showing the hierarchical clustering of different biological replicates sequenced based on the Pearson correlation coefficient obtained from all log2(TPM + 1) values across all the protein coding genes. The two groups of cell lines can be clearly observed.
Fig. S9. Analysis of differentially expressed genes. Venn diagram showing the (A) overexpressed and (B) under‐expressed genes selected for qPCR validation. (C‐F) Gene expression levels with ΔΔCT value being relative to the respective parental line.
Fig. S10. Summary of genes assessed to identify regulators of metastatic colonisation by comparative genomics. Flow chart showing the number of genes obtained throughout the different stages of our analysis to identify regulators of metastatic colonisation in melanoma.
Fig. S11. Number of overlapping and concordant genes on random simulated samples. The null distribution of overlapping genes observed across 1000 samples in a set of randomisation tests with sample sizes of (A) n = 1290, (B) n = 388. The dashed red line shows the number of genes observed in our main analysis. The probability of obtaining the same number of overlapping genes as the ones observed in the real data is shown.
Fig. S12. Validation of reduced Lfng expression in B16‐BL6 cells and plasmid constructs used to generate Lfng‐deficient B16‐F0 cells. (A) Fold change in expression of Lfng in B16‐BL6 cells against B16‐F0 cells as measured by qPCR, whiskers shows the standard error and P‐value was calculated using two tailed t test from 3 biological replicates. (B) Schematics of the different plasmids used.
Fig. S13. Lfng targeting and validation of g2d1 clone. (A) Diagram showing the targeting location of the gRNA (Lfng_g2) used in the single targeting experiment. (B) Expression analysis of g2d1 by quantitative RT‐PCR. Fold change in expression of Lfng in g2d1 cells against control cells as measured by qPCR, whiskers shows the standard error and P‐value was calculated using two tailed t test from 3 biological replicates. This frameshift mutation, although disrupting the gene, appears to cause an upregulation of Lfng mRNA expression although the expression difference is not statistically significant. (C) Pairwise alignment using CLUSTALX 2.1 between mouse Lfng protein (from Transcript ENSMUST00000031555) and the resulting predicted protein in clone (g2d1) mutated Lfng alleles. The single base insertion at the Lfng locus causes a frameshift that introduces a stop codon 36 amino acids downstream of the mutation site.
Fig. S14. Lfng targeting and validation of L1 clone. (A) Diagram showing the targeting location of the gRNAs (Lfng_g2 and Lfng_g3) used in the double targeting experiment. (B) Fold change in expression of Lfng in L1 cells against control cells as measured by quantitative RT‐PCR, whiskers show the standard error and P‐value was calculated using two tailed t test from 3 biological replicates. IGV screenshot showing mapped reads from the whole exome sequencing data generated from the Lfng KO clone (L1). Forward reads are shown in blue and reverse reads are shown in pink. Mismatched bases in comparison with the reference genome are highlighted above the read. The position of the targeting sites for gRNAs (C) Lfng_g2 gRNA and (D) Lfng_g3 gRNA are highlighted with a red box.
Table S1. Patient cohort demographic information. Demographic information for the two patient cohorts analysed where available.
Table S2. Predictors of patient outcome in both melanoma patient cohorts. Gene name, hazard ratios, confidence intervals, P‐value and corrected P‐values for all the genes with a P‐val < 0.1 after applying the FDR correction.
Table S3. Survival predictors gene set functional annotation enrichment. Results of the functional annotation enrichment analysis performed with DAVID with the list of gene expression predictors of outcome.
Table S4. Summary of chromosomal aberrations identified by spectral karyotyping of the murine melanoma cell lines. Summary of chromosomal aberrations detected by spectral karyotyping in the different mouse melanoma cell lines.
Table S5. Summary of Copy Number Variants identified on the mouse melanoma cell line genomes. Copy number variants (CNV) calls identified in the cell line genomes for the parental lines (B16‐F0 and K1735‐P). Somatic CNVs are reported for the metastatic lines (B16‐F10, B16‐BL6 and K1735‐M2).
Table S6. Differentially Expressed genes identified across the comparisons of all the murine melanoma cell lines. Information on the 1430 genes that were differentially expressed throughout all the comparisons.
Table S7. Oligos and gRNA sequences.
Acknowledgements
MDCV‐H, AOS, LvdW, and DJA were supported by Cancer Research UK, the ERC Synergy (Combat Cancer) programme and the Wellcome Trust. We thank Sandra Gomes Pereira and Fengtang Yang from the Sanger Institute Cytogenetics Facility for performing FISH, James Hewinson for assistance with cloning the Piggy_gRNAScaffold plasmid and the staff of the Research Support Facility (RSF) for animal care. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007‐2013) / ERC synergy grant agreement n° 319661 COMBATCANCER.
References
- Aktary Z and Pasdar M (2013) Plakoglobin represses SATB1 expression and decreases in vitro proliferation, migration and invasion. PLoS ONE 8, e78388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik‐Zainal S and Stratton MR (2015) Clock‐like mutational processes in human somatic cells. Nat Genet 47, 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik‐Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen‐Dale AL et al ; Australian Pancreatic Cancer Genome I ; Consortium IBC ; Consortium IMS ; PedBrain I (2013) Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT and Huber W (2015) HTSeq‐a Python framework to work with high‐throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravindaram K, Yu HH, Lan CW, Wang PH, Chen YH, Chen HM, Yagita H and Yang NS (2009) Transgenic expression of human gp100 and RANTES at specific time points for suppression of melanoma. Gene Ther 16, 1329–1339. [DOI] [PubMed] [Google Scholar]
- Asemissen AM, Scheibenbogen C, Letsch A, Hellstrand K, Thoren F, Gehlsen K, Schmittel A, Thiel E and Keilholz U (2005) Addition of histamine to interleukin 2 treatment augments type 1 T‐cell responses in patients with melanoma in vivo: immunologic results from a randomized clinical trial of interleukin 2 with or without histamine (MP 104). Clin Cancer Res 11, 290–297. [PubMed] [Google Scholar]
- Bachmann SB, Frommel SC, Camicia R, Winkler HC, Santoro R and Hassa PO (2014) DTX3L and ARTD9 inhibit IRF1 expression and mediate in cooperation with ARTD8 survival and proliferation of metastatic prostate cancer cells. Mol Cancer 13, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Park J and Kim JS (2014) Cas‐OFFinder: a fast and versatile algorithm that searches for potential off‐target sites of Cas9 RNA‐guided endonucleases. Bioinformatics 30, 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez‐Ramos D, Kohlmeyer J, Riesenberg S, van den Boorn‐Konijnenberg D, Homig‐Holzel C et al (2014) Ultraviolet‐radiation‐induced inflammation promotes angiotropism and metastasis in melanoma. Nature 507, 109–113. [DOI] [PubMed] [Google Scholar]
- Boeva V, Zinovyev A, Bleakley K, Vert JP, Janoueix‐Lerosey I, Delattre O and Barillot E (2011) Control‐free calling of copy number alterations in deep‐sequencing data using GC‐content normalization. Bioinformatics 27, 268–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray SJ (2016) Notch signalling in context. Nat Rev Mol Cell Biol 17, 722–735. [DOI] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M and van Steensel B (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2015) Genomic classification of cutaneous melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL and Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Chen GD, Cheng YB, Zhang ZZ, Martinka M and Li G (2011) Cytoplasmic Skp2 expression is increased in human melanoma and correlated with patient survival. PLoS ONE 6, e17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky WE, Rosenbaum LE and Bosenberg M (2010) Decoding melanoma metastasis. Cancers 3, 126–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST et al ; Genomes Project Analysis G (2011) The variant call format and VCFtools. Bioinformatics 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E and Huber W (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont AM (2016) Adjuvant ipilimumab in stage III melanoma: new landscape, new questions. Eur J Cancer 69, 39–42. [DOI] [PubMed] [Google Scholar]
- Fidler IJ (1970) Metastasis: quantitative analysis of distribution and fate of tumor embolilabeled with 125 I‐5‐iodo‐2’‐deoxyuridine. J Natl Cancer Inst 45, 773–782. [PubMed] [Google Scholar]
- Fidler IJ (1973) Selection of successive tumour lines for metastasis. Nat New Biol 242, 148–149. [DOI] [PubMed] [Google Scholar]
- Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3, 453–458. [DOI] [PubMed] [Google Scholar]
- Fu TY, Wu CN, Sie HC, Cheng JT, Lin YS, Liou HH, Tseng YK, Shu CW, Tsai KW, Yen LM et al (2016) Subsite‐specific association of DEAD box RNA helicase DDX60 with the development and prognosis of oral squamous cell carcinoma. Oncotarget 7, 85097–85108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring JS, Fischer B, Lawrence M and Huber W (2015) SomaticSignatures: inferring mutational signatures from single‐nucleotide variants. Bioinformatics 31, 3673–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan T, Messer AR, Amitai‐Lange A, Melamed Z, Ohana R, Bell RE, Kapitansky O, Lerman G, Greenberger S, Khaled M et al (2015) Interactions of melanoma cells with distal keratinocytes trigger metastasis via notch signaling inhibition of MITF. Mol Cell 59, 664–676. [DOI] [PubMed] [Google Scholar]
- Gregory RWBB, Bonebakker L, Gentleman R, Huber W, Liaw Y, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M et al (2013). gplots: Various R programming tools for plotting data, R package version 2.11.3 ed.
- Hannani D, Vetizou M, Enot D, Rusakiewicz S, Chaput N, Klatzmann D, Desbois M, Jacquelot N, Vimond N, Chouaib S et al (2015) Anticancer immunotherapy by CTLA‐4 blockade: obligatory contribution of IL‐2 receptors and negative prognostic impact of soluble CD25. Cell Res 25, 208–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson M, Hermodsson S, Brune M, Mellqvist UH, Naredi P, Betten A, Gehlsen KR and Hellstrand K (1999) Histamine protects T cells and natural killer cells against oxidative stress. J Interferon Cytokine Res 19, 1135–1144. [DOI] [PubMed] [Google Scholar]
- Hao JM, Chen JZ, Sui HM, Si‐Ma XQ, Li GQ, Liu C, Li JL, Ding YQ and Li JM (2010) A five‐gene signature as a potential predictor of metastasis and survival in colorectal cancer. J Pathol 220, 475–489. [DOI] [PubMed] [Google Scholar]
- Herrero J, Muffato M, Beal K, Fitzgerald S, Gordon L, Pignatelli M, Vilella AJ, Searle SM, Amode R, Brent S et al (2016). Ensembl comparative genomics resources. Database (Oxford) 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT and Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jafarnejad SM, Wani AA, Martinka M and Li G (2010) Prognostic significance of Sox4 expression in human cutaneous melanoma and its role in cell migration and invasion. Am J Pathol 177, 2741–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeltsch M, Jha SK, Tvorogov D, Anisimov A, Leppanen VM, Holopainen T, Kivela R, Ortega S, Karpanen T and Alitalo K (2014) CCBE1 enhances lymphangiogenesis via A disintegrin and metalloprotease with thrombospondin motifs‐3‐mediated vascular endothelial growth factor‐C activation. Circulation 129, 1962–1971. [DOI] [PubMed] [Google Scholar]
- Jonsson G, Busch C, Knappskog S, Geisler J, Miletic H, Ringner M, Lillehaug JR, Borg A and Lonning PE (2010) Gene expression profiling‐based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res 16, 3356–3367. [DOI] [PubMed] [Google Scholar]
- Kakuda S and Haltiwanger RS (2017) Deciphering the fringe‐mediated notch code: identification of activating and inhibiting sites allowing discrimination between ligands. Dev Cell 40, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M et al (2011) Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna P, Chung CY, Neves RI, Robertson GP and Dong C (2014) CD82/KAI expression prevents IL‐8‐mediated endothelial gap formation in late‐stage melanomas. Oncogene 33, 2898–2908. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R and Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Yang Y, Lee MS, Yoo YD, Lee HG and Lim JS (2008) NDRG2 gene expression in B16F10 melanoma cells restrains melanogenesis via inhibition of Mitf expression. Pigment Cell Melanoma Res 21, 653–664. [DOI] [PubMed] [Google Scholar]
- Kitago M, Martinez SR, Nakamura T, Sim MS and Hoon DS (2009) Regulation of RUNX3 tumor suppressor gene expression in cutaneous melanoma. Clin Cancer Res 15, 2988–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike‐Yusa H, Li Y, Tan EP, Del CastilloVelasco‐Herrera M and Yusa K (2014) Genome‐wide recessive genetic screening in mammalian cells with a lentiviral CRISPR‐guide RNA library. Nat Biotechnol 32, 267–273. [DOI] [PubMed] [Google Scholar]
- Koral K, Paranjpe S, Bowen WC, Mars W, Luo J and Michalopoulos GK (2015) Leukocyte‐specific protein 1: a novel regulator of hepatocellular proliferation and migration deleted in human hepatocellular carcinoma. Hepatology 61, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kripke ML (1979) Speculations on the role of ultraviolet radiation in the development of malignant melanoma. J Natl Cancer Inst 63, 541–548. [DOI] [PubMed] [Google Scholar]
- Kripke ML, Gruys E and Fidler IJ (1978) Metastatic heterogeneity of cells from an ultraviolet light‐induced murine fibrosarcoma of recent origin. Can Res 38, 2962–2967. [PubMed] [Google Scholar]
- LeBon L, Lee TV, Sprinzak D, Jafar‐Nejad H and Elowitz MB (2014) Fringe proteins modulate Notch‐ligand cis and trans interactions to specify signaling states. Elife 3, e02950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Genome Project Data Processing Subgroup (2009) The Sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W and Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P and Cunningham F (2016) The ensembl variant effect predictor. Genome Biol 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikova VO, Bolshakov SV, Walker C and Ananthaswamy HN (2004) Genomic alterations in spontaneous and carcinogen‐induced murine melanoma cell lines. Oncogene 23, 2347–2356. [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS et al (2000) Fringe is a glycosyltransferase that modifies Notch. Nature 406, 369–375. [DOI] [PubMed] [Google Scholar]
- Nsengimana J, Laye J, Filia A, Walker C, Jewell R, Van den Oord JJ, Wolter P, Patel P, Sucker A, Schadendorf D et al (2015) Independent replication of a melanoma subtype gene signature and evaluation of its prognostic value and biological correlates in a population cohort. Oncotarget 6, 11683–11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T et al (2004) A multigene assay to predict recurrence of tamoxifen‐treated, node‐negative breast cancer. New Engl J Med 351, 2817–2826. [DOI] [PubMed] [Google Scholar]
- Poste G, Doll J, Hart IR and Fidler IJ (1980) In vitro selection of murine B16 melanoma variants with enhanced tissue‐invasive properties. Can Res 40, 1636–1644. [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA and Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD and Livak KJ (2008) Analyzing real‐time PCR data by the comparative C‐T method. Nat Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Soltani MH, Pichardo R, Song Z, Sangha N, Camacho F, Satyamoorthy K, Sangueza OP and Setaluri V (2005) Microtubule‐associated protein 2, a marker of neuronal differentiation, induces mitotic defects, inhibits growth of melanoma cells, and predicts metastatic potential of cutaneous melanoma. Am J Pathol 166, 1841–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Robson T, Doig T, Brenn T, Mathers M, Brown ER, Doherty V, Bartlett JMS, Anderson N and Melton DW (2013) DNA repair and replication proteins as prognostic markers in melanoma. Histopathology 62, 343–350. [DOI] [PubMed] [Google Scholar]
- Talmadge JE and Fidler IJ (1982) Enhanced metastatic potential of tumor cells harvested from spontaneous metastases of heterogeneous murine tumors. J Natl Cancer Inst 69, 975–980. [PubMed] [Google Scholar]
- Turajlic S and Swanton C (2016) Metastasis as an evolutionary process. Science 352, 169–175. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van ‘t Veer LJ, Dai H, Hart AAM, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ et al (2002) A gene‐expression signature as a predictor of survival in breast cancer. New Engl J Med 347, 1999–2009. [DOI] [PubMed] [Google Scholar]
- Wardwell‐Ozgo J, Dogruluk T, Gifford A, Zhang Y, Heffernan TP, van Doorn R, Creighton CJ, Chin L and Scott KL (2013) HOXA1 drives melanoma tumor growth and metastasis and elicits an invasion gene expression signature that prognosticates clinical outcome. Oncogene 33, 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weyden L, Arends MJ, Campbell AD, Bald T, Wardle‐Jones H, Griggs N, Velasco‐Herrera MD, Tuting T, Sansom OJ, Karp NA et al ; Sanger Mouse Genetics Project (2017) Genome‐wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 541, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Usary J, Kousis PC, Prat A, Wang DY, Adams JR, Wang W, Loch AJ, Deng T, Zhao W et al (2012) Lunatic fringe deficiency cooperates with the Met/Caveolin gene amplicon to induce basal‐like breast cancer. Cancer Cell 21, 626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Li ZN, Fang QG, Zhang X, Yang LL, Sun CF and Liu FY (2015) The role of Pyk2 in the CCR7‐mediated regulation of metastasis and viability in squamous cell carcinoma of the head and neck cells in vivo and in vitro . Oncol Rep 34, 3280–3287. [DOI] [PubMed] [Google Scholar]
- Zhang S, Chung WC, Wu G, Egan SE and Xu K (2014a) Tumor‐suppressive activity of Lunatic Fringe in prostate through differential modulation of Notch receptor activation. Neoplasia 16, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chung WC and Xu K (2016) Lunatic Fringe is a potent tumor suppressor in Kras‐initiated pancreatic cancer. Oncogene 35, 2485–2495. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zheng Z, Yingji S, Kim H, Jin R, Renshu L, Lee DY, Roh MR and Yang S (2014b) Downregulation of glutathione peroxidase 3 is associated with lymph node metastasis and prognosis in cervical cancer. Oncol Rep 31, 2587–2592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Spectral Karyotyping of the B16 cell lines. Spectral karyotype analysis of ten different metaphases from (A) B16‐F0, (B) B16‐F10 and (C) B16‐BL6 cells. High levels of polyploidy, multiple chromosomal aberrations and at least one event of whole genome amplification can be observed.
Fig. S2. Spectral Karyotyping of the K1735 cell lines. Spectral karyotype analysis of ten different metaphases from (A) K1735‐P and (B) K1735‐M2. High levels of polyploidy, multiple chromosomal aberrations and at least one event of whole genome amplification can be observed.
Fig. S3. Cell line somatic variant calling and filtering strategy. Diagram showing the multiple steps followed to call single nucleotide variants and short indels from whole genome data of the murine lines in the absence of a matched normal sample from the same mouse.
Fig. S4. Somatic variants in murine melanoma cell lines (A) Total number of SNV and indel variants identified in each cell line. (B) Mean number SNVs identified in each mouse melanoma cell line genome. (C) Bar plot showing the mutational spectra of base substitutions identified in the lines according to the 96‐substitution type and genomic context classification.
Fig. S5. Variation in highly metastatic mouse cell lines. (A) Circos plot showing from the innermost track; somatic short indels and SNVs identified uniquely in the B16‐BL6 cell line genome, the CNVs identified in the B16‐BL6 cell line against the B16‐F0 genome, and the CNVs identified in the B16‐F0. (B) Circos plot showing from the innermost track somatic short indels, SNVs identified uniquely in the K1735‐M2 cell line genome, the CNVs identified in the K1735‐M2 cell line against the K1735‐P and the CNVs identified in the K1735‐P parental line against the C3H/HeN genome.
Fig. S6. Orthogonal validation of SNVs identified in the murine melanoma lines. A total of 262 variants were tested; 146 from the B16 cell line group and 116 from the K1735 lines; using three biological replicates per cell line. (A) Bar plot showing the proportion of SNVs that were validated using the Sequenom technology across three different replicates per cell line. (B) Box and whisker plot showing the proportion of validated SNVs per cell line across the three replicates, whiskers represent the upper and lower quartiles and solid thick line represents the mean.
Fig. S7. Cdkn2a genomic deletions. (A) Screenshot from the integrated genomics viewer showing the coverage of the Cdkn2a locus, from top to bottom, on the C57BL/6 genome data from (Keane et al., 2011), the B16‐F0, B16‐F10 and B16‐BL6 cell line genomes. (B) Screenshot from the integrated genomics viewer showing the coverage of the Cdkn2a locus, from top to bottom, on the C3H/HeJ genome data from (Keane et al., 2011), the K1735‐P and K1735‐M2 cell line genomes.
Fig. S8. Hierarchical clustering of the murine cell line RNA‐seq data. Heat map showing the hierarchical clustering of different biological replicates sequenced based on the Pearson correlation coefficient obtained from all log2(TPM + 1) values across all the protein coding genes. The two groups of cell lines can be clearly observed.
Fig. S9. Analysis of differentially expressed genes. Venn diagram showing the (A) overexpressed and (B) under‐expressed genes selected for qPCR validation. (C‐F) Gene expression levels with ΔΔCT value being relative to the respective parental line.
Fig. S10. Summary of genes assessed to identify regulators of metastatic colonisation by comparative genomics. Flow chart showing the number of genes obtained throughout the different stages of our analysis to identify regulators of metastatic colonisation in melanoma.
Fig. S11. Number of overlapping and concordant genes on random simulated samples. The null distribution of overlapping genes observed across 1000 samples in a set of randomisation tests with sample sizes of (A) n = 1290, (B) n = 388. The dashed red line shows the number of genes observed in our main analysis. The probability of obtaining the same number of overlapping genes as the ones observed in the real data is shown.
Fig. S12. Validation of reduced Lfng expression in B16‐BL6 cells and plasmid constructs used to generate Lfng‐deficient B16‐F0 cells. (A) Fold change in expression of Lfng in B16‐BL6 cells against B16‐F0 cells as measured by qPCR, whiskers shows the standard error and P‐value was calculated using two tailed t test from 3 biological replicates. (B) Schematics of the different plasmids used.
Fig. S13. Lfng targeting and validation of g2d1 clone. (A) Diagram showing the targeting location of the gRNA (Lfng_g2) used in the single targeting experiment. (B) Expression analysis of g2d1 by quantitative RT‐PCR. Fold change in expression of Lfng in g2d1 cells against control cells as measured by qPCR, whiskers shows the standard error and P‐value was calculated using two tailed t test from 3 biological replicates. This frameshift mutation, although disrupting the gene, appears to cause an upregulation of Lfng mRNA expression although the expression difference is not statistically significant. (C) Pairwise alignment using CLUSTALX 2.1 between mouse Lfng protein (from Transcript ENSMUST00000031555) and the resulting predicted protein in clone (g2d1) mutated Lfng alleles. The single base insertion at the Lfng locus causes a frameshift that introduces a stop codon 36 amino acids downstream of the mutation site.
Fig. S14. Lfng targeting and validation of L1 clone. (A) Diagram showing the targeting location of the gRNAs (Lfng_g2 and Lfng_g3) used in the double targeting experiment. (B) Fold change in expression of Lfng in L1 cells against control cells as measured by quantitative RT‐PCR, whiskers show the standard error and P‐value was calculated using two tailed t test from 3 biological replicates. IGV screenshot showing mapped reads from the whole exome sequencing data generated from the Lfng KO clone (L1). Forward reads are shown in blue and reverse reads are shown in pink. Mismatched bases in comparison with the reference genome are highlighted above the read. The position of the targeting sites for gRNAs (C) Lfng_g2 gRNA and (D) Lfng_g3 gRNA are highlighted with a red box.
Table S1. Patient cohort demographic information. Demographic information for the two patient cohorts analysed where available.
Table S2. Predictors of patient outcome in both melanoma patient cohorts. Gene name, hazard ratios, confidence intervals, P‐value and corrected P‐values for all the genes with a P‐val < 0.1 after applying the FDR correction.
Table S3. Survival predictors gene set functional annotation enrichment. Results of the functional annotation enrichment analysis performed with DAVID with the list of gene expression predictors of outcome.
Table S4. Summary of chromosomal aberrations identified by spectral karyotyping of the murine melanoma cell lines. Summary of chromosomal aberrations detected by spectral karyotyping in the different mouse melanoma cell lines.
Table S5. Summary of Copy Number Variants identified on the mouse melanoma cell line genomes. Copy number variants (CNV) calls identified in the cell line genomes for the parental lines (B16‐F0 and K1735‐P). Somatic CNVs are reported for the metastatic lines (B16‐F10, B16‐BL6 and K1735‐M2).
Table S6. Differentially Expressed genes identified across the comparisons of all the murine melanoma cell lines. Information on the 1430 genes that were differentially expressed throughout all the comparisons.
Table S7. Oligos and gRNA sequences.