

Abstract

We describe a new 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU)-mediated coupling reaction to produce 2-imino benzo[e]-1,3-oxazin-4-ones from salicylic acids and anilines. Mechanistic studies support a reaction pathway in which HATU mediates carbon transfer to the initially formed salicylanilides to form in succession reactive tetramethylisouronium and N-acyl(dimethyl)isouronium intermediates, which then undergo imine–iminium exchange to generate the desired oxazinones.
Introduction
Formation of amides through the use of coupling reagents is one of the most valuable methods in medicinal chemistry. Some of the most widely used coupling reagents are carbodiimides, phosphonium, and immonium salts. 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), a commonly used immonium salt, is a powerful coupling reagent because of its high efficiency and fast reaction rate.1−3 We envisioned that salicylanilide 3a (Scheme 1) could be generated via HATU-mediated coupling of salicylic acid (1a) and aniline (2a). When the reaction (Scheme 1) failed to produce 3a in a satisfactory yield (15%), we discovered that the major product was the unexpected 2-imino benzo[e]-1,3-oxazin-4-one 4a (39%). In view of this intriguing reaction course and the widespread use of HATU in the synthesis of amides and peptides, we investigated the mechanism by which HATU mediates a carbon transfer to 3a to give the cyclic product 4a.
Scheme 1. Initially Observed Reaction.

Results and Discussion
First, we attempted to identify all of the reaction intermediates and products by running the reaction with 4-bromoaniline (2b) in place of 2a as we envisioned that the presence of the bromine atom could facilitate product detection by gas chromatography–mass spectrometry (GC–MS) and NMR. As can be seen in Scheme 2, the major product was dibromo 2-imino benzo[e]-1,3-oxazin-4-one 4b (40%), along with guanidinium salt 5, salicylurea 6, and benzo[e][1,3]oxazine-2,4(3H)-dione 7. The formation of 7 led us to formulate a reaction pathway from 3a to 4a that would involve multiple isouronium intermediates (vide infra).
Scheme 2. Reaction of 1a and 2b with HATU.

Clearly, the observed distribution of reaction products suggests that HATU-mediated coupling is not well-suited for the synthesis of salicylanilides. To assess if 3a was a precursor of 4a, we conducted a reaction using 3a as the starting material, 1 equiv of 2a, and 1.5 equiv of HATU. As anticipated, 4a was produced in a 35% yield; thus, it appears that amide formation is likely the initial step in this transformation. However, substituting 2b for 2a in the same reaction produced a 1:1 ratio of monobromo 2-imino benzo[e]-1,3-oxazin-4-one regioisomers 4c (expected) and 4d (unexpected) in a combined yield of 38% (Scheme 3, reaction 1). 1D and 2D NMR data verified the structures of each isomer (see Supporting Information for details). When we ran the same reaction using N-(4-bromophenyl)-2-hydroxybenzamide (3b) and 2a (Scheme 3, reaction 2) as a complementary set of starting materials, we obtained the unexpected 4a in 29% yield and only trace amounts of 4c and 4d.
Scheme 3. Formation of 2-Imino Benzo[e]-1,3-oxazin-4-ones via Transamidation.

Yield determined by GC–MS.
Although the formation of unexpected product 4d added complexity to this reaction, it led us to propose a three-step mechanism (Scheme 4) to account for the conversion of salicylanilides 3 to 2-imino benzo[e]-1,3-oxazin-4-ones 4: (1) guanidinium exchange with HATU to form the linchpin 1,1,3,3-tetramethyl-2-phenylisouronium intermediate A and HOAt; (2) cyclization of tetramethylisouronium intermediates A and B (formed from A by transamidation) to form a pair of N-acyl(dimethyl)isouronium benzo[e]-1,3-oxazin-4-ones C; and (3) imine–iminium exchange to generate 2-imino benzo[e]-1,3-oxazin-4-ones 4. This mechanism also accounts for the formation of reaction products 6 and 7 (Scheme 2) as a result of two modes of hydrolysis of intermediates C.
Scheme 4. Proposed Mechanism for the Formation of Benzo[e]-1,3-oxazin-4-ones from Reactions of Salicylic Acids and Anilines with HATU.

Formation of the unexpected product 4a (Scheme 3, reaction 2) can be explained by a transamidation process where the more nucleophilic 2a can displace the less nucleophilic 2b to form the more stable intermediate C. Importantly, the more reactive aniline would also speed up the imine–iminium exchange in step 3. For the reaction described in Scheme 3, reaction 1, the 1,1,3,3-tetramethyl-2-phenylisouronium intermediates A and B are apparently formed in a balanced ratio.
We next asked the question whether alkylamines would undergo a similar transformation to form the corresponding 2-imino benzo[e]-1,3-oxazin-4-ones as they are more nucleophilic than anilines and would consequently be expected to facilitate salicylamide formation and step 3 to a greater extent. Thus, similar reactions were conducted with cyclohexylamine (2c) and benzylamine (2d) using the same conditions as those in Scheme 1.
To our surprise, in the reaction between 1a and 2d, the corresponding dibenzyl benzo[e]-1,3-oxazin-4-one was not observed by GC–MS and NMR. Consistent with the existing data,4 the major product (45% yield) was the benzyl guanidinium hexafluorophosphate salt in addition to trace amounts of N-benzyl salicylamide (3d). However, reaction between 1a and 2c produced the desired benzo[e]-1,3-oxazin-4-one 4e in 10% yield accompanied by N-cyclohexylsalicylamide (3c) detected by GC–MS (Scheme 5). It appears that alkylamines are poor substrates for this type of transformation as they appear to form more stable guanidinium salts.
Scheme 5. Reaction with Cyclohexylamine.

Yield determined by GC–MS.
To further probe our proposed reaction mechanism, we conducted two HATU reactions with a complementary set of starting materials: 3d with 2b and 3b with 2d (Scheme 6, reactions 1 and 2). For both the reactions, 2-imino benzo[e]-1,3-oxazin-4-one 4f was the major product formed, implying that cyclization in step 2 can still occur with the more stable N-alkyl salicylamides. The reaction outcome also indicates that 2d does not undergo the iminium–imine exchange (Scheme 4, step 3). We suggest that arylamines are favored over alkylamines in the last step because of the conjugative stabilization provided by the aryl system. 1D and 2D NMR data for 4f (see Supporting Information for details) are consistent with the structure of the major product. The heteronuclear multiple bond correlation (HMBC) of the C-18 hydrogen atoms with the C-2, C-4, and N-3 atoms provided key structural assignments.
Scheme 6. Effect of Alkylamines on Product Formation.

Yield determined by GC–MS.
The structure of 4f was also unambiguously confirmed via single crystal X-ray analysis (Figure 1).
Figure 1.
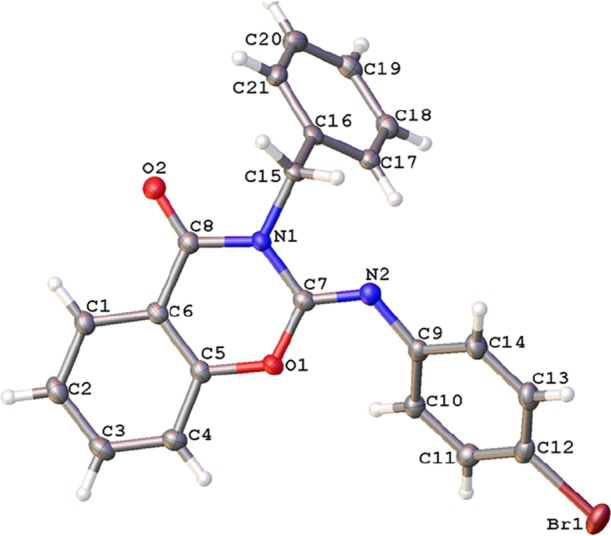
Crystal structure of 4f determined from single-crystal X-ray diffraction (displacement ellipsoids given at 50% probability level).
We were able to find only a single study4 with a slightly similar mechanism: the synthesis of 3-substituted-4(3H)-quinazolinones via HATU-mediated coupling of 4-hydroxyquinazolines with amines. In this reaction, HATU functions as a guanidine-forming agent, whereas in our new reaction, HATU serves as a carbon transfer reagent. To support their proposed reaction mechanism, the authors4 isolated reaction intermediate 8 (Scheme 7). Out of curiosity, we resynthesized 8 and obtained the same 1H and 13C NMR spectra. However, we suggest that 1D, 2D (COSY, HMBC HSQC), and 19F NMR data (Supporting Information), most notably, key signals at 12.19 (s, NH-1) and 7.78 (d, J = 12.7 Hz, CH-17), are more consistent with the structure of the hexafluorophosphate salt of its regioisomer 10. We propose a Dimroth-type attack of cyclohexylamine on an initially formed N-acylguanidinium intermediate 9 to form 10, which then undergoes intramolecular transamidation to form 11 (Scheme 7).
Scheme 7. Dimroth-Type Mechanism for Intermediate 10.

In view of this new HATU carbon transfer reaction to form the relatively unexplored 2-imino benzo[e]-1,3-oxazin-4-one class of heterocycles, we then sought to optimize the reaction conditions. We first surveyed two other coupling reagents using the original reaction conditions: 1a (1 equiv), 2a (2 equiv), triethylamine (TEA, 2.1 equiv), coupling reagent (2.5 equiv), CH3CN, rt, 24 h. Dichloromethylene-dimethyliminium chloride produced a large quantity of byproducts and a low yield of 4a (8%). In contrast, 1-[(dimethylamino) (morpholino)methylene]-1H-[1,2,3]triazolo[4,5-b]pyridine-1-ium 3-oxide hexafluorophosphate afforded a yield of 4a (40%), similar to that of HATU (39%). We decided to use HATU for the remaining optimization experiments because it is less expensive and more widely available.
A brief study of the effect of solvents and bases is shown in Table 1. At room temperature in dimethylacetamide (DMA), a slightly lower yield was obtained compared to the same reaction in CH3CN. When using CH2Cl2 (entry 2), reflux temperatures were required to completely dissolve all of the starting materials but the yield was no better than using CH3CN at rt. Indeed, the reaction yield decreased in refluxing CH3CN (entry 5). The reaction was relatively insensitive to the solvent as long as the starting materials were soluble. Yields were lower with pyridine (entry 7) compared to aliphatic weak bases such as TEA, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and N,N-diisopropylethylamine (DIPEA) (entries 1–3). On the basis of these data, CH3CN, rt, and TEA, DBU, or DIPEA appear to be the optimal combination of solvent, temperature, and base. Because stopping the reaction after 1 h lowered the yield to 12%, we opted to continue to use a 24 h reaction time. It is important to note that the addition of base is required for the transformation to proceed (entry 8). Increasing the equiv of 2a from the stoichiometric 2 to 6 had no significant effect on the yield of cyclized product 4a (40%).
Table 1. Effect of Solvents and Basesa.
![]()
| entry | solvent | base | yield (%) of 4ab |
|---|---|---|---|
| 1 | DMA | TEA | 35 |
| 2 | CH2Cl2 | DIPEA | 39c |
| 3 | CH3CN | DBU | 38 |
| 4 | CH3CN | TEA | 39 |
| 5 | CH3CN | TEA | 14c |
| 6 | CH3CN | DIPEA | 38 |
| 7 | CH3CN | pyridine | 25 |
| 8 | CH3CN | 0 |
Conditions: salicylic acid (1 equiv), aniline (2 equiv), base (2.1 equiv), HATU (2.5 equiv), rt, 24 h.
Isolated yield after chromatographic purification.
Reaction heated to reflux.
Using these optimized conditions, we began to explore the scope and limitations of this transformation by studying the reaction between various anilines 1 and salicylic acids 2 to form oxazinones 4, the results of which are presented in Table 2. Reactions proceeded in moderate yields for a range of salicylic acids and anilines (entries 1, 2, and 5–8) and failed only for reactions with anilines bearing strong electron-withdrawing groups (entries 3 and 4). Interestingly, the reaction of 5-chlorosalicylic acid 1b with 2a and HATU gave the highest yield (65%), possibly because of the activation of phenol by the chloro group. Surprisingly, the reaction to form 4o failed in CH3CN but proceeded reasonably well (40% yield) when it was run in DMA. As shown earlier, 4a and 4b were obtained in 39 and 40% yields using the same conditions. Some reaction yields that otherwise might have been higher were significantly diminished because of difficult chromatographic purification.
Table 2. Scope and Limitations of the One-Pot Synthesis of 2-Imino benzo[e]-1,3-oxazin-4-ones 4.

| entry | product 4h–o | yielda (%) |
|---|---|---|
| 1 | 4h X = 4-F, Y = H | 32 |
| 2 | 4i X = 4-Me, Y = H | 40 |
| 3 | 4j X = 4-CN, Y = H | 0 |
| 4 | 4k X = 3,5-(CF3)2, Y = H | 0 |
| 5 | 4l X = 3,4,5-(OMe)3, Y = H | 43 |
| 6 | 4m X = H, Y = 5-Cl | 65 |
| 7 | 4n X = X = 3,4-(Cl)2, Y = H | 50 |
| 8 | 4o X = 3,4,5-(OMe)3, Y = 4,5-(OMe)2 | 40b |
Isolated Yield.
Reaction done using DMA as a solvent; CH3CN gave a trace yield.
Few reports describe the synthesis of 2-imino benzo[e]-1,3-oxazin-4-ones. For example, Larksarp and Alper5 synthesized 2-imino benzo[e]-1,3-oxazin-4-ones in good to excellent yields via cyclocarbonylation of o-iodophenols with carbodiimides in the presence of 1,4-bis(diphenylphosphino)butane (dppb), Pd, and pressurized carbon monoxide. Ziegler et al.8 also described a method to synthesize 2-imino benzo[e]-1,3-oxazin-4-ones using 2-hydroxybenzoyl chloride. Although our new reaction gives low to moderate yields of benzo[e]-1,3-oxazin-4-ones, it enables fast access to these compounds under mild conditions.
Conclusions
In summary, we discovered a new reaction to obtain 2-imino benzo[e]-1,3-oxazin-4-ones from salicylic acids, anilines, and HATU. On the other hand, this work reveals potential drawbacks in using HATU or similar immonium-type coupling reagents in the synthesis of amides from salicylic acids because of the production of 2-imino benzo[e]-1,3-oxazin-4-ones and other byproducts. The carbon transfer reaction mechanism appears to occur through successive formation of reactive tetramethylisouronium and N-acyl(dimethyl)isouronium reaction intermediates, which then undergo imine–iminium exchange to generate the oxazinone reaction products. While the reaction yields are not ideal, the combination of mild reaction conditions, flexible substitution patterns, and commercial availability of reagents may make this protocol a practical route to 2-imino benzo[e]-1,3-oxazin-4-ones.
Experimental Section
General Information
All commercially available reagents and solvents were used as received. Melting points are uncorrected. 1D 1H and 13C NMR spectra were recorded on either a 400, 500, or 600 MHz spectrometer using CDCl3 or deuterated dimethyl sulfoxide (DMSO-d6) as solvents. 2D NMR spectra were recorded on a 600 MHz spectrometer. All chemical shifts are reported in parts per million (ppm) and are relative to internal (CH3)4Si (0 ppm) for 1H and CDCl3 (77.2 ppm) or DMSO-d6 (39.5 ppm) for 13C NMR. Electron ionization GC–MS data were obtained using a quadrupole mass spectrometer with 30 m DB-XLB type columns and a He flow rate of 1 mL/min. Silica gel (particle size 32–63 μm) was used for all flash column chromatography. Reported reaction temperatures are those of the oil bath. High-resolution mass spectrometry (HRMS) analysis was obtained from the University of Nebraska Medical Center Mass Spectrometry and Proteomics Core Facility.
General Procedure A for the Synthesis of 2-Imino Benzo[e]-1,3-oxazin-4-ones
A mixture of salicylic acid 1 (2 mmol), aniline 2 (4 mmol), and TEA (4.2 mmol) in CH3CN (15 mL) was stirred at rt for 5 min. HATU (5 mmol) was then added in one portion, and the reaction was covered with a rubber septum. After 24 h, the CH3CN was removed in vacuo and the residue was dissolved in CH2Cl2 (20 mL). The organic layer was washed with water (20 mL) and separated; the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (4:1 hexanes/EtOAc).
General Procedure B for the Synthesis of 2-Imino Benzo[e]-1,3-oxazin-4-ones
A mixture of salicylanilide 3 (2 mmol) and TEA (4 mmol) in CH3CN (15 mL) was stirred at rt for 5 min. HATU (3 mmol) was then added in one portion, and the reaction was covered with a rubber stopper. After 24 h, the CH3CN was removed in vacuo and the residue was dissolved in CH2Cl2 (20 mL). The organic layer was washed with water (20 mL) and separated; the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (4:1 hexanes/EtOAc).
(Z)-3-Phenyl-2-(phenylimino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4a)
Using general procedure A, 4a was isolated as a white microcrystalline solid, 245 mg, 39% yield. mp 161–163 °C. 1H NMR (500 MHz, CDCl3): δ 6.99 (d, J = 7.1 Hz, 2H), 7.06 (t, J = 7.4 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 7.26–7.33 (m, 3H), 7.39 (d, J = 7.2 Hz, 2H), 7.45 (t, J = 7.5 Hz, 1H), 7.53 (t, J = 7.8 Hz, 2H), 7.62 (m, 1H), 8.10 (dd, J = 1.7, 7.8 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 114.92, 115.81, 122.68, 123.54, 124.66, 128.33, 128.56, 128.59, 128.69, 129.42, 135.73, 136.09, 142.15, 144.61, 153.07, 159.83. HRMS (ESI-Orbitrap) m/z: calcd for C20H15N2O2 [M + H]+, 315.1134; found, 315.1127.
(Z)-3-(4-Bromophenyl)-2-((4-bromophenyl)imino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4b)
Using general procedure A, 4b was isolated as a white microcrystalline solid, 378 mg, 40% yield. mp 173–175 °C. 1H NMR (500 MHz, CDCl3): δ 6.86 (d, J = 8.7 Hz, 2H), 7.11 (d, J = 8.3 Hz, 1H), 7.25 (d, J = 8.5 Hz, 2H), 7.32 (t, J = 7.6 Hz, 1H), 7.39 (d, J = 8.7 Hz, 2H), 7.65 (d, J = 11.3 Hz, 3H), 8.09 (dd, J = 1.7, 7.9 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 114.65, 115.82, 116.69, 122.89, 124.49, 125.01, 128.40, 130.29, 131.67, 132.75, 134.85, 136.08, 142.40, 143.45, 152.86, 159.48. HRMS (ESI-Orbitrap) m/z: calcd for C20H1379Br2N2O2 [M + H]+, 470.9344; found, 470.9338.
N-(Bis(dimethylamino)methylene)-4-bromobenzenaminium hexafluorophosphate(V) (5)
Using general procedure A, 5 was isolated as a white microcrystalline solid, 166 mg, 20% yield. mp 147–149 °C.·1H NMR (500 MHz, DMSO-d6): δ 2.90 (s, 12H), 6.99 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H), 9.96 (s, 1H). 13C NMR (125 MHz, DMSO-d6): δ 39.98, 116.65, 122.44, 132.39, 137.52, 157.43. HRMS (ESI-Orbitrap) m/z: calcd for C11H1779BrN3 [M]+, 270.0600; found, 270.0597.
N-(4-Bromophenyl)-N-(dimethylcarbamoyl)-2-hydroxybenzamide (6)
Using general procedure A, 6 was isolated as a white microcrystalline solid, 87 mg, 12% yield. mp 154–156 °C. 1H NMR (500 MHz, CDCl3): δ 3.01 (s, 3H), 3.10 (s, 3H), 7.12 (d, J = 8.1 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 7.40–7.56 (m, 5H), 7.74 (d, J = 9.5 Hz, 1H), 8.68 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 36.73, 36.92, 116.82, 121.08, 123.31, 126.39, 130.00, 130.36, 131.97, 132.02, 137.29, 148.06, 155.38, 164.33. HRMS (ESI-Orbitrap) m/z: calcd for C16H1679BrN2O3 [M + H]+, 363.0344; found, 363.0338.
3-(4-Bromophenyl)-2H-benzo[e][1,3]oxazine-2,4(3H)-dione (7)
Using general procedure A, 7 was isolated as a white microcrystalline solid, 64 mg, 10% yield. mp 252–254 °C. 1H NMR (500 MHz, DMSO-d6): δ 7.41 (d, J = 8.5 Hz, 2H), 7.44–7.54 (m, 2H), 7.73 (d, J = 8.5 Hz, 2H), 7.87 (d, J = 7.1 Hz, 1H), 8.00 (dd, J = 1.6, 7.8 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): δ 114.77, 116.42, 121.94, 125.35, 127.43, 130.90, 132.08, 134.58, 136.41, 147.41, 152.55, 160.57. HRMS (ESI-Orbitrap) m/z: calcd for C14H979BrNO3 [M + H]+, 317.9766; found, 317.9763.
(Z)-2-((4-Bromophenyl)imino)-3-phenyl-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4c)
Using general procedure B, 4c was isolated as a tan microcrystalline solid, 142 mg, 18% yield. mp 160–162 °C. 1H NMR (600 MHz, CDCl3): δ 6.86 (d, J = 8.6 Hz, 2H), 7.10 (d, J = 8.3 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 7.37 (t, J = 8.58 Hz, 4H), 7.45 (t, J = 7.4 Hz, 1H), 7.53 (t, J = 7.8 Hz, 2H), 7.61–7.66 (m, 1H), 8.09 (dd, J = 1.6, 7.8 Hz, 1H). 13C NMR (150 MHz, CDCl3): δ 114.81, 115.74, 116.43, 124.51, 124.87, 128.35, 128.47, 128.81, 129.47, 131.56, 135.85, 135.87, 142.67, 143.76, 152.88, 159.65. HRMS (ESI-Orbitrap) m/z: calcd for C20H1479BrN2O2 [M + H]+, 393.0239; found, 393.0230.
(Z)-3-(4-Bromophenyl)-2-(phenylimino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4d)
Using general procedure B, 4d was isolated as a tan microcrystalline solid, 142 mg, 18% yield. mp 171–173 °C. 1H NMR (600 MHz, CDCl3): δ 6.98 (d, J = 7.4 Hz, 2H), 7.02–7.15 (m, 2H), 7.15–7.35 (m, 5H), 7.52–7.70 (m, 3H), 8.07 (dd, J = 1.7, 7.8 Hz, 1H). 13C NMR (150 MHz, CDCl3): δ 114.64, 115.84, 122.61, 122.71, 123.71, 124.77, 128.28, 128.65, 130.34, 132.65, 135.02, 135.92, 141.86, 144.25, 152.95, 159.61. HRMS (ESI-Orbitrap) m/z: calcd for C20H1479BrN2O2 [M + H]+, 393.0239; found, 393.0230.
N-(4-Bromophenyl)-2-hydroxybenzamide (3b)
Salicylic acid (4.741 g, 34.3 mmol), SOCl2 (12.242 g, 103 mmol), DMF (0.5 mL), and CH2Cl2 (15 mL) were heated to reflux under Ar for 24 h. The solvent was removed in vacuo, and the residue was dissolved in tetrahydrofuran (THF, 10 mL). 4-Bromoaniline (6.505 g, 37.8 mmol) in THF (10 mL) was added in portions. The solution was heated to reflux under Ar for 24 h. The solvent was removed and EtOAc (20 mL) was added followed by 1 N NaOH (20 mL). The organic layer was removed and the aq layer was extracted with EtOAc (2 × 20 mL) and acidified with concd HCl to pH 1. The precipitated solid was filtered and dried in vacuo to afford 3b as a tan solid with no further purification, 942 mg, 68%. 1H NMR (400 MHz, CDCl3): δ 6.93 (ddd, J = 1.2, 7.2, 8.2 Hz, 1H), 7.04 (dd, J = 1.2, 8.4 Hz, 1H), 7.36–7.85 (m, 7H), 7.92 (s, 1H), 11.82 (s, 1H). All recorded data were in accordance with those previously reported.6
(Z)-3-Cyclohexyl-2-(cyclohexylimino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4e)
Using general procedure A, 4e was isolated as a white microcrystalline solid, 65 mg, 10% yield. mp 90–92 °C. 1H NMR (400 MHz, CDCl3): δ 1.15–1.47 (m, 8H), 1.51–1.88 (m, 10H), 2.58 (qd, J = 3.7, 12.4 Hz, 2H), 3.80 (m, 1H), 4.87 (tt, J = 3.7, 12.1 Hz, 1H), 7.05 (d, J = 8.2 Hz, 1H), 7.17 (t, J = 7.6 Hz, 1H), 7.52 (ddd, J = 1.7, 7.4, 8.6 Hz, 1H), 7.98 (dd, J = 1.7, 7.8 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 24.37, 25.57, 26.07, 26.49, 28.30, 33.98, 53.26, 55.60, 114.86, 115.40, 123.53, 128.00, 134.57, 138.53, 153.22, 160.37. HRMS (ESI-Orbitrap) m/z: calcd for C20H27N2O2 [M + H]+, 327.2073; found, 327.2049.
N-Benzyl-2-hydroxybenzamide (3c)7
Salicylic acid (2.037 g, 14.7 mmol), SOCl2 (5.247 g, 44.1 mmol), DMF (0.5 mL), and CH2Cl2 (15 mL) were heated to reflux under Ar for 24 h. The solvent was removed in vacuo, and the residue was dissolved in CH2Cl2 (10 mL) and added dropwise to a stirring solution of benzylamine (1.618 g, 15.1 mmol) and TEA (2.997 g, 29.6 mmol) in CH2Cl2 (15 mL), at 0 °C under Ar. The solution was allowed to warm to rt and stir for 24 h. The solvent was removed, and CH2Cl2 (20 mL) was added followed by water (20 mL). The organic layer was removed and the aq layer was extracted with CH2Cl2 (3 × 20 mL), washed with brine, and dried over MgSO4. The solvent was removed, and the residue was recrystallized (EtOH/H2O), filtered, and dried in vacuo to afford 3c. 1H NMR (400 MHz, CDCl3): δ 4.65 (d, J = 5.6 Hz, 2H), 6.55 (br s, 1H), 6.83 (ddd, J = 1.2, 7.3, 8.2 Hz, 1H), 7.00 (dd, J = 1.2, 8.4 Hz, 1H), 7.30–7.45 (m, 7H), 12.29 (s, 1H). All recorded data were in accordance with those previously reported.7
(Z)-3-Benzyl-2-((4-bromophenyl)imino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4f)
Using general procedure B, 4f was isolated as white needles, 293 mg, 36% yield. mp 123–124 °C. 1H NMR (600 MHz, CDCl3): δ 5.37 (s, 2H), 6.95 (d, J = 8.6 Hz, 1H), 7.00 (dd, J = 0.9, 8.3 Hz, 1H), 7.23–7.28 (m, 2H), 7.32 (t, J = 7.3 Hz, 2H), 7.43 (d, J = 8.6 Hz, 1H), 7.55 (ddd, J = 1.7, 7.3, 8.6 Hz, 1H), 7.59 (d, J = 7.0 Hz, 1H), 8.06 (dd, J = 1.7, 7.9 Hz, 1H). 13C NMR (150 MHz, CDCl3): δ 45.77, 114.59, 115.52, 116.47, 124.64, 124.74, 127.72, 128.11, 128.32, 129.33, 131.71, 135.53, 136.48, 141.57, 143.76, 152.53, 159.42. HRMS (ESI-Orbitrap) m/z: calcd for C21H1679BrN2O2 [M + H]+, 407.0395; found, 407.0376.
(Z)-3-(4-Fluorophenyl)-2-((4-fluorophenyl)imino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4h)
Using general procedure A, 4h was isolated as white needles, 224 mg, 32% yield. mp 164–166 °C. 1H NMR (500 MHz, CDCl3): δ 6.91–7.02 (m, 4H), 7.11 (d, J = 8.4 Hz, 1H), 7.21 (t, J = 8.4 Hz, 2H), 7.28–7.38 (m, 3H), 7.64 (d, J = 7.1 Hz, 1H), 8.09 (d, J = 7.7 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 114.75, 115.29 (d, J = 22.2 Hz), 115.78, 116.53 (d, J = 23.0 Hz), 124.00 (d, J = 7.9 Hz), 124.89, 128.39, 130.34 (d, J = 8.8 Hz), 131.75 (3.5 Hz), 135.95, 140.37 (d, J = 3.0 Hz), 142.48, 152.91, 159.42 (d, J = 241.0 Hz), 159.76, 162.40 (d, J = 247.0 Hz). HRMS (ESI-Orbitrap) m/z: [M + H]+ calcd for C20H13F2N2O2, 351.0945; found, 351.0921.
(Z)-3-(p-Tolyl)-2-(p-tolylimino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4i)
Using general procedure A, 4i was isolated as a white crystalline solid, 274 mg, 40% yield. mp 166–167 °C. 1H NMR (500 MHz, CDCl3): δ 2.31 (s, 3H), 2.40 (s, 3H), 6.89 (d, J = 7.9 Hz, 2H), 7.02–7.12 (m, 3H), 7.20–7.36 (m, 5H), 7.59 (t, J = 2.2 Hz, 1H), 8.08 (d, J = 7.7 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 20.90, 21.32, 114.94, 115.74, 122.56, 124.52, 128.19, 128.28, 129.13, 130.13, 132.86, 133.45, 135.60, 138.48, 141.99, 142.14, 153.07, 159.92. HRMS (ESI-Orbitrap) m/z: [M + H]+ calcd for C22H19N2O2, 343.1447; found, 343.1421.
(Z)-3-(3,4,5-Trimethoxyphenyl)-2-((3,4,5-trimethoxyphenyl)imino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4l)
Using general procedure A, 4l was isolated as a white crystalline solid, 425 mg, 43% yield. mp 236–238 °C. 1H NMR (500 MHz, CDCl3): δ 3.83 (s, 3H), 3.84 (s, 6H), 3.88 (s, 6H), 3.91 (s, 3H), 6.27 (s, 2H), 6.60 (s, 2H), 7.15 (d, J = 8.4 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 7.66 (t, J = 7.98 Hz, 1H), 8.11 (dd, J = 1.6, 7.8 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 56.11, 56.19, 60.84, 61.01, 100.30, 105.90, 114.83, 115.74, 124.84, 128.40, 131.49, 134.54, 135.93, 138.19, 140.52, 142.32, 153.01, 153.20, 153.81, 159.85. HRMS (ESI-Orbitrap) m/z: [M + H]+ calcd for C26H27N2O8, 495.1767; found, 495.1745.
(Z)-7-Chloro-3-phenyl-2-(phenylimino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4m)
Using general procedure A, 4m was precipitated from the reaction solution after 24 h, filtered, washed with CH3CN, and dried in vacuo to yield a white crystalline solid, 453 mg, 65%. mp 185–187 °C. 1H NMR (500 MHz, CDCl3): δ 6.97 (d, J = 7.7 Hz, 2H), 7.03–7.10 (m, 2H), 7.28 (t, J = 7.7 Hz, 2H), 7.37 (d, J = 7.7 Hz, 2H), 7.45 (t, J = 7.5 Hz, 1H), 7.50–7.61 (m, 3H), 8.05 (d, J = 2.5 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 116.11, 117.48, 122.56, 123.74, 127.74, 128.45, 128.64, 128.86, 129.49, 130.26, 135.70, 135.81, 141.49, 144.26, 151.48, 158.76. HRMS (ESI-Orbitrap) m/z: [M + H]+ calcd for C20H14ClN2O2, 349.0744; found, 349.0720.
(Z)-3-(3,4-Dichlorophenyl)-2-((3,4-dichlorophenyl)imino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4n)
Using general procedure A, 4n was precipitated from the reaction solution after 24 h, filtered, washed with CH3CN, and dried in vacuo to yield a white crystalline solid, 452 mg, 50% yield. mp 223–224 °C. 1H NMR (500 MHz, CDCl3): δ 6.85 (dd, J = 2.4, 8.7 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 7.23 (dd, J = 2.4, 8.5 Hz, 1H), 7.30–7.39 (m, 2H), 7.48 (d, J = 2.3 Hz, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.69 (ddd, J = 1.7, 7.4, 8.7 Hz, 1H), 8.10 (dd, J = 1.7, 7.9 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 114.44, 115.93, 122.44, 124.70, 125.34, 127.32, 128.11, 128.48, 130.28, 130.80, 131.18, 132.32, 133.43, 133.44, 134.78, 136.38, 142.82, 143.74, 152.71, 159.23. HRMS (ESI-Orbitrap) m/z: [M + H]+ calcd for C20H11Cl4N2O2, 450.9575; found, 450.9571.
(Z)-6,7-Dimethoxy-3-(3,4,5-trimethoxyphenyl)-2-((3,4,5-trimethoxyphenyl)imino)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (4o)
Using general procedure A with DMA as a solvent, water (30 mL) was added to the reaction mixture after 24 h and 4o was precipitated, filtered, washed with water (3 × 20 mL), recrystallized (EtOH/H2O), and dried to yield a white microcrystalline solid, 444 mg, 40% yield. mp 230 °C dec. 1H NMR (600 MHz, DMSO-d6): δ 3.62 (s, 3H), 3.73 (s, 3H), 3.73 (s, 6H), 3.76 (s, 6H), 3.82 (s, 3H), 3.88 (s, 3H), 6.26 (s, 2H), 6.82 (s, 2H), 6.84 (s, 1H), 7.29 (s, 1H). 13C NMR (150 MHz, DMSO-d6): δ 55.63, 55.89, 55.97, 56.58, 59.86, 59.93, 99.01, 99.83, 105.85, 106.67, 107.00, 132.38, 133.16, 136.93, 140.91, 142.82, 146.00, 148.36, 152.70, 152.92, 155.35, 158.92. HRMS (ESI-Orbitrap) m/z: [M]+ calcd for C28H31N2O10, 554.1900; found, 554.1883.
Acknowledgments
We acknowledge the U.S. National Institutes of Health (AI116723-01) for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01824.
The authors declare no competing financial interest.
Supplementary Material
References
- Carpino L. A. J. Am. Chem. Soc. 1993, 115, 4397–4398. 10.1021/ja00063a082. [DOI] [Google Scholar]
- Albericio F.; Bofill J. M.; El-Faham A.; Kates S. A. J. Org. Chem. 1998, 63, 9678–9683. 10.1021/jo980807y. [DOI] [Google Scholar]
- Valeur E.; Bradley M. Chem. Soc. Rev. 2009, 38, 606–631. 10.1039/b701677h. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Yang M. G.; Li P.; Carter P. H. Org. Lett. 2009, 11, 1421–1424. 10.1021/ol802946p. [DOI] [PubMed] [Google Scholar]
- Larksarp C.; Alper H. J. Org. Chem. 1999, 64, 9194–9200. 10.1021/jo991256u. [DOI] [Google Scholar]
- Lu C.-R.; Zhao B.; Jiang Y.-P.; Ding H.; Yang S. Synth. Commun. 2011, 41, 1257–1266. 10.1080/00397911.2010.481745. [DOI] [Google Scholar]
- Balkrishna S. J.; Kumar S. Synthesis 2012, 44, 1417–1426. 10.1055/s-0031-1289755. [DOI] [Google Scholar]
- Ziegler E.; Kollenz G.; Kappe T. Monatsh. Chem. 1969, 100, 1722–1725. 10.1007/bf00900193. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.