


Abstract
Alignment-free genome and metagenome comparisons are increasingly important with the development of next generation sequencing (NGS) technologies. Recently developed state-of-the-art k-mer based alignment-free dissimilarity measures including CVTree, 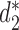 and
and 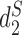 are more computationally expensive than measures based solely on the k-mer frequencies. Here, we report a standalone software, aCcelerated Alignment-FrEe sequence analysis (CAFE), for efficient calculation of 28 alignment-free dissimilarity measures. CAFE allows for both assembled genome sequences and unassembled NGS shotgun reads as input, and wraps the output in a standard PHYLIP format. In downstream analyses, CAFE can also be used to visualize the pairwise dissimilarity measures, including dendrograms, heatmap, principal coordinate analysis and network display. CAFE serves as a general k-mer based alignment-free analysis platform for studying the relationships among genomes and metagenomes, and is freely available at https://github.com/younglululu/CAFE.
are more computationally expensive than measures based solely on the k-mer frequencies. Here, we report a standalone software, aCcelerated Alignment-FrEe sequence analysis (CAFE), for efficient calculation of 28 alignment-free dissimilarity measures. CAFE allows for both assembled genome sequences and unassembled NGS shotgun reads as input, and wraps the output in a standard PHYLIP format. In downstream analyses, CAFE can also be used to visualize the pairwise dissimilarity measures, including dendrograms, heatmap, principal coordinate analysis and network display. CAFE serves as a general k-mer based alignment-free analysis platform for studying the relationships among genomes and metagenomes, and is freely available at https://github.com/younglululu/CAFE.
INTRODUCTION
Sequence comparison is widely used to study the relationship among molecular sequences. The dominant tools for sequence comparison are alignment-based methods, including global (1) and local (2) sequence alignments. With the advent of alignment-based tools such as BLAST (3) and sequence databases such as RefSeq (4), alignment-based methods are widely used in a broad range of applications. Despite their extensive applications, alignment-based methods are not appropriate in some situations. First, gene regulatory regions are generally not highly conserved making alignment-based approaches difficult to identify related regulatory regions that are bound by similar transcription factors (5). Second, next generation sequencing (NGS) technologies generate large amounts of short reads and it is challenging to assemble them for both genomic and metagenomic studies. Without long assembled contigs across many samples, it is challenging for alignment-based methods to compare genomes and metagenomes (6,7). Third, viruses are more likely to infect bacterial hosts having similar word pattern usage (8,9), and thus, the hosts of viruses can potentially be inferred based on their word pattern usages. However, alignment based methods are usually not applicable for studying virus-host infectious associations.
Alignment-free sequence comparison methods serve as attractive alternatives for studying the relationships among sequences when alignment based methods are not appropriate or too time consuming to be implemented in practice (10,11). Several types of alignment free approaches are available including those based on the counts of k-mers, longest common subsequences, shortest absent patterns, etc. that have recently been reviewed in a special issue of Briefing in Bioinformatics (12). Here we concentrate on alignment-free statistics using k-mer counts. These approaches project each sequence into k-mer (or equivalently k-tuple, k-gram) counts feature space, where sequence information is transformed into numerical values such as k-mer frequency. We do not consider dissimilarity measures using spaced k-mers due to the added computational complexity counting spaced k-mers. The recently developed statistics 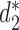 and
and 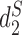 have been shown to perform well theoretically (13) as well as in many applications including the comparison of gene regulatory regions (11), whole genome sequences (14), metagenomes (7) and virus-bacteria host infectious associations (8). Despite their excellent performance in many applications, the original implementation of these statistics are relatively slow due to the requirement of calculating the expected k-mer counts and thus limits their usage.
have been shown to perform well theoretically (13) as well as in many applications including the comparison of gene regulatory regions (11), whole genome sequences (14), metagenomes (7) and virus-bacteria host infectious associations (8). Despite their excellent performance in many applications, the original implementation of these statistics are relatively slow due to the requirement of calculating the expected k-mer counts and thus limits their usage.
CAFE significantly speeds up the calculation of recently developed measures based on background adjusted k-mer counts, such as CVTree (15), 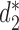 (13) and
(13) and 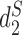 (13), with reduced memory requirement. In addition, CAFE integrates 10 conventional measures based on k-mer counts such as Chebyshev (Ch), Euclidean (Eu), Manhattan (Ma), d2 dissimilarity (16), Jensen-Shannon divergence (JS) (17), feature frequency profiles (FFP) (18) and Co-phylog (19). CAFE also offers 15 measures based on presence/absence of k-mers, such as Jaccard and Hamming distances. We further demonstrate the value of alignment-free dissimilarity measures using CAFE on real datasets, ranging from primate, vertebrate and microbial genomic sequences, to metagenomic sequence reads.
(13), with reduced memory requirement. In addition, CAFE integrates 10 conventional measures based on k-mer counts such as Chebyshev (Ch), Euclidean (Eu), Manhattan (Ma), d2 dissimilarity (16), Jensen-Shannon divergence (JS) (17), feature frequency profiles (FFP) (18) and Co-phylog (19). CAFE also offers 15 measures based on presence/absence of k-mers, such as Jaccard and Hamming distances. We further demonstrate the value of alignment-free dissimilarity measures using CAFE on real datasets, ranging from primate, vertebrate and microbial genomic sequences, to metagenomic sequence reads.
MATERIALS AND METHODS
Workflows
CAFE works with sequence data, both assembled genomic sequences and unassembled shotgun sequence reads from NGS technologies and counts k-mers by JELLYFISH (20), a fast and memory-efficient k-mer counting tool. JELLYFISH produces compressed databases containing all k-mer counts given the query sequences in parallel. CAFE subsequently loads the databases and generates necessary transformed information with respect to various dissimilarity measures. For example, measures based on presence/absence of k-mers binarize k-mer counts into presence/absence indicators. Most conventional measures normalize k-mer counts into the k-mer frequencies. Besides, expected k-mer counts are involved in recently developed measures based on background adjusted k-mer counts, such as CVTree,  and
and  . In such cases, the Markov models for the sequences are assumed as the underlying generative models, with the parameters estimated from the sequence data accordingly. The Markov order can be either manually set or automatically chosen using the Bayesian information criterion (BIC) (21).
. In such cases, the Markov models for the sequences are assumed as the underlying generative models, with the parameters estimated from the sequence data accordingly. The Markov order can be either manually set or automatically chosen using the Bayesian information criterion (BIC) (21).
The resulting pairwise dissimilarities among the sequences form a symmetric matrix. CAFE can directly output the dissimilarity matrix in a standard PHYLIP format. Alternatively, CAFE provides four types of built-in downstream visualized analyses, including clustering the sequences into dendrograms using the UPGMA algorithm, heatmap visualization of the matrix, projecting the matrix to a 2D space using principal coordinate analysis (PCoA) and network display. A graphical illustration of CAFE workflow is shown in Figure 1.
Figure 1.

The workflow of CAFE. The JELLYFISH software parses the input sequence files (in Fasta format), counts k-mers and saves compressed information into separate databases. CAFE subsequently loads the databases and constructs a symmetric dissimilarity matrix among the inputs. CAFE also integrates four types of visualized downstream analysis, including dendrograms, heatmap, principal coordinate analysis (PCoA) and network display.
Graphical user interface
The CAFE user interface consists of four major tools—data selection toolbar, dissimilarity setting toolbar, image toolbar and visualized analyses. The data selection toolbar enables users to browse and add/delete genome sequences or NGS shotgun reads of the file extension ‘.fasta’, ‘.fa’ or ‘.fna’. The selected files are shown in the input data list. The data selection toolbar also supports loading pre-computed results in a standard PHYLIP format.
The dissimilarity setting toolbar determines the choice of dissimilarity measures as well as the involved parameter configuration, including the k-mer length, the order of potential Markov model, the cutoff of the minimum k-mer occurrences and whether to consider the reverse complement of each k-mer, a common practice in dealing with NGS shotgun reads. When a certain parameter is unnecessary for particular dissimilarity measures, the corresponding configuration is disabled. In the cases of CVTree, 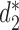 and
and  , usually the proper order of Markov model remains unclear to the user. A simple yet time-consuming way is to set ‘-1’, which will infer the order automatically using the BIC (21).
, usually the proper order of Markov model remains unclear to the user. A simple yet time-consuming way is to set ‘-1’, which will infer the order automatically using the BIC (21).
After the ‘Run’ button is pressed, the CAFE workflow starts, and consolidated dissimilarity results are saved in a standard PHYLIP format, together with the run-time information trackable from the console. Meanwhile, four types of built-in analyses are provided in tabbed windows, including dendrograms, heatmap, PCoA and network display.
The view of the visualized analyses can be adjusted by using the ‘zoom-in’ and ‘zoom-out’ buttons located in the image toolbar. CAFE also supports downloading the visualized results for publication. To access this function, users can either use the ‘save’ button in the image toolbar or right-click on the figure directly. A screenshot of the CAFE user interface is shown in Figure 2.
Figure 2.

Screenshot of CAFE user interface based on a toy example. The user interface layout divides into six parts in terms of functionality: (i) data selection toolbar (top left), (ii) dissimilarity setting toolbar (top middle), (iii) image toolbar (top right), (iv) input data list (middle left), (v) run-time information console (bottom left) and (vi) visualized analyses (bottom right).
Design
CAFE is designed for extensibility and reusability, following the software engineering paradigm. For example, users can specify a threshold to filter out k-mers whose counts are below the threshold. In this case, the Iterator hides the details of filtering, wrapping the enumeration of qualified k-mer counts or frequencies uniformly. Also, some dissimilarity measures do not need the expected k-mer counts. Hence the Proxy provides the calculation of expected k-mer counts as a service on demand. Moreover, the dissimilarity measures are encapsulated in Strategy, enabling users to integrate customized dissimilarity measures into CAFE easily as plug-in.
RESULTS
Application to primate and vertebrate genomic sequences
We compared various alignment-free dissimilarity measures using CAFE on three real genomic datasets. We first investigated the evolutionary relationship of 21 primates whose complete genome sequences are available in the NCBI database (22). For each dissimilarity measure, the calculated pairwise dissimilarity measures are directly compared against the corresponding evolutionary distances calculated by Ape (An R package) (23) as the benchmark, in terms of Spearman correlations. Comparison using Pearson correlations between the estimated alignment-free dissimilarity and the evolutionary distances, and normalized Robinson-Foulds distance (24) between the clustering tree using UPGMA and the standard phylogenetic tree are also available in the supplementary material. Similarly, we investigated the evolutionary relationship of 28 vertebrate species and compared the alignment-free dissimilarity measures with the pairwise evolutionary distances given in (25). Finally, we combined the two datasets to see how the alignment-free dissimilarity measures relate to evolutionary distances calculated based on maximum likelihood approach from a large number of genomic regions.
The comparison involves three dissimilarity measures based on background adjusted k-mer counts including CVTree, 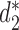 and
and 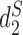 , 10 conventional measures based on k-mer counts, including Canberra, Ch, Cosine, Co-phylog, d2, Eu, FFP, JS, Ma and Pearson, and 15 measures based on presence/absence of k-mers including Anderberg, Antidice, Dice, Gower, Hamman, Hamming, Jaccard, Kulczynski, Matching, Ochiai, Phi, Russel, Sneath, Tanimoto and Yule. We used k = 14 as in (14). The results are illustrated in Figure 3. The Markov order 12 is used for
, 10 conventional measures based on k-mer counts, including Canberra, Ch, Cosine, Co-phylog, d2, Eu, FFP, JS, Ma and Pearson, and 15 measures based on presence/absence of k-mers including Anderberg, Antidice, Dice, Gower, Hamman, Hamming, Jaccard, Kulczynski, Matching, Ochiai, Phi, Russel, Sneath, Tanimoto and Yule. We used k = 14 as in (14). The results are illustrated in Figure 3. The Markov order 12 is used for 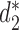 ,
, 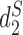 and JS as most of the sequences have estimated order 12 based on BIC (21). Consistent with previous studies, the background adjusted dissimilarity measures outperform markedly the non-background adjusted measures.
and JS as most of the sequences have estimated order 12 based on BIC (21). Consistent with previous studies, the background adjusted dissimilarity measures outperform markedly the non-background adjusted measures.
Figure 3.

The Spearman correlation of various dissimilarity measures with the evolutionary distances using maximum likelihood approach across many genomic regions based on 21 primate species (top), 28 vertebrate species (middle) and the combination of both (bottom).
We then evaluate the computational speed of CAFE compared to the original implementation for  in (14). We calculate the dissimilarity using
in (14). We calculate the dissimilarity using 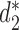 measure (
measure (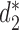 and
and 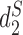 share highly similar formulation) on random pairs of genome sequences. As shown in Figure 4, CAFE achieves 24.0× speedup with 55.3% peak memory consumption on average.
share highly similar formulation) on random pairs of genome sequences. As shown in Figure 4, CAFE achieves 24.0× speedup with 55.3% peak memory consumption on average.
Figure 4.
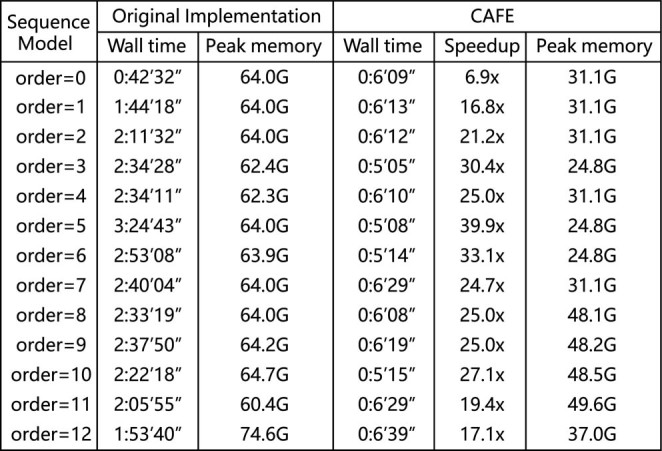
Wall time, peak memory usage and speedup ratio comparison between CAFE and the original implementation to calculate 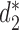 dissimilarity between a pair of genomes for k = 14.
dissimilarity between a pair of genomes for k = 14.
Application to microbial genomic sequences
We applied CAFE to analyze 27 E. coli and Shigella genomes dataset as in (26). These genomes can be assigned to 6 E. coli reference (ECOR) groups: A, B1, B2, D, E and S. We investigated how well various alignment-free dissimilarity measures can identify these groups. For each dissimilarity measure, we used UPGMA to cluster the samples based on the calculated pairwise dissimilarity matrix. The Markov order 1 is used for 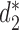 and
and  as most of the sequences have estimated order 1 based on BIC (21).
as most of the sequences have estimated order 1 based on BIC (21).
We used k = 14 for the comparison. The comparison involves three dissimilarity measures based on background adjusted k-mer counts including CVTree, 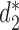 and
and 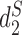 , and the results are illustrated in Figure 5. The results using the other 10 conventional measures based on 14-mer counts as well as 15 measures based on presence/absence of 14-mers, are given in the supplementary material. Consistent with previous studies, for
, and the results are illustrated in Figure 5. The results using the other 10 conventional measures based on 14-mer counts as well as 15 measures based on presence/absence of 14-mers, are given in the supplementary material. Consistent with previous studies, for 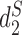 , each ECOR is monophyletic except A and B2. The normalized Robinson-Foulds distances (24) between the estimated clustering tree and the standard phylogenetic tree are available in the Supplementary Data.
, each ECOR is monophyletic except A and B2. The normalized Robinson-Foulds distances (24) between the estimated clustering tree and the standard phylogenetic tree are available in the Supplementary Data.
Figure 5.

The clustering results of 27 Escherichia coli and Shigella genomes using measures based on background adjusted 14-mer counts: 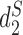 ,
, 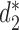 and CVTree. The Markov order of the sequences were set at 1. The colors indicate the six different E. Coli reference groups.
and CVTree. The Markov order of the sequences were set at 1. The colors indicate the six different E. Coli reference groups.
Application to metagenomic samples
We used CAFE to analyze a mammalian gut metagenomic dataset (7) comprised of NGS short reads from 28 samples. These samples further split into 3 groups: 8 hindgut-fermenting herbivores, 13 foregut-fermenting herbivores and 7 simple-gut carnivores. We investigated how well various alignment-free dissimilarity measures can identify these groups. For each dissimilarity measure, we used UPGMA to cluster the samples based on the calculated pairwise dissimilarity matrix.
We used k = 5 as in (7). The comparison involves three dissimilarity measures based on background adjusted k-mer counts including CVTree,  and
and 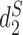 , and the results are illustrated in Figure 6. The results based on nine conventional measures based on k-mer counts are given in the Supplementary Data. Other measures are not applicable because k = 5 is not large enough. The Markov order 0 is used in
, and the results are illustrated in Figure 6. The results based on nine conventional measures based on k-mer counts are given in the Supplementary Data. Other measures are not applicable because k = 5 is not large enough. The Markov order 0 is used in 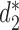 and
and 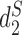 as in (7). Consistent with previous studies,
as in (7). Consistent with previous studies, 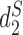 achieves clear separations among the three groups.
achieves clear separations among the three groups.
Figure 6.

The clustering results of the mammalian gut samples using measures based on background adjusted k-mer counts: 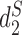 ,
, 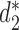 and CVTree.
and CVTree.
DISCUSSION
We have developed a fast and user-friendly alignment-free analyses platform, CAFE, for studying the relationships among genomes and metagenomes. With reduced memory usage, CAFE speeds up the calculation of the state-of-the-art alignment-free measures that perform well theoretically and practically. For easy usage, CAFE not only integrates 28 dissimilarity measures extensively but also integrates four types of downstream visualized analyses. CAFE will make the usage of alignment-free methods more accessible to researchers. We encourage users to contribute their own dissimilarity measures to CAFE as plug-ins.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NSF [DMS-1518001]; OCE [1136818]; NIH [R01GM120624]; Gordon and Betty Moore Foundation Marine Microbiology Initiative [GBMF3779]. Funding for open access charge: NSF [DMS-1518001].
Conflict of interest statement. None declared.
REFERENCES
- 1. Needleman S.B., Wunsch C.D.. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970; 48:443–453. [DOI] [PubMed] [Google Scholar]
- 2. Smith T.F., Waterman M.S.. Identification of common molecular subsequences. J. Mol. Biol. 1981; 147:195–197. [DOI] [PubMed] [Google Scholar]
- 3. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 4. Pruitt K.D., Tatusova T., Maglott D.R.. NCBI reference sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005; 33(Suppl. 1):D501–D504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leung G., Eisen M.B.. Identifying cis-regulatory sequences by word profile similarity. PLoS One. 2009; 4:e6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Willner D., Vega T.R., Rohwer F.. Metagenomic signatures of 86 microbial and viral metagenomes. Environ. Microbiol. 2009; 11:1752–1766. [DOI] [PubMed] [Google Scholar]
- 7. Jiang B., Song K., Ren J., Deng M., Sun F., Zhang X.. Comparison of metagenomic samples using sequence signatures. BMC Genomics. 2012; 13:730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahlgren N.A., Ren J., Young L.Y., Fuhrman J.A., Sun F.. Alignment-free d2* oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences. Nucleic Acids Res. 2017; 45:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roux S., Hallam S.J., Woyke T., Sullivan M.B.. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. Elife. 2015; 4:e08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vinga S., Almeida J.. Alignment-free sequence comparison-a review. Bioinformatics. 2003; 19:513–523. [DOI] [PubMed] [Google Scholar]
- 11. Song K., Ren J., Reinert G., Deng M., Waterman M.S., Sun F.. New developments of alignment-free sequence comparison: measures, statistics and next-generation sequencing. Brief. Bioinformtics. 2014; 15:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vinga S. Editorial: Alignment-free methods in computational biology. Brief. Bioinform. 2014; 15:341–342. [DOI] [PubMed] [Google Scholar]
- 13. Reinert G., Chew D., Sun F., Waterman M.S.. Alignment-free sequence comparison (I): statistics and power. J. Comput. Biol. 2009; 16:1615–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ren J., Song K., Deng M., Reinert G., Cannon C.H., Sun F.. Inference of Markovian properties of molecular sequences from NGS data and applications to comparative genomics. Bioinformatics. 2016; 32:993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qi J., Luo H., Hao B.. CVTree: a phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res. 2004; 32:Suppl. 2W45–W47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blaisdell B.E. A measure of the similarity of sets of sequences not requiring sequence alignment. Proc. Natl. Acad. Sci. 1986; 83:5155–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jun S.R., Sims G.E., Wu G.A., Kim S.H.. Whole-proteome phylogeny of prokaryotes by feature frequency profiles: an alignment-free method with optimal feature resolution. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sims G.E., Jun S.R., Wu G.A., Kim S.H.. Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:2677–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yi H., Jin L.. Co-phylog: an assembly-free phylogenomic approach for closely related organisms. Nucleic Acids Res. 2013; 41:e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marçais G., Kingsford C.. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011; 27:764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Narlikar L., Mehta N., Galande S., Arjunwadkar M.. One size does not fit all: on how Markov model order dictates performance of genomic sequence analyses. Nucleic Acids Res. 2013; 41:1416–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perelman P., Johnson W.E., Roos C., Seuánez H.N., Horvath J.E., Moreira M.A.M., Kessing B., Pontius J., Roelke M., Rumpler Y. et al. A molecular phylogeny of living primates. PLoS Genet. 2011; 7:e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paradis E., Claude J., Strimmer K.. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004; 20:289–290. [DOI] [PubMed] [Google Scholar]
- 24. Robinson D.F., Foulds L.R.. Comparison of phylogenetic trees. Math. Biosci. 1981; 53:131–147. [Google Scholar]
- 25. Miller W., Rosenbloom K., Hardison R.C., Hou M., Taylor J., Raney B., Burhans R., King D.C., Baertsch R., Blankenberg D. et al. 28-way vertebrate alignment and conservation track in the UCSC genome browser. Genome Res. 2007; 17:1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bernard G., Chan C.X., Ragan M.A.. Alignment-free microbial phylogenomics under scenarios of sequence divergence, genome rearrangement and lateral genetic transfer. Sci. Rep. 2016; 6:28970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.