

Abstract
Reversible protein phosphorylation is one of the major mechanisms in the regulation of protein expression and protein activity, controlling physiological functions of the important human pathogen Staphylococcus aureus. Phosphorylations at serine, threonine and tyrosine are known to influence for example protein activity in central metabolic pathways and the more energy-rich phosphorylations at histidine, aspartate or cysteine can be found as part of two component system sensor domains or mediating bacterial virulence. In addition to these well-known phosphorylations, the phosphorylation at arginine residues plays an essential role. Hence, the deletion mutant S. aureus COL ΔptpB (protein tyrosine phosphatase B) was studied because the protein PtpB is assumed to be an arginine phosphatase. A gel-free approach was applied to analyze the changes in the phosphoproteome of the deletion mutant ΔptpB and the wild type in growing cells, thereby focusing on the occurrence of phosphorylation on arginine residues. In order to enhance the reliability of identified phosphorylation sites at arginine residues, a subset of arginine phosphorylated peptides was chemically synthesized. Combined spectral libraries based on phosphoenriched samples, synthetic arginine phosphorylated peptides and classical proteome samples provide a sophisticated tool for the analysis of arginine phosphorylations. This way, 212 proteins phosphorylated on serine, threonine, tyrosine or arginine residues were identified within the mutant ΔptpB and 102 in wild type samples. Among them, 207 arginine phosphosites were identified exclusively within the mutant ΔptpB, widely distributed along the whole bacterial metabolism. This identification of putative targets of PtpB allows further investigation of the physiological relevance of arginine phosphorylations and provides the basis for reliable quantification of arginine phosphorylations in bacteria.
Staphylococcus aureus has emerged as an important human pathogen and is the causative agent of several nosocomial infections (1). It overcomes not only the challenging stress and starvation conditions of the ever-changing environment of bacterial habitats, but also host defense mechanisms and antibiotic treatments (2, 3). Furthermore, Staphylococcus aureus (S. aureus)1 can express a high number of different virulence factor proteins that boost pathogenicity by complex mechanisms (4). Basic research of staphylococcal protein regulation is therefore required to decipher molecular and cellular mechanisms that underlie pathogenesis and virulence.
Among the most important topics of these mechanisms are the elements of prokaryotic signal transduction, for example proteins modified by post-translational modifications (PTMs). Protein activity can be regulated by several PTMs. One of the most important groups within reversible protein modifications are phosphorylations on different amino acid residues (5). Because of their negative charge, phosphorylations can induce conformational changes to enable effects on protein structure, protein-protein interactions, protein activity or changes in substrate specificity and subcellular localization (6–8). To date, phosphorylation events on four different groups of amino acid residues are known: (1) the phosphorylation on hydroxyl groups to form phosphoesters (9, 10), (2) the phosphorylation on acidic groups to form phosphoanhydrides (11), (3) the phosphorylation on cysteine moieties to form thioesters (12), and (4) the phosphorylation on amide groups to form phosphoamidates (13, 14).
To provide evidence that reversible protein phosphorylation events occur in living bacteria and fulfil important functional roles in the regulation of protein activity, different methods were developed to analyze the more stable phosphoesters (15, 16) and applied to characterize the phosphoproteome of several prokaryotes (17–24).
Only recently, the existence and physiological relevance of phosphorylations on arginine residues was shown for the Gram-positive model organism Bacillus subtilis (25–27). The analysis of arginine phosphorylations by mass spectrometric approaches, however, comes along with substantial challenges concerning the stability of the phosphoamidates (28) and the reliability of mass spectrometric results (29, 30). Methodical improvements in this field were addressed, but still, further optimization is needed to consider the specific chemical properties of protein-N-phosphorylation. Because of the destabilization of the phosphoamidate bond by nitrogen protonation, N-phosphates in general are not stable under acidic conditions (28, 31). The stability of phosphoarginine decreases in a pH range below pH 3 (26) and on hot alkali (32).
First approaches to analyze phosphorylated arginine residues therefore relied on the detection of radiolabeled [γ-32P] in combination with Edman degradation (33). Only recently, a few other methods became available. The most important among them are enrichment techniques using either TiO2 beads (34) or a phosphatase trap mutant (27). In addition, an anti-phosphoarginine-specific antibody was designed (31).
On the level of data analysis, several software tools were developed in order to determine not only phosphorylated proteins and peptides, but also the phosphosite localization (35–37). Mann et al. and Goldstrohm et al., however, showed that the identification of phosphorylations should be reviewed by manual evaluation of all spectra and therefore relies on subjective judgements (38, 39). One example is the altered enzymatic specificity of trypsin, which is no longer supposed to cleave after phosphorylated arginine residues. That means that only peptides with a missed cleavage site and without terminal phosphorylated arginine residues can be truly positive, which is not considered by all data analysis software.
Although data analysis can be performed using different classical database search approaches (40, 41), spectral libraries are reliable tools for MS/MS based peptide identification (42) and can be used for complex proteomic samples or in combination with synthetic phosphopeptides to provide more detailed information on phospho-specific identification and localization features (43, 44).
In B. subtilis, the protein McsB was identified as phosphoarginine kinase having the protein YwlE as counteracting phosphatase (25, 45). Although staphylococcal McsB is known to influence stress tolerance and virulence, there is currently no proof of a possible arginine kinase activity (46). As putative counteracting arginine phosphatases, S. aureus COL possesses the two low-molecular-mass phosphotyrosine protein phosphatases PtpA and PtpB, described as tyrosine phosphatases with so far unknown substrates and metabolic functions (47, 48). In this study, the gene ptpB was exchanged with a spectinomycin resistance cassette to generate a mutant that is deficient in the phosphatase activity of PtpB. This mutant was compared with the wild type and investigated for qualitative changes in the phosphoproteome in exponential growing cells to scrutinize the putative phosphoarginine phosphatase or phosphoarginine phosphotransferase activity of PtpB. Hence, a gel-free phosphoenrichment protocol adapted to the specific challenges of arginine phosphorylations and subsequent LC-MS/MS measurements was applied. Raw data were analyzed with a combination of classical database search, spectral libraries of synthetic arginine phosphorylated peptides and spectral libraries of experimental data. This provides the basis for further investigation of physiological questions mediated by phosphorylations on arginine residues in S. aureus.
EXPERIMENTAL PROCEDURES
Mutant Construction
We constructed the isogenic deletion mutant ΔptpB in Staphylococcus aureus COL (49) using the pMAD mutant construction system (50). Briefly, a fusion product, which consists of ptpB upstream DNA, a spectinomycin resistance cassette and ptpB downstream DNA was ligated to the pMAD plasmid for homologous recombination and deletion of ptpB in S. aureus COL (see supplemental Table S1 for used primer sequences).
Bacterial Growth and Sample Preparation
S. aureus COL wild type (49) was grown in Luria-Bertani (LB) or a chemically defined medium (34); constant shaking at 37 °C was applied unless otherwise stated. Bacteria were cultivated under different stress conditions (supplemental Table S2) and harvested during exponential growth or in the stationary growth phase (supplemental Table S3) by centrifugation (5 min, 10,016 × g, 4 °C). Cells were used to prepare cytosolic and enriched membrane fractions according to Becher et al. (51) and Dreisbach et al. (52); Briefly, cell pellets were washed three times with TE buffer (50 mm Tris/HCl pH 7.5, 10 mm EDTA) and disrupted by bead beating. After removal of cell debris by centrifugation, samples were subjected to ultracentrifugation (100,000 × g, 60 min, 4 °C) and the supernatant was kept as cytosolic fraction. The enriched membrane fraction was obtained by alternate washing and centrifugation steps using (1) high salt buffer (20 mm Tris/HCl, 10 mm EDTA, 1 m NaCl, pH adjusted to 7.5), (2) carbonate buffer (100 mm Na2CO3, 10 mm EDTA, 10 mm NaCl, pH adjusted to 11), and (3) TE buffer. Afterward, the pellet was resolved in HTH buffer (8 m Urea, 2 m Thiourea). Both cytosolic and enriched membrane fractions were subjected to GeLC-MS/MS analysis according to Bonn et al. (53). Briefly, samples were subjected to SDS-PAGE and gels were stained with Coomassie. The gel lanes were cut into ten pieces, washed with gel wash buffer (0.2 m NH4HCO3, 30% (v/v) ACN, 37 °C) and desiccated. In-gel digest was performed overnight at 37 °C using 2 μg/ml trypsin. Elution was performed by ultrasonication and final samples were concentrated to a volume of 10 μl. Extracellular samples have been prepared according to Bonn et al. (53). Shortly, the culture supernatant was incubated with 20 μl primed StrataClean beads. Beads were subsequently sedimented by centrifugation, washed, resuspended in 1 ml TE buffer, dried and resolved in 30 μl TEAB buffer (50 mm triethylammonium bicarbonate, 20 μl RapiGest 0.2% (w/v)). After disulfide reduction and cysteine alkylation, in-solution digest was performed for six hours (0.25 μg activated trypsin, 37 °C, second addition of 0.25 μg trypsin after three hours). After centrifugation, beads were washed twice with elution buffer (30% (v/v) ACN, 0.1% (v/v) acetic acid). All three supernatants were pooled and samples were purified using C18-ZipTips (Merck Millipore, Billerica, USA). In short, ZipTips were wetted and equilibrated before peptides were loaded. Loaded tips were washed twice and eluted peptide samples were desiccated and resolved in buffer A (99.9% (v/v) acetic acid) before subjected to LC-MS/MS analysis. If no phosphoenrichment was performed, these cytosolic, enriched membrane and extracellular fractions were considered as “classical proteome samples.”
Preparation of Phosphoproteome Samples
For phosphoproteome experiments, the wild type and the mutant ΔptpB were grown in LB medium, mutant samples supplemented with 200 μg/ml spectinomycin. Each sample (n = 4) was cultivated and harvested during exponential growth (OD540 nm 0.5–0.6). Harvested cells were washed, subsequently incubated in lysis buffer (Tris/HCl 50 mm, pH 7.5, supplemented with 5 mg/ml lysozyme, 5 mm C3H7O6PNa2, 5 mm Na4P2O7, 5 mm Na3VO4 and 10 mm NaF) and disrupted as described for the cytosolic “classical proteome samples.” The suspension was afterward treated with 1× Nuclease Mix (GE Healthcare, Little Chalfont, United Kingdom; 10 min, RT) and 1% (w/v) Octyl β-d-glucopyranoside (Sigma-Aldrich, St. Louis, USA; 5 min, RT) before cell debris was removed by two centrifugation steps (5 min and 30 min, 15,800 × g, 4 °C). The protein concentration was determined using Roti®-Nanoquant (Carl Roth GmbH & Co. KG, Karlsruhe, Germany) according to the manufacturer's instructions to ensure the precipitation of 50 mg protein for each sample. Acetone precipitation was followed by sample digestion, strong cation exchange chromatography (SCX) and titanium dioxide bead based phosphopeptide enrichment according to Bäsell et al. (34) with slight modifications to respect the lower stability of arginine phosphorylations (Table I). Briefly, precipitated samples were resolved in denaturation buffer (6 m Urea, 2 m Thiourea in 10 mm Tris/HCl pH 8.0) and disulfide reduction as well as cysteine alkylation were performed. Samples were diluted and digested for twelve hours using 2 μg trypsin per mg sample. After sample acidification to pH 3.30 - 3.37 with TFA, SCX was performed according to Olsen et al. (54), adjusting the pH values of the SCX buffers with TFA to pH 3.2 to favor arginine phosphorylations. Peptides were loaded with buffer A (5 mm KH2PO4, 30% (v/v) ACN, pH 3.2) and eluted with a linear gradient of 0 - 30% of buffer B (5 mm KH2PO4, 350 mm KCl, 30% (v/v) ACN, pH 3.2). TiO2 enrichment was performed directly afterward, incubating each fraction for 30 min with 5 mg of DHB-pretreated beads at pH 3.0–4.5. Afterward, beads were washed twice with each wash solution (wash solution 1: 30% (v/v) ACN, adjusted to pH 3.30–pH 3.37 with TFA; wash solution 2: 80% (v/v) ACN, adjusted to pH 3.30 - pH 3.37 with TFA) and eluted three times (elution buffer: 40% (v/v) NH4OH, 60% (v/v) ACN, pH > 10.5) using C8-microcolumns (C8-StageTips (Thermo Fisher Scientific, Waltham, MA), packed with 3–4 × 1 mm2 pieces of 3 m Empore C8 material). All samples were measured by MS (see supplemental data for details).
Table I. Modified incubation times and temperatures.
For phosphoproteome experiments, samples were prepared according to Bäsell et al. (34) with the slight modifications listed in this table. LysC = Lysyl Endopeptidase (Wako Pure Chemical Industries Ltd., Osaka, Japan).
| Incubation step | Incubation conditions according to Bäsell et al. | Incubation conditions of adapted protocol |
|---|---|---|
| Centrifugation after harvest | 20 min, 8000 × g, 4 °C | 10 min, 10,016 × g, 4 °C |
| Cell wash | 2×, Tris/HCl 50 mm, samples freezed afterwards | 3×, Tris/HCl 50 mm, no freezing |
| Cell lysis | 15 min, 37 °C | 10 min, RT |
| Nuclease digestion | 15 min, 37 °C | 10 min, RT |
| Acetone precipitation | Overnight (app. 16 hours), −20 °C | Four volumes, 6 hours, −20 °C |
| Centrifugation after precipitation | 1 × 45 min, 8000 × g; 1 × 15 min, 10,000 × g, 4 °C | 1 × 30 min, 10,016 × g; 1 × 10 min, 15,800 × g, 4 °C |
| Evaporation of acetone rests | 30 min under fume cupboard | 6.5 min with concentrator plus (Eppendorf AG, Hamburg, Germany) |
| Enzymatic digestion | 8 hours LysC, Trypsin overnight (app. 14–15 hours) | 12 hours, Trypsin only |
| Acidification | 15 min, RT, pH 2.70 | 10 min, 12 °C, pH 3.30 − 3.37 |
| Centrifugation prior to SCX | 1 × 15 min, RT | 2 × 15 min, 12 °C |
| SCX | pH 2.70, fractions freezed afterwards | pH 3.2, no freezing |
| Wash during TiO2 enrichment | 1×, pH < 1 | 2×, pH 3.30 − 3.37 |
| Acidification prior to MS analysis | pH < 1 | pH 3.30 − 3.37 |
Synthetic Arginine Phosphorylated Peptides
For synthetic spectral libraries, 39 sequences of putative arginine phosphorylated peptides were selected from MaxQuant search results. These peptide sequences were synthesized by Pepscan (Lelystand, The Netherlands). Peptides were solved and diluted to 5–500 pmol/μl according to their physicochemical properties (supplemental Table S4). Instable arginine phosphorylations were analyzed additionally by offline measurements coupled to the MS via direct infusion.
Enrichment of Synthetic Arginine Phosphorylated Peptides with TiO2
To test the TiO2 enrichment protocol for its influence on peptide stability and reproducibility of replicates, the physicochemical properties of all synthetic arginine phosphorylated peptides were analyzed, focusing on sequence specific stability at different temperatures. A subset of five synthetic arginine phosphorylated peptides of medium stability reflecting highly different properties (supplemental Table S5) was then subjected to TiO2 enrichment and C8 purification followed by LC-MS/MS analysis as described for complex samples. To determine peptide stability, recovery rate, enrichment efficiency and reproducibility of sample preparation, integrated peptide peak areas were used for all fractions examined. In total, seven different fractions (supplemental Table S6) were compared regarding the amount as well as the ratio of phosphorylated and nonphosphorylated isoforms. Experiments were carried out in three replicates.
Classical Data Analysis
For classical database search, spectra were analyzed with the software MaxQuant (version 1.5.3.30, Max Planck Institute of Biochemistry, Martinsried, Germany) (55); peak lists were searched by the integrated search engine Andromeda (56) against the database from Uniprot (release May 2015), containing 2680 proteins of S. aureus COL (common laboratory contaminants and all reversed sequences added). Parameters for mass deviation were set to 6 ppm at Full scan level and 0.5 Da for CID spectra or 20 ppm for HCD spectra at MS/MS level. A false discovery rate (FDR) of 0.01 on protein, peptide and phosphosite level was applied as well as a minimum peptide length of seven amino acids and two peptides per protein. Full tryptic specificity with a maximum of two missed cleavage sites was applied. Variable modifications were oxidation on methionine (+15.9949 Da), carbamidomethylation on cysteine (+57.0214 Da) and phosphorylation on serine, threonine, tyrosine or arginine (+79.9663 Da). Phosphorylated peptides were then subjected to manual validation and the spectrum was only considered to be of sufficient quality and kept for further steps if all validation criteria were fulfilled (Table II).
Table II. Criteria for manual validation of phosphospectra. Phosphorylated peptides were subjected to manual validation and the spectrum was only considered to be of sufficient quality and kept for further steps if the following validation criteria were fulfilled.
| Filter criteria | Source |
|---|---|
| Phosphosite localization probability ≥ 0.75 | Bäsell et al. (34) |
| Precursor mass deviation < 5 ppm | Olsen et al. (80) |
| Identification of at least three phosphorylated fragment ions with sufficient signal to noise level | Varied after Mann et al. (38) |
| b/y Ion coverage confirming the phosphorylation site must not be impaired by peak clusters | This study |
| Intensity of precursor ion ≠ 0 | This study |
| Ion series coverage must be able to locate the arginine phosphorylation unambiguouslya if the sequence contains additional STY residues | This study |
| No terminal arginine phosphorylation is allowed (only protein C-terminus) | This study |
| No multiple phosphorylation is allowed | This study |
aunambiguous identification = Based on probability and fragment ion coverage neither software based evaluation nor manual validation of spectra indicate adjacent STY residues as possible phosphosites.
Construction of Spectral Libraries and Spectral Library Search
For spectral library construction, 1095 .raw files (supplemental Table S7) were converted to mzXML (MSConvert GUI, ProteoWizard (57)) and processed using the Trans Proteomics Pipeline (58) (TPP, v. 4.8.0) (see supplemental data for details).
For spectral library search, 376 .raw files of the wild type and the mutant ΔptpB (n = 4) were subjected to SpectraST library search. Search results were combined to interact.pep.xml, filtered for maximum absolute precursor mass tolerance of less than 0.005 Da and with dot value cut-off enabled for CID data. To determine the respective cut-off value, raw files were first searched against a SpectraST library only containing consensus spectra of phosphorylated peptides. Search results were subsequently filtered for the minimum dot value, which reflected no false positives on the level of phosphospectra. Subsequently, raw files were searched against the SpectraST library consisting of phosphorylated as well as nonphosphorylated peptides and the previously determined cut-off value was applied for filtering. For HCD data, probability filter was enabled.
Experimental Design and Statistical Rationale
For spectral library construction, proteome samples of S. aureus COL wild type were prepared after cultivation under eleven different growth conditions (supplemental Table S2) without biological or technical replicates, generating samples for cytosolic, enriched membrane and extracellular fraction (1095 .raw files, origin is provided in supplemental Table S7). Spectral libraries for nonphosphorylated peptides considered hits with probability ≥ 0.95. All phosphoenrichment fractions of S. aureus COL wild type and mutant were analyzed with MaxQuant (FDR = 0.01 on protein, peptide and phosphosite level, probability of phosphosite localization ≥ 0.75 according to PTMscore implemented in MaxQuant software) and phosphospectra passing filter criteria and manual validation (Table II) were added to the library. For data analysis based on spectral libraries, four biological replicates (n = 4) of the wild type and the mutant ΔptpB were then subjected to SpectraST search (376 .raw files). SpectraST search results were filtered afterward for probability ≥ 0.9 (HCD samples) or dot values (CID samples). By means of decoy hits, the cut-off for the dot value was set to the value reflecting no false positives on the level of phosphospectra. For the experiment regarding reproducibility among technical replicates, 18 phosphoenrichment fractions of ΔptpB (grown in LB) were prepared according to the protocol described above. Each fraction was then separated into four immediately prior to the MS measurement. This way, four sets of data, each containing 18 .raw files, were subjected to MaxQuant and SpectraST library search. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (59) via the PRIDE partner repository (60) with the data set identifier PXD007167.
RESULTS
Slight Modifications Within the Workflow Greatly Enhance the Identification of Arginine Phosphorylations
The analysis of arginine phosphorylations is challenging because of the lower stability of protein-N-phosphorylations (28), the occurrence of phosphoshifts from arginine to serine or threonine residues (29) and the chemical properties under acidic conditions (26). Therefore, the applicability of the gel-free, site-specific method for reliable identification of O-phosphorylation established by Olsen et al. (54) to arginine phosphorylation was analyzed. A subset of five synthetic arginine phosphorylated peptides was enriched with TiO2 to assess recovery, retention and conversion capacity as well as the influence of TiO2 on the stability of arginine phosphorylated peptides. To assess peptide stability, a directly measured peptide mixture was compared with a peptide mixture stored at room temperature for the same time the TiO2 enrichment took place. It can be seen that 55–66% of the phosphorylations were lost after 7.5 h at room temperature (Table III). To address the retention and conversion capacity of C8 purification, the untreated samples were compared with samples purified with C8 material. In the result, phosphorylated peptides were almost completely recovered whereas 14–19% of the nonphosphorylated peptide fraction was captured by C8. To investigate the binding capacity of TiO2 in a further step, untreated samples were compared with the loading fraction after TiO2 enrichment. This resulted in 79–92% of synthetic peptides that were bound to TiO2, proving the general binding capacity of TiO2. Interestingly, under the given conditions, also nonphosphorylated peptides were bound to TiO2, additionally emphasizing the need to adapt the protocol to the complexity of the samples. This experiment was performed with five synthetic arginine phosphorylated peptides instead of a total protein extract of S. aureus to have a known initial concentration of phosphorylated peptide isoforms. The lower complexity resulted in an oversupply of binding sites that can be occupied by nonphosphorylated peptides as well. To finally assess the retention and conversion capacity of TiO2, loading, wash and elution fraction were compared. Results depended on the amino acid sequence, but between 60 and 99% of the peptides were recovered within the elution fraction. Finally, the washing steps were compared with the elution fraction regarding the amount and ratio of phosphorylated and nonphosphorylated peptide forms. It can clearly be stated that the washing procedure primarily contained nonphosphorylated peptide forms. Results, however, depended on the initial amount of phosphorylated peptides. Whereas low abundant peptides were almost only found in their nonphosphorylated forms within the washing fractions, high abundant peptides were also partly found in their phosphorylated form. However, the washing fraction always contained a higher amount of nonphosphorylated isoforms. It can therefore be concluded that the general enrichment procedure using TiO2 can be applied for arginine phosphorylations under the given conditions, but optimal results depend on the amount and ratio of TiO2 beads in comparison to the complexity and concentration of the phosphorylated peptide mixture. To estimate the benefits of the adapted sample preparation protocol for the enrichment of arginine phosphorylated peptides, two replicates of a mutant sample were prepared according to either the original protocol of Olsen et al. (54) or the adapted protocol described in the methods section. Notably, the number of arginine phosphosites greatly increased when using the adapted method (Fig. 1). These findings support the notion that slight adaptations considering the lower stability enable the analysis of arginine phosphorylations with formerly established protocols.
Table III. Synthetic arginine phosphorylated peptides subjected to TiO2 enrichment. The following peptides were selected according to the criteria displayed in supplemental Table S5. Length displays the number of amino acids. MW = molecular weight in Da. RT = retention time in 30 minutes gradient (see methods section). Displayed is the average retention time (average values between all replicates) in minutes of the phosphorylated isoform. Stability is determined in % of remaining phosphorylated isoform after 7.5 hours incubation at 25 °C.
Fig. 1.
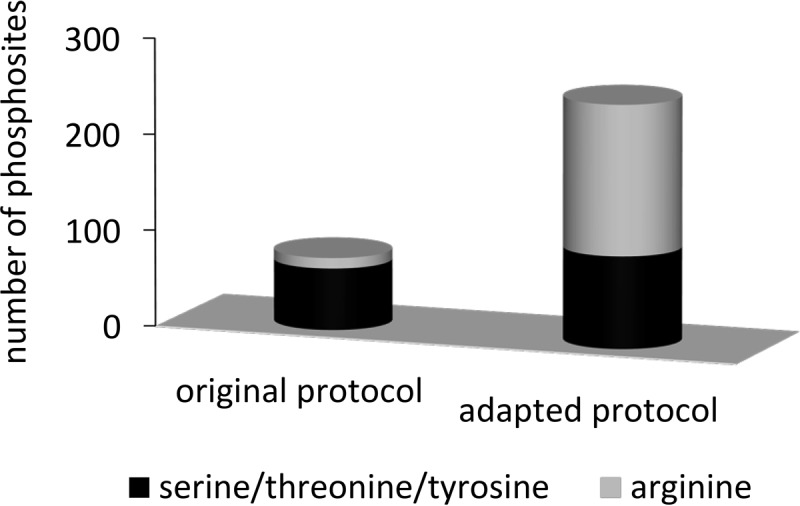
Comparison of original and adapted method. Comparison of two replicates of ΔptpB indicating methodical improvements. Results show the number of identified phosphosites after sample preparation with the protocol of Olsen et al. (54) in comparison with the number of phosphosites after sample preparation with the adapted protocol described in the methods section. The abundance of phosphosites on arginine residues is colored gray, the number of phosphosites on serine, threonine or tyrosine residues is depicted in black.
Combined Spectral Libraries Provide an Additional Tool for Reliable Identification of Arginine Phosphorylations in S. aureus COL
All collected data of classical proteomic and phosphoenriched samples as well as synthetic peptides were included into spectral libraries to improve protein identification in phosphoproteome analyses by matching true fragmentation patterns of peptides instead of theoretical spectra generated during classical database search. The workflow for construction of spectral libraries is depicted in Fig. 2. Briefly, all phosphoenriched samples were searched with MaxQuant and phosphorylated spectra were filtered and manually validated. In parallel, phosphoenriched samples were searched with Comet, X! Tandem and Sequest and combined to raw spectral libraries containing all phosphorylated peptides irrespective of their quality metrics. In the following step, only spectra of high quality were kept in the phosphopeptide spectral library, providing 1,688 consensus spectra reflecting 960 phosphopeptides (396 peptides are phosphorylated on arginine residues). To improve the spectral quality of arginine phosphopeptides, a subset of 39 arginine phosphopeptides was synthesized chemically (supplemental Table S4). 38 synthesized peptides were reliably identified by spectral library search and subsequent manual validation, leaving out a single false positive. To additionally provide a broad range of proteomic data for nonphosphorylated proteins, classical proteomic data were additionally analyzed with the TPP. This resulted in a high quality spectral library containing 108,438 consensus spectra of 2257 proteins of S. aureus COL, which account for 84% of the theoretically predicted proteome. 2071 proteins (77%) were found with at least two unique peptides and 385 phosphoproteins are part of the library, 191 with at least one arginine phosphosite. This study therefore provides a dataset of S. aureus COL proteins reflecting the highest proteome coverage reported to date. Finally, all consensus libraries were combined to two spectral libraries (one for CID and one for HCD), considering equal spectrum numbers for phosphopeptide species of experimental and synthetic origin. These combined spectral libraries based on Andromeda search results, synthetic arginine phosphorylated peptides and classical proteomic experiments provide a valuable tool to facilitate elucidation of crucial physiological questions from S. aureus COL. The spectral libraries have been deposited to the ProteomeXchange Consortium (59) via the PRIDE partner repository (60) with the data set identifier PXD007167.
Fig. 2.

Workflow to construct combined spectral libraries considering arginine phosphorylations, Andromeda based identifications, synthetic phosphopeptides and classical proteomic data. 1095 .raw files were searched by different search engines, combined, validated and used for construction of raw spectral libraries. Phosphoenriched samples were searched using the search engine Andromeda implemented in the software MaxQuant. All spectra passing the manual validation were considered for implementation in the spectral library. For technical reasons, the raw library was divided into two in order to separate STY phosphorylations from arginine phosphorylations. Classical proteomic data were subjected to Comet and X! Tandem and final interact.ipro.pep.xml files underwent iRT alignment before raw spectral libraries were cleared of false positive hits, human contaminants and phosphorylated peptides and filtered for probability > 0.95. Synthetic spectral libraries were cleared of false positives, human contaminants and filtered for probability > 0.9. In addition, each consensus spectrum of the synthetic spectral library was validated manually. Afterward, all three libraries were combined, considering equal spectrum numbers for comparable phosphopeptide species of the experimental and the synthetic spectral library. The Voronoi treemap (79) displays the functional categories of all theoretical proteins of S. aureus (2680 proteins, colored dark gray). Each cell displays a single protein. Light gray colored proteins are part of the spectral library and if one or more phosphopeptides are part of the library, the respective protein is depicted in light blue. Proteins containing at least one arginine phosphorylation are depicted in dark blue. Different levels of the treemap are additionally provided as separate high quality figures (supplemental Fig. S6A–S6C).
451 Phosphosites Were Identified in S. aureus COL
The high-quality data of the spectral library can be used to validate spectral data from subsequent MS analyses. The phosphoproteome of the deletion mutant ΔptpB, which lacks the ability to express the predicted phosphatase PtpB, was compared with the wild type in exponential growing cells (cultivated in LB medium) regarding the pattern of arginine protein phosphorylations. Raw files of wild type and mutant (n = 4) were subjected to spectral library search. This way, phosphosite, -peptide and -protein identifications were compared between wild type and mutant. Overall, 167 phosphosites on 102 proteins were identified in the wild type (supplemental Table S8) and 421 phosphosites on 212 proteins within the mutant (supplemental Table S9, Fig. 3). Considering all possible phosphorylated amino acids, it became obvious that deletion of ptpB led to a higher number of identified phosphosites (supplemental Table S10) and phosphopeptides (supplemental Fig. S1), respectively. Eight arginine phosphosites were identified in wild type and mutant, but another 207 arginine phosphosites were exclusively identified in the mutant ΔptpB. Remarkably, although the number of tyrosine phosphorylations is comparable between wild type and mutant, the number of phosphosites at serine and threonine residues is about 30% higher in the mutant than within the wild type. To address substrate specificity and possible protein side effects, in vitro experiments with recombinant PtpB were performed. PtpB was incubated with synthetic peptides phosphorylated on either arginine or adjacent serine or threonine phosphorylations demonstrating that PtpB solely dephosphorylated arginine phosphorylated substrates (supplemental Table S12). In addition, this experiment showed that no dephosphorylation was observed after adding the same phosphatase inhibitors that have been used for sample preparation (see supplemental data for details).
Fig. 3.

Number of identified phosphoproteins. Comparison of the wild type and the mutant concerning the number of identified phosphoproteins.
To further address substrate specificity, motif analyses were performed. Unfortunately, no preferred sequence motifs were identified. This is not surprising considering the fact that Schmidt et al. performed extensive analyses for B. subtilis with similar results (26). However, it is at least possible to identify some amino acid residues, which are more likely to be found close to arginine phosphorylations. Indeed, several peptides with more than two serial glycine residues near arginine phosphorylations (± ten amino acids) were identified. Furthermore, we identified a higher abundance of the amino acids glutamine and asparagine as well as glutamic acid and aspartic acid about identified arginine phosphorylations. In the case of glutamine, this is consistent to observations of Schmidt et al. In contrast to Schmidt et al., however, we did not find an accumulation of serine residues next to arginine phosphorylations. We therefore conclude that no preferred sequence motifs exist for arginine phosphorylation, but the accumulation of several amino acids residues possibly provides the basis for subsequent studies.
To analyze possible co-occurrence for proteins with several phosphosites, results were additionally filtered for multiple phosphorylations as well as phosphosites in close neighborhood. This left out 12% of proteins containing arginine and serine, threonine or tyrosine phosphosites next to each other. Only for these proteins, co-occurrence of arginine and serine or threonine residues might be an issue. Although co-occurrence might not affect the results of this study regarding the physiological relevance of arginine phosphorylations because of the low number of cases, multiple phosphopeptides were excluded from detailed physiological analyses in order to prevent consideration of putatively mislocalized phosphosites.
Identification as well as Reproducibility Were Highly Increased with Spectral Library Search
To compare database with spectral library search, samples were subjected to a classical database search, as well as to an identification search using a combined spectral library. Although database search allowed 382 phosphopeptide identifications (serine, threonine, tyrosine or arginine residues) on 195 phosphoproteins, the identifications obtained by spectral library search were significantly higher belonging to 459 phosphopeptides of 228 phosphoproteins. The intersection accounted for 74.8% on the level of phosphoproteins. In addition, spectral library search exclusively identified 13 arginine phosphorylated proteins as for example another transcriptional regulator (SACOL1065).
Besides the total number of identifications, focus was laid on the reproducibility of results. One sample was measured four times to generate technical replicates. After subjection to classical database search, the number of arginine phosphorylations was investigated. Although the absolute number of identifications was comparable (43.8 ± 5.7 arginine phosphopeptides), the overlap among all four replicates was about 15%. To yield sufficient reliability of identification results, at least four biological replicates were therefore considered as necessary for each experiment and only results that were found in at least two biological replicates are supposed to be considered for further analyses. On the contrary, the same raw data were subjected to a SpectraST library search only containing consensus spectra of phosphorylated peptides resulting in 47 ± 2.6 arginine phosphopeptides and an intriguingly higher overlap of more than 41%.
Phosphorylations Are Widely Distributed Within the Bacterial Metabolic Pathways
In a further step, global impact and physiological relevance of arginine phosphorylations were addressed. According to TIGRFAM (61) or KEGG based annotation (62), each phosphorylated protein was classified to functional groups of proteins. This protein family classification scheme was used to sort identified phosphorylations in metabolic pathways or functional groups. It can be demonstrated that phosphorylations on serine, threonine, tyrosine and arginine are widely distributed along the whole metabolism. Most of the identified phosphoproteins (phosphorylated on arginine, serine, threonine or tyrosine residues) cover all displayed functional groups to a similar extent, but carbohydrate metabolism, transcription, protein synthesis and energy metabolism stand out (Fig. 4). Indeed, nearly 43% of all identified phosphoproteins belong to one of these four groups. If the distribution of metabolic pathways and functional groups is regarded only for arginine phosphorylations, a similar pattern was observed (nearly 48% belong to these four groups, supplemental Fig. S2). It can therefore be seen that phosphorylations in general as well as arginine phosphorylations are widely distributed along the entire bacterial metabolism of S. aureus COL.
Fig. 4.

Functional groups of identified phosphoproteins of the mutant ΔptpB. Each phosphoprotein was matched to one or more functional groups, according to TIGRfam or KEGG based annotation. The green line shows the absolute number of identified phosphoproteins for the main functional groups whereas the orange line represents the percentage coverage of identified phosphoproteins with respect to the theoretical number of proteins belonging to this group. Highlighted with a red box are the functional groups that stand out.
Although phosphorylations were found in distinct physiological classes, a closer look to protein synthesis showed that an intriguingly high number of phosphorylated proteins belonged to the translation machinery, subdivided into ribosomal proteins, tRNA ligases and translation factors (Fig. 5A). The same inclination was noted when investigated for arginine phosphorylations only. Strikingly, the distribution of the phosphoproteins respecting the phosphorylation on different amino acids showed that for example five out of six phosphorylated translation factors were found with phosphosites at arginine and serine/threonine/tyrosine residues (Fig. 5B).
Fig. 5.

Functional classification of arginine phosphorylated proteins of the mutant ΔptpB. Among the protein biosynthesis, a high number of phosphorylated proteins belonging to the functional classes of ribosomal proteins, tRNA ligases and translation factors was identified within the mutant. A, Number of phosphoprotein identifications belonging to functional classes of protein synthesis Absolute as well as relative values of phosphoproteins identified in only one (colored light gray) or at least two biological replicates (colored dark gray) of the theoretically annotated proteins (colored white). The group “theoretically annotated proteins” comprises all proteins belonging to the respective functional group according to TIGRFAM and KEGG based annotation. B, Phosphoprotein identifications differentiated according to amino acid Detailed view on the distribution of the phosphoproteins in respect of the phosphorylation on different amino acids. Considered are only phosphorylations that are found in at least two biological replicates. The sum of the number of proteins phosphorylated on arginine residues (colored dark gray) and the number of proteins phosphorylated on serine, threonine or tyrosine residues (colored light gray) is higher than the total number of proteins (black bar), indicating that a lot of phosphoproteins were found with more than one phosphosite.
Arginine Phosphorylations Are Part of Important Regulons
To evaluate the impact of protein arginine phosphorylation on the regulation of cellular processes, results were searched for regulator proteins. Within the mutant ΔptpB, three regulator proteins and a sigma factor were phosphorylated on arginine residues: CtsR, MgrA, SigA, and SACOL1065, two of them regulating a high number of proteins likewise phosphorylated on arginine (ten for MgrA, four for CtsR) or STY residues (eleven for MgrA, five for CtsR). In addition, although the regulator CggR itself was not found with phosphorylated residues, four out of five proteins of the respective regulon were found to be phosphorylated on arginine and/or serine/threonine/tyrosine residues. A similar observation was made for the regulons of HrcA, PerR and SigB. As to HrcA, three out of six proteins belonging to this regulon (63) show at least two arginine phosphosites. This agrees with former observations in B. subtilis, investigating the role of protein arginine phosphorylation under oxidative stress conditions (26, 64). In addition, the transcriptional regulator SarA as well as 23 proteins belonging to this regulon were found phosphorylated, 14 with at least one phosphosite on arginine residues and many of them associated to putative roles under infection related or virulence conditions (65).
DISCUSSION
Spectral Library Based Analyses Complement Classical Database Search
Protein identification can be improved by the use of spectral libraries because true fragmentation patterns of peptides are matched instead of theoretical spectra generated during classical database search. As spectral library based identification is limited to spectra formerly added to the library, spectral library based analyses complement classical database search hazarding the consequence that completely new spectra and their respective peptides cannot be found. To investigate this effect on our data, classical database search based on MaxQuant was compared with spectral library search. The intersection accounted for 74.8% on the level of phosphoproteins. Thus, protein identifications specific for the database search contributed only to a small proportion to the total sum of identifications (5.8%). Depke et al. showed similar results comparing classical database search using Mascot and SpectraST library search (3).
These identifications in spectral library searches but not in database searches illustrate the advantage of applying the spectral library from this study to phosphoproteome samples of S. aureus COL.
Comparison of Combined Libraries with Different Numbers of Spectra Used for Construction of Consensus Spectra
During the construction of combined spectral libraries, a high difference between the numbers of spectra originating from experimental and synthetic data was observed: Phosphoproteins are often of low abundance and there is only a small number of phosphorylated amino acids per phosphoprotein. Moreover, it is possible that only some fractions of one protein species are phosphorylated under the given cultivation condition. Together with the challenges imposed by the complexity of proteome samples and the mass spectrometric analysis of especially arginine phosphorylations, this leads to a high number of cases where only a very limited number of high quality phosphospectra were identified for the respective arginine phosphosite. Aside from that, the chemical synthesis of artificial arginine phosphorylated peptides provides the respective peptide in a high concentration and allows the generation of hundreds or thousands of high quality spectra representing the same peptide sequence and mostly all relevant charge states. This resulted in 84% of phosphopeptides with a ratio between spectra of synthetic and experimental origin of > 50:1 in the present study. For this reason, different ways of combining experimental and synthetic spectral libraries regarding the number of identifications as well as the quality of resulting consensus spectra were tested. Eleven arginine phosphopeptides cannot be found when data were searched against the combined library with disparate number of spectra used for consensus instead of a search against a library only containing spectra of experimental origin. Ten of them, however, can again be found when equal numbers of spectra of synthetic and experimental origin were combined. This could be explained by the fact that experimental libraries were constructed from phosphoenriched samples and spectra of phosphoenriched samples searched against these libraries are more similar than their synthetic homologues. For libraries with disparate numbers, the consensus spectrum is generated from a high number or solely out of spectra of synthetic origin, which might suppress special features of spectra of experimental origin. In addition, it was tested whether the generation of independent data to subject to the spectral libraries would influence the above results. Hence, all spectra originating from a fifth biological replicate were removed from the library so that the libraries did not contain any spectra of this sample. This sample was then searched against the libraries. Results showed nevertheless the same tendencies as described above (data not shown). It is therefore advised to generate combined libraries with equal numbers of spectra used for the construction of consensus spectra, combining the benefits of clear and reasonable, nearly perfect spectra of synthetic peptides together with characteristic features of experimental spectra originating from the same type of data than the samples. To support this, the quality of the different spectral libraries was compared and measured by dot values and probabilities. When data of four biological replicates of ΔptpB were searched against spectral libraries of synthetic peptides, the mean probability values were approximate to 1 (supplemental Fig. S3A). This could be explained by the fact that spectra of chemically synthesized peptides provide a nearly perfect match of the respective peptide species. Because the phosphospectra used for generation of experimental spectral libraries were implemented after validation on MaxQuant level and manual validation, but did not have to pass probability filters of TPP search engines, the probabilities for experimental libraries of arginine phosphorylation cannot be compared in a reliable way. They differ within the experimental library between 0.001 and 1 (supplemental Fig. S3B). Searches against combined libraries with equal numbers of spectra, however, nearly achieved probabilities as high as searches against synthetic libraries for comparable phosphopeptide species. In addition, when dot values of the same samples were compared among all spectral libraries (experimental library, combined library with equal and disparate numbers and synthetic library), analyses revealed that highest values were identified for experimental libraries and lowest values for synthetic ones, again providing the best compromise with combined libraries with equal numbers of spectra (supplemental Fig. S3C). To conclude, combined libraries with equal numbers of spectra used for consensus provide the best repository of arginine phosphorylated spectra.
SpectraST Search Results of CID Measurements Were Filtered for Dot Values
In addition, when CID measurements are subjected to SpectraST library search, the appropriate dot value cut-off had to be determined to enable reliable filtering of search results. If samples were searched against the library containing spectra of phosphorylated as well as nonphosphorylated peptides, the cut-off to reflect no false positives on the level of phosphospectra resulted in a mean value of 0.88 (standard deviation = 0.02). However, when samples were filtered against a cut-off this strict, a high number of synthetic arginine phosphorylated peptides cannot be found within the remaining search results indicating a high number of false negatives. This is in accordance with recent studies of Hart-Smith et al., which indicated that global FDR estimates influence the reliability of results for modified peptides (66). To decrease the number of false negatives and to determine both the correct cut-off as well as the number of remaining decoy hits after filtering with less strict criteria, samples were subjected to a SpectraST library search against a spectral library only containing consensus spectra of phosphorylated peptides. This provided a mean cut-off value of 0.783 (standard deviation = 0.005). This cut-off was then applied to the search results against the whole library resulting in about 0.05% of remaining decoy hits within the final search results. These data indicate that an appropriate cut-off can be determined if samples were first subjected to a SpectraST library search only containing consensus spectra of phosphorylated peptides, decreasing the otherwise very high number of false negatives. This cut-off can in turn be used for the search against the complete library. However, it would not be advisable to apply the same filter criteria when nonphosphorylated peptides are of interest.
For the results of HCD measurements, however, the standard probability filter of 0.9 was sufficient to yield high quality results. Indeed, if results of HCD measurements are filtered the same way than CID measurements, this results in a relatively high number of remaining decoys (data not shown).
PtpB Acts as Phosphoarginine Phosphatase
S. aureus contains two low-molecular-mass phosphotyrosine protein phosphatases, which are known to be more common in Gram-negative bacteria (47) and were formerly characterized in vitro as acid low-molecular-mass phosphotyrosine protein phosphatases PtpA and PtpB (47). Those in vitro studies reported substrate specific activity of PtpB to release inorganic phosphate from o-phosphotyrosines with no effect on o-phosphoserines or o-phosphothreonines. However, an arginine phosphate containing substrate has never been tested for these studies. In vitro experiments performed within this study using recombinant purified PtpB and synthetic arginine phosphorylated peptides proved assumed phosphatase activity under the given conditions.
Additionally, the phosphotyrosine phosphatase activity of PtpB in S. aureus can be proved by structural motifs typical for LMWPTPases, which are highly conserved among different staphylococcal strains and even species and play pivotal roles in the catalytic cleavage mechanism (48).
In the present study, 207 arginine phosphorylations were identified exclusively within ΔptpB, revealing putative targets of PtpB. Although the number of tyrosine phosphorylations is comparable between wild type and ΔptpB, almost no phosphosites were identified within the wild type in terms of arginine phosphorylations, whereas the number of identified arginine phosphosites of the mutant ΔptpB comprises more than 50% of all identified phosphorylations of the mutant. Thirteen tyrosine phosphosites were identified for wild type samples and 15 tyrosine phosphosites were found within ΔptpB samples, providing an intersection of ten phosphopeptides. As no significant differences were found regarding phosphotyrosine results, these phosphosites might be targets of the second acid low-molecular-mass phosphotyrosine protein phosphatase PtpA, which is showing a higher enzymatic activity in vitro (47). It should further be noted that the respective homologous arginine phosphatase YwlE in B. subtilis only exhibits tyrosine phosphatase activity under acidic pH conditions (67) while targeting exclusively arginine phosphorylations under neutral or physiological conditions (27). Indeed, in vitro studies targeting the enzymatic activity of PtpB also describe the highest tyrosine phosphatase activity of PtpB under slightly acidic conditions (47). Besides, S. aureus contains CapC, another tyrosine specific phosphoesterase, belonging to the Polymerase and Histidinol Phosphatase family (PHP family) that is more common in Gram-positive bacteria (47). CapC could maybe target the phosphotyrosine peptides of this study. Further, it is known that the deletion of the ptpA or ptpB gene does not affect growth of S. aureus under every condition (68). Together with the fact that the same sequence motifs of the respective protein homologs are used for catalysis in Yersinia pestis, some studies suggest that the phosphotyrosine phosphatase is only active under infection conditions (68–70). Our hypothesis, based on the literature (71), that the protein PtpB possesses arginine phosphatase or arginine phosphotransferase activity in S. aureus COL is further supported by the fact that all eight arginine phosphosites identified in the wild type were also found in ΔptpB, but additional 207 arginine phosphosites were exclusively found in ΔptpB. In addition, in vitro studies addressing phosphatase activity and substrate specificity of recombinant purified PtpB showed significant phosphatase activity for exemplary synthetic arginine phosphorylated peptides, which have been identified as putative targets of PtpB. Additionally, the assay did not show enzymatic activity for the respective synthetic peptides phosphorylated on adjacent serine or threonine phosphosites (supplemental Table S12).
Moreover, arginine phosphosite identifications of this study were distributed within most metabolic pathways. Recent studies in B. subtilis, suggesting that protein arginine phosphorylation in bacteria marks proteins for degradation (45), could explain the wide distribution in S. aureus as well. Taken together, the current study did not confirm the phosphotyrosine activity of the protein PtpB, but identified a possible arginine phosphatase or phosphotransferase activity.
Comparison with Further Phosphoproteome Studies in S. aureus COL
This study was compared with former results of phosphoproteome analyses in S. aureus COL. Gel-free TiO2 enrichments of Bäsell et al. (34) identified 30 proteins phosphorylated on STY residues (multiple phosphopeptides excluded). 20 of these proteins were also found in wild type samples of this study and another three proteins were found in ΔptpB samples. Moreover, 78 additional proteins phosphorylated on STY residues for wild type samples were exclusively identified within this study. These results could be explained by the fact that Bäsell et al. cultivated under differing conditions (Bäsell et al. cultivated the late exponential and transient growth phase in chemical defined synthetic medium; this study investigated exponential growing cells cultivated in LB medium) and used a slightly different sample preparation protocol as well as an older MaxQuant version. The overlap is also limited because of technical restrictions because SpectraST search can only identify phosphopeptides that were added to the library first. Some phosphorylations of Bäsell et al. which could not be identified in this study, however, were not part of the spectral library. MaxQuant and SpectraST assigned the respective spectra to different peptide sequences and these spectra had to be removed from the library for this reason. To minimize these restrictions, all phosphospectra of the study of Bäsell et al. (34) providing sufficient spectral quality were added to the spectral library. Besides, six out of seven phosphorylations on arginine residues identified within the former study were again identified in the wild type or mutant samples of this study and 209 additional arginine phosphosites were identified in the mutant. Hence, our findings complemented the study of Bäsell et al. (34).
Comparison with Studies on Arginine Phosphorylations in B. subtilis
Protein arginine phosphorylation plays an important role in the regulation of cellular processes, but only recently putative target proteins as well as physiological connections to different stress conditions were investigated in B. subtilis (25, 26, 64). It is therefore interesting to compare the results of this study with phosphorylations on arginine residues with putative or evident target proteins of former studies in B. subtilis. Elsholz et al. (25) and Schmidt et al. (26) identified 190 proteins phosphorylated on arginine residues. 39 of the respective homologous proteins were found phosphorylated on arginine residues within the mutant samples of this study (supplemental Fig. S4). For another 17 proteins, no protein homologue exists in B. subtilis. Comparing the level of phosphosites in B. subtilis and S. aureus, it became obvious that 69% of all arginine phosphoproteins were only identified within S. aureus. Additionally, 17.5% of proteins are not only phosphorylated on arginine residues within both organisms but were identified with at least one identical or adjacent arginine phosphosite (supplemental Fig. S5).
Moreover, Elsholz et al. (25) co-purified 264 proteins together with the PtpB homologous protein phosphatase YwlE. For 23% of these co-purifications, homologous proteins were found with phosphorylations on arginine residues within the current study. The findings of Schmidt et al. (26), identifying the highest number of arginine phosphorylations in proteins belonging to protein synthesis pathways and TCA cycle also fit with the identifications in this study. Taken together, despite the different cultivation conditions, different harvesting time points and even the use of another organism, arginine phosphorylation seems to be widely conserved at least for Gram-positive bacteria.
Arginine Phosphorylations Could Play a Role for Staphylococcal Virulence
Several studies suggest a possible role of the protein PtpB for staphylococcal pathogenesis and virulence although evidence and target proteins remained elusive. Furthermore, Wozniak et al. (46) described the impact of the respective arginine kinase McsB in S. aureus for staphylococcal pathogenesis and virulence. The phosphorylation on arginine residues of regulators such as MgrA and a high number of the proteins belonging to the regulon of MgrA or SarA might bridge the gap among these former suggestions (72, 73). Aside from that, PerR and CtsR influence the ability of S. aureus COL to adapt to stress conditions, for example in host pathogen interactions and virulence modulating effects of SigB were studied (74–78). Further studies targeting these stress conditions, as for example stress induced by antibiotics, ROS or infection related conditions, could therefore shed light on possible links between arginine phosphorylations and staphylococcal virulence.
CONCLUSION
This study provides a complex and comprehensive protein repository of high proteome coverage of S. aureus COL including identification of serine/threonine/tyrosine as well as arginine phosphorylations, which will facilitate further analyses of this important human pathogen. Slight modifications within the workflow, and the use of combined spectral libraries based on phosphoenriched samples, synthetic arginine phosphorylated peptides and classical proteome samples of a high number of different growth and cultivation conditions highly improved reliable and reproducible identification of arginine phosphorylations. 207 arginine phosphosites were identified as putative targets of PtpB, widely distributed along the whole metabolism of S. aureus. Findings of an intriguingly high number of arginine phosphosites belonging to energy metabolism, protein synthesis, transcription and stress regulons suggest a very broad regulatory potential of this modification to impact protein expression in S. aureus on a global scale. In conclusion, our data provide the basis for reliable analysis of arginine phosphorylations in pathogenic bacteria, for example under stress conditions. This way, the analysis of arginine phosphorylations in human pathogenic bacteria provides first hints on the global impact and physiological role of protein arginine phosphorylation for bacterial virulence.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (59) via the PRIDE partner repository (60) with the data set identifier PXD007167.
Supplementary Material
Acknowledgments
We thank Jürgen Bartel and Sebastian Grund for excellent technical assistance.
Footnotes
* This article was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft, SFB/TRR34-3 and GRK 1870).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- S. aureus
- Staphylococcus aureus
- B. subtilis
- Bacillus subtilis
- DHB
- 2,3- dihydroxybenzoic acid
- FDR
- false discovery rate
- GUI
- graphical user interface
- HCD
- higher energy collisional dissociation
- HTH
- Urea/Thiourea
- iRT
- indexed Retention Time
- LMWPTPase
- Low-molecular-weight phosphotyrosine phosphatase
- PTM
- post-translational modification
- ROS
- reactive oxygen species
- RT
- room temperature
- SCX
- strong cation exchange chromatography
- STY
- serine/threonine/tyrosine
- TEAB
- triethylammonium bicarbonate
- TE
- Tris EDTA
- TPP
- Trans-Proteomic Pipeline.
REFERENCES
- 1. Hecker M., Becher D., Fuchs S., and Engelmann S. (2010) A proteomic view of cell physiology and virulence of Staphylococcus aureus. Int. J. Med. Microbiol. 300, 76–87 [DOI] [PubMed] [Google Scholar]
- 2. Vu C. H., Kolata J., Stentzel S., Beyer A., Salazar M. G., Steil L., Pané-Farré J., Rühmling V., Engelmann S., Götz F., van Dijl J. M., Hecker M., Mäder U., Schmidt F., Völker U., and Bröker B. M. (2016) Adaptive immune response to lipoproteins of Staphylococcus aureus in healthy subjects. Proteomics 16, 2667–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Depke M., Michalik S., Rabe A., Surmann K., Brinkmann L., Jehmlich N., Bernhardt J., Hecker M., Wollscheid B., Sun Z., Moritz R. L., Völker U., and Schmidt F. (2015) A peptide resource for the analysis of Staphylococcus aureus in host-pathogen interaction studies. Proteomics 15, 3648–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonar E., Wójcik I., and Wladyka B. (2015) Proteomics in studies of Staphylococcus aureus virulence. Acta Biochim. Pol. 62, 367–381 [DOI] [PubMed] [Google Scholar]
- 5. Grangeasse C., Stülke J., and Mijakovic I. (2015) Regulatory potential of post-translational modifications in bacteria. Front. Microbiol. 6, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Derouiche A., Cousin C., and Mijakovic I. (2012) Protein phosphorylation from the perspective of systems biology. Curr. Opin. Biotechnol. 23, 585–590 [DOI] [PubMed] [Google Scholar]
- 7. Mijakovic I., and Maček B. (2012) Impact of phosphoproteomics on studies of bacterial physiology. FEMS Microbiol. Rev. 36, 877–892 [DOI] [PubMed] [Google Scholar]
- 8. Maček B., and Mijakovic I. (2011) Site-specific analysis of bacterial phosphoproteomes. Proteomics 11, 3002–3011 [DOI] [PubMed] [Google Scholar]
- 9. Eymann C., Becher D., Bernhardt J., Gronau K., Klutzny A., and Hecker M. (2007) Dynamics of protein phosphorylation on Ser/Thr/Tyr in Bacillus subtilis. Proteomics 7, 3509–3526 [DOI] [PubMed] [Google Scholar]
- 10. Ohlsen K., and Donat S. (2010) The impact of serine/threonine phosphorylation in Staphylococcus aureus. Int. J. Med. Microbiol. 300, 137–141 [DOI] [PubMed] [Google Scholar]
- 11. Shankar M., Mohapatra S. S., Biswas S., and Biswas I. (2015) Gene regulation by the LiaSR two-component system in Streptococcus mutans. PLoS ONE 10, e0128083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun F., Ding Y., Ji Q., Liang Z., Deng X., Wong C. C. L., Yi C., Zhang L., Xie S., Alvarez S., Hicks L. M., Luo C., Jiang H., Lan L., and He C. (2012) Protein cysteine phosphorylation of SarA/MgrA family transcriptional regulators mediates bacterial virulence and antibiotic resistance. Proc. Natl. Acad. Sci. U.S.A. 109, 15461–15466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Medzihradszky K. F., Phillipps N. J., Senderowicz L., Wang P., and Turck C. W. (1997) Synthesis and characterization of histidine-phosphorylated peptides. Protein Sci. 6, 1405–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oslund R. C., Kee J.-M., Couvillon A. D., Bhatia V. N., Perlman D. H., and Muir T. W. (2014) A phosphohistidine proteomics strategy based on elucidation of a unique gas-phase phosphopeptide fragmentation mechanism. J. Am. Chem. Soc. 136, 12899–12911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mann M., Ong S.-E., Grønborg M., Steen H., Jensen O. N., and Pandey A. (2002) Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends Biotechnol. 20, 261–268 [DOI] [PubMed] [Google Scholar]
- 16. Maček B., Mann M., and Olsen J. V. (2009) Global and site-specific quantitative phosphoproteomics: principles and applications. Annu. Rev. Pharmacol. Toxicol. 49, 199–221 [DOI] [PubMed] [Google Scholar]
- 17. Bendt A. K., Burkovski A., Schaffer S., Bott M., Farwick M., and Hermann T. (2003) Towards a phosphoproteome map of Corynebacterium glutamicum. Proteomics 3, 1637–1646 [DOI] [PubMed] [Google Scholar]
- 18. Maček B., Mijakovic I., Olsen J. V., Gnad F., Kumar C., Jensen P. R., and Mann M. (2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis*. Mol. Cell. Proteomics 6, 697–707 [DOI] [PubMed] [Google Scholar]
- 19. Soufi B., Gnad F., Jensen P. R., Petranovic D., Mann M., Mijakovic I., and Maček B. (2008) The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics 8, 3486–3493 [DOI] [PubMed] [Google Scholar]
- 20. Sun X., Ge F., Xiao C.-L., Yin X.-F., Ge R., Zhang L.-H., and He Q.-Y. (2010) Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J. Proteome Res. 9, 275–282 [DOI] [PubMed] [Google Scholar]
- 21. Maček B., Gnad F., Soufi B., Kumar C., Olsen J. V., Mijakovic I., and Mann M. (2008) Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial ser/thr/tyr phosphorylation*. Mol. Cell. Proteomics 7, 299–307 [DOI] [PubMed] [Google Scholar]
- 22. Manteca A., Ye J., Sánchez J., and Jensen O. N. (2011) Phosphoproteome analysis of Streptomyces development reveals extensive protein phosphorylation accompanying bacterial differentiation. J. Proteome Res. 10, 5481–5492 [DOI] [PubMed] [Google Scholar]
- 23. Spät P., Maček B., and Forchhammer K. (2015) Phosphoproteome of the cyanobacterium Synechocystis sp. PCC 6803 and its dynamics during nitrogen starvation. Front. Microbiol. 6, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin M. H., Sugiyama N., and Ishihama Y. (2015) Systematic profiling of the bacterial phosphoproteome reveals bacterium-specific features of phosphorylation. Sci. Signal 8, rs10. [DOI] [PubMed] [Google Scholar]
- 25. Elsholz A. K. W., Turgay K., Michalik S., Hessling B., Gronau K., Oertel D., Mäder U., Bernhardt J., Becher D., Hecker M., and Gerth U. (2012) Global impact of protein arginine phosphorylation on the physiology of Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 109, 7451–7456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmidt A., Trentini D. B., Spiess S., Fuhrmann J., Ammerer G., Mechtler K., and Clausen T. (2014) Quantitative phosphoproteomics reveals the role of protein arginine phosphorylation in the bacterial stress response*. Mol. Cell. Proteomics 13, 537–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trentini D. B., Fuhrmann J., Mechtler K., and Clausen T. (2014) Chasing phosphoarginine proteins: development of a selective enrichment method using a phosphatase trap*. Mol. Cell. Proteomics 13, 1953–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sickmann A., and Meyer H. E. (2001) Phosphoamino acid analysis. Proteomics 1, 200–206 [DOI] [PubMed] [Google Scholar]
- 29. Schmidt A., Ammerer G., and Mechtler K. (2013) Studying the fragmentation behavior of peptides with arginine phosphorylation and its influence on phospho-site localization. Proteomics 13, 945–954 [DOI] [PubMed] [Google Scholar]
- 30. Fuhrmann J., Mierzwa B., Trentini D. B., Spiess S., Lehner A., Charpentier E., and Clausen T. (2013) Structural basis for recognizing phosphoarginine and evolving residue-specific protein phosphatases in gram-positive bacteria. Cell Rep. 3, 1832–1839 [DOI] [PubMed] [Google Scholar]
- 31. Fuhrmann J., Subramanian V., and Thompson P. R. (2015) Synthesis and use of a phosphonate amidine to generate an anti-phosphoarginine-specific antibody. Angew. Chemie Int. Ed. 54, 14715–14718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cieœla J., Frączyk T., and Rode W. (2011) Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim. Pol. 58, 137–148 [PubMed] [Google Scholar]
- 33. Wakim B. T., and Aswad G. D. (1994) Ca2+-calmodulin-dependent phosphorylation of arginine in histone 3 by a nuclear kinase from mouse leukemia cells*. J. Biol. Chem. 269, 2722–2727 [PubMed] [Google Scholar]
- 34. Bäsell K., Otto A., Junker S., Zühlke D., Rappen G.-M., Schmidt S., Hentschker C., Maček B., Ohlsen K., Hecker M., and Becher D. (2014) The phosphoproteome and its physiological dynamics in Staphylococcus aureus. Int. J. Med. Microbiol. 304, 121–132 [DOI] [PubMed] [Google Scholar]
- 35. Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., and Mechtler K. (2011) Universal and confident phosphorylation site localization Using phosphoRS. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 36. Wiese H., Kuhlmann K., Wiese S., Stoepel N. S., Pawlas M., Meyer H. E., Stephan C., Eisenacher M., Drepper F., and Warscheid B. (2014) Comparison of alternative MS/MS and bioinformatics approaches for confident phosphorylation site localization. J. Proteome Res. 13, 1128–1137 [DOI] [PubMed] [Google Scholar]
- 37. Ravikumar V., Macek B., and Mijakovic I. (2016) Resources for assignment of phosphorylation sites on peptides and proteins. Methods Mol Biol 1355, 293–306 [DOI] [PubMed] [Google Scholar]
- 38. Mann K., and Edsinger E. (2014) The Lottia gigantea shell matrix proteome: re-analysis including MaxQuant iBAQ quantitation and phosphoproteome analysis. Proteome Sci. 12, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goldstrohm D. A., Broeckling C. D., Prenni J. E., and Curthoys N. P. (2011) Importance of manual validation for the identification of phosphopeptides using a linear ion trap mass spectrometer. J. Biomol. Tech. 22, 10–20 [PMC free article] [PubMed] [Google Scholar]
- 40. Refsgaard J. C., Munk S., and Jensen L. J. (2016) Search databases and statistics: pitfalls and best practices in phosphoproteomics. Methods Mol. Biol. 1355, 323–339 [DOI] [PubMed] [Google Scholar]
- 41. Xue Y., Liu Z., Cao J., Ma Q., Gao X., Wang Q., Jin C., Zhou Y., Wen L., and Ren J. (2011) GPS 2. 1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 24, 255–260 [DOI] [PubMed] [Google Scholar]
- 42. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., and Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 43. Hu Y., and Lam H. (2013) Expanding tandem mass spectral libraries of phosphorylated peptides: advances and applications. J. Proteome Res. 12, 5971–5977 [DOI] [PubMed] [Google Scholar]
- 44. Marx H., Lemeer S., Schliep J. E., Matheron L., Mohammed S., Cox J., Mann M., Heck A. J. R., and Küster B. (2013) A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol. 31, 557–564 [DOI] [PubMed] [Google Scholar]
- 45. Trentini D. B., Suskiewicz M. J., Heuck A., Kurzbauer R., Deszcz L., Mechtler K., and Clausen T. (2016) Arginine phosphorylation marks proteins for degradation by the ClpCP protease. Nature 539, 48–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wozniak D. J., Tiwari K. B., Soufan R., and Jayaswal R. K. (2012) The mcsB gene of the clpC operon is required for stress tolerance and virulence in Staphylococcus aureus. Microbiology 158, 2568–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Soulat D., Vaganay E., Duclos B., Genestier A.-L., Etienne J., and Cozzone A. J. (2002) Staphylococcus aureus contains two low-molecular-mass phosphotyrosine protein phosphatases. J. Bacteriol. 184, 5194–5199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mukherjee S., Dhar R., and Das A. K. (2009) Analyzing the catalytic mechanism of protein tyrosine phosphatase PtpB from Staphylococcus aureus through site-directed mutagenesis. Int. J. Biol. Macromol. 45, 463–469 [DOI] [PubMed] [Google Scholar]
- 49. Shafer W. M., and Iandolo J. J. (1979) Genetics of staphylococcal enterotoxin B in methicillin-resistant isolates of Staphylococcus aureus. Infect. Immun. 25, 902–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Arnaud M., Chastanet A., and Débarbouillé M. (2004) New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70, 6887–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Becher D., Hempel K., Sievers S., Zühlke D., Pané-Farré J., Otto A., Fuchs S., Albrecht D., Bernhardt J., Engelmann S., Völker U., van Dijl J. M., and Hecker M. (2009) A proteomic view of an important human pathogen - towards the quantification of the entire Staphylococcus aureus proteome. PLoS ONE 4, e8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dreisbach A., Otto A., Becher D., Hammer E., Teumer A., Gouw J. W., Hecker M., and Völker U. (2008) Monitoring of changes in the membrane proteome during stationary phase adaptation of Bacillus subtilis using in vivo labeling techniques. Proteomics 8, 2062–2076 [DOI] [PubMed] [Google Scholar]
- 53. Bonn F., Bartel J., Büttner K., Hecker M., Otto A., and Becher D. (2014) Picking vanished proteins from the void: how to collect and ship/ share extremely dilute proteins in a reproducible and highly efficient manner. Anal Chem 86, 7421–7427 [DOI] [PubMed] [Google Scholar]
- 54. Olsen J. V., and Maček B. (2009) High accuracy mass spectrometry in large-scale analysis of protein phosphorylation. Methods Mol Biol 492, 131–142 [DOI] [PubMed] [Google Scholar]
- 55. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 56. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 57. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M.-Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Paape R., Suckau D., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., and Mallick P. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A., and Aebersold R. (2010) A guided tour of the trans-proteomic pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P.-A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H.-J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally co-ordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vizcaíno J. A., Csordas A., Del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.-W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Haft D. H., Loftus B. J., Richardson D. L., Yang F., Eisen J. A., Paulsen I. T., and White O. (2001) TIGRFAMs: a protein family resource for the functional identification of proteins. Nucleic Acids Res. 29, 41–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tanabe M., and Kanehisa M. (2012) Using the KEGG database resource. Curr. Protoc. Bioinforma., Ch.1, Unit 1.12 [DOI] [PubMed] [Google Scholar]
- 63. Michel A., Agerer F., Hauck C. R., Herrmann M., Ullrich J., Hacker J., and Ohlsen K. (2006) Global Regulatory Impact of ClpP Protease of Staphylococcus aureus on regulons involved in virulence, oxidative stress response, autolysis, and DNA repair. J. Bacteriol. 188, 5783–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fuhrmann J., Subramanian V., Kojetin D. J., and Thompson P. R. (2016) Activity-based profiling reveals a regulatory link between oxidative stress and protein arginine phosphorylation. Cell Chem. Biol. 23, 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li L., Cheung A., Bayer A. S., Chen L., Abdelhady W., Kreiswirth B. N., Yeaman M. R., and Xiong Y. Q. (2016) The global regulon sarA regulates β-lactam antibiotic mrsa in vitro and in endovascular infections resistance in methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 214, 1421–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hart-Smith G., Yagoub D., Tay A. P., Pickford R., and Wilkins M. R. (2016) Large scale mass spectrometry-based identifications of enzyme-mediated protein methylation are subject to high false discovery rates*. Mol. Cell. Proteomics 15, 989–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Musumeci L., Bongiorni C., Tautz L., Edwards R. A., Osterman A., Perego M., Mustelin T., and Bottini N. (2005) Low-molecular-weight protein tyrosine phosphatases of Bacillus subtilis. J. Bacteriol. 187, 4945–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vega C., Chou S., Engel K., Harrell M. E., Rajagopal L., and Grundner C. (2012) Structure and substrate recognition of the Staphylococcus aureus protein tyrosine phosphatase PtpA. J. Mol. Biol. 413, 24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang Z.-Y., Wang Y., Wu L., Fauman E. B., Stuckey J. A., Schubert H. L., Saper M. A., and Dixon J. E. (1994) The Cys(X)5Arg catalytic motif in phosphoester hydrolysis. Biochemistry 33, 15266–15270 [DOI] [PubMed] [Google Scholar]
- 70. Zhang Z.-Y., Wang Y., and Dixon J. E. (1994) Dissecting the catalytic mechanism of protein-tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 91, 1624–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fuhrmann J., Schmidt A., Spiess S., Lehner A., Turgay K., Mechtler K., Charpentier E., and Clausen T. (2009) McsB is a protein arginine kinase that phosphorylates and inhibits the heat-shock regulator CtsR. Science 324, 1323–1327 [DOI] [PubMed] [Google Scholar]
- 72. Truong-Bolduc Q. C., and Hooper D. C. (2010) Phosphorylation of MgrA and its effect on expression of the NorA and NorB efflux pumps of Staphylococcus aureus. J. Bacteriol. 192, 2525–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Truong-Bolduc Q. C., Ding Y., and Hooper D. C. (2008) Posttranslational modification influences the effects of MgrA on norA expression in Staphylococcus aureus. J. Bacteriol. 190, 7375–7381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ji C.-J., Kim J.-H., Won Y.-B., Lee Y.-E., Choi T.-W., Ju S.-Y., Youn H., Helmann J. D., and Lee J.-W. (2015) Staphylococcus aureus PerR is a hypersensitive hydrogen peroxide sensor using iron-mediated histidine oxidation. J. Biol. Chem. 290, 20374–20386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Frees D., Gerth U., and Ingmer H. (2014) Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus. Int. J. Med. Microbiol. 304, 142–149 [DOI] [PubMed] [Google Scholar]
- 76. Elsholz A. K. W., Gerth U., and Hecker M. (2010) Regulation of CtsR activity in low GC, gram+ bacteria. Adv. Microb. Physiol. 57, 119–144 [DOI] [PubMed] [Google Scholar]
- 77. Lorenz U., Hüttinger C., Schäfer T., Ziebuhr W., Thiede A., Hacker J., Engelmann S., Hecker M., and Ohlsen K. (2008) The alternative sigma factor sigma B of Staphylococcus aureus modulates virulence in experimental central venous catheter-related infections. Microbes Infect. 10, 217–223 [DOI] [PubMed] [Google Scholar]
- 78. Pané-Farré J., Jonas B., Förstner K., Engelmann S., and Hecker M. (2006) The σB regulon in Staphylococcus aureus and its regulation. Int. J. Med. Microbiol. 296, 237–258 [DOI] [PubMed] [Google Scholar]
- 79. Bernhardt J., Funke S., Hecker M., and Siebourg J. (2009) Visualizing gene expression data via Voronoi Treemaps. 6th Int. Symp. Vor. Diagrams Sci. Eng. ISVD 233–241, 2009 [Google Scholar]
- 80. Olsen J. V., Blagoev B., Gnad F., Maček B., Kumar C., Mortensen P., and Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (59) via the PRIDE partner repository (60) with the data set identifier PXD007167.