

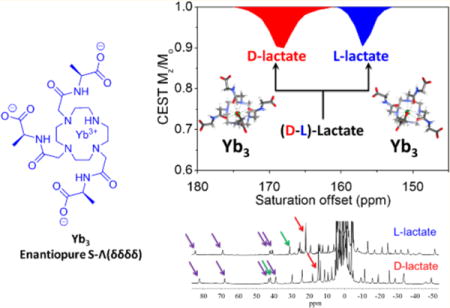
Abstract
A previous report demonstrated that EuDO3A could be used as an NMR shift reagent for imaging extracellular lactate produced by cancer cells using CEST imaging. In this work, a series of heptadentate macrocyclic YbDO3A-trisamide complexes with δ-chiral carbons in the three pendant side-arms were examined as shift reagents for lactate detection. High resolution 1H NMR spectra and DFT calculations provided evidence for the formation of stereoselective lactate·YbDO3A-trisamide complexes each with a different CEST signature. This stereoselectivity allowed discrimination of D-versus L-lactate by both high-resolution NMR and CEST. This work demonstrates that lanthanide-based paramagnetic shift reagents can be designed to detect important metabolites by CEST MRI selectively.
Graphical abstract
1. INTRODUCTION
The binding of biologically important molecules to paramagnetic lanthanide DO3A-type complexes offers new avenues for molecular imaging of such species.1–4 Lactate, for example, is overproduced in most cancers yet methods to quantify lactate production in vivo lack especially methods that can distinguish intracellular lactate from extracellular lactate.5,6 Recently, Zhang et al. reported a strategy to image extracellular lactate produced by cancer cells by using the shift reagent (SR), EuDO3A.7 This SR forms a bidentate complex with lactate even in the presence of a variety of competing biological anions. Once bound to EuDO3A, lactate was easily detected via CEST-MRI (chemical exchange saturation transfer magnetic resonance imaging) by selective activation of the highly shifted, exchanging lactate–OH proton. Two major lactate-EuDO3A species of equal population as detected by CEST suggested that the metal ion-bound L-lactate lifts the magnetic degeneracy of the two diastereomers, Λ(δδδδ) and Δ(λλλλ), known to be present in solution. Evidence supporting this hypothesis was reported by Dickins et al. in a study of a chiral heptadentate lanthanide complex which also forms a stable ternary complex with L-lactate.8,9 In that case, the two diasteriomeric ternary complexes were resolved by 1H NMR yet only a single Λ(δδδδ) crystal was isolated for crystallographic studies. Somewhat later, Terreno et al.10 also found that a variety of α-hydroxy-carboxylate molecules form ternary complexes with heptadentate YbDO3AM-type complexes and some result in rather specific stereochemical discrimination of complex isomers. This suggests that bidentate coordination of a α-hydroxy-carboxylate ligand such as lactate to a LnDO3A-type complex can result in isomer discrimination likely on the basis of differences in affinity. This is not terribly surprising given that complex isomer discrimination has even been reported for octadentate systems such as EuDOTP even by forming strong ion-pair complexes.11,12
The current study was initiated to investigate this phenomenon in more detail by using various YbDO3A-derivatives as shift reagents for L-lactate with the overarching goal of improving the sensitivity of CEST for detection of lactate. Previous studies of various YbDOTA-tetraamide complexes with chiral centers on the pendent arms often result in a single species in solution due to the steric and torsional strain of placing larger groups in pseudoaxial positions.13–15 One exception reported by Mani et al.16 was that the chirality introduced by sec-butyl amide substituents resulted in complexes where up to four diastereoisomeric coordination isomers could be observed by 1H NMR. Given our previous observation that addition of lactate to EuDO3A results in discrimination of lactate·EuDO3A stereoisomers by CEST but not high resolution 1H NMR,7 we were interested in knowing (1) whether introduction of chiral centers onto the shift reagent side-arms results in improved stereoisomer discrimination to yield a single CEST-active species and (2) whether the proton exchange rate between the bound lactate–OH proton and solvent water is sensitive to such structural changes in the SR. One of our overarching goals is to optimize the lactate–OH proton exchange rate to achieve maximum CEST sensitivity.
2. RESULTS AND DISCUSSION
The ligand designs shown in Figure 1 were chosen to vary in (1) overall charge and affinity for lactate, (2) position and stereochemistry of the δ chiral carbons on the side-arms, and (3) capability to form stable heptadentate LnDO3A-type complexes. Carboxylates and carboxylate esters were used as bulky groups on the δ chiral carbon centers, and Yb(III) was selected among the lanthanide series for its greater charge density, small ionic radius, and ability to induce large hyperfine NMR shifts without extensive line-broadening. Heptadentate macrocyclic complexes with Yb3+ are known to increase the binding affinity to lactate by 1 order of magnitude over the corresponding Eu3+ complexes.10 The synthetic details are either given in the Supporting Information (SI) or described elsewhere.17
Figure 1.
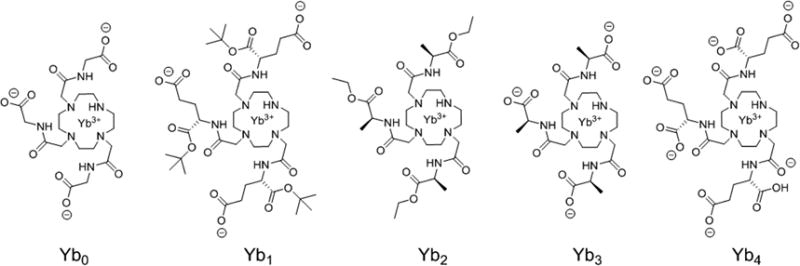
Structures of the YbDO3A derivatives used in this study. All chiral molecules are drawn as the (S)-configuration.
Octadentate LnDOTA-type chelates are known to provide four isomeric structures determined by the orientation of the ethylene groups in the macrocycle (positive = δδδδ, negative = λλλλ) or arm rotation (positive = Δ, negative = Λ) relative to the four nitrogen atoms, see Figure 2). In the absence of chiral centers, Δ(δδδδ)/Λ(λλλλ) and Λ(δδδδ)/Δ(λλλλ) form enantiomeric pairs so only two isomers are typically detected by 1H NMR.16,18 Heptadentate Ln-complexes also exist in solution as multiple species,10 but a report has shown that positively charged Yb3+-complexes favor a single SAP isomer.8 The presence of chiral centers in the ligand framework increases the number of isomers that can be potentially detected by NMR. With chiral lactate bound as a bidentate ligand, many more structures are possible since lactate can bind Yb3+ with the either the hydroxyl or carboxylate group at the capping position of the square antiprism (SAP) or twisted square antiprism (TSAP) coordination polyhedron.7 The 1H NMR shifts observed for Yb3+ complexes are largely pseudocontact in origin and produce large lanthanide induced shifts (LIS) with negligible contact contributions. These properties also make Yb3+ a preferred lanthanide for structural elucidations of metal-complexes by NMR. The hyperfine NMR shifts induced by the paramagnetic Yb3+ in these complexes are somewhat smaller compared to those observed for the analogous octadentate YbDOTA-type complexes.19
Figure 2.
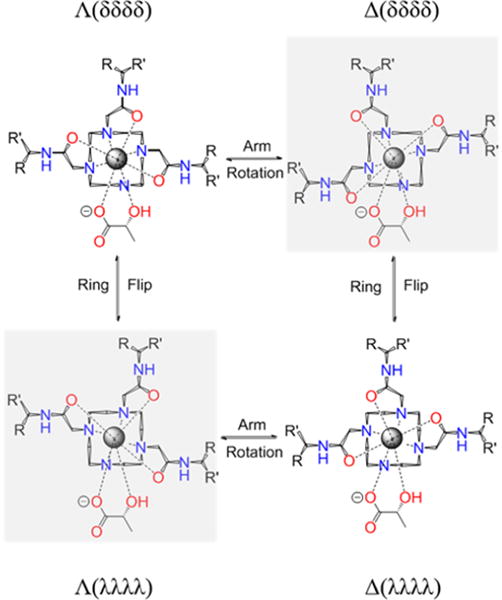
Schematic representation of the isomeric forms of heptadentate Yb3+ complexes with one equivalent of lactate. Arm rotation results in interconversion between enantiomers while ring flips result in interconversion between diastereoisomers. The arrangement is described through (λλλλ)/(δδδδ) and the side arm orientation, leading to Λ/Δmetal coordination.
The high resolution 1H spectra of Yb1, Yb3, and Yb4 show multiple, broad overlapping proton resonances in the absence of lactate (Figure S2) but become much sharper and well-resolved after addition of one equivalent of lactate (Figure 3). The one exception is Yb2 which shows moderately sharp 1H resonances even in the absence of lactate (Figure S2). This indicates that the interconversion rate between the multiple isomers present in solution is moderately fast in the free complexes but much slower once lactate is bound. The assignment of most highly downfield shifted 1H resonances, commonly referred to as the axial H4 ethylene protons, were made by comparison with analogous YbDOTMA, YbDOTTA, and YbDTMA complexes.20–23 The H4 resonances in the more compact square antiprism isomers often appear around 150 ppm for YbDOTA-type complexes (carboxylate donors) and around 100 ppm or less for LnDOTAM-type complexes (amide donors). In comparison, the H4 proton resonances in the less-compacted twisted square antiprism isomers are less highly shifted, typically <40 ppm. For axially symmetric LnDOTAM-tetraamide complexes, the H4 protons are magnetically equivalent and appear as a single resonance24 while the four H4 resonances in asymmetric YbDO3A-type complexes shown in Figure 3 show four magnetically nonequivalent resonances appearing in the chemical shift range of ∼50–100 ppm. Thus, one can conclude that these complexes exist in solution largely as SAP coordination isomers.
Figure 3.
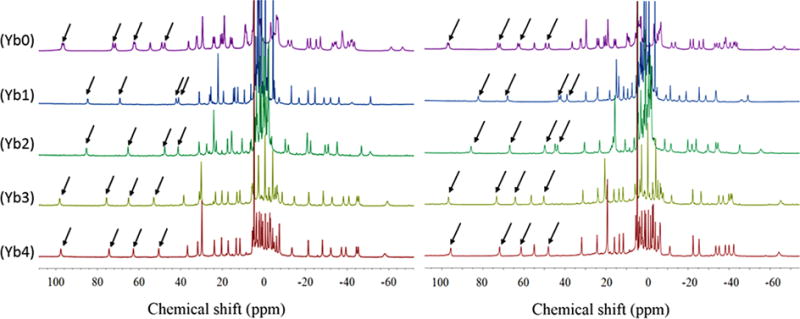
1H NMR spectra of Ybx complexes containing 1:1 L-lactate (left) or 1:1 D-lactate (right). All NMR spectra were recorded at 25 °C in D2O at 9.4 T. pD = 6.5. Arrows point to the H4 proton resonances.
Among these five complexes, a striking difference was observed in the 1H NMR spectra of Yb0, the only complex lacking a chiral carbon. Here, each H4 proton resonance appears as two equally intense peaks with very similar chemical shifts while the lactate CH and CH3 resonances also appear as two resonances of equal intensity separated by ∼15 and ∼10 ppm, respectively. This suggests that lactate binds to both SAP isomers, Λ(δδδδ) and Δ(λλλλ), of Yb0 and that the protons in these two diastereoisomeric L-lactate·Yb0 complexes are not magnetically equivalent. The 1H spectrum of Yb0 in the presence of one equivalent of D-lactate was identical (Figure 3). In comparison, the 1H NMR spectra of D- or L-lactate with the four complexes containing chiral carbons in their side-arms (Yb1–4) each displayed four sharp ethylene proton resonances (Figure 3) and single lactate CH and CH3 resonances. This suggests that the lactate Yb1–4 complexes either exist in solution as a single species or inversion/rotation between these diastereomers is much faster than that seen for the lactate Yb0 complexes. Close inspection of these spectra shows that the chemical shifts of the ethylene protons and lactate protons differed in all four L-lactate·Yb1–4 complexes. Furthermore, the chemical shifts of the protons in the D-lactate·Yb1–4 complexes differed from those seen in spectra of the L-lactate·Yb1–4 complexes (see Table 1). This suggests that a unique single stereoisomer, either Λ(δδδδ) or Δ(λλλλ), is formed in each of these complexes.
Table 1.
Chemical Shifts of the Lactate–OH and Methyl Proton Resonances for Each of the D- or L-Lactate·Ybx Complexes, Proton Exchange Rate of Bound L and D Lactate at 298 and 310 K and CEST Amplitude at 298 and 310 K with B1 of 600 Hz
| lactate type | Yb0
|
Yb1
|
Yb2
|
Yb3
|
Yb4
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| L-lac | D-lac | L-lac | D-lac | L-lac | D-lac | L-lac | D-lac | L-lac | D-lac | |
| δ-OH at 298 and 310 K | 164/149 | 164/149 | 136 | 145 | 129 | 145 | 157 | 168 | 155 | 166 |
| 152/140 | 152/140 | 125 | 132 | 120 | 133 | 143 | 154 | 142 | 153 | |
| CH-lac (ppm) | 54.9/36.5 | 54.9/36.5 | 31.1 | 42.0 | 31.2 | 44.8 | 38.5 | 56.4 | 36.7 | 54.9 |
| CH3-lac (ppm)a | 29.4/19.2 | 29.4/19.2 | 22.1 | 14.9 | 24.1 | 15.7 | 30.1 | 20.6 | 29.8 | 19.3 |
| kex 298 K (s−1)b | 1900/1800 | 2200/2000 | 1800 | 1700 | 2600 | 2600 | 2000 | 2000 | 1500 | 1400 |
| kex 310 K (s−1) | 3000/2900 | 2800/2700 | 2800 | 2800 | 4100 | 4200 | 2600 | 2300 | 1800 | 1800 |
| CESTeff % (298 K)c | 7.0/7.8 | 8.2/9.0 | 8.5 | 12 | 20 | 20 | 14 | 12 | 6.6 | 4.4 |
| CESTeff% (310 K) | 19/18 | 19/18 | 19 | 22 | 35 | 34 | 30 | 27 | 16 | 16 |
1H NMR spectra were collected in D2O.
Omega plot was performed with 50 mM complex plus 50 mM lactate. Presaturation pulse of 3s with B1 from 100 to 1000 Hz was applied.
CEST experiment was performed with 50 mM complex plus 50 mM lactate. A presaturation pulse of 3s with B1 of 600 Hz was applied at 298 and 310 K, respectively. Proton exchange rate and CEST amplitude are in 10% error range.
To examine these effects further, 2D EXSY NMR (Exchange Spectroscopy) spectra were collected on samples of L-lactate·Yb0 and L-lactate·Yb4 (Figure 4). The EXSY mixing time was varied and optimized to obtain optimal exchange peaks for L-lactate·Yb0 and this same value was then used for the spectrum of L-lactate·Yb4. As seen in Figure 4, the EXSY spectrum of L-lactate·Yb0 shows exchange cross-peaks between the axial and equatorial proton resonances and between the nonequivalent CH and CH3 lactate resonances characteristic of interconversion between the two diastereotopic L-lactate·Yb0 complexes while the EXSY spectrum of L-lactate·Yb4 showed no cross peaks under identical experimental conditions. This supports our earlier conclusion that that the later complex exists as a single species in solution. An alternative explanation could be that interconversion rate between two diastereomers in L-lactate·Yb4 is much faster than that seen in L-lactate·Yb0. However, this is highly unlikely because interconversion between diastereomers requires side arm rotation and his would not be expected to occur more rapidly in the bulkier L-lactate·Yb4 complex. Thus, the only reasonable conclusion is that L-lactate·Yb4 exists as a single species in solution and, by comparison, the remaining L-lactate·Yb1–4 likely do as well. Unfortunately, none of the lactate-Yb1–4 complexes formed crystals likely due to their high solubility in polar protic solvents. One of the complexes, Yb3, crystallized as a dimer where one of the carboxylate groups of the ligand acted as a bridge between the two metal ions (Figure S3). Lactate was not present in this structure. This behavior has been reported previously in another similar lanthanide complex.25 In the structure shown in Figure S3, the Yb3 complex was present as the S-Δ(λλλλ) isomer in both metal centers.
Figure 4.
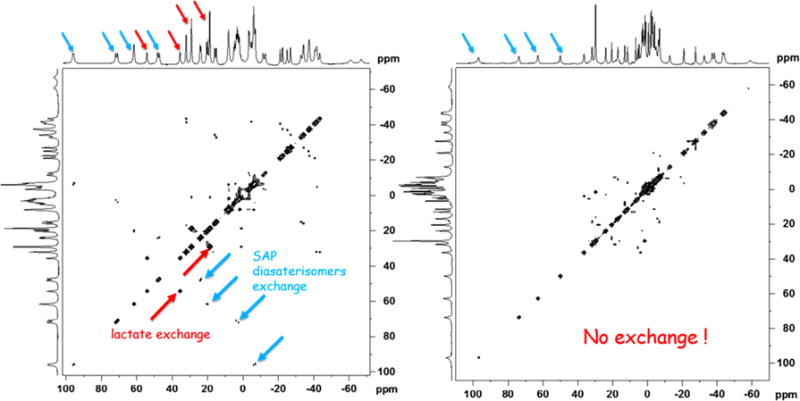
(Left). 2D EXSY NMR spectrum of Yb0·L-lactate (1:1) in D2O using a mixing time of 15 ms. (Right). 2D EXSY NMR spectrum of Yb4·L-lactate (1:1) in D2O using a mixing time of 15 ms. The red arrows reflect exchange of the lactate CH and CH3 resonances while the blue arrows reflect exchange of the axial proton resonances between the two SAP diasterisomers.
The chemical shifts of the bound lactate protons in these complexes are also informative (Table 1). Most notable is that the chemical shift differences between the D- and L-lactate complexes are much larger (∼10 ppm) in comparison to the ethylene proton resonances. These large chemical shift differences between D- and L-lactate observed here by 1H NMR are much larger than those observed using nonaqueous chiral agents.26–28 This indicates that the geometrical positions of the CH and CH3 protons in D-lactate·Ybx versus L-lactate·Ybx are quite different concerning the magnetic axes governing the Yb-induced pseudocontact shifts. To investigate further the origin of these differences, a complete NMR assignment of all proton resonances in spectra of the Yb3 complexes with D- and L-lactate, was undertaken by analysis of their 1H,1H–COSY spectra (Figure S4). Cross-peaks in these spectra were observed for (i) the axial and equatorial protons of the cyclen structure; (ii) the axial protons of the cyclen unit separated by three bonds; (iii) the geminal protons of the methylenic groups of the pendant arms; (iv) the CH and methyl groups of the pendant arms; and (v) the CH and methyl groups of lactate (Table S2).
A detailed analysis of the pseudocontact shifts induced by Yb was performed using the following expressions:29
| (1) |
where
| (2) |
In these equations x, y, and z are the Cartesian coordinates of nuclei i relative to the location of the paramagnetic ion, and χpk are the components of the magnetic susceptibility tensor (p, k = x, y, or z). In a principal magnetic axis system, χxy = χxz = χyz = 0, so that only the first two terms of eq 1 remain, representing the axial and rhombic contributions to the pseudocontact shifts. Equation 1 was used to analyze the pseudocontact shifts by following the methodology of Forsberg,30 which involves a least-squares fit procedure including the axial [χzz − 1/3(χxx + χyy + χzz)] and rhombic (χxx − χyy) anisotropies of the magnetic susceptibility tensor χ as fitting parameters, as well as three Euler angles that relate the orientation of the Cartesian axes of the input structure and the principal magnetic axis system.31 As structural models, we used molecular geometries obtained with DFT calculations following standard procedures (see computational details, SI).32,33 These calculations provided four local energy minima corresponding to the S-Λ(δδδδ), S-Δ(λλλλ), S-Δ(δδδδ), and S-Λ(λλλλ) diastereoisomers. According to DFT, the minimum energy conformation corresponds to the S-Λ(δδδδ) isomer for both L-lactate·Yb3 and D-lactate·Yb3. A similar structure was observed in the solid state for a lactate-YbDO3A complex with chiral D-phenyl groups.8 The orientation of the lactate anion is such that the carboxylate group occupies one of the coordination positions in the upper plane of the SAP coordination polyhedron, while the hydroxyl group of lactate coordinates at the apical position. DFT calculations indicated that this coordination mode was far more favorable (>9.0 kcal mol−1) than the reverse situation in which the apical position is occupied by the carboxylate group. This likely reflects the preference of the weaker hydroxyl donor atom to occupy the more hindered capping position.34 The analysis of the pseudocontact shifts confirmed the predictions of DFT, as the chemical shifts calculated with eq 1 showed excellent agreement with the experimental ones, with deviations <5.5 ppm for L-lactate·Yb3 and <4.9 ppm for D-lactate·Yb3 (Table S2). Much larger deviations were observed by using the geometry of the S-Δ(λλλλ) isomer, particularly for the CH protons of the side arms (up to 15–17 ppm, SI).
Figure 5 shows the orientation of the molecular geometries obtained for L-lactate·Yb3 and D-lactate·Yb3 in the principal magnetic axis system. The analysis shows that the principal magnetic axis (the z axis) is not placed perpendicular to the ideal plane defined by the four nitrogen atoms of the macrocycle, but parallel to that plane. This surprising result is however in agreement with recent studies reported for DyDOTA.35 Furthermore, the analysis provides virtually identical orientations of the magnetic axes in L-lactate·Yb3 and D-lactate·Yb3 so the slightly different chemical shifts observed for the lactate resonances in L-lactate·Yb3 and D-lactate·Yb3 cannot be attributed to the formation of a different isomer, or to a different magnetic anisotropy of the complex (the axial and rhombic anisotropies are very similar, Table S2). Instead, the origin of the different chemical shifts is related to the different position of lactate proton nuclei with respect to the magnetic axes (Figure 5). Indeed, the CH proton of lactate presents virtually identical distances to the paramagnetic center in both L-lactate·Yb3 and D-lactate·Yb3 (r = 3.860 and 3.820 Å, respectively), but different z coordinates (1.160 and 1.755 Å), which leads to somewhat different axial pseudocontact shifts (−14.3 and −8.4 ppm). However, the different position of the lactate protons in the xy plane is the main reason for the different chemical shifts observed for L-lactate·Yb3 and D-lactate·Yb3, leading to rhombic contributions of −15.3 and −45.3 ppm (Table S2).
Figure 5.
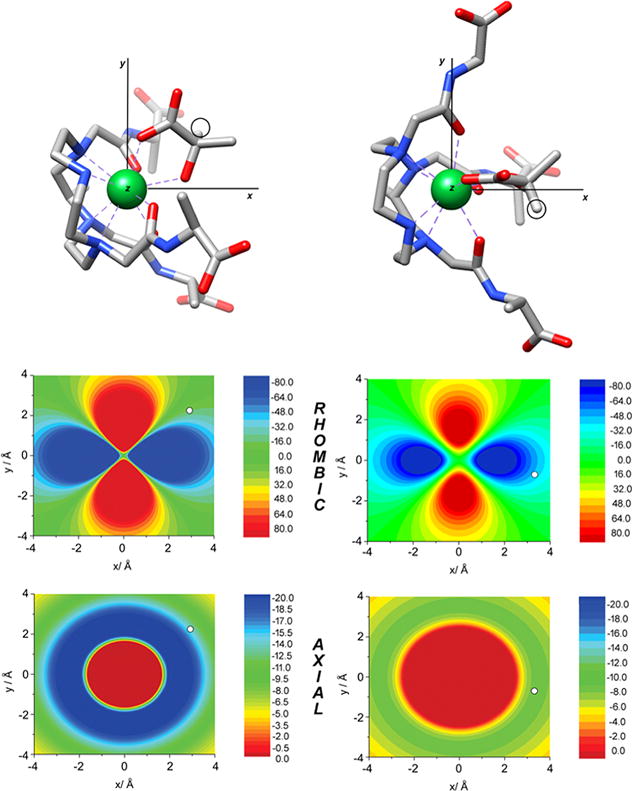
Top: Molecular geometries of the L-lactate·Yb3 and D-lactate·Yb3 systems obtained with DFT showing the orientation of the magnetic axes. Circles highlight the position of lactate CH protons. Middle: Contour plots showing the rhombic contribution of the pseudocontact shifts (ppm) in the plane parallel to the x and y axis containing the lactate CH proton. Bottom: Axial contribution to the pseudocontact shifts (ppm) in the same plane. White circles indicate the position of lactate CH protons. Negative pseudocontact shifts correspond to shifts to lower fields.
CEST Spectra
To investigate the utility of these Ybx complexes for imaging lactate by CEST,7 we collected CEST spectra of samples containing each complex (Yb0–4) with one equivalent of L-lactate or D-lactate. As shown in Figure 6, the chemical shifts of the exchanging lactate–OH proton in the Ybx complexes are substantially larger (∼130–170 ppm) compared to the shifts observed when using EuDO3A as a shift reagent.7 For the achiral complex, Yb0, two sharp CEST peaks at 149 and 164 ppm were detected that are likely attributable to two the equally favored diasteroisomeric complexes, Λ(δδδδ) and Δ(λλλλ). This is supported by DFT calculations, which provide two virtually isoenergetic minima for the Λ(δδδδ) and Δ(λλλλ) isomers of the L-lactate·Yb0 and d-lactate·Yb0 adducts. Identical CEST exchange peaks were observed when either L-lactate or D-lactate was added to a sample of Yb0 similar to that seen in the 1H NMR spectra. Upon addition of either D- or L-lactate to any one of the chiral complexes, Yb1–4, only a single CEST peak was observed, (Table 1) but again, the chemical shifts of the CEST exchange peaks for D-versus L-lactate complexes varied from 9 to 16 ppm. This observation is again consistent with enantiomeric discrimination of D- or L-lactate by the Yb1–4 complexes (Table 1).
Figure 6.
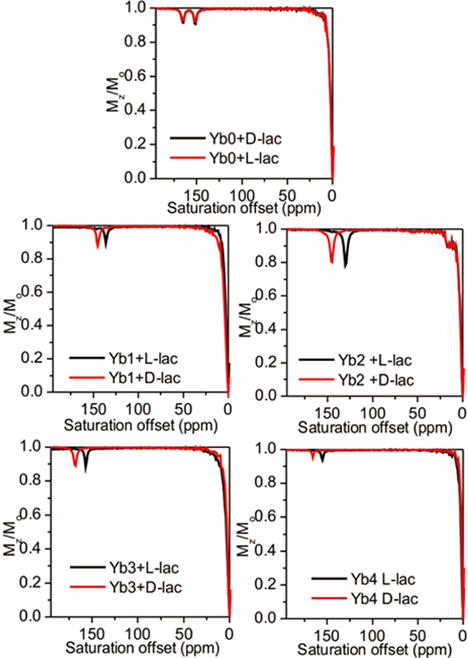
CEST spectra of Yb(III)-complexes with either L-lactate or D-lactate in H2O. (50 mM complex with 50 mM D-lactate or L-lactate). Presaturation pulse of 4 s with B1 of 14.1 μT was applied at 298 K using a 9.4 T NMR spectrometer. The pH was adjusted to 7.4 in all samples just prior to data collection. CEST spectra were only shown the downfield part for clarification.
The amplitude of the CEST exchange peaks for the D-versus L-lactate in each complex varied to some extent. For example, the CEST intensities of the D-lactate–OH exchange peaks were larger than the corresponding L-lactate–OH exchange peaks in the Yb1 complexes, about equal in the Yb3 complexes, and smaller in the Yb2 and Yb4 complexes. The corresponding 1H spectra show that the concentrations of the D-versus L-lactate species are identical so these intensity differences must reflect differences in proton exchange rates. With a temperature increase to 310 K, larger CEST amplitudes were observed for all Yb1–4–lactate complexes. The proton exchange rates were evaluated using the Omega plot method at both temperatures and, indeed, faster proton exchange rates were observed at 310 K than at 298 K, as would be expected. The proton exchange rates at 310 K were close to optimal for CEST and hence should benefit experiments performed at physiological temperatures. In each complex, the D-lactate–OH exchange peak was shifted further downfield compared to the analogous L-lactate–OH exchange peak. From the results described above, this can be attributed to slightly different positions of the OH protons with respect to the orientation of the magnetic axes.
3. CONCLUSIONS
In this study, we investigated whether the substitution of a δ chiral carbon in the pendant arms of YbDO3A-amide type complexes can limit the number of stereoisomers present in solution and thereby enhance detection of lactate by CEST NMR. Our results show that with different substitutions at the δ-position on the pendant arms, a single energetically favorable isomer is formed. This was supported by both high resolution 1H NMR and CEST data. In the presence of (D or L)-lactate, the achiral Yb0 complex forms two equally populated Λ(δδδδ) and Δ(λλλλ) isomers with lactate whereas the Ybx complexes having δ chiral centers yield a single isomer (shown to be the S-Λ(δδδδ) isomer for the Yb3 complexes by analysis of the pseudocontact shifts). The second finding of this study was the observation of enantiomeric discrimination of D- and L-lactate by the chiral DO3A-derived Ybx complexes. In summary, our study shows that heptadentate macrocyclic Yb-complexes with δ chiral centers serve as key predictors of the conformational orientation of lanthanide complexes with macrocyclic ligands and powerful discriminators of enantiomeric substrates such as D- and L-lactate. Ternary complexes are formed with the lactate OH group bound to the Yb3+ ion in an apical position as reported previously for lactate·EuDO3A.7 The lactate OH proton is highly shifted downfield in the presence of the Yb-complexes as seen by CEST NMR. The approach used magnifies the chemical shift of lactate OH CEST which may ultimately prove useful in the design of metabolite-specific shift reagents for functional MRI. L-lactate is considered the only form produced by cancer cells while D-lactate, although rarely produced by human cells, is a metabolic product of some essential bacteria that exist in the human microbiota. Consequently, D-lactate acidosis has been associated with several diseases including bowel syndrome and D-lactate encephalopathy.36,37 Given that the development of enantiopure agents is of interest in molecular imaging, this work is a positive step forward toward optimizing inorganic responsive agents (shift reagents) for selective identification of important biomarkers and metabolic profiles.
Supplementary Material
Acknowledgments
The authors acknowledge partial financial support for this work from the National Institutes of Health (CA-115531, EB-01598, EB-00482), Harold C. Simmons Cancer Center through an NCI Cancer Center Support Grant, 1P30 CA142543, and the Robert A. Welch Foundation (AT-584). The authors C. P.-I. and D. E.-G. thank Ministerio de Economía y Competitivad (CTQ2016-76756-P) and Centro de Supercomputación de Galicia (CESGA) for providing the computer facilities.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08292.
Synthetic details for preparing the Yb(III) complexes, the experimental details of the CEST experiments, experimental and calculated 1H NMR shifts, computational details, and high-resolution NMR of the Yb complexes (PDF)
ORCID
A. Dean Sherry: 0000-0001-7150-8301
Notes
The authors declare no competing financial interest.
References
- 1.Terreno E, Botta M, Fedeli F, Mondino B, Milone L, Aime S. Inorg Chem. 2003;42:4891. doi: 10.1021/ic034321y. [DOI] [PubMed] [Google Scholar]
- 2.Aime S, Delli Castelli D, Fedeli F, Terreno E. J Am Chem Soc. 2002;124:9364. doi: 10.1021/ja0264044. [DOI] [PubMed] [Google Scholar]
- 3.Botta M, Aime S, Barge A, Bobba G, Dickins RS, Parker D, Terreno E. Chem - Eur J. 2003;9:2102. doi: 10.1002/chem.200204475. [DOI] [PubMed] [Google Scholar]
- 4.Terreno E, Botta M, Boniforte P, Bracco C, Milone L, Mondino B, Uggeri F, Aime S. Chem - Eur J. 2005;11:5531. doi: 10.1002/chem.200500129. [DOI] [PubMed] [Google Scholar]
- 5.Heiden MGV, Cantley LC, Thompson CB. Science. 2009;324:1029. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rato L, Alves MG, Socorro S, Carvalho RA, Cavaco JE, Oliveira PF. Biosci Rep. 2012;32:61. doi: 10.1042/BSR20110030. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Martins AF, Mai Y, Zhao P, Funk AM, Clavijo Jordan MV, Zhang S, Chen W, Wu Y, Sherry AD. Chem - Eur J. 2017;23:1752. doi: 10.1002/chem.201604558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickins RS, Love CS, Puschmann H. Chem Commun. 2001:2308. [PubMed] [Google Scholar]
- 9.Dickins RS, Aime S, Batsanov AS, Beeby A, Botta M, Bruce JI, Howard JAK, Love CS, Parker D, Peacock RD, Puschmann H. J Am Chem Soc. 2002;124:12697. doi: 10.1021/ja020836x. [DOI] [PubMed] [Google Scholar]
- 10.Terreno E, Botta M, Fedeli F, Mondino B, Milone L, Aime S. Inorg Chem. 2003;42:4891. doi: 10.1021/ic034321y. [DOI] [PubMed] [Google Scholar]
- 11.Wenzel TJ, Morin CA, Brechting AA. J Org Chem. 1992;57:3594. [Google Scholar]
- 12.Jiang J, Li A-H, Jang S-Y, Chang L, Melman N, Moro S, Ji X, Lobkovsky EB, Clardy JC, Jacobson KA. J Med Chem. 1999;42:3055. doi: 10.1021/jm980688e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howard JAK, Kenwright AM, Moloney JM, Parker D, Woods M, Howard JAK, Port M, Navet M, Rousseau O. Chem Commun. 1998:1381. [Google Scholar]
- 14.Vipond J, Woods M, Zhao P, Tircsó G, Ren J, Bott SG, Ogrin D, Kiefer GE, Kovacs Z, Sherry AD. Inorg Chem. 2007;46:2584. doi: 10.1021/ic062184+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mani T, Opina ACL, Zhao P, Evbuomwan OM, Milburn N, Tircso G, Kumas C, Sherry AD. JBIC J Biol Inorg Chem. 2014;19:161. doi: 10.1007/s00775-013-1031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mani T, Tircsó G, Zhao P, Sherry AD, Woods M. Inorg Chem. 2009;48:10338. doi: 10.1021/ic9017008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Winter P, Wu K, Sherry AD. J Am Chem Soc. 2001;123:1517. doi: 10.1021/ja005820q. [DOI] [PubMed] [Google Scholar]
- 18.Delli Castelli D, Terreno E, Aime S. Angew Chem, Int Ed. 2011;50:1798. doi: 10.1002/anie.201007105. [DOI] [PubMed] [Google Scholar]
- 19.Aime S, Botta M, Ermondi G. Inorg Chem. 1992;31:4291. [Google Scholar]
- 20.Brittain HG, Desreux JF. Inorg Chem. 1984;23:4459. [Google Scholar]
- 21.Aime S, Barge A, Bruce JI, Botta M, Howard JAK, Moloney JM, Parker D, de Sousa AS, Woods M. J Am Chem Soc. 1999;121:5762. [Google Scholar]
- 22.Rohovec J, Kývala M, Vojtísěk P, Hermann P, Lukeš I. Eur J Inorg Chem. 2000;2000:195. [Google Scholar]
- 23.Aime S, Botta M, Garda Z, Kucera BE, Tircso G, Young VG, Woods M. Inorg Chem. 2011;50:7955. doi: 10.1021/ic2012827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dickins RS, Parker D, Bruce JI, Tozer DJ. Dalton Trans. 2003:1264. [Google Scholar]
- 25.Kang SI, Ranganathan RS, Emswiler JE, Kumar K, Gougoutas JZ, Malley MF, Tweedle MF. Inorg Chem. 1993;32:2912. [Google Scholar]
- 26.Ema T, Tanida D, Sakai T. J Am Chem Soc. 2007;129:10591. doi: 10.1021/ja073476s. [DOI] [PubMed] [Google Scholar]
- 27.Ema T, Tanida D, Sakai T. Org Lett. 2006;8:3773. doi: 10.1021/ol0613665. [DOI] [PubMed] [Google Scholar]
- 28.Iwaniuk DP, Wolf C. J Org Chem. 2010;75:6724. doi: 10.1021/jo101426a. [DOI] [PubMed] [Google Scholar]
- 29.Piguet C, Geraldes CFGC. In: Handbook on the Physics and Chemistry of Rare Earths. Gschneidner KA, Bünzli J-C, Pecharsky V, editors. Elsevier; United Kingdom: 2003. pp. 353–463. [Google Scholar]
- 30.Forsberg JH, Delaney RM, Zhao Q, Harakas G, Chandran R. Inorg Chem. 1995;34:3705. [Google Scholar]
- 31.Regueiro-Figueroa M, Bensenane B, Ruscsák E, Esteban-Gómez D, Charbonnière LJ, Tircsó G, Tóth I, de Blas A, Rodríguez-Blas T, Platas-Iglesias C. Inorg Chem. 2011;50:4125. doi: 10.1021/ic2001915. [DOI] [PubMed] [Google Scholar]
- 32.Regueiro-Figueroa M, Platas-Iglesias C. J Phys Chem A. 2015;119:6436. doi: 10.1021/acs.jpca.5b01728. [DOI] [PubMed] [Google Scholar]
- 33.Esteban-Gómez D, de Blas A, Rodríguez-Blas T, Helm L, Platas-Iglesias C. Chem Phys Chem. 2012;13:3640. doi: 10.1002/cphc.201200417. [DOI] [PubMed] [Google Scholar]
- 34.Rodríguez-Rodríguez A, Regueiro-Figueroa M, Esteban-Gómez D, Rodríguez-Blas T, Patinec V, Tripier R, Tircsó G, Carniato F, Botta M, Platas-Iglesias C. Chem - Eur J. 2017;23:1110. doi: 10.1002/chem.201604390. [DOI] [PubMed] [Google Scholar]
- 35.Cucinotta G, Perfetti M, Luzon J, Etienne M, Car P-E, Caneschi A, Calvez G, Bernot K, Sessoli R. Angew Chem, Int Ed. 2012;51:1606. doi: 10.1002/anie.201107453. [DOI] [PubMed] [Google Scholar]
- 36.de Bari L, Moro L, Passarella S. FEBS Lett. 2013;587:467. doi: 10.1016/j.febslet.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Kowlgi NG, Chhabra L. Gastroenterol Res Pract. 2015;2015:e476215. doi: 10.1155/2015/476215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.