

The counterbalance between coactivator and co-repressor complexes at enhancers instructs stem cell pluripotency transition.
Abstract
Chromatin regulators control cellular differentiation by orchestrating dynamic developmental gene expression programs, and hence, malfunctions in the regulation of chromatin state contribute to both developmental disorders and disease state. Mll4 (Kmt2d), a member of the COMPASS (COMplex of Proteins ASsociated with Set1) protein family that implements histone H3 lysine 4 monomethylation (H3K4me1) at enhancers, is essential for embryonic development and functions as a pancancer tumor suppressor. We define the roles of Mll4/COMPASS and its catalytic activity in the maintenance and exit of ground-state pluripotency in murine embryonic stem cells (ESCs). Mll4 is required for ESC to exit the naive pluripotent state; however, its intrinsic catalytic activity is dispensable for this process. The depletion of the H3K4 demethylase Lsd1 (Kdm1a) restores the ability of Mll4 null ESCs to transition from naive to primed pluripotency. Thus, we define an opposing regulatory axis, wherein Lsd1 and associated co-repressors directly repress Mll4-activated gene targets. This finding has broad reaching implications for human developmental syndromes and the treatment of tumors carrying Mll4 mutations.
INTRODUCTION
In eukaryotic cells, nucleosomes at active promoters are trimethylated on histone H3 lysine 4 (H3K4), whereas enhancers are enriched with H3K4 mono-methylated nucleosomes (1, 2). Although H3K4 methylation recruits transcription activators such as the chromatin remodeler CHD1 (3, 4), NURF subunit BPTF (5, 6), and basal transcription factor TFIID (7), the direct functional link between H3K4 methylation and transcription activation remains unclear. All three states of H3K4 methylation are deposited by SET1/COMPASS (COMplex of Proteins ASsociated with Set1) in yeast (8, 9), whereas in mammals, there are six COMPASS family members with distinct specificities in implementing methylation marks (1): Mammalian Set1/COMPASS is responsible for global H3K4me3 levels in mammalian cells (10–12). Mll1 and Mll2 implement H3K4me2 and H3K4me3 at promoters and enhancers in a locus-specific manner (13–17). In contrast, Mll3 and Mll4 catalyze H3K4me1 at enhancers, suggesting a role of this branch of COMPASS family at distal cis-regulatory elements (18, 19). We have previously demonstrated that Mll4, in lieu of Mll3, is the major H3K4 monomethyltransferase in murine embryonic stem cells (ESCs) (20). Nevertheless, the functions of H3K4me1 at enhancers remain elusive.
Accumulating evidence demonstrates that Mll4/COMPASS plays a crucial role in development and disease. Mouse embryos null for Mll4 die during embryogenesis (21), and loss-of-function mutations in human Mll4 cause the developmental disorder Kabuki syndrome (22). Mll4 mutations are frequently detected in various human cancers including non-Hodgkin’s lymphoma, bladder cancer, and breast cancer (23–27). Moreover, genetic studies in mice reveal that Mll4 acts as a tumor suppressor in the development of B cell lymphoma (28, 29), highlighting the instructive role of Mll4 in malignant transformation. Recent reports demonstrate that H3K4me1 implemented by Mll3 and Mll4 in ESCs has a modest impact on transcription and that H3K4me1 catalyzed by the Mll3/Mll4 ortholog Trr is dispensable for Drosophila viability (30, 31). However, the extent to which the biological functions of Mll4 in mammalian development and during cancer pathogenesis rely on its catalytic function is unknown.
ESCs are pluripotent stem cells that are capable of contributing to all adult tissues through cellular differentiation. Recent advances in cell culture techniques enable the maintenance of ESCs in the ground/naive pluripotent state (32), which resembles the inner cell mass of embryos at the blastocyst stage. The hallmarks of ground-state pluripotency include homogeneous expression of naive pluripotency factors such as Nanog and Klf4, X chromosome activation in female cells, and genome-wide DNA and H3K27 hypomethylation (33). Naive pluripotent cells can be induced to exit the ground state and transition into primed pluripotency (34), which has characteristics of post-implantation epiblasts (35). The transcriptome and epigenome are markedly transformed during the transition of these distinct pluripotency states (36, 37). However, the mechanisms governing the exit from naive pluripotency are not fully understood. It was recently demonstrated that ESCs null for both Mll3 and Mll4 are defective in embryoid body and teratoma formation (38), suggesting a role of these methyltransferases in cellular differentiation. Nevertheless, whether Mll4 or its catalytic activity has an impact on the transition from naive to primed pluripotency, a critical step in lineage specification, remains elusive.
Here, we set out to examine the functional link between H3K4me1 and active transcription by disrupting Mll4 and its catalytic function in ESCs. We have found that the total loss of Mll4 protein leads to enhancer decommissioning and failure to exit the naive pluripotent state, whereas Mll4 catalytic inactivation has minimal effect on this process. Furthermore, the effects of Mll4 loss on ESC differentiation can be rescued by the depletion of the H3K4 demethylase Lsd1, suggesting that active demethylation and deacetylation by Lsd1 and its associated co-repressor complexes (CoREST, CtBP, and NuRD) play a role in this process. We demonstrate that Lsd1 is responsible for the repression of genes that are dependent on Mll4 for their activation. Together, our results reveal novel mechanisms underlying the regulation of enhancer function through an epigenetic balance in cell fate transitions and define an Mll4/COMPASS-Lsd1 regulatory axis that has implications for human developmental disorders, such as Kabuki syndrome and many cancer types that frequently harbor Mll4 mutations.
RESULTS
Mll4 is dispensable for the maintenance of ESC self-renewal
To understand the role of Mll4 in transcriptional regulation and stem cell pluripotency, we generated Mll4 knockout (Mll4KO) ESCs by deleting the second plant homeodomain (PHD) finger clusters of Mll4 using CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR-associated 9)–guided gene editing (fig. S1A). Sanger sequencing and RNA sequencing (RNA-seq) demonstrated deletion of the intended genomic DNA region and transcript of the Mll4 (Kmt2d) gene, respectively (fig. S1B and Fig. 1A). Mll4 protein was undetectable in Mll4KO cells by Western blotting using previously characterized antibodies against the N terminus (NT) or C terminus (CT) of Mll4 (Fig. 1B and fig. S1A) (19). Unlike wild-type (WT) and the previously generated Mll4 SET [Su(var)3-9, enhancer-of-zeste, trithorax] domain–deleted (Mll4ΔSET) cells (20), Mll4KO ESC clones display enlarged intercellular gaps (fig. S1C). Nevertheless, these cells are alkaline phosphatase–positive and exhibit homogeneous expression of Oct4 and Nanog comparable to WT and Mll4ΔSET cells (fig. S1D and Fig. 1C), indicating that Mll4 loss does not impair the self-renewal capability of ESCs.
Fig. 1. Mll4 depletion results in transcription deregulation but does not impair self-renewal of ESCs.
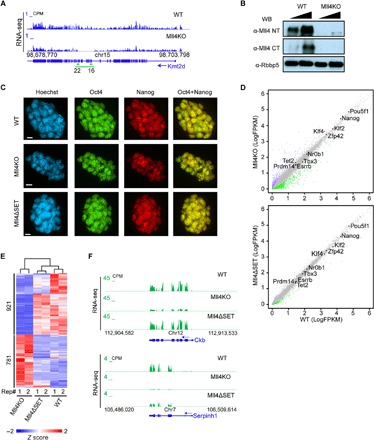
(A) University of California at Santa Cruz (UCSC) genome browser view of RNA-seq signals of WT and Mll4KO ESCs showing successful deletion of the indicated exons (green arrows) at the Kmt2d gene. The green line indicates the deleted genomic region in Mll4KO ESCs. CPM, counts per million mapped reads. (B) Western blotting assay with antibodies against Mll4 and Rbbp5 using nuclear extracts of WT and Mll4KO ESCs. (C) Immunostaining of pluripotency factors Oct4 and Nanog in WT, Mll4KO, and Mll4ΔSET ESCs. Scale bars, 10 μm. (D) Correlation analysis of gene expression levels between WT and respective Mll4KO (top) and Mll4ΔSET (bottom) ESCs. Plots were generated with two biological replicates of RNA-seq experiments from two cell clones for each genotype. Significantly down-regulated genes (compared with WT, adjusted P < 0.01, logFC > |1|) are labeled green, and up-regulated ones are labeled purple. The major pluripotency marker genes are marked on the plot with significantly changed ones labeled in red. Other unchanged genes are labeled as gray dots. FPKM, fragments per kilobase of transcript per million mapped reads. (E) K-means clustering analysis of expression levels of the 1702 differentially expressed genes (DEGs) in Mll4KO cells comparing WT, Mll4KO, and Mll4ΔSET ESCs. Z scores were used to generate the heat map. Numbers below the heat map denote the two biological replicates of each genotype. (F) UCSC genome browser view of RNA-seq signals of WT, Mll4KO, and Mll4ΔSET ESCs at Ckb (top) and Serpinh1 (bottom) loci.
To examine the impact of Mll4 depletion on the ESC transcriptome, we performed RNA-seq analyses of WT, Mll4KO, and Mll4ΔSET cells and found that key pluripotency genes, including Pou5f1 and Nanog, are not perturbed by either Mll4 mutation (Fig. 1D and fig. S1E), further demonstrating that Mll4 is dispensable for ESC self-renewal. There are 921 genes significantly down-regulated in Mll4KO ESCs, whereas only 215 genes are significantly down-regulated in Mll4ΔSET ESCs (fig. S1F). Unsupervised hierarchical clustering of RNA-seq signals revealed that the transcriptomes of WT and Mll4ΔSET cells are grossly similar, whereas gene expression in Mll4KO cells is distinct (Fig. 1E). Mll4 target genes are enriched for factors involved in regulating guanosine triphosphatase activity and cell-cell adhesion (fig. S1G). For instance, creatine kinase (Ckb) expression is reduced in Mll4KO cells but is unchanged in Mll4ΔSET cells (Fig. 1F). In contrast, collagen-binding protein gene Serpinh1 expression is strongly diminished in cells lacking Mll4, whereas specific deletion of the SET domain elicits only a moderate reduction in transcript levels. (Fig. 1F). These data suggest that the role of Mll4 in transcription regulation in ESCs is largely independent of its catalytic activity and that additional transcriptional regulatory functions reside outside of the SET domain.
Mll4 regulates enhancers independent of its methyltransferase activity
To understand how Mll4 regulates gene expression in ESCs, we performed chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) in WT and Mll4KO cells to map the genome-wide occupancy of Mll4. We identified Mll4-specific enriched regions using ChIP-seq signals in Mll4KO cells as a background control (fig. S2A). About 91% of Mll4 peaks are located at introns and intergenic regions (Fig. 2A). Motif analysis revealed that the DNA binding motifs of key pluripotency factors Klf4, Esrrb, Oct4, and Sox2 are enriched in Mll4 peak regions (Fig. 2B), suggesting that Mll4 is enriched at super-enhancers in ESCs. Mll4 binds regions containing large domains of H3K27ac (Fig. 2, C and F), a hallmark of super-enhancers (39). Mll4 ChIP-seq in Mll4ΔSET cells indicated that the occupancy of the mutant Mll4 is comparable to its WT counterpart (Fig. 2C), suggesting that the SET domain is largely dispensable for Mll4 recruitment to chromatin.
Fig. 2. Mll4 is responsible for enhancer activity independent of its catalytic function.
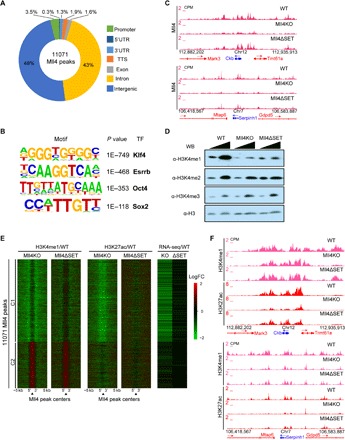
(A) Donut diagram showing the genome-wide distribution of Mll4-enriched regions. (B) Top transcription factors motifs at Mll4-enriched regions. (C) UCSC genome browser view of Mll4 ChIP-seq signals of WT, Mll4KO, and Mll4ΔSET ESCs at Ckb (top) and Serpinh1 (bottom) loci. ChIP-seq experiments were performed using the CT antibody. (D) Western blotting assay of WT, Mll4KO, and Mll4ΔSET ESC lysates using antibodies against H3K4me1, H3K4me2, H3K4me3, and H3. (E) Heat maps showing the log2 fold change (LogFC) in the levels of H3K4me1, H3K27ac, and gene expression at Mll4-enriched regions between Mll4KO, or Mll4ΔSET, and WT cells. Clusters were generated by K-means clustering, and the nearest gene to each Mll4 peak was used to generate the RNA-seq heat map. Signals 5 kb up- and downstream of Mll4 peak regions were included. (F) UCSC genome browser view of H3K4me1 and H3K27ac ChIP-seq signals of WT, Mll4KO, and Mll4ΔSET ESCs at Ckb (top) and Serpinh1 (bottom) loci.
The Mll3/Mll4 branches of the COMPASS family are responsible for H3K4me1 at enhancers (18, 19, 40). We previously demonstrated that Mll4 is the major methyltransferase catalyzing H3K4me1 in ESCs comparing WT and Mll4ΔSET cells (20). Western blotting analysis demonstrated that H3K4me1 levels are reduced as the result of Mll4 deletion or catalytic inactivation (Fig. 2D). We consistently observed lower H3K4me1 levels in Mll4KO cells compared to Mll4ΔSET cells (Fig. 2D), suggesting that mechanisms beyond the loss of Mll4 catalytic activity determine the total H3K4me1 levels. ChIP-seq of H3K4me1 and the active enhancer mark H3K27ac in WT, Mll4KO, and Mll4ΔSET ESCs indicated that H3K4me1 levels around Mll4 binding sites are globally reduced in Mll4KO and Mll4ΔSET cells compared with WT, except for the redistribution of H3K4me1 signals at cluster 2 Mll4 peaks (Fig. 2E and fig. S2B). Note that cluster 2 Mll4 peaks in WT cells harbor higher H3K4me3, harbor lower H3K4me1, and are more enriched with transcription start site (TSS) proximal regions compared with cluster 1 Mll4 peaks (fig. S2, B to D), reminiscent of promoter regions that gain H3K4me1 in Trr-depleted Drosophila cells (18). On a genome-wide scale, total Mll4 protein loss leads to lower H3K4me1 levels than Mll4 SET deletion, consistent with our analysis of bulk H3K4me1 levels by Western blotting (Fig. 2E and fig. S2B). Furthermore, H3K27ac levels were markedly reduced at Mll4-enriched regions in Mll4KO but not Mll4ΔSET cells, correlating with the down-regulation of nearby genes (Fig. 2E and fig. S2B). In addition, levels of H3K4me2 and H3K4me3 were slightly reduced at Mll4 peaks in Mll4KO cells (fig. S2D). We also observed these epigenetic changes at Mll4 target Ckb and Serpinh1 loci (Fig. 2F and fig. S2E). These results indicate that the context of Mll4 protein rather than its catalytic activity within the SET domain is indispensable for the activity of Mll4 bound enhancers.
Lsd1 mediates enhancer decommissioning in Mll4KO ESCs
Histone demethylase Lsd1, which targets mono- and dimethylated H3K4 (41), has been shown to bind enhancers and is required for ESC differentiation (42, 43). To explore whether Lsd1 colocalizes with Mll4 on chromatin, we performed Lsd1 ChIP-seq in ESCs and found that 78% of Lsd1 peaks are located at introns and intergenic regions (fig. S3A). Moreover, Lsd1 occupancy at Mll4 target loci had a similar pattern to Mll4 (Fig. 3A), and Lsd1 binding signals were elevated at Mll4 peak centers (Fig. 3B), suggesting the genome-wide colocalization of Mll4 and Lsd1. Seventy-nine percent of Mll4-enriched regions overlapped with Lsd1 peaks (Fig. 3C), suggesting that the two proteins co-regulate the activity of a common group of enhancers. These findings and the result that Mll4KO ESCs harbor lower H3K4me1 levels than Mll4ΔSET cells prompted us to hypothesize that the loss of Mll4 recruitment at Mll4 target enhancers is accompanied with the removal of H3K4 methylation by Lsd1.
Fig. 3. Lsd1 is required for the full deactivation of enhancers caused by Mll4 loss.
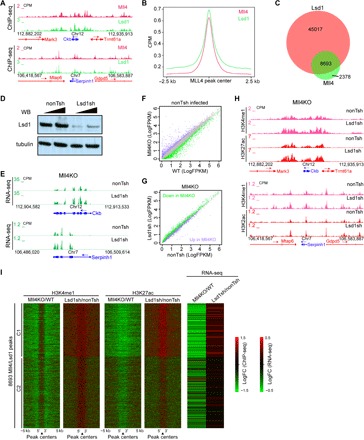
(A) UCSC genome browser view of Mll4 and Lsd1 ChIP-seq signals at Ckb (top) and Serpinh1 (bottom) loci. (B) Metaplot showing Mll4 and Lsd1 ChIP-seq signals at 2.5 kb up- and downstream of Mll4 peaks. (C) Venn diagram showing the overlap between Mll4 and Lsd1 peak regions. (D) Western blotting of Lsd1 and tubulin in ESCs infected with control virus (nonTsh) and virus containing shRNA-targeting Lsd1 (Lsd1sh). (E) UCSC genome browser view of RNA-seq signals of control and Lsd1-depleted ESCs at Ckb (top) and Serpinh1 (bottom) loci. (F) Correlation analysis of gene expression levels between nonTsh-infected WT and Mll4KO ESCs. Significantly down-regulated genes are labeled green, and up-regulated ones are labeled purple. Unchanged genes are labeled as gray dots. (G) Correlation analysis of the expression levels of DEGs in (F) between nonTsh- and Lsd1sh-infected Mll4KO ESCs. Significantly down-regulated genes by Mll4 depletion in (F) are labeled green, and up-regulated ones are labeled purple. (H) UCSC genome browser view of H3K4me1 and H3K27ac ChIP-seq signals of nonTsh- and Lsd1sh-infected Mll4KO ESCs at Ckb (top) and Serpinh1 (bottom) loci. (I) Heat maps showing the log2 fold change in the levels of H3K4me1, H3K27ac, and gene expression at Mll4/Lsd1 co-bound regions for Mll4KO versus WT cells and nonTsh-infected versus Lsd1sh-infected Mll4KO cells. Clusters were generated by K-means clustering and the nearest gene to each peak was used to generate the RNA-seq heat map. Signals 5 kb up- and downstream of Lsd1/Mll4 co-bound peaks were included.
To test this hypothesis, we depleted Lsd1 in Mll4KO ESCs using short hairpin RNA (shRNA) (Fig. 3D). The expression levels of Mll4 target genes Ckb and Serpinh1 were elevated upon Lsd1 depletion (Fig. 3E). RNA-seq analysis further demonstrated that the set of genes down-regulated by Mll4 deletion is largely up-regulated by Lsd1 knockdown. In addition, genes with elevated expression levels in Mll4KO cells were down-regulated upon Lsd1 depletion (Fig. 3, F and G). A counterbalance between Lsd1 and Mll4 in transcriptional regulation could also be observed for Lsd1 target genes (fig. S3, B and C). Specifically, 41% of genes up-regulated by Lsd1 knockdown were down-regulated by Mll4 deletion (fig. S3D). Mll4 targets rescued by Lsd1 depletion are enriched for genes involved in membrane organization and adhesion (fig. S3E). The restoration of transcriptional defects caused by Mll4 loss suggests that Lsd1 is at least partially responsible for the repression of Mll4 target genes in Mll4KO ESCs.
We next performed ChIP-seq in Lsd1-depleted Mll4KO ESCs and found that H3K4me1 and H3K27ac levels at Mll4 target genes were increased by Lsd1 depletion (Fig. 3H). K-means clustering analysis indicated that Lsd1 depletion restores H3K4me1 and H3K27ac levels at Mll4 and Lsd1 colocalized regions as well as the expression of genes near these enhancers (Fig. 3I and fig. S3F). Because most of the Mll4 and Lsd1 co-occupied regions are far from TSS and decorated with H3K27ac and H3K4me1 (fig. S3F and Fig. 3G), our data indicate that Lsd1 and potentially its associated co-repressors contribute to the silencing of Mll4 target enhancers and their associated genes in Mll4KO ESCs.
Mll4 is essential for the exit from naive pluripotency
During development, the inner cell mass of blastocyst stage embryos exits from naive pluripotency into primed pluripotency and is subsequently induced to commit to specific lineages by various signals (44, 45). To examine whether Mll4 is required for the exit from naive pluripotency, we switched the culture conditions of ESCs from 2i/LIF to medium containing fetal bovine serum and leukemia inhibitory factor (FBS/LIF). As expected, WT ESCs lost the dome-shaped morphology in FBS/LIF (Fig. 4A) and underwent marked transcriptional changes during this transition, including down-regulation of naive state markers and up-regulation of primed state markers (Fig. 4B and fig. S4A). WT and Mll4ΔSET cell clones became flat upon switching to FBS/LIF, whereas Mll4KO cells exhibit a similar morphology to naive pluripotent cells (Fig. 4A). Although the key pluripotency gene Pou5f1 remained unchanged by Mll4 deletion in FBS/LIF cultures (Fig. 4E), multiple naive markers were up-regulated, and primed markers were down-regulated in Mll4KO cells compared with WT cells (Fig. 4, C and F), suggesting that Mll4 is required for exiting naive pluripotency in ESCs. Genes highly expressed in 2i/LIF were mostly up-regulated by Mll4 deletion in FBS/LIF cultures, whereas genes highly expressed in FBS/LIF were largely down-regulated in Mll4KO cells grown in FBS/LIF (Fig. 4D). However, the expression levels of most genes remained unchanged in Mll4ΔSET cells compared with WT cells in FBS/LIF (fig. S4, B and C, and Fig. 4F), indicating that the role of Mll4 in the transition between pluripotency states is independent of its catalytic function.
Fig. 4. Mll4 deletion impairs the exit of naive pluripotency.
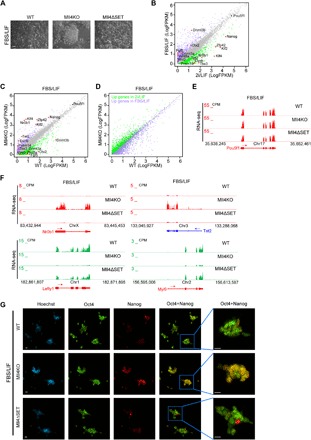
(A) Phase images of WT, Mll4KO, and Mll4ΔSET ESCs grown in FBS/LIF condition. Scale bar, 100 μm. (B) Correlation analysis of gene expression levels between ESCs grown in FBS/LIF and 2i/LIF conditions. Genes significantly up-regulated in FBS/LIF are labeled purple, whereas genes significantly increased in 2i/LIF are labeled green. The major naive pluripotency and primed pluripotency markers are marked, with red dots denoting significantly differentially expressed ones. (C) Correlation analysis of gene expression levels between WT and Mll4KO ESCs grown in FBS/LIF condition. Genes significantly down-regulated in Mll4KO cells are labeled green, whereas up-regulated ones are labeled purple. The major naive pluripotency and primed pluripotency markers are marked, with red dots denoting significantly changed ones. (D) Correlation analysis of the expression levels of DEGs in (B) between WT and Mll4KO ESCs grown in FBS/LIF condition. Genes significantly up-regulated in FBS/LIF are labeled purple, whereas genes significantly increased in 2i/LIF are labeled green. (E) UCSC genome browser view of RNA-seq signals of WT, Mll4KO, and Mll4ΔSET ESCs cultured in FBS/LIF condition at the Pou5f1 gene. (F) UCSC genome browser view of RNA-seq signals of WT, Mll4KO, and Mll4ΔSET ESCs cultured in FBS/LIF condition at Nr0b1, Tet2, Lefty1, and Myl9 genes. (G) Immunostaining of pluripotency factors Oct4 and Nanog in WT, Mll4KO, and Mll4ΔSET ESCs cultured in FBS/LIF condition. Scale bars, 10 μm.
Homogeneous expression of the pluripotency factor Nanog is one of the major hallmarks of the naive pluripotent state (Fig. 1C) (33). Upon exiting naive pluripotency, the expression pattern of Nanog becomes heterogeneous (Fig. 4G). Mll4ΔSET cells grown in FBS/LIF displayed a similar Nanog expression pattern to WT cells (Fig. 4G). However, Nanog was ubiquitously expressed in Mll4KO cells after the medium switch (Fig. 4G), supporting the notion that Mll4 loss impairs the exit of naive pluripotency in ESCs. Moreover, ChIP-seq analysis demonstrated that levels of H3K4me1 and H3K27ac are decreased at enhancers of FBS/LIF up-regulated genes in Mll4KO ESCs cultured with FBS/LIF (fig. S4, D and E), suggesting that Mll4 activates enhancers required for primed pluripotency during cell fate transition. Collectively, these results demonstrate that a lack of Mll4 protein locks ESCs into a naive-like pluripotent state.
Lsd1 depletion restores the pluripotency transition defects of Mll4 null ESCs
To determine whether the epigenetic balance of Lsd1 and Mll4 influences the pluripotency transition, we depleted Lsd1 in FBS/LIF-cultured Mll4KO cells and performed RNA-seq. Our results demonstrate that the naive pluripotency markers Nr0b1 and Tet2 are down-regulated, whereas primed pluripotency markers Lefty1 and Myl9 are elevated, upon Lsd1 knockdown in Mll4KO cells (Fig. 5A and fig. S5A). Lsd1 depletion largely restored transcriptional misregulation in Mll4KO ESCs cultured in FBS/LIF, indicating that the transcription defects in pluripotency transition caused by Mll4 deletion are dependent on Lsd1 function (Fig. 5, B and C). The restorative effects of Lsd1 depletion are most apparent at genes that depend on Mll4 for their activation (Fig. 5C), consistent with Lsd1 acting predominantly as a transcriptional repressor at Mll4-activated targets. Lsd1 depletion in Mll4KO cells cultured in FBS/LIF reverted the ubiquitous expression of Nanog to a heterogeneous pattern as evidenced by immunostaining (Fig. 5D), further demonstrating that Lsd1 is responsible for the failure in exiting naive pluripotency caused by Mll4 loss. ChIP-seq analysis revealed that Lsd1 depletion leads to increased levels of H3K4me1 and H3K27ac at primed pluripotency loci (fig. S5B), suggesting that Lsd1 silences these genes in Mll4KO cells during the exit of naive pluripotency. Furthermore, enhancers of Mll4-activated genes in FBS/LIF displayed elevated H3K4me1 and H3K27ac upon removal of Lsd1 (fig. S5C), corroborating the derepression of these genes by Lsd1 depletion.
Fig. 5. Lsd1 depletion rescues the pluripotency transition defects in Mll4KO ESCs.
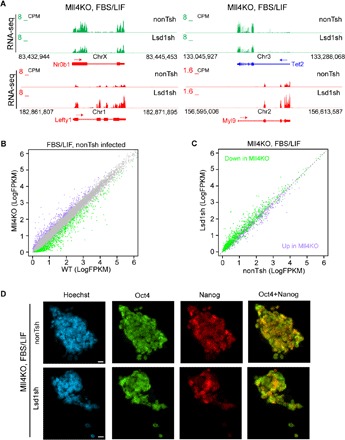
(A) UCSC genome browser view of RNA-seq signals of nonTsh- and Lsd1sh-infected Mll4KO ESCs cultured in FBS/LIF condition at Nr0b1, Tet2, Lefty1, and Myl9 genes. (B) Correlation analysis of gene expression levels between nonTsh-infected WT and Mll4KO ESCs grown in FBS/LIF condition. Genes significantly down-regulated in Mll4KO cells are labeled green, whereas up-regulated ones are labeled purple. (C) Correlation analysis of the expression levels of DEGs in (B) between nonTsh- and Lsd1sh-infected Mll4KO ESCs cultured in FBS/LIF condition. Significantly down-regulated genes by Mll4 depletion in (B) are labeled green, and up-regulated ones are labeled purple. (D) Immunostaining of pluripotency factors Oct4 and Nanog in nonTsh- and Lsd1sh-infected Mll4KO ESCs cultured in FBS/LIF condition. Scale bars, 10 μm.
The NT of Mll4 is essential for gene regulation and pluripotency transition
To further explore the role of Mll4 in regulating gene expression and cell fate transition, we generated Mll4 NT–deleted ESCs (hereafter named Mll4ΔNT) by deleting 1195 amino acids, which include the first PHD finger cluster, from the NT of Mll4 using CRISPR/Cas9 (fig. S1A). This in-frame deletion is predicted to produce a mutant Mll4 protein deficient in chromatin binding because of the role of PHD fingers as histone-binding modules (46, 47). RNA-seq results demonstrated the desired deletion of exons 4 to 12 of Mll4 transcript in Mll4ΔNT cells (Fig. 6A), which was confirmed by Sanger sequencing (fig. S6A). Similar to Mll4KO ESCs, Mll4ΔNT cells grown in 2i/LIF medium exhibit increased intercellular gaps (Fig. 6B). NT-deleted Mll4 can coimmunoprecipitate with Rbbp5 (fig. S6B), a common subunit of all COMPASS, indicating that the deletion of NT does not impair the appropriate assembly of Mll4/COMPASS. In contrast, the SET domain–deleted Mll4 mutant could not bind Rbbp5 (fig. S6B), suggesting that the integrity of Mll4/COMPASS is not essential for the regulation of transcription and enhancer activity in ESCs.
Fig. 6. Deletion of Mll4 NT leads to enhancer decommissioning and pluripotency transition defects.
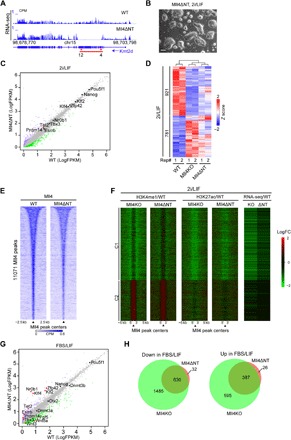
(A) UCSC genome browser view of RNA-seq signals of WT and Mll4ΔNT ESCs showing successful deletion of the indicated exons (red arrows) at Kmt2d gene. The red line indicates the deleted genomic region in Mll4ΔNT ESCs. (B) A representative phase image of Mll4ΔNT ESCs grown in 2i/LIF condition. Scale bar, 100 μm. (C) Correlation analysis of gene expression levels between WT and Mll4ΔNT ESCs cultured in 2i/LIF. Significantly down-regulated genes by Mll4 NT deletion are labeled green, and up-regulated ones are labeled purple. The major pluripotency marker genes are marked on the plot with significantly changed ones labeled in red. Other unchanged genes are labeled as gray dots. (D) K-means clustering analysis of expression levels of the 1702 DEGs in Mll4KO cells in 2i/LIF culture conditions. Z scores of WT, Mll4KO, and Mll4ΔNT ESCs of the genes are shown. Numbers below the heat map denote the two biological replicates of each genotype. (E) Heat map showing Mll4 occupancy at the 11,071 Mll4 peaks and the surrounding 5-kb regions in WT and Mll4ΔNT ESCs. The profiles are sorted in a descending order of Mll4 occupancy in WT ESCs. (F) Heat maps showing the log2 fold change in the levels of H3K4me1, H3K27ac, and gene expression at Mll4 peaks and 10-kb surrounding regions between Mll4KO, or Mll4ΔNT, and WT cells. Cluster division and the order within the clusters are the same as Fig. 2E. (G) Correlation analysis of gene expression levels between WT and Mll4ΔNT ESCs grown in FBS/LIF condition. Genes significantly down-regulated in Mll4ΔNT cells are labeled green, whereas up-regulated ones are labeled purple. The major naive pluripotency and primed pluripotency markers are marked, with red dots denoting significantly changed ones. (H) Venn diagrams showing the overlap of DEGs in Mll4KO and Mll4ΔNT ESCs cultured in FBS/LIF. Down-regulated genes (versus WT) are plotted on the left, and up-regulated genes are plotted on the right.
RNA-seq analysis comparing the transcriptome of WT and Mll4ΔNT cells under naive pluripotency conditions demonstrated that pluripotency marker expression is not impaired by this mutation (Fig. 6C). Nevertheless, we observed transcriptional changes of more than 900 genes in Mll4ΔNT cells, and most of these DEGs were also perturbed by Mll4 deletion (fig. S6C), suggesting that the transcriptomes of Mll4KO and Mll4ΔNT cells are similar. Unsupervised clustering of RNA-seq signals demonstrated a similarity between the transcriptomes of these cells (Fig. 6D). Many Mll4 target genes were down-regulated in Mll4ΔNT ESCs, including Ckb (fig. S6D). As expected, we observed a decrease in Mll4 occupancy at the Ckb locus in Mll4ΔNT cells (fig. S6E). Moreover, a genome-wide reduction in chromatin binding was observed for the Mll4ΔNT mutant protein (Fig. 6E). We next examined the levels of H3K4me1, H3K4me2, and H3K4me3 in WT, Mll4ΔNT, and Mll4KO cells by Western blotting. Although the catalytic domain of Mll4 was not affected in Mll4ΔNT cells, H3K4me1 was reduced compared with WT ESCs (fig. S6F), highlighting the importance of Mll4 recruitment on H3K4me1 deposition. ChIP-seq analysis showed decreased H3K4me1 and H3K27ac at the Mll4 target Ckb, and genome-wide at Mll4 bound regions (fig. S6G and Fig. 6F), supporting the notion that the impaired Mll4 recruitment to chromatin rather than the loss of its catalytic activity is the main cause of enhancer decommissioning in Mll4KO ESCs.
To investigate whether Mll4ΔNT ESCs display similar failures in exiting naive pluripotency as Mll4KO cells, we switched the culture medium from 2i/LIF to FBS/LIF (fig. S6H) and performed RNA-seq of WT and Mll4ΔNT cells. Our results showed that multiple naive pluripotency markers were up-regulated by Mll4 NT deletion (Fig. 6G). Similar to Mll4KO cells, the expression level of Lefty1 was reduced, whereas the expression of Nr0b1 was elevated in Mll4ΔNT cells (fig. S6I). Moreover, 95% of genes misregulated in FBS/LIF-cultured Mll4ΔNT cells overlapped with DEGs in Mll4KO cells (Fig. 6H). In summary, these data strongly argue that the recruitment of Mll4 to chromatin is critical for enhancer activation, gene expression, and the exit from naive pluripotency.
DISCUSSION
Despite the importance of Mll4 in normal development and during disease pathogenesis, the molecular function of Mll4 underlying these biological processes is unclear. The results presented in this study provide evidence that Mll4 protein is indispensable for enhancer activation and for the exit from naive pluripotency in ESCs, whereas the catalytic function of Mll4 is not required for these processes. Lsd1 participates in enhancer decommissioning and skewing of pluripotency transition in Mll4KO cells. Moreover, we demonstrated that the recruitment of Mll4 protein to chromatin is required for enhancer activation and pluripotency transition (Fig. 7).
Fig. 7. A working model for the regulation of enhancer function by Mll4/COMPASS and Lsd1.
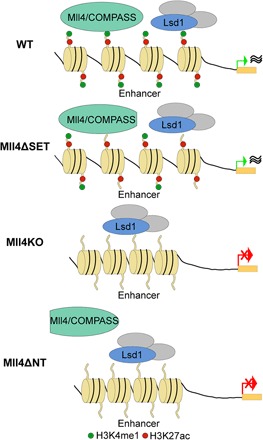
Mll4/COMPASS and Lsd1 co-occupy a large cohort of enhancers in WT ESCs. In Mll4ΔSET ESCs, the levels of H3K4me1 are reduced at enhancers without affecting transcription. Upon the deletion (Mll4KO) or loss of recruitment of Mll4 (Mll4ΔNT), Lsd1 and its associated co-repressors decommission enhancers, which results in repression of transcription and failures in pluripotency transition.
Mll4 is dispensable for ESC self-renewal but regulates more than 1700 genes (Fig. 1). In contrast, there are only 300 misregulated genes in Mll4ΔSET cells compared with WT, suggesting that the primary functions of Mll4 in transcriptional regulation are catalytic-independent. This is consistent with our recent finding of the Mll3/Mll4 ortholog Trr in Drosophila, Set1A in mouse ESCs, and Set1B in human triple-negative breast cancer (12, 20, 31, 48). We recently reported that the catalytically inactive Trr can rescue the embryonic lethality of Trr null alleles, demonstrating for the first time that enhancer H3K4me1 catalyzed by Trr is dispensable in the context of metazoan development (31). In addition, we demonstrated that the catalytic SET domain of Set1A is dispensable for ESC self-renewal although Set1A protein is required for ESC survival and proliferation (12). Similarly, Set1B, but not its SET domain, is responsible for maintaining the survival of triple-negative breast cancer cells (48). Collectively, these data point to novel catalytic-independent functions of the COMPASS family of methyltransferases in normal development and in human diseases. Catalytic-independent functions have also been reported for other chromatin regulatory proteins such as the PRC1 component Ring1b (49). Mouse embryos expressing catalytically inactive Ring1b develop much further than Ring1b null embryos (50, 51), demonstrating that the ubiquitin ligase activity of Ring1b is dispensable for early mouse development. However, unlike Ring1b catalytic mutants, which exhibit losses of H2A ubiquitination similar to Ring1b KO ESCs, Mll4ΔSET ESCs have evidently higher H3K4me1 levels than Mll4KO ESCs (Fig. 2, D and E). The reason for this difference in H3K4me1 is currently undetermined; however, it is possible that Mll4 lacking its SET domain is still capable of mediating recruitment of other COMPASS family members or unknown methyltransferases to chromatin, whereas Mll4 null alleles completely abrogate this recruitment. Small differences in H3K4me1 levels at enhancers may contribute to the distinct enhancer activities and transcriptomes of Mll4KO and Mll4ΔSET ESCs. H3K27ac levels at Mll4 target regions are reduced by Mll4 depletion but not by deletion of its catalytic domain, although Mll3 is WT, suggesting that the H3K4me1 remaining in Mll4ΔSET cells may play an important role in the maintenance of enhancer activities. The reduction of H3K27ac levels at Mll4 bound enhancers in Mll4KO ESCs may be attributed to reduced recruitment of P300, as recent studies indicated that P300 recruitment is impaired by Mll4 depletion and that Mll4 and P300 synergistically activate transcription (38, 52). Note that in Drosophila, the stability of UTX is dependent on Trr, the sole Drosophila ortholog of Mll3/Mll4 (18). Therefore, the potential reduction of UTX in Mll4-depleted cells may contribute to enhancer deactivation as well through increase of H3K27 trimethylation levels. It is also possible that histone deacetylases (HDACs) facilitate the removal of enhancer H3K27ac and transcription repression upon Mll4 depletion. Future studies of the interplay between Mll4 and other major coactivators and co-repressors will elucidate how epigenetic modifications are regulated at enhancers and how they modulate enhancer activity.
Lsd1 depletion is capable of restoring, at least in part, the decommissioned enhancers and the skewed transcriptome in Mll4-depleted ESCs (Fig. 3). Lsd1 binds to active enhancers in ESCs (42) and demethylates H3K4me1 (41). We observed that most of the Mll4 binding regions in ESCs are also bound by Lsd1 (Fig. 3C), which has been found in multiple repressive complexes such as CoREST, CtBP, and NuRD (53–55). It has also been shown that many enhancers in ESCs are enriched with Lsd1 and NuRD (42, 56). We believe that Lsd1 and associated co-repressor complexes may erase the remaining H3K4me1 at enhancers, abolish enhancer-associated H3K27ac, and facilitate the silencing of associated genes in Mll4-depleted cells. The mechanistic basis of how removal of H3K4me1 leads to enhancer decommissioning is currently unknown. We speculate that because H3K4me1 at promoters can be antagonistic to the recruitment of co-repressors ING1 and Sin3A (57), the reduction of H3K4me1 at enhancers in Mll4KO cells may enhance the recruitment of the Sin3A-HDAC complex to enhancers. Despite having a lower affinity compared with H3K4me3, the chromatin remodeler CHD1 can also bind H3K4me1 (3, 4). Moreover, the recently characterized H3K4me1/H3K4me3 binding protein BRWD2 associates with the chromatin remodeler CHD4 (58). Therefore, it is possible that nucleosome occupancy at Mll4 bound enhancers is altered by Mll4 depletion, which may contribute to enhancer deactivation.
We observed that de novo DNA methyltransferases DNMT3a and DNMT3b are significantly down-regulated in Mll4KO ESCs cultured in FBS/LIF (Fig. 4C). DNMT3a and DNMT3b have been shown to maintain enhancer activity in epidermal stem cells (59). We surmise that the levels of DNA methylation at enhancers are critical for their activity during pluripotency transition. It would be interesting to measure DNA methylation levels at enhancers in Mll4 mutant cells in future studies to dissect the relationship between histone modification and DNA methylation at enhancers.
Note that enhancer H3K4me1 is not completely lost in Mll4KO ESCs. One or several of the remaining COMPASS family members may be responsible for the implementation of the residual H3K4me1 at these enhancers. We have previously shown that Mll2 and Set1A bind distal cis-regulatory regions in ESCs and that depletion of Mll3 in Mll4ΔSET ESCs results in further reduction of H3K4me1 levels (16, 20). Unidentified histone methyltransferases could also affect the deposition of H3K4me1 at enhancers. Future studies with genome-wide screening using CRISPR or shRNA libraries would facilitate the discovery of novel factors involved in this process.
Mll4 null ESCs are more resistant to the medium switch from 2i/LIF to FBS/LIF compared with WT or Mll4 catalytically inactive cells, suggesting that the loss of Mll4 protein causes failures in exiting naive pluripotency. Activation of signaling pathways, such as FGF2/MAPK and Activin/Nodal pathways, facilitates the transition from naive to primed pluripotency, whereas the activation of WNT and LIF/STAT3 pathways supports the maintenance of naive pluripotency (44). We surmise that Mll4 acts downstream of signaling pathways that promote the naive to primed transition by activating transcription factors that shape the primed pluripotency transcriptome. Thus, Mll4 loss may disrupt signaling cascades, resulting in a failure to exit naive pluripotency. Mll4 depletion leads to the down-regulation of key primed pluripotency factors such as Otx2, Zic2, Pou3f1, and Fgf5 in FBS/LIF. Otx2 depletion in ESCs leads to deregulation of primed pluripotency markers during the transition from ESCs to epiblast-like cells (36), and Zic2 has been shown to be required for ESC specification (56). It is likely that the down-regulation of these critical transcription factors by Mll4 depletion prohibits ground-state ESCs from leaving naive pluripotency.
Although Mll4 catalytic activity is largely dispensable for enhancer activation and pluripotency transition, deletion of 1195 amino acids from Mll4 NT (MLL4ΔNT), which includes the first PHD finger cluster, severely abrogates the ability of Mll4 to associate with chromatin and phenocopies the Mll4KO phenotype, leading to transcriptome reconfiguration, enhancer decommissioning, and pluripotency transition impairment of ESCs. The second Mll4 PHD finger cluster has been shown to be important for Mll4 to bind histone tails and methylate H3K4 (60), supporting the notion that the recruitment of Mll4 to chromatin is critical for its function at enhancers. Furthermore, Rbbp5 binds to the NT-truncated Mll4 mutant but not SET domain–deleted Mll4 mutant, suggesting that the formation of intact Mll4/COMPASS is not required to maintain global transcription levels and enhancer activities in ESCs. It is possible that the recruitment of Mll4 to its target enhancers is sufficient to protect them from being deactivated by Lsd1 and associated co-repressors. Catalytically inactive Mll4 could achieve the epigenetic balance with Lsd1, supporting the notion that the chromatin recruitment ability of Mll4 is critical for its functions in enhancer regulation and ESC pluripotency.
In summary, our results define the requirements for the Mll4 protein and its catalytic activity in the maintenance and exit from ESC ground-state pluripotency. Moreover, we establish an antagonistic regulatory axis between Mll4/COMPASS and the co-repressor Lsd1. Our finding that Lsd1 depletion restores the aberrant gene expression in Mll4 mutant cells has important implications for human disease and points to Lsd1 inhibitors as a potential therapy for cancers harboring mutations in Mll4.
MATERIALS AND METHODS
ESC culture, shRNA knockdown, and CRISPR/Cas9-guided gene editing
V6.5 ESCs were grown in N2B27 medium supplemented with two inhibitors and LIF, as described previously (20). For induction of the exit of naive pluripotency, 2i/LIF-cultured ESCs were passaged to 0.1% gelatin (STEMCELL Technologies)–coated plates and cultured for more than two passages in FBS/LIF medium containing Dulbecco’s modified Eagle’s medium (Corning) supplemented with 15% FBS (Gemini), 1× penicillin-streptomycin (Life Technologies), 1× GlutaMAX (Life Technologies), 1× minimum essential medium nonessential amino acids (Life Technologies), 1× β-mercaptoethanol (Life Technologies), and LIF (1.8 × 103 U/ml; Gemini). Knockdown was performed as described by Cao et al. (20). The lentiviral construct containing shRNA against Lsd1 (TRCN0000071373) was purchased from Open BioSystems.
Plasmids containing desired guide RNAs (gRNAs) were generated as previously described (20). ESCs were transfected and selected with puromycin (2 μg/ml) for 48 hours and grown in 2i/LIF medium without puromycin until the cell clones were ready to be picked. gRNA sequences used in this study are listed as follows: Mll4KO, TGGGGATGGACAGCCCGACG (left), GGTATAATCAATCCGTCCTT (right); Mll4ΔNT AGGAGGTAAACGGTTCACCC (left), CAGAGAGCACAACGCCGTGC (right).
Antibodies
The following antibodies were used in this study: anti-H3K4me1 (generated in-house), anti-H3K4me2 (generated in-house), anti-H3K4me3 (generated in-house), anti-H3K27ac (Cell Signaling, 8173), anti-H3 (generated in-house), anti-Mll3 (generated in-house), anti-Mll4 NT (generated in-house), anti-Mll4 CT (generated in-house), anti-Rbbp5 (Bethyl Laboratories, A300-109A), anti-Oct4 (Santa Cruz Biotechnology, 5279), anti-Nanog (eBioscience, 14-5761-80), anti-Lsd1 (Abcam, 17721), and anti-tubulin (Developmental Studies Hybridoma Bank, E7).
Immunostaining
Cells were grown on 1% gelatin–coated glass coverslips, fixed with 4% paraformaldehyde for 20 min, and blocked with 10% FBS solutions before immunofluorescence staining. The following primary antibodies were used: mouse anti-Oct4 (1:50) and rat anti-Nanog (1:100). The following secondary antibodies were used: Alexa Fluor 488–coupled donkey anti-mouse antibodies or Alexa Fluor 568–coupled goat anti-rat antibodies (1:400, Molecular Probes). Nuclei were counterstained with Hoechst (1:1000, Thermo Fisher). Images were collected at 60× on a Nikon Eclipse Ts2R inverted microscope with a Nikon DS-Qi2 camera.
ChIP-seq and RNA-seq
ChIP and ChIP-seq library generation were performed as previously described (20). TRIzol reagent (Life Technologies) was used to extract RNA. Deoxyribonuclease I treatment and further RNA purification were performed as previously described (20). ChIP-seq and RNA-seq libraries were sequenced on the NextSeq 500 sequencer (Illumina) for 50 cycles. BCL to FASTQ conversion, reads trimming, and alignment were performed as described by Cao et al. (20). Raw read counts from ChIP-seq and RNA-seq were normalized to total read counts per million and visualized in the UCSC genome browser as bigWig-formatted coverage tracks. Next-generation sequencing data were deposited at Gene Expression Omnibus under accession number GSE99022.
Next-generation sequencing data analysis
ChIP peaks were called using model-based analysis of ChIP-seq (MACS) v1.4.2 with default parameters (61) using corresponding input samples as controls. Mll4 peaks were called using Mll4 ChIP-seq in Mll4KO cells as the control. Metaplots and heat maps were generated using ngsplot (62). K-means clustering was also performed using ngsplot, and nearest-gene log fold changes in gene expression corresponding to the clustered peaks in the heat maps were determined using in-house scripts and visualized with Java TreeView (61, 63).
For RNA-seq, gene count tables were used as input for edgeR 3.0.8 (64). Genes with Benjamini-Hochberg–adjusted P values less than 0.01 and log fold changes >|1| were considered to be differentially expressed. Custom perl and R scripts were used to generate the correlation plots.
Supplementary Material
Acknowledgments
We thank the members of the Shilatifard laboratory for helpful suggestions and discussions. Funding: E.R.S. is supported by the National Cancer Institute (grant R50CA211428). This study was supported in part by the Outstanding Investigator Award from the National Cancer Institute (grant R35CA197569) to A.S. Author contributions: K.C. and A.S. designed the research. K.C., M.A.M., S.A.M., and E.J.R. performed experiments. K.C., C.K.C., S.A.M., P.A.O., and E.R.S. analyzed the data. K.C., M.A.M., E.R.S., and A.S. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaap8747/DC1
fig. S1. Characterization of Mll4KO ESCs.
fig. S2. Enhancer inactivation by Mll4 depletion.
fig. S3. Epigenetic balance between Lsd1 and Mll4/COMPASS.
fig. S4. Mll4 deletion impairs enhancer activation during pluripotency transition.
fig. S5. Lsd1 is indispensable for enhancer decommissioning in FBS/LIF-cultured Mll4KO cells.
fig. S6. Characterization of Mll4ΔNT ESCs.
REFERENCES AND NOTES
- 1.Shilatifard A., The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 81, 65–95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heintzman N. D., Stuart R. K., Hon G., Fu Y., Ching C. W., Hawkins R. D., Barrera L. O., Van Calcar S., Qu C., Ching K. A., Wang W., Weng Z., Green R. D., Crawford G. E., Ren B., Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Flanagan J. F., Mi L.-Z., Chruszcz M., Cymborowski M., Clines K. L., Kim Y., Minor W., Rastinejad F., Khorasanizadeh S., Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 438, 1181–1185 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Sims R. J. III, Chen C.-F., Santos-Rosa H., Kouzarides T., Patel S. S., Reinberg D., Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J. Biol. Chem. 280, 41789–41792 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wysocka J., Swigut T., Xiao H., Milne T. A., Kwon S. Y., Landry J., Kauer M., Tackett A. J., Chait B. T., Badenhorst P., Wu C., Allis C. D., A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442, 86–90 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Li H., Ilin S., Wang W., Duncan E. M., Wysocka J., Allis C. D., Patel D. J., Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442, 91–95 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vermeulen M., Mulder K. W., Denissov S., Pijnappel W. W. M. P., van Schaik F. M. A., Varier R. A., Baltissen M. P., Stunnenberg H. G., Mann M., Timmers H. T. M., Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Miller T., Krogan N. J., Dover J., Erdjument-Bromage H., Tempst P., Johnston M., Greenblatt J. F., Shilatifard A., COMPASS: A complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. U.S.A. 98, 12902–12907 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krogan N. J., Dover J., Khorrami S., Greenblatt J. F., Schneider J., Johnston M., Shilatifard A., COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J. Biol. Chem. 277, 10753–10755 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Wu M., Wang P. F., Lee J. S., Martin-Brown S., Florens L., Washburn M., Shilatifard A., Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell. Biol. 28, 7337–7344 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bledau A. S., Schmidt K., Neumann K., Hill U., Ciotta G., Gupta A., Torres D. C., Fu J., Kranz A., Stewart A. F., Anastassiadis K., The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development 141, 1022–1035 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Sze C. C., Cao K., Collings C. K., Marshall S. A., Rendleman E. J., Ozark P. A., Chen F. X., Morgan M. A., Wang L., Shilatifard A., Histone H3K4 methylation-dependent and-independent functions of Set1A/COMPASS in embryonic stem cell self-renewal and differentiation. Genes Dev. 31, 1732–1737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang P., Lin C., Smith E. R., Guo H., Sanderson B. W., Wu M., Gogol M., Alexander T., Seidel C., Wiedemann L. M., Ge K., Krumlauf R., Shilatifard A., Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 29, 6074–6085 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rickels R., Hu D., Collings C. K., Woodfin A. R., Piunti A., Mohan M., Herz H. M., Kvon E., Shilatifard A., An Evolutionary Conserved Epigenetic Mark of Polycomb Response Elements Implemented by Trx/MLL/COMPASS. Mol. Cell 63, 318–328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu D., Garruss A. S., Gao X., Morgan M. A., Cook M., Smith E. R., Shilatifard A., The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 20, 1093–1097 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu D., Gao X., Cao K., Morgan M. A., Mas G., Smith E. R., Volk A. G., Bartom E. T., Crispino J. D., Di Croce L., Shilatifard A., Not all H3K4 methylations are created equal: Mll2/COMPASS dependency in primordial germ cell specification. Mol. Cell 65, 460–475.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denissov S., Hofemeister H., Marks H., Kranz A., Ciotta G., Singh S., Anastassiadis K., Stunnenberg H. G., Stewart A. F., Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 141, 526–537 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Herz H.-M., Mohan M., Garruss A. S., Liang K., Takahashi Y.-h., Mickey K., Voets O., Verrijzer C. P., Shilatifard A., Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 26, 2604–2620 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu D., Gao X., Morgan M. A., Herz H.-M., Smith E. R., Shilatifard A., The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol. 33, 4745–4754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao K., Collings C. K., Marshall S. A., Morgan M. A., Rendleman E. J., Wang L., Sze C. C., Sun T., Bartom E. T., Shilatifard A., SET1A/COMPASS and shadow enhancers in the regulation of homeotic gene expression. Genes Dev. 31, 787–801 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J.-E., Wang C., Xu S., Cho Y.-W., Wang L., Feng X., Baldridge A., Sartorelli V., Zhuang L., Peng W., Ge K., H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2, e01503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng S. B., Bigham A. W., Buckingham K. J., Hannibal M. C., McMillin M. J., Gildersleeve H. I., Beck A. E., Tabor H. K., Cooper G. M., Mefford H. C., Lee C., Turner E. H., Smith J. D., Rieder M. J., Yoshiura K., Matsumoto N., Ohta T., Niikawa N., Nickerson D. A., Bamshad M. J., Shendure J., Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42, 790–793 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morin R. D., Mendez-Lago M., Mungall A. J., Goya R., Mungall K. L., Corbett R. D., Johnson N. A., Severson T. M., Chiu R., Field M., Jackman S., Krzywinski M., Scott D. W., Trinh D. L., Tamura-Wells J., Li S., Firme M. R., Rogic S., Griffith M., Chan S., Yakovenko O., Meyer I. M., Zhao E. Y., Smailus D., Moksa M., Chittaranjan S., Rimsza L., Brooks-Wilson A., Spinelli J. J., Ben-Neriah S., Meissner B., Woolcock B., Boyle M., McDonald H., Tam A., Zhao Y., Delaney A., Zeng T., Tse K., Butterfield Y., Birol I., Holt R., Schein J., Horsman D. E., Moore R., Jones S. J. M., Connors J. M., Hirst M., Gascoyne R. D., Marra M. A., Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476, 298–303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasqualucci L., Trifonov V., Fabbri G., Ma J., Rossi D., Chiarenza A., Wells V. A., Grunn A., Messina M., Elliot O., Chan J., Bhagat G., Chadburn A., Gaidano G., Mullighan C. G., Rabadan R., Dalla-Favera R., Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 43, 830–837 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sze C. C., Shilatifard A., MLL3/MLL4/COMPASS Family on Epigenetic Regulation of Enhancer Function and Cancer. Cold Spring Harb. Perspect. Med. 6, a026427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan J., Ong C. K., Lim W. K., Ng C. C., Thike A. A., Ng L. M., Rajasegaran V., Myint S. S., Nagarajan S., Thangaraju S., Dey S., Nasir N. D., Wijaya G. C., Lim J. Q., Huang D., Li Z., Wong B. H., Chan J. Y., McPherson J. R., Cutcutache I., Poore G., Tay S. T., Tan W. J., Putti T. C., Ahmad B. S., Iau P., Chan C. W., Tang A. P. H., Yong W. S., Madhukumar P., Ho G. H., Tan V. K., Wong C. Y., Hartman M., Ong K. W., Tan B. K. T., Rozen S. G., Tan P., Tan P. H., Teh B. T., Genomic landscapes of breast fibroepithelial tumors. Nat. Genet. 47, 1341–1345 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Gui Y., Guo G., Huang Y., Hu X., Tang A., Gao S., Wu R., Chen C., Li X., Zhou L., He M., Li Z., Sun X., Jia W., Chen J., Yang S., Zhou F., Zhao X., Wan S., Ye R., Liang C., Liu Z., Huang P., Liu C., Jiang H., Wang Y., Zheng H., Sun L., Liu X., Jiang Z., Feng D., Chen J., Wu S., Zou J., Zhang Z., Yang R., Zhao J., Xu C., Yin W., Guan Z., Ye J., Zhang H., Li J., Kristiansen K., Nickerson M. L., Theodorescu D., Li Y., Zhang X., Li S., Wang J., Yang H., Wang J., Cai Z., Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 43, 875–878 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ortega-Molina A., Boss I. W., Canela A., Pan H., Jiang Y., Zhao C., Jiang M., Hu D., Agirre X., Niesvizky I., Lee J.-E., Chen H. T., Ennishi D., Scott D. W., Mottok A., Hother C., Liu S., Cao X.-J., Tam W., Shaknovich R., Garcia B. A., Gascoyne R. D., Ge K., Shilatifard A., Elemento O., Nussenzweig A., Melnick A. M., Wendel H.-G., The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 21, 1199–1208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J., Dominguez-Sola D., Hussein S., Lee J. E., Holmes A. B., Bansal M., Vlasevska S., Mo T., Tang H., Basso K., Ge K., Dalla-Favera R., Pasqualucci L., Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med. 21, 1190–1198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dorighi K. M., Swigut T., Henriques T., Bhanu N. V., Scruggs B. S., Nady N., Still C. D. II, Garcia B. A., Adelman K., Wysocka J., Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol. Cell 66, 568–576.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rickels R., Herz H.-M., Sze C. C., Cao K., Morgan M. A., Collings C. K., Gause M., Takahashi Y.-h., Wang L., Rendleman E. J., Marshall S. A., Krueger A., Bartom E. T., Piunti A., Smith E. R., Abshiru N. A., Kelleher N. L., Dorsett D., Shilatifard A., Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet. 49, 1647–1653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ying Q.-L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A., The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Los Angeles A., Ferrari F., Xi R., Fujiwara Y., Benvenisty N., Deng H., Hochedlinger K., Jaenisch R., Lee S., Leitch H. G., Lensch M. W., Lujan E., Pei D., Rossant J., Wernig M., Park P. J., Daley G. Q., Hallmarks of pluripotency. Nature 525, 469–478 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Hayashi K., Ohta H., Kurimoto K., Aramaki S., Saitou M., Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146, 519–532 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Brons I. G., Smithers L. E., Trotter M. W. B., Rugg-Gunn P., Sun B., Chuva de Sousa Lopes S. M., Howlett S. K., Clarkson A., Ahrlund-Richter L., Pedersen R. A., Vallier L., Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Buecker C., Srinivasan R., Wu Z., Calo E., Acampora D., Faial T., Simeone A., Tan M., Swigut T., Wysocka J., Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell 14, 838–853 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tesar P. J., Chenoweth J. G., Brook F. A., Davies T. J., Evans E. P., Mack D. L., Gardner R. L., McKay R. D., New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Wang C., Lee J.-E., Lai B., Macfarlan T. S., Xu S., Zhuang L., Liu C., Peng W., Ge K., Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci. U.S.A. 113, 11871–11876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whyte W. A., Orlando D. A., Hnisz D., Abraham B. J., Lin C. Y., Kagey M. H., Rahl P. B., Lee T. I., Young R. A., Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piunti A., Shilatifard A., Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352, aad9780 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Shi Y., Lan F., Matson C., Mulligan P., Whetstine J. R., Cole P. A., Casero R. A., Shi Y., Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Whyte W. A., Bilodeau S., Orlando D. A., Hoke H. A., Frampton G. M., Foster C. T., Cowley S. M., Young R. A., Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foster C. T., Dovey O. M., Lezina L., Luo J. L., Gant T. W., Barlev N., Bradley A., Cowley S. M., Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol. Cell. Biol. 30, 4851–4863 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinberger L., Ayyash M., Novershtern N., Hanna J. H., Dynamic stem cell states: Naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 17, 155–169 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Arnold S. J., Robertson E. J., Making a commitment: Cell lineage allocation and axis patterning in the early mouse embryo. Nat. Rev. Mol. Cell Biol. 10, 91–103 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Bienz M., The PHD finger, a nuclear protein-interaction domain. Trends Biochem. Sci. 31, 35–40 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Sanchez R., Zhou M. M., The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 36, 364–372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L., Collings C. K., Zhao Z., Cozzolino K. A., Ma Q., Liang K., Marshall S. A., Sze C. C., Hashizume R., Savas J. N., Shilatifard A., A cytoplasmic COMPASS is necessary for cell survival and triple-negative breast cancer pathogenesis by regulating metabolism. Genes Dev. 31, 2056–2066 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eskeland R., Leeb M., Grimes G. R., Kress C., Boyle S., Sproul D., Gilbert N., Fan Y., Skoultchi A. I., Wutz A., Bickmore W. A., Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell 38, 452–464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Illingworth R. S., Moffat M., Mann A. R., Read D., Hunter C. J., Pradeepa M. M., Adams I. R., Bickmore W. A., The E3 ubiquitin ligase activity of RING1B is not essential for early mouse development. Genes Dev. 29, 1897–1902 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voncken J. W., Roelen B. A., Roefs M., de Vries S., Verhoeven E., Marino S., Deschamps J., van Lohuizen M., Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc. Natl. Acad. Sci. U.S.A. 100, 2468–2473 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang S.-P., Tang Z., Chen C.-W., Shimada M., Koche R. P., Wang L.-H., Nakadai T., Chramiec A., Krivtsov A. V., Armstrong S. A., Roeder R. G., A UTX-MLL4-p300 transcriptional regulatory network coordinately shapes active enhancer landscapes for eliciting transcription. Mol. Cell 67, 308–321 e306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi Y.-J., Matson C., Lan F., Iwase S., Baba T., Shi Y., Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 19, 857–864 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Shi Y., Sawada J., Sui G., Affar E. B., Whetstine J. R., Lan F., Ogawa H., Luke M. P.-S., Nakatani Y., Shi Y., Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Wang Y., Zhang H., Chen Y., Sun Y., Yang F., Yu W., Liang J., Sun L., Yang X., Shi L., Li R., Li Y., Zhang Y., Li Q., Yi X., Shang Y., LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138, 660–672 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Luo Z., Gao X., Lin C., Smith E. R., Marshall S. A., Swanson S. K., Florens L., Washburn M. P., Shilatifard A., Zic2 is an enhancer-binding factor required for embryonic stem cell specification. Mol. Cell 57, 685–694 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng J., Blum R., Bowman C., Hu D., Shilatifard A., Shen S., Dynlacht B. D., A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol. Cell 53, 979–992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morgan M. A. J., Rickels R. A., Collings C. K., He X., Cao K., Herz H. M., Cozzolino K. A., Abshiru N. A., Marshall S. A., Rendleman E. J., Sze C. C., Piunti A., Kelleher N. L., Savas J. N., Shilatifard A., A cryptic Tudor domain links BRWD2/PHIP to COMPASS-mediated histone H3K4 methylation. Genes Dev. 31, 2003–2014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rinaldi L., Datta D., Serrat J., Morey L., Solanas G., Avgustinova A., Blanco E., Pons J. I., Matallanas D., Von Kriegsheim A., Di Croce L., Benitah S. A., Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell 19, 491–501 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Dhar S. S., Lee S.-H., Kan P.-Y., Voigt P., Ma L., Shi X., Reinberg D., Lee M. G., Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 26, 2749–2762 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen L., Shao N., Liu X., Nestler E., ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saldanha A. J., Java Treeview—Extensible visualization of microarray data. Bioinformatics 20, 3246–3248 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaap8747/DC1
fig. S1. Characterization of Mll4KO ESCs.
fig. S2. Enhancer inactivation by Mll4 depletion.
fig. S3. Epigenetic balance between Lsd1 and Mll4/COMPASS.
fig. S4. Mll4 deletion impairs enhancer activation during pluripotency transition.
fig. S5. Lsd1 is indispensable for enhancer decommissioning in FBS/LIF-cultured Mll4KO cells.
fig. S6. Characterization of Mll4ΔNT ESCs.