

Abstract
Non-visual arrestins interact with hundreds of different G protein-coupled receptors (GPCRs). Here we show that by introducing mutations into elements that directly bind receptors, the specificity of arrestin-3 can be altered. Several mutations in the two parts of the central “crest” of the arrestin molecule, middle-loop and C-loop, enhanced or reduced arrestin-3 interactions with several GPCRs in receptor subtype and functional state-specific manner. For example, the Lys139Ile substitution in the middle-loop dramatically enhanced the binding to inactive M2 muscarinic receptor, so that agonist activation of the M2 did not further increase arrestin-3 binding. Thus, the Lys139Ile mutation made arrestin-3 essentially an activation-independent binding partner of M2, whereas its interactions with other receptors, including the β2-adrenergic receptor and the D1 and D2 dopamine receptors, retained normal activation dependence. In contrast, the Ala248Val mutation enhanced agonist-induced arrestin-3 binding to the β2-adrenergic and D2 dopamine receptors, while reducing its interaction with the D1 dopamine receptor. These mutations represent the first example of altering arrestin specificity via enhancement of the arrestin-receptor interactions rather than selective reduction of the binding to certain subtypes.
Keywords: Arrestin, GPCRs, Receptor specificity, Protein-protein interactions, Protein engineering
1. Introduction
Dysfunction of G protein-coupled receptor (GPCR) signaling plays an important role in the pathogenesis of numerous human diseases [1,2]. For example, gain-of-function mutations that result in excessive activity or elevated ligand production that over-stimulates normal receptors can each cause endocrinological disorders and malignant tumors [1,3,4]. Because receptor activity is crucial for normal cell function, it is tightly controlled by several regulatory mechanisms. Active receptors are phosphorylated by specific GPCR kinases, whereupon arrestin proteins bind active phosphoreceptors [5]. Conceivably, the introduction of strong negative regulators into pathological cells, such as enhanced arrestins that bind unphosphorylated GPCRs, can suppress excessive signaling and potentially correct these problems.
Wild type (WT) arrestins bind active phosphorylated GPCRs [6,7], thereby terminating G protein signaling [8], and facilitating receptor internalization [9]. Thus, enhanced arrestins that do not require receptor phosphorylation can potentially be used in compensational therapy aiming at suppression of excessive G protein-mediated signaling [10,11]. Vertebrates have> 800 GPCRs [12,13] and only four different arrestins [14]. Arrestin-1 and -4 are expressed in the visual system [15], whereas arrestin-2 and -3 (also known as β-arrestin1 and 2) are ubiquitously expressed, and each regulates hundreds of receptors. Previous work established that arrestins can be “pre-activated” by mutations destabilizing their basal conformation, thereby increasing their binding to GPCRs [16–18]. Enhanced arrestin-1 was shown to partially compensate for defects of rhodopsin phosphorylation in vivo [11]. Similarly, enhanced non-visual arrestins could reduce the signaling of hyperactive GPCRs. But as arrestin-2 and -3 are fairly promiscuous [19], enhanced non-visual arrestins would likely have unwanted side effects as they would simultaneously affect the function of all receptors expressed in the same cell. Thus, to make enhanced non-visual arrestins useful as therapeutic agents, their receptor specificity must be dramatically increased.
The feasibility of engineering arrestin-2 and -3 with enhanced receptor selectivity is suggested by strict selectivity of visual arrestin-1, exclusively expressed in photoreceptor cells, for photopigments. Arrestin-1 binds to phosphorylated light-activated (P-Rh*) rhodopsin much better than other receptors [20,21]. Earlier studies identified elements [21] and individual residues on the receptor-binding surface [22] responsible for receptor preference of arrestins. The exchange of ten non-identical residues on the receptor-binding surface can change arrestin-1 preference to that of arrestin-2, and vice versa [22]. Point mutations at these positions were shown to increase the preference of arrestin-3 for some cognate receptors over others up to 4-fold, whereas double mutations yielded 50-fold differential in the binding to M2 muscarinic over β2-adrenergic receptor [23]. Comparison of crystal structures [24,25] and primary sequences of arrestin-1 and -2 additionally identifies a key valine residue in arrestin-1 that stabilizes its N-domain, and likely contributes to its high selectivity for rhodopsin, as demonstrated by a dramatic increase in the binding to non-cognate M2 muscarinic receptor of arrestin-1 with a single Val83Ser mutation [24]. However, mutations of the ten “receptor-discriminator” residues identified in the arrestin-1/arrestin-2 chimeras did not affect arrestin-3 binding to some of the GPCRs tested, including the D1 dopamine [23] or Y1 neuropeptide [26] receptors, while many of the same mutations dramatically change arrestin-3 binding to D2 dopamine and Y2 neuropeptide receptors. Here we took advantage of the recent crystal structure of arrestin-1 in complex with rhodopsin [27], which identified additional elements, including the middle-loop and C-loop, both of which engage the receptor and therefore might play a role in arrestin selectivity. We used four model GPCRs (M2 muscarinic, β2-adrenergic, D1 dopaminergic, and D2 dopaminergic) and show that mutations in these regions of arrestin-3 differentially affect its basal and agonist-induced binding.
2. Material and methods
2.1. Materials
Restriction endonucleases and other DNA modifying enzymes were from New England Biolabs (Ipswich, MA). Cell culture reagents and media were from Mediatech-Corning (Manassas, VA), Life-technologies (Carlsbad, CA), or PAA Laboratories GmbH (Pasching, Austria). Luciferase substrate coelenterazine h was from NanoLight Technology (Pinetop, AZ). DNA purification kits were from Zymo Research (Irvine, CA). All other reagents were from Amresco (Solon, OH) or Sigma-Aldrich (St Louis, MO).
2.2. Mutagenesis and plasmid construction
Plasmids encoding short splice variant of bovine arrestin-3 [28] with unique restriction sites introduced by silent mutations were used to introduce mutations on the background of Ala87Val base mutant (which is expected to be predisposed to higher receptor selectivity), as described [23,26]. To generate all mutants, oligonucleotides harboring the desired substitutions were used as forward primers and an oligonucleotide downstream from the far restriction site was used as a reverse primer for PCR. Resulting fragments of various lengths and an appropriate primer upstream of the near restriction site were then used as reverse and forward primers, respectively, for the second round of PCR. Restriction sites BamHI and BsiWI were used to introduce indicated Lys139 substitutions and BsiWI and XbaI to introduce mutations in the C-domain. The resulting fragments were purified, digested with the respective enzymes, and subcloned into the suitably digested pGEM-2 plasmid (Promega; Madison, WI) encoding WT bovine arrestin-3 with engineered restriction sites [23].
2.3. Direct in vitro binding assay
In vitro transcription, translation, preparation of light-activated phosphorylated rhodopsin (P-Rh*), and in vitro direct binding assay were performed, as previously described [22,29].
2.4. Bioluminescence resonance energy transfer (BRET) assay
COS-7 cells were transfected using Lipofectamine 2000 (2 μL Lipofectamine/1 μg DNA) in 6 well plates. We have previously determined the amounts of plasmid DNA to produce sufficient excess of arrestin-3 over receptor and therefore saturate the BRET signal [23,30]. 48 h after transfection, the appropriate ligands (agonist carbachol (carbamoylcholine) and inverse agonist atropine for M2R, agonists isoproterenol for β2AR, dopamine for D1R, and quinpirole for D2R) were used at 10 μM. After stimulation, 5 μM luciferase substrate coelenterazine h was added, and BRET was measured between RLuc8-tagged receptors, and Venus-tagged arrestins, as previously described [23,26,30]. BRET measurements for Fig. 5 were performed with Varioskan Flash Multimode Reader (Thermo Scientific, Waltham, MA).
Fig. 5.
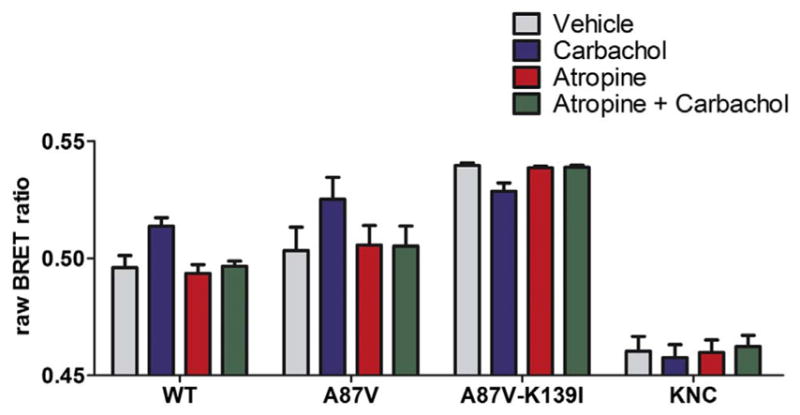
Lys139Ile mutant binds inactive M2R. COS-7 cells co-expressing M2R-RLuc8 and indicated form of Venus-arrestin-3. Cells were pretreated for 5 min with vehicle or 10 μM inverse agonist atropine [50], and stimulated with carbamylcholine as in Fig. 2. Raw BRET ratios are shown. Basal arrestin-3 binding was not altered by atropine treatment. *, p < 0.05 (vs. vehicle control), analyzed with one-way repeated measures ANOVA, followed by Dunnett’s post-hoc test. Atropine prevented the effects of carbachol. #, p < 0.05 (significant interaction between the two treatments), analyzed with two-way repeated measures ANOVA, followed by Bonferroni post-hoc test.
2.5. Co-immunoprecipitation
HEK293 arrestin-2/3 KO cells [31] were co-transfected with HA-M2R-RLuc8 (0.5 μg) and a Venus-Arrestin construct (0.25–1 μg) using Trans-Hi™ DNA transfection reagent (1 μg DNA: 3 μL reagent) in 6-well plates. At 48 h post-transfection, the cells were incubated in either serum-free DMEM or serum-free DMEM with 10 μM carbachol (carbamoylcholine) for 15 min. After stimulation, the cells were lysed in IP buffer (50 mM Tris-HCl pH 7.5, 2 mM EDTA, 250 mM NaCl, 10% glycerol, 0.5% NP-40, 20 mM NaF, and 1 mM NaVO3) and incubated at 4 °C for 1 h. The lysates were then centrifuged (max speed, 15 min) and the supernatants were pre-cleared by incubation for 1 h with 25 μL protein G agarose beads. The beads were pelleted by centrifugation, a 30 μL aliquot was removed for input analysis, and the remaining lysates were incubated overnight at 4 °C with 1 μg of anti-HA antibody (Sigma-Aldrich, 3F10). The next morning 50 μL protein G agarose beads were added and incubated for an additional 2 h. The beads were then washed three times with 1 mL of ice-cold IP buffer before elution with 35 μL of SDS sample buffer (Sigma-Aldrich, Laemmli 2×) at room temperature. Samples were analyzed by Western blot. Co-immunoprecipitated arrestins were quantified on blots using the QuantityOne Software (BioRad). The results from three independent experiments were statistically analyzed using one-way ANOVA followed by Dunnett’s post-hoc test (p < 0.05 was considered significant).
2.6. Data analysis and statistics
GraphPad Prism 5.0 was used to plot the results of BRET experiments (Figs. 2–3), direct binding assay (Fig. 4), and to statistically analyze them, as described [23]. Protein sequences for multiple sequence alignments were taken from OMA database [32]. To characterize the level of residue conservation, entropy was calculated for each amino acid position.
Fig. 2.

Basal and agonist-induced binding of Lys139 mutants to GPCRs. A. Basal (agonist-independent) association of the indicated luciferase-tagged receptor and wild type or mutant Venus-Arrestin-3 in COS-7 cells at 10 min of vehicle treatment. Nonspecific (bystander) BRET was measured using non-receptor-binding arrestin-3 mutant (Arrestin3-KNC [23,30] ). Net BRET ratio was calculated by subtracting non-specific BRET from the raw BRET data in each experiment. B. The agonist-induced arrestin-3 recruitment to indicated receptors. The BRET change (ΔBRET) was determined by the difference between the BRET ratio between ligand and vehicle treated cells (nonspecific BRET was subtracted from both). Means ± S.E. of at least three independent experiments are shown. Each experiment was performed in quadruplicate. Statistical significance was determined by one-way ANOVA, followed by Dunnett’s post-hoc test: *, p < 0.05; **, p < 0.01; ***, p < 0.001, as compared with A87V base mutant binding to each receptor.
Fig. 3.

The effect of C-loop mutations on basal and agonist-induced receptor binding. A. Basal BRET (net BRET ratio) and B. agonist-induced BRET change (delta BRET) between indicated luciferase-tagged receptors and Venus-tagged arrestin-3C-loop mutants was calculated, as described in the legend to Fig. 2. The cells were stimulated for 10 min with the appropriate agonist. BRET ratio obtained with negative control (arrestin-3 KNC [26,83] ) was subtracted. Means ± S.E. of at least three independent experiments are shown. Each experiment was performed in quadruplicate. Statistical significance was determined by one-way ANOVA: *, p < 0.05; **, p < 0.01; ***, p < 0.001, as compared with A87V base mutant binding to each receptor.
Fig. 4.

Direct binding of arrestin-3 mutants to light-activated phosphorylated rhodopsin. WT and mutant forms of arrestin-3 produced in cell-free translation in the presence of radiolabeled leucine (2 nM) were incubated with 0.3 μg of light-activated phosphorylated rhodopsin (P-Rh*) for 5 min at 37 °C. Free and rhodopsin-bound arrestin was separated by gel filtration on 2-ml Sepharose 2B–CL columns. The amount of bound arrestin eluting with rhodopsin-containing membranes was quantified by scintillation counting. Non-specific binding (in the absence of rhodopsin) was subtracted. Means ± S.D. of three independent experiments performed in duplicate are shown. The data were analyzed by one-way ANOVA with arrestin type as the main factor, followed by Dunnett’s post-hoc test.
3. Results
3.1. Highly conserved lysine in the middle-loop differentially affects basal receptor binding
All four vertebrate arrestins exhibit remarkable conservation of their structures, consisting of two “cup-like” domains [24,25,33–36] (Fig. 1A). Within this context, the receptor-binding surface of arrestins comprises the concave sides of the N- and the C-domains, as well as the central loops [22,37–41]. The “middle-loop” [42], also called “139-loop” in arrestin-1 [43,44], is a flexible region, connecting β-strands VIII and IX in the N-domain, forming part of the central crest on the receptor interaction surface (Fig. 1A, blue). It has been reported that the middle-loop plays an important role in the high selectivity of visual arrestin-1 for P-Rh* [43,45], but its functional role in non-visual arrestins has not been reported in the literature.
Fig. 1.

Structure and sequence of the middle and C-loops of arrestins. A. Crystal structure of rhodopsin-bound arrestin-1 (Protein Data Bank entry 4ZWJ [27] ). The middle and C-loops are shown in blue and pink, respectively. Residues mutated in this study are shown as CPK models. B. Multiple sequence alignment of arrestin-1 (Arr1), arrestin-3 (Arr3) and arrestin homologs (Arr) from invertebrate species. Residues mutated in this study are highlighted. Bar graph under the alignment shows the extent of residue conservation at each position. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Multiple sequence alignment of the middle-loop region shows that lysine139 (in bovine arrestin-3) is highly conserved among all four arrestin subtypes in vertebrates (Fig. 1B). However, various residues (Ile, Ala, Asp, Gln, Arg) are found in invertebrate arrestins in position homologous to Lys139 in arrestin-3 (Fig. 1B; see also [14] ). Available structures suggest that a putative salt bridge involving this conserved lysine anchors the middle-loop to the receptor-binding “finger loop” in the basal state of vertebrate arrestins [25,45]. To test the role of lysine139 in arrestin-3, we substituted it with residues found in the equivalent position in non-vertebrate species. The wide variety of homologues provides interesting substitutions: uncharged (Gln, Ile, Ala), acidic (Asp), and other positively charged (Arg) residues have all appeared at the corresponding position during evolution, among which Ile and Ala represent the least conservative substitutions according to substitution matrices [46].
To measure arrestin interactions with GPCRs co-expressed in the same cell, we used bioluminescence resonance energy transfer (BRET) between receptors C-terminally tagged with Renilla luciferase and arrestins N-terminally tagged with Venus, as described [10,23,26,30]. The middle-loop mutations were introduced in arrestin-3 carrying an Ala87Val mutation, which is expected to increase its receptor selectivity [23]. This Val residue is involved in multiple hydrophobic interactions in arrestin-1, and thus responsible for the rigidity of the N-domain, which is proposed to ensure selective rhodopsin binding. The Ala87Val base mutant was used because it was expected to predispose arrestin-3 to higher receptor selectivity, by making its N-domain less flexible. As a negative control, we used the arrestin-3-KNC mutant, in which 12 key receptor-binding residues are replaced with alanines. Arrestin-3-KNC does not demonstrate basal or agonist-induced binding to any GPCR tested so far [23,26,30]. It is the most appropriate control for bystander BRET, as it has the same molecular weight and therefore is expected to diffuse at the same rate as other arrestin-3 variants.
We tested several Class A GPCRs: β2-adrenergic (β2AR), D1 (D1R) and D2 (D2R) dopamine, and M2 muscarinic (M2R) receptors. In the case of visual arrestin-1, certain mutations in the middle-loop “loosened up” the basal state of the molecule and facilitated arrestin-1 binding to P-Rh* and non-preferred forms of rhodopsin, light-activated unphosphorylated (Rh*) and inactive phosphorylated (P–Rh) [45,47]. Therefore, we first investigated possible “pre-docking” of the arrestin-3 middle-loop mutants to the receptors without agonist stimulation (Fig. 2A). In the case of β2AR, Lys139 substitutions did not significantly affect basal arrestin binding, whereas we observed receptor-specific changes with the other three GPCRs. The most dramatic were the effects of Lys139Ile substitution, which increased pre-docking to M2R> 3-fold, while decreasing basal interactions with D1R and D2R (Fig. 2A). Interestingly, while none of the other Lys139 substitutions affected basal binding of arrestin-3 to M2R, D2R, or β2AR, most of them reduced basal binding to D1R to varying degrees (Fig. 2A).
The effects of Lys139 substitutions on agonist-induced binding to the tested receptors were remarkably different. The Lys139Ile mutation, which greatly increased the binding to inactive M2, essentially eliminated agonist-induced increase in binding to this receptor (Fig. 2B). Apparently, the interaction between basal M2R and arrestin3-Lys139Ile mutant reached the maximum possible extent, so that the presence of M2 agonist could not further increase binding. None of the other Lys139 substitutions appreciably affected agonist-induced increase of M2 interaction. The conservative Lys139Arg substitution enhanced agonist-induced binding to the β2-adrenergic receptor, whereas other mutations did not significantly affect it. The interactions with activated D1R were not perturbed by Lys139 substitutions, whereas the effect of D2R agonist quinpirole was reduced by the Lys139Ile mutation (Fig. 2B).
3.2. The effects of the perturbation of arrestin-3C-loop
A recent crystal structure of the visual arrestin-1-rhodopsin complex revealed a direct interaction between the C-loop of arrestin-1 (Fig. 1A, pink) and the receptor [27,48]. The C-loop connects two β-strands in the C domain of all arrestins, and is found in close proximity to the middle-loop in the basal conformation [24,25,33,34]. To test the role of this loop in receptor binding, we generated single mutations in the C-loop at three selected positions: Ser246, A248 and Q249. These residues were replaced with residues in homologous positions of invertebrate arrestins [14] in the context of the Ala87Val arrestin-3.
Gln249 is the least conservative position of the C-loop (Fig. 1B). While Gln dominates in non-visual arrestins, Tyr and Phe are found at the corresponding place in visual arrestin-1. The Gln249His substitution somewhat increased the basal binding to β2AR and D1R, but the increase of BRET signal upon isoproterenol or dopamine stimulation were not different from WT arrestin-3.
The replacement of Ala248 with a larger hydrophobic residue Val, yielded enhanced receptor binding upon activation in case of two class A receptors out of four tested (Fig. 3): β2AR and D2R both bound Ala248Val mutant better than WT arrestin-3 or base mutant Ala87Val. However, the basal arrestin-3 binding to these receptors was not affected by Ala248 mutation. The Ala248Val substitution reduced arrestin-3 interaction with inactive D1R.
Interestingly, neither glutamate nor glutamine substitution of Ser246 in the same loop where Ala248 and Gln249 are localized affected the binding to the four receptors tested (Fig. 3).
3.3. Middle and C-loop of arrestin-3 participate in rhodopsin binding
The crystal structure of the rhodopsin-arrestin-1 complex identifies global conformational changes in receptor-bound arrestin-1 as compared to basal arrestin-1, including an increase in the distance between the middle-loop and C-loop. This particular conformational rearrangement forms cleft that allows binding of the second intracellular loop of rhodopsin [27]. Arrestin-3 is not a cognate interaction partner of rhodopsin, but can bind to P-Rh*, although it yields lower binding than arrestin-1 [17,20]. To test whether the same interaction interface drives non-visual arrestin binding to rhodopsin, we incorporated radiolabeled leucine into untagged arrestin-3 mutants [29,49] and performed in vitro binding to rhodopsin [17,49]. We found that the Lys139Ile mutation of arrestin-3 significantly reduced P-Rh* binding, whereas arrestin-3 harboring other substitutions exhibited binding that was not statistically different from WT (Fig. 4). Intriguingly, a recent report indicates that when the analogous residue in mouse arrestin-1 (Lys 142) is substituted, the conservative Lys142Arg and, to a lesser extent, Lys142His mutation enhanced binding to both P-Rh*, and Rh*, whereas alanine and glutamine substitutions did not affect the binding [45]. Our test of arrestin-3 mutants yielded similar results (Fig. 4). Among C-loop mutations, Ala248Val enhanced arrestin-3 binding, whereas the others did not (Fig. 4).
3.4. Agonist-independent receptor recruitment of arrestin-3-Lys139Ile mutant
Muscarinic acetylcholine receptors display measurable constitutive activity in heterologous expression systems [50]. This raises the possibility that the high basal binding of arrestin-3-Lys139Ile is due to its interaction with a spontaneously active pool of M2 receptors. Therefore, we tested whether the mutant binds the truly inactive receptors, by incubating cells with atropine, a known inverse agonist of the M2 receptor [50]. We found that atropine pretreatment did not decrease the binding of arrestin-3-Lys139Ile, and the agonist-independent binding of the WT arrestin-3 or the A87V base-mutant were not altered, either (Fig. 5). As expected, atropine pretreatment prevented all carbachol-induced BRET changes. These data support the conclusion that the Lys139Ile mutant binds the inactive conformation of M2R.
As high agonist-independent binding of arrestin-3-Lys139Ile mutant (Fig. 2) is fairly unusual, we tested it using co-immunoprecipitation as an independent method (Fig. 6). To this end, we transfected HEK293 cells made deficient for both non-visual arrestins using CRIPR/Cas9 [31] to exclude endogenous arrestin competition, with the same HA-M2R-RLuc8 and Venus-arrestin-3-Lys139Ile constructs used for BRET (Fig. 2), immunoprecipitated the receptor with anti-HA antibody, and blotted for Venus. In these experiments we used WT arrestin-3 as a positive control and arrestin-3-KNC that does not bind receptors [23,26,30], as a negative control (Fig. 5). We found that arrestin-3-Lys139Ile binds M2 muscarinic receptors in the absence of agonists, and the addition of saturating agonist 10 μM carbamylcholine does not further increase its binding (Fig. 5). In full agreement with BRET data (Fig. 2), if anything, the addition of the agonist slightly suppresses the interaction of this mutant with M2R, although in neither case this trend rose to statistical significance (Fig. 5).
Fig. 6.

Co-immunoprecipitation of Venus-arrestin constructs with M2R. HEK arrestin-2/3 KO cells [31] were co-transfected with Venus-arrestin constructs and HA-M2R-RLuc8. After 48 h, the cells were incubated in either serum-free media alone, or serum-free media with 10 μM carbamylcholine for 15 min. The cells were lysed in IP buffer (50 mM Tris-HCl pH 7.5, 2 mM EDTA, 250 mM NaCl, 10% glycerol, 0.5% NP-40, 20 mM NaF, and 1 mM NaVO3) and the supernatant was cleared by centrifugation at max speed for 15 min, then pre-cleared with protein G agarose. The supernatant was immunoprecipitated using a rat HA antibody against HA-M2R-Rluc8. The input (IB) and immunoprecipitated material (IP) were subjected to Western blotting to detect HA and GFP, as indicated. The GFP blot of the immunoprecipitated samples was analyzed using Versadoc. The results were statistically analyzed using one-way ANOVA followed by Dunnett’s post-hoc test. The difference with KNC is shown (*, p < 0.05, n = 3). The effect of agonist treatment on Venus-arrestin-3 (K139I) was not statistically significant (n.s.).
4. Discussion
Arrestins preferentially bind active phosphorylated GPCRs, precluding their coupling to G proteins, facilitating receptor internalization, and/or facilitating G protein-independent signaling [51–53]. Vertebrates express only four arrestin subtypes [14], as compared to hundreds of different GPCRs [12,13]. Arrestin-1 and arrestin-4 are specialized visual proteins, and are expressed at fairly high levels in photoreceptor cells, where they bind photopigments [15,54,55]. The two non-visual subtypes, arrestin-2 and -3, are ubiquitously expressed and bind hundreds of different GPCRs [14,19,51,56–59]. Non-visual arrestins also interact with numerous non-receptor signaling proteins [60], orchestrating a second, G protein-independent wave of GPCR signaling [14,61,62].
Arrestin-3 is the most promiscuous of the two non-visual subtypes: it interacts with a wide variety of GPCRs [19,34,59]. Previously, by manipulating the residues on the receptor-binding surface that determine receptor specificity of arrestins [21,22], we constructed arrestin-3 mutants with up to 50-fold preference for some GPCR subtypes over others [23]. In the first set of point mutants the effects of substitutions on agonist-independent “pre-docking” and agonist-induced increase in arrestin binding to β2AR, M2R, D1R and D2R appeared to correlate [23]. More extensive mutagenesis of the same regions yielded variants of arrestin-3 that showed virtually no “pre-docking”, but normally responded to agonist challenge of neuropeptide Y receptors Y1 and Y2 [26]. None of the mutations tested affected arrestin-3 interactions with D1R and Y1 receptor significantly [23,26], suggesting that additional residues might be involved. The crystal structure of the arrestin-1-rhodopsin complex [27] revealed direct contacts between rhodopsin and two loops in the central “crest” of the arrestin-1 molecule (Fig. 1A), the middle loop [42] and the C-loop [27].
Pre-activating mutations destabilize the basal conformation of arrestin, suggesting that fully active arrestin conformation can only be stable in its receptor-bound state. The first crystal structure of pre-activated arrestin-1 in complex with a constitutively active rhodopsin mutant has recently been solved [27,48], revealing new aspects of the arrestin-receptor interaction. The structure showed that several distinct contact surfaces mediate arrestin-1 binding to rhodopsin [27]. One of the critical interaction sites in the centre of arrestin molecule is created by the middle and the C-loop, coming from the N- and C-domain, respectively. Receptor binding is accompanied by global conformational changes in arrestin, including ~20° rotation between the N- and C-domain [27,42,62].
The middle loop is highly flexible both in free arrestin-1 [25,35] and in the rhodopsin complex [27]. Intramolecular distance measurements in arrestin-1 revealed large movement of the middle loop [47]. Deletions in this element (called “139-loop” in arrestin-1) enhanced arrestin-1 binding to phosphorylated, light activated rhodopsin (P-Rh*) and non-preferred forms, light-activated unphosphorylated, inactive phosphorylated rhodopsin, and phospho-opsin [45,47]. Perturbations in this region also dramatically reduced thermal stability of the protein [45]. These results suggest that the presence of this loop contributes to the high selectivity of arrestin-1 for P-Rh* and stabilizes the basal conformation. Structural data also suggest the existence in the basal state of a salt bridge between Lys142 in the middle-loop and Asp72 in the finger loop (residue numbers correspond to bovine arrestin-1), which might contribute to the stability of the basal conformation.
Here we tested nine mutations in the middle and C-loops of arrestin-3 (Figs. 2–4). The results are consistent with the hypothesis that these elements directly participate in the binding to non-phosphorylated parts of GPCRs, in agreement with the structure of the arrestin-1-rhodopsin complex, where these elements directly contact the receptor [27]. Various middle loop and C-loop mutations increase (Lys139Arg, Ala248Val) or decrease (Lys139Ile) GPCR binding in receptor-specific manner (Figs. 2–4). Based on the current model of arrestin activation [6,63], arrestin binding to the receptor can be increased via two distinct mechanisms: destabilization of the basal conformation [11,45,47,64–66] or changing the residues that contact the receptor [38,67]. The mutations of the first type tend to increase the binding to all GPCRs indiscriminately [17,18]. Since the effects of the mutations tested here are receptor subtype-specific (Figs. 2–4), it is likely that targeted residues are in the arrestin-receptor interface, similar to previously characterized mutants [23,26].
However, there is a difference. All earlier constructed mutants with enhanced receptor specificity demonstrated lower than WT arrestin-3 binding to some receptors. The specificity was achieved by large reduction of binding to some GPCRs with minimal to no reduction of the interactions with others [23,26]. Several mutants tested here demonstrate an increase in binding to certain GPCRs: Lys139Ile dramatically enhances the binding to inactive M2R, to the level where agonist stimulation of this receptor does not produce any effect (Fig. 2). Interestingly, the same mutation reduces arrestin-3 binding to D1 and D2 dopaminergic receptors (Figs. 2,4). While none of the previously tested mutations appreciably affected arrestin-3 binding to D1R, one mutation in this series, Gln249His, enhanced it, whereas two others, Lys139Ile and Ala248Val, decreased it (Figs. 2,3). In fact, Ala248Val selectively reduced the binding to D1R, while increasing the binding to rhodopsin, D2R, and β2AR (Figs. 3,4). Thus, manipulation of these residues, which are likely engaged by the receptor directly, yielded arrestin-3 variants where receptor preference was achieved by selective increase in the binding to some GPCRs, rather than solely by receptor-specific reduction of the binding (Figs. 2,3,7). For the sake of clarity, we presented receptor preference of the biased arrestin mutants as radar charts, which show that some point mutations yield 2–5-fold selectivity toward certain receptors over others (Fig. 7). Importantly, the magnitude and direction of bias is strongly dependent on the activation state of the receptors (Fig. 7).
Fig. 7.

Receptor selectivity of arrestin-3 mutants. Each receptor pair is provided as radially arranged axis that starts from the centre. The relative binding of each arrestin-3 mutant was plotted along all axes, and the connected values create a polygon. The binding ratio of WT arrestin-3 was set at 1 for each receptor pair. If the relative binding is> 1, the indicated arrestin has a preference for the first member of the receptor pair (receptor1/receptor2). Panel A shows the basal arrestin interactions. The green regular hexagon indicates WT arrestin. Only those mutants are highlighted that have at least a 1.5-fold bias against one receptor. Panel B shows the selectivity of the arrestin3 mutants after ligand treatment. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Cellular signaling is regulated by a complex network of protein-protein interactions [68–70]. The majority of the therapeutically used agents are small molecules, which have limited ability to interact with flat or disordered protein elements mediating these interactions [71,72]. Manipulation of these interactions by changing expression levels of proteins involved affects every function of proteins in question, not the one function that needs to be targeted [73]. Custom-designed signaling proteins can overcome this limitation, and selectively enhance or disrupt individual protein-protein interactions [23,74–76]. Arrestins play a role in many critical cellular functions, including cell death and survival [14,61,77–79]. All of their effects are mediated through direct binding to other proteins, which gives arrestins exceptional potential in protein-based therapy [23]. The putative usefulness of an enhanced arrestin molecule in compensational gene therapy has already been proven with an enhanced form of arrestin-1, which prolonged photoreceptor survival and improved rod function in rhodopsin kinase-deficient mice [11]. Non-visual arrestins can be pre-activated by homologous mutations [17,18,80,81]. Enhanced versions of non-visual arrestins can quench hyperactive GPCRs in all cell types where excessive receptor signaling underlies disease state [1,2]. However, broad receptor specificity of non-visual arrestins does not allow specific targeting of the receptor of interest. Our data demonstrate that there are many ways of narrowing receptor specificity of even the most promiscuous non-visual subtype, arrestin-3, paving the way to the construction of arrestin mutants with therapeutic potential.
5. Conclusions
Here we show that the middle-loop (called 139-loop in arrestin-1) and the C-loop are important parts of the receptor-binding surface of arrestin-3, and that the manipulation of these elements yields subtype-selective changes of non-visual arrestin interactions with different GPCRs. One of the introduced mutations, Lys139Ile, has resulted in high-affinity basal (agonist-independent) binding of arrestin-3 to M2 muscarinic receptor, while reducing the agonist-induced interaction with D2 dopamine receptor. Subtype-specific increase in receptor binding described here can enhance the specificity of non-visual arrestins in a different way than in previous studies, where a decrease of arrestin-3 binding to certain receptor subtypes enhanced the receptor specificity of arrestin-3.
Acknowledgments
Funded by Hungarian National Research, Development and Innovation Fund (K 116954 to LH) and NIH grants GM109955 and GM077561 (VVG), GM129569 (TMI) and DA043680 (TMI/VVG).
6. Abbreviations
- arrestin-3 KNC
arrestin-3 Lys11Ala, Lys12Ala, Leu49Ala, Asp51Ala, Arg52Ala, Leu69Ala, Tyr239Ala, Asp241Ala, Cys252Ala, Pro253Ala, Asp260Ala, and Gln262Ala
- BRET
Bioluminescence Resonance Energy Transfer
- GPCR
G protein-coupled receptor
- β2AR
β2-adrenergic receptor
- D1R, D2R
D1 dopamine receptor, D2 dopamine receptor
- M2R
M2 muscarinic receptor
References
- 1.Schoneberg T, Schulz A, Biebermann H, Hermsdorf T, Rompler H, Sangkuhl K. Pharmacol Ther. 2004;104:173–206. doi: 10.1016/j.pharmthera.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 2.Stoy H, Gurevich VV. Genes Dis. 2015;2:108–132. doi: 10.1016/j.gendis.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russo D, Arturi F, Schlumberger M, Caillou B, Monier R, Filetti S, Suarez HG. Oncogene. 1995;11:1907–1911. [PubMed] [Google Scholar]
- 4.Hebrant A, van Staveren WC, Maenhaut C, Dumont JE, Leclere J. Eur J Endocrinol. 2011;164:1–9. doi: 10.1530/EJE-10-0775. [DOI] [PubMed] [Google Scholar]
- 5.Carman CV, Benovic JL. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/s0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- 6.Gurevich VV, Gurevich EV. Trends Pharmacol Sci. 2004;25:105–111. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Kuhn H, Hall SW, Wilden U. FEBS Lett. 1984;176:473–478. doi: 10.1016/0014-5793(84)81221-1. [DOI] [PubMed] [Google Scholar]
- 8.Wilden U, Hall SW, Kühn H. Proc Natl Acad Sci. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim YM, Benovic JL. J Biol Chem. 2002;277:30760–30768. doi: 10.1074/jbc.M204528200. [DOI] [PubMed] [Google Scholar]
- 10.Gimenez LE, Vishnivetskiy SA, Gurevich VV. Handb Exp Pharmacol. 2014;219:153–170. doi: 10.1007/978-3-642-41199-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song X, Vishnivetskiy SA, Gross OP, Emelianoff K, Mendez A, Chen J, Gurevich EV, Burns ME, Gurevich VV. Curr Biol. 2009;19:700–705. doi: 10.1016/j.cub.2009.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 13.Bockaert J, Pin JP. EMBO J. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurevich EV, Gurevich VV. Genome Biol. 2006;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikonov SS, Brown BM, Davis JA, Zuniga FI, Bragin A, Pugh EN, Jr, Craft CM. Neuron. 2008;59:462–474. doi: 10.1016/j.neuron.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gurevich VV. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- 17.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 18.Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. J Biol Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 19.Barak LS, Ferguson SS, Zhang J, Caron MG. J Biol Chem. 1997;272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- 20.Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, Benovic JL. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 21.Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. J Biol Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 22.Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, Gurevich VV. J Biol Chem. 2011;286:24288–24299. doi: 10.1074/jbc.M110.213835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gimenez LE, Vishnivetskiy SA, Baameur F, Gurevich VV. J Biol Chem. 2012;287:29495–29505. doi: 10.1074/jbc.M112.366674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 25.Hirsch JA, Schubert C, Gurevich VV, Sigler PB. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 26.Gimenez LE, Babilon S, Wanka L, Beck-Sickinger AG, Gurevich VV. Cell Signal. 2014;26:1523–1531. doi: 10.1016/j.cellsig.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MH, Zhang C, Moeller A, West GM, Pascal BD, Van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino-Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy-Chowdhury S, Conrad CE, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Howe N, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JC, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov V, Melcher K, Xu HE. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sterne-Marr R, Gurevich VV, Goldsmith P, Bodine RC, Sanders C, Donoso LA, Benovic JL. J Biol Chem. 1993;268:15640–15648. [PubMed] [Google Scholar]
- 29.Gurevich VV, Benovic JL. J Biol Chem. 1992;267:21919–21923. [PubMed] [Google Scholar]
- 30.Gimenez LE, Kook S, Vishnivetskiy SA, Ahmed MR, Gurevich EV, Gurevich VV. J Biol Chem. 2012;287:9028–9040. doi: 10.1074/jbc.M111.311803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, Milligan G. J Biol Chem. 2016;291:27147–27159. doi: 10.1074/jbc.M116.754887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altenhoff AM, Skunca N, Glover N, Train CM, Sueki A, Pilizota I, Gori K, Tomiczek B, Muller S, Redestig H, Gonnet GH, Dessimoz C. Nucleic Acids Res. 2015;43:D240–D249. doi: 10.1093/nar/gku1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 34.Zhan X, Gimenez LE, Gurevich VV, Spiller BW. J Mol Biol. 2011;406:467–478. doi: 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Granzin J, Wilden U, Choe HW, Labahn J, Krafft B, Buldt G. Nature. 1998;391:918–921. doi: 10.1038/36147. [DOI] [PubMed] [Google Scholar]
- 36.Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. Biochemistry. 2002;41:3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- 37.Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Proc Natl Acad Sci U S A. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanson SM, Gurevich VV. J Biol Chem. 2006;281:3458–3462. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohguro H, Palczewski K, Walsh KA, Johnson RS. Protein Sci. 1994;3:2428–2434. doi: 10.1002/pro.5560031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pulvermuller A, Schroder K, Fischer T, Hofmann KP. J Biol Chem. 2000;275:37679–37685. doi: 10.1074/jbc.M006776200. [DOI] [PubMed] [Google Scholar]
- 41.Szczepek M, Beyriere F, Hofmann KP, Elgeti M, Kazmin R, Rose A, Bartl FJ, von Stetten D, Heck M, Sommer ME, Hildebrand PW, Scheerer P. Nat Commun. 2014;5:4801. doi: 10.1038/ncomms5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, Paduch M, Tripathi-Shukla P, Koide A, Koide S, Weis WI, Kossiakoff AA, Kobilka BK, Lefkowitz RJ. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim M, Vishnivetskiy SA, Van Eps N, Alexander NS, Cleghorn WM, Zhan X, Hanson SM, Morizumi T, Ernst OP, Meiler J, Gurevich VV, Hubbell WL. Proc Nat Acad Sci USA. 2012;109:18407–18412. doi: 10.1073/pnas.1216304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhuang T, Chen Q, Cho MK, Vishnivetskiy SA, Iverson TI, Gurevich VV, CRS Proc Nat Acad Sci USA. 2013;110:942–947. doi: 10.1073/pnas.1215176110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vishnivetskiy SA, Baameur F, Findley KR, Gurevich VV. J Biol Chem. 2013;288:11741–11750. doi: 10.1074/jbc.M113.450031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henikoff S, Henikoff JG. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim M, Vishnivetskiy SA, Van Eps N, Alexander NS, Cleghorn WM, Zhan X, Hanson SM, Morizumi T, Ernst OP, Meiler J, Gurevich VV, Hubbell WL. Proc Natl Acad Sci U S A. 2012;109:18407–18412. doi: 10.1073/pnas.1216304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou XE, Gao X, Barty A, Kang Y, He Y, Liu W, Ishchenko A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Suino-Powell KM, Boutet S, Williams GJ, Wang M, Li D, Caffrey M, Chapman HN, Spence JC, Fromme P, Weierstall U, Stevens RC, Cherezov V, Melcher K, Xu HE. Sci Data. 2016;3:160021. doi: 10.1038/sdata.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gurevich VV, Benovic JL. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 50.Nelson CP, Nahorski SR, Challiss RA. J Pharmacol Exp Ther. 2006;316:279–288. doi: 10.1124/jpet.105.094383. [DOI] [PubMed] [Google Scholar]
- 51.Gurevich VV, Gurevich EV. Pharmacol Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walther C, Ferguson SS. Prog Mol Biol Transl Sci. 2013;4(118):93–113. doi: 10.1016/B978-0-12-394440-5.00004-8. [DOI] [PubMed] [Google Scholar]
- 53.Luttrell LM, Gesty-Palmer D. Pharmacol Rev. 2010;62:305–330. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gurevich VV, Hanson SM, Song X, Vishnivetskiy SA, Gurevich EV. Prog Retin Eye Res. 2011;30:405–430. doi: 10.1016/j.preteyeres.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV. Neuroscience. 2011;174:37–49. doi: 10.1016/j.neuroscience.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 57.Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, Lefkowitz RJ. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- 58.Lohse MJ, Andexinger S, Pitcher J, Trukawinski S, Codina J, Faure JP, Caron MG, Lefkowitz RJ. J Biol Chem. 1992;267:8558–8564. [PubMed] [Google Scholar]
- 59.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 60.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, Lefkowitz RJ. Proc Natl Acad Sci U S A. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 62.Shenoy SK, Lefkowitz RJ. Trends Pharmacol Sci. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim YJ, Hofmann KP, Ernst OP, Scheerer P, Choe HW, Sommer ME. Nature. 2013;497:142–146. doi: 10.1038/nature12133. [DOI] [PubMed] [Google Scholar]
- 64.Gurevich VV, Gurevich EV. Curr Protoc Pharmacol. 2014;67 doi: 10.1002/0471141755.ph0210s67. 2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. J Biol Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- 66.Smith WC, Milam AH, Dugger D, Arendt A, Hargrave PA, Palczewski K. J Biol Chem. 1994;269:15407–15410. [PubMed] [Google Scholar]
- 67.Pulvermuller A, Maretzki D, Rudnicka-Nawrot M, Smith WC, Palczewski K, Hofmann KP. Biochemistry. 1997;36:9253–9260. doi: 10.1021/bi970772g. [DOI] [PubMed] [Google Scholar]
- 68.Vishnivetskiy SA, Chen Q, Palazzo MC, Brooks EK, Altenbach C, Iverson TM, Hubbell WL, Gurevich VV. J Biol Chem. 2013;288:11741–11750. doi: 10.1074/jbc.M112.445437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elowitz M, Lim WA. Nature. 2010;468:889–890. doi: 10.1038/468889a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lim WA. Nat Rev Mol Cell Biol. 2010;11:393–403. doi: 10.1038/nrm2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peisajovich SG, Garbarino JE, Wei P, Lim WA. Science. 2010;328:368–372. doi: 10.1126/science.1182376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gurevich EV, Gurevich VV. Handb Exp Pharmacol. 2014;219:1–12. doi: 10.1007/978-3-642-41199-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shoemaker BA, Portman JJ, Wolynes PG. Proc Natl Acad Sci U S A. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gurevich VV, Gurevich EV. Crit Rev Biochem Mol Biol. 2015;50:440–452. doi: 10.3109/10409238.2015.1067185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gurevich EV, Gurevich VV. Br J Pharmacol. 2015;172:3229–3241. doi: 10.1111/bph.13066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SSG, Caron MG, Barak LS. Proc Natl Acad Sci U S A. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meng D, Lynch MJ, Huston E, Beyermann M, Eichhorst J, Adams DR, Klusmann E, Houslay MD, Baillie GS. J Biol Chem. 2009;284:11425–11435. doi: 10.1074/jbc.M806395200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Song X, Seo J, Baameur F, Vishnivetskiy SA, Chen Q, Kook S, Kim M, Brooks EK, Altenbach C, Hong Y, Hanson SM, Palazzo MC, Chen J, Hubbell WL, Gurevich EV, Gurevich VV. Cell Signal. 2013;25:2613–2624. doi: 10.1016/j.cellsig.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kook S, Zhan X, Cleghorn WM, Benovic JL, Gurevich VV, Gurevich EV. Cell Death Differ. 2014;21:172–184. doi: 10.1038/cdd.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prossnitz ER. Life Sci. 2004;75:893–899. doi: 10.1016/j.lfs.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 81.Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- 82.Breitman M, Kook S, Gimenez LE, Lizama BN, Palazzo MC, Gurevich EV, Gurevich VV. J Biol Chem. 2012;287:19653–19664. doi: 10.1074/jbc.M112.358192. [DOI] [PMC free article] [PubMed] [Google Scholar]