

SUMMARY
Dopamine (DA) loss in Parkinson’s disease (PD) alters the function of striatal projection neurons (SPNs) and causes motor deficits, but DA replacement can induce further abnormalities. A key pathological change in animal models and patients is SPN hyperactivity; however, the role of glutamate in altered DA responses remains elusive. We tested the effect of locally applied AMPAR or NMDAR antagonists on glutamatergic signaling in SPNs of parkinsonian primates. Following a reduction in basal hyperactivity by antagonists at either receptor, DA inputs induced SPN firing changes that were stable during the entire motor response, in clear contrast with the typically unstable effects. The SPN activity reduction over an extended putamenal area controlled the release of involuntary movements in the “on” state and therefore improved motor responses to DA replacement. These results demonstrate the pathophysiological role of upregulated SPN activity and support strategies to reduce striatal glutamate signaling for PD therapy.
In Brief
Singh et al. analyze the role of SPN hyperactivity in abnormal responses to dopamine replacement in parkinsonian primates with selective local blockade of glutamate signaling. Lowering basal firing stabilizes the SPN response to dopamine and normalizes motor responses.
INTRODUCTION
Motor failure in Parkinson’s disease (PD) is caused primarily by progressive neurodegeneration of the substantia nigra pars compacta. The loss of nigral dopamine (DA) cells has usually reached a considerable level by the time motor deficits develop (Lang and Lozano, 1998). The central role of DA is also demonstrated by the effectiveness of DA replacement to improve motor symptoms in all stages of the disease. However, our understanding of the pathophysiology of motor control in PD is far from clear, particularly with respect to the response to DA replacement. Adding DA to the system does not restore normal movement but rather induces a partial and short recovery that is further complicated by involuntary movements called dyskinesias (Obeso et al., 2000). Indeed, in experiments that are controlled for pharmacological variables, the effective DA stimulation is not yet followed by the expected restitution of normal function (Bravi et al., 1994; Nutt et al., 2000).
DA modulates the excitability of striatal projection neurons (SPNs), which express DA D1 receptors (D1R) or DA D2 receptors (D2R), forming the direct and indirect striatal output pathways, respectively (Gerfen and Surmeier, 2011). Direct SPNs (dSPNs) and indirect SPNs (iSPNs) undergo multiple functional and morphological changes following nigrostriatal denervation that may be involved in altered responses to dopaminergic stimulation (Surmeier et al., 2014). One of the salient changes is the increased spontaneous SPN activity that has been found across animal models and patients. From activity levels usually below 2 Hz in the normal condition, the average firing frequency increases variably in rodent models to 5–12 Hz under anesthesia (Tseng et al., 2001) and to more than 20 Hz in alert, advanced parkinsonian primates and patients with PD (Liang et al., 2008; Singh et al., 2016). These large SPN activity increases in primates and humans were not yet identified in cells segregated into specific output pathways. In line with classic views of the functional model of PD, the use of optogenetics in transgenic mouse models has suggested that iSPNs are most likely the upregulated units after DA denervation (Kravitz et al., 2010). However, further studies disputed the classic views of the model, demonstrating the cooperative activity of both striatal pathways for basal ganglia outputs and movement initiation (Cui et al., 2013; Freeze et al., 2013). In addition, the primate studies show few low-activity units and opposite responses to DA among the recorded SPNs. These observations are difficult to reconcile with the idea of recordings limited to one SPN subpopulation in the primate and thereby call into question previous assumptions on the distribution of hyperactive SPNs (Beck et al., 2017). Yet crude single-cell recordings in primates and patients critically show that there are large firing increases in the active SPNs in the absence of DA. Such a state of high basal activity likely may interfere with the strength of DA signaling to modulate SPN excitability. Congruent with this premise, dopaminergic stimulation induces unstable changes in SPN firing frequency that are associated with dyskinesias in primates with advanced parkinsonism (Liang et al., 2008; Singh et al., 2015). Thus, SPN hyperactivity may play a primary role in the altered responses to DA replacement.
Glutamate inputs from cortical and thalamic terminals provide the excitatory drive of the SPN and likely contribute to the hyper-activity developed in PD. The cumulative evidence supports upregulation of corticostriatal signals (Gubellini et al., 2002; Ingham et al., 1998), but recent data also show changes in the strength of thalamostriatal synapses after DA loss (Parker et al., 2016). Glutamatergic synaptic contacts undergo significant reorganization due to morphological changes of the SPN dendritic arborization (Day et al., 2006; Villalba and Smith, 2017). Notably, spine loss and dendrite changes are differentially developed in dSPNs and iSPNs, indicating that various adaptations may remodel glutamatergic synapses. In the same line, ex vivo recordings following DA depletion demonstrate increased excitability and loss of long-term potentiation and depression at corticostriatal synapses (Bagetta et al., 2011; Shen et al., 2008). Although some of these changes can be reversed by DA replacement, corticostriatal synaptic connectivity, strength, and plasticity remain altered (Fieblinger et al., 2014; Picconi et al., 2003). At the level of glutamate receptors, both DA loss and replacement are associated with changes in the expression, subunit composition, and ratio of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) to N-methyl-D-aspartate receptor (NMDAR) (Bagetta et al., 2012; Mellone et al., 2015). Altogether these data suggest that an intervention to regulate the glutamate signals tuning SPN activity may enable more effective DA modulation.
Here, we used the primate model of advanced PD to study the SPN firing changes related to the motor response induced by dopaminergic stimulation under the control of effective glutamate inputs on the recorded SPN by applying selective NMDAR or AMPAR antagonists. Prior to these tests in the primate, we determined (1) the selectivity of the drug at the concentration applied locally using patch-clamp recordings in cultured cells, (2) the expected concentration of the drug reached in vivo using in silico prediction of diffusion following intraparenchymal injection, and (3) the corresponding doses of the NMDAR and AMPAR antagonists for an equivalent effect reducing high SPN activity in vivo. We also tested the motor effects of glutamate-controlled SPN activity over the primate putamen. Our data show that a decrease of either AMPAR or NMDAR signaling that substantially reduced the basal SPN firing rate results in effective stabilization of the neuronal response to DA during the whole period of reversal of parkinsonian symptoms, referred to as the “on” state. This effect at the single-cell level translated into motor behavioral effects following the NMDAR antagonist infusion over an extended striatal area. The drug-induced reduction of activity across SPNs markedly decreased the dyskinesias caused by L-DOPA. These data support a pivotal role of striatal glutamatergic excitation and SPN hyperactivity in the response to DA replacement in PD.
RESULTS
Selectivity of NMDAR and AMPAR Antagonists for In Vivo Microinjection Assays
The impact of reduced glutamatergic signaling on the dysregulated SPN activity in parkinsonian monkeys was determined with single-cell recordings after intrastriatal injection of high concentrations of selective NMDAR and AMPAR antagonists. Because the rapid dilution and diffusion of antagonists in vivo can be determined quantitatively, we experimentally confirmed the antagonist selectivity in vitro by establishing the concentration-effect relationship for inhibition by each antagonist on currents mediated by recombinant isoforms of NMDAR and AMPAR found in the SPN. We evoked these currents using rapid glutamate applications in order to mimic the time course of the glutamate concentration profile in the synaptic cleft during synaptic transmission (Clements et al., 1992). Pre-application of the NMDAR-selective antagonist LY235959 for 1 s antagonized AMPAR currents only at concentrations much higher than those predicted to be reached with intraparenchymal injection following diffusion and dilution (see below). Specifically, the NMDAR-selective antagonist LY235959 inhibited recombinant AMPAR (GluA1/stargazin) currents by 80% ± 2% at 9 mM and 50% ± 6% at 900 μM but only 13% ± 7% at 90 μM; the half maximal inhibitory concentration (IC50) of LY235959 was 1.1 mM. By contrast, the IC50 of NMDAR antagonist LY235959 at GluN1/GluN2A NMDARs activated by 1 mM glutamate was 12 μM (Figure 1A), indicating that LY235959 is at least 100-fold selective for NMDARs over AMPARs activated by concentrations of glutamate reached in the synaptic cleft. We also evaluated the selectivity of the AMPAR competitive antagonist NBQX by determining non-selective inhibition of recombinant GluN1/GluN2A NMDAR currents. NBQX inhibited NMDAR current responses to 1 mM glutamate by 50% ± 0.3% at 1 mM and 36% ± 0.7% at 300 μM but only 14% ± 0.6% at 100 μM; the fitted IC50 for NBQX non-selective inhibition of GluN1/GluN2A NMDARs was 0.9 mM. By contrast, the IC50 for NBQX inhibition of GluA1/stargazin AMPARs activated by rapid application of 1 mM glutamate was 2.7 μM (Figure 1B). Thus, NBQX is 300-fold selective for AMPAR over NMDAR activated by synaptic-like rapid exposure to glutamate. We expect both competitive antagonists to be even more potent at blocking their target receptors when these are activated by brief glutamate pulses in the synaptic cleft, which are less likely to displace any pre-bound antagonist molecules.
Figure 1. In Vitro Assessment of Drug Concentration-Effect Relationship, Prediction of Brain Dilution, and Recording Timeline.

(A) The effects of increasing concentrations of LY235959 on responses to rapid application of 1 mM glutamate from GluA1/stargazin receptors expressed in HEK cells measured using patch-clamp recordings (black symbols; five HEK cells per concentration). The effects of LY235959 on GluN1/GluN2A receptor responses activated by 1 mM glutamate and 10 μM glycine expressed in oocytes held under two-electrode voltage clamp (red symbols; five oocytes). Error bars indicate SEM.
(B) Concentration-response data for NBQX inhibition of GluN1/GluN2A responses to 1 mM glutamate and 10 μM glycine for receptors expressed in Xenopus oocytes measured using two-electrode voltage clamp (black symbols; four oocytes). NBQX concentration response data for GluA1/stargazin receptors activated by 1 mM glutamate obtained using patch-clamp recordings (red symbols; four HEK cells). Error bars indicate SEM.
(C) Schematic representation of the injectrode used to apply aSCF, LY235959, or NBQX to the putamen of primates.
(D) Peak LY235959 concentration (y axis) reached at various distances from the injection site (x axis) 1, 10, and 30 min following injections. The color-coded traces refer to data obtained when injecting 1, 3, or 9 mM LY235959. The gray shaded area between 100 and 200 μm represents the location from which the SPN activity was recorded.
(E) As in (D) for 0.5, 1, or 3 mM NBQX.
(F) Schematic drawing of the injection site depicting the injectrode with recording electrode in the putamen (left) and a coronal brain section with a small scar at the site of guide cannula penetration. The dashed blue line represents the injectrode trajectory (right).
(G) Timeline of the continuous single-cell recording showing data storage before (“pre,” black box) and after the local injection (L.I.) of antagonist or vehicle (“post,” red box), and again after transitioning to the “on” state and the dyskinesia state (blue boxes) following the s.c. L-DOPA injection.
The striatum of a rhesus macaque is approximately 1,000 mm3 (Yin et al., 2009), which is equivalent to 1 mL in volume. Thus, injection of 1 μL of antagonist into this brain area, at steady state, would lead to an approximate 1,000-fold dilution. The time dependence of diffusion is governed by Fick’s laws and depends on the complex geometry of cell processes forming barriers and hindering diffusion in the brain tissue. We estimated the peak concentration of AMPAR and NMDAR antagonists at different distances and at different time intervals after being injected into brain parenchyma (Figure 1C). Briefly, we described their space-time concentration profile from an instantaneous point source using the classic solution of diffusion from a point source in an isotropic medium, with parameters that describe the diffusion properties of small molecules in the brain neuropil (Savtchenko and Rusakov, 2005; Zheng et al., 2008). The diffusion analysis showed that a more than 1,000-fold dilution of LY235959 or NBQX occurs within less than 1 min from the injection (Figures 1D and 1E). These data suggest that injection of millimolar concentrations of antagonists will rapidly produce sub-micromolar concentrations in the brain tissue surrounding the injection site. These simulations, together with voltage-clamp recordings of the non-equilibrium receptor response to the rapid application of glutamate at concentrations that approach those in the synapse, suggest that our in vitro estimation of diffusion and potency can be used to predict in vivo properties and that the antagonists reached concentrations in brain parenchyma that were selective following microinjection.
Dose Selection of NMDAR and AMPAR Antagonists
On the basis of the results of antagonists’ specificity in vitro and on available estimates of drug dilution in brain tissue, we performed repeated striatal microinjections of 1–9 mM LY235959 and 0.5–3 mM NBQX in the parkinsonian primate to select doses that are equally effective in reducing cell activity for application in subsequent experiments of L-DOPA administration. A dose-dependent decrease in spontaneous SPN firing frequency was observed with each antagonist (Figures S1 and S2; a total of 69 SPNs, 8–10 SPNs per antagonist dose). In order to avoid reaching the extremes of the concentration-response relationship (variable subtle effects and firing silence), the antagonist dose that reduced activity by approximately 50% (45%–55% of the baseline firing rate) was the selected dose. On average, microinjection of 9 mM LY235959 and 1 mM NBQX produced 44% (10 SPNs; p < 0.001) and 47% (10 SPNs; p < 0.01) reductions in firing frequency, respectively. Therefore, these doses had equivalent inhibitory actions on SPN activity and were injected at the site of recording in all subsequent single-cell recording experiments to analyze changes in the SPN response to DA input.
Effect of Reduced NMDAR and AMPAR Signaling on the SPN Response to DA in Parkinsonian Primates
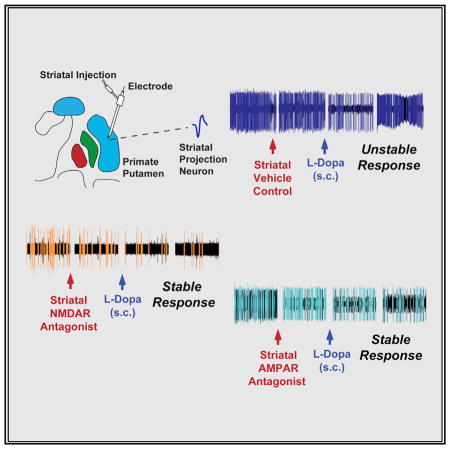
The SPN activity in parkinsonian primates (n = 5; Table S1) was recorded continuously throughout the local application of artificial cerebrospinal fluid (aCSF) (control tests), LY235959, or NBQX, and s.c. L-DOPA injection that induced motor changes (Figures 1F and 1G). Recordings started in the “off” state from before to after local injection and continued through the L-DOPA-induced “on” and dyskinesia states. Thus, neuronal activity data were stored for offline analysis at the following time points: (1) before local injection (“pre”), (2) after local injection (“post”), (3) “on” state (onset of reversal of parkinsonism induced by L-DOPA; “on”), and (4) dyskinesias (L-DOPA-induced dyskinesias at the peak-dose effect; “dys”). Following local aCSF application (control test) that had no effect or produced very brief changes of SPN activity (pre to post), L-DOPA induced the typical responses of increased or decreased firing frequency (post to on; Figures 2A–2D) (Singh et al., 2015). In these control tests, SPN activity increased in 14 units and decreased in the other 15 units, and these firing changes correlated with the behavioral changes indicating that the animal had transitioned to the “on” state. Also, aCSF application had no effect on the magnitude of responses. The firing frequency in the “on” state increased by 106% and decreased by 36% (average changes in SPNs grouped by their response to DA), comparable with previously reported data. At the peak of L-DOPA effect, the firing rate changes were maintained or continued to develop (stable responses) in 41% of SPNs (on to dys; Figures 2A and 2C). In contrast, at the peak effect responses reversed in 59% of SPNs producing the inverted firing rate changes during the “on” state that correlate with the appearance of peak-dose dyskinesias in advanced parkinsonian primates (Figures 2B, 2D, 2I, and 2J) (Liang et al., 2008).
Figure 2. NMDAR Antagonist Injection at the Site of SPN Recording Reduced the Basal Activity and Stabilized DA Responses in the Parkinsonian Primate.

(A–D) Control tests. Firing frequency changes of each SPN at baseline (pre), after local injection of aCSF (post), and after transitioning to “on” (on) and dyskinesias states (dys) following s.c. injection of L-DOPA. In each panel, each colored curve (top graph) represents an individual SPN grouped according to the type of DA response (increase, A and B, or decrease, C and D, in the “on” state followed by stable, A and C, or unstable, B and D, response in the dyskinesia state), and the averaged firing frequency change for the group is shown as percentage (bottom graph). In each SPN, differences between post and on are significant at p < 0.01 (A–D). Responses were classified as unstable by significant changes in the dyskinesia state (p < 0.01). Total SPNs, 29; units with activity increase, 14; units with activity decrease, 15.
(E–H) NMDAR antagonist tests. The firing frequency changes of each SPN as described above for control tests are shown in (E) and (G) after local injection of LY235959. In each SPN, differences between pre and post and between post and on are significant at p < 0.01. Differences between on and dys were non-significant (see also Figure S3). SPN stable responses after LY235959 are compared with control tests in (F) and (H). Total SPNs, 39; units with activity increase in the “on” state, 24; units with activity decrease, 15. In each group analysis, ^p < 0.01 versus baseline, *p < 0.01 versus post aCSF or LY235959 injection, and +p < 0.01 versus the “on” state (ANOVAs for repeated-measure followed by Bonferroni correction). In (F) and (H), *p < 0.05 between LY235959 and aCSF unstable response (one way ANOVAs). Error bars indicate SEM (n = 5 primates; see Table S1).
(I–L) Examples of SPN unstable responses in aCSF tests (I and J) or stable responses in LY235959 tests (K and L). Short (5 s) spike trains and spectrograms for the segment duration (180 s) are shown for each segment (pre, post, on, and dys). The unit activity is colored after spike sorting as the corresponding curve in the frequency graphs (B), (D), (E), and (G).
Following reduction of the baseline (“off” state) SPN activity by LY235959 application, L-DOPA administration produced changes in SPN firing associated with the “on” state that were similar to those found in control experiments (i.e., activity increase or decrease from post to on). However, the firing frequency changes during the “on” state were stable in the large majority of SPNs (Figures 2E–2H, 2K, and 2L). As dyskinesias remain the same with the limited effect of local microinjection of LY235959, the stabilized SPN activity in response to DA was caused by the NMDAR antagonist. Similar effects were obtained with NBQX application (Figure 3). Thus, both NMDAR and AMPAR antagonists at selective doses that induced nearly 50% reduction of SPN activity equally prevented the inversion of frequency changes at the peak of the L-DOPA response (concomitant with dyskinesias), contrasting with the control experiments with application of aCSF alone that resulted in a large number of inverted (unstable) responses. The stability of changes during the “on” state in experiments of local application of NMDAR or AMPAR antagonist was found regardless of the type of response to L-DOPA (i.e., increase or decrease of the firing rate during the “on” state). Following LY235959 application, 39 of 43 SPNs had stable responses to L-DOPA, and following NBQX application, 43 of 45 SPNs also had stable responses, reaching a total of 93% of the recorded SPNs with stable responses (Figures 4 and S3). Therefore, the blockade of NMDAR or AMPAR transmission that effectively reduced the SPN hyperactivity of the parkinsonian state restored full, stable responsiveness to DA signaling.
Figure 3. AMPAR Antagonist Injection at the Site of SPN Recording Reduced the Basal Activity and Stabilized DA Responses in the Parkinsonian Primate.

(A–D) The same control data as presented in Figures 2A–2D, respectively, for comparison with results obtained with the AMPAR antagonist. See Figure 2 for details.
(E–H) AMPAR antagonist tests. Firing frequency changes of each SPN as described for control tests are shown in (E) and (G) after local injection of NBQX. In each SPN, differences between Pre and Post, and between Post and On are significant at p < 0.01. Differences between On and Dys were non-significant (see also Figure S3). SPN stable responses after NBQX are compared to control tests in (F) and (H). Total SPNs, 43; units with activity increase in the “on” state, 26; units with activity decrease, 17.
In each group analysis, ^p < 0.01 versus baseline, *p < 0.01 versus Post aCSF or NBQX injection, and +p < 0.01 versus the “on” state (ANOVAs for repeated measure followed by Bonferroni’s correction). In (F) and (H), *p < 0.05 between NBQX and aCSF unstable response (one way ANOVAs). Error bars indicate SEM (n = 5 primates; see Table S1).
(I–L) Examples of SPN unstable responses in aCSF tests ([I and J], same examples as in Figures 2I and 2J for comparison) or stable responses in NBQX tests (K and L). Short (5 s) spike trains and spectrograms for the segment duration (180 s) are shown for each segment, Pre, Post, On, and Dys. The unit activity is colored after spike sorting as the corresponding curve in the frequency graphs B, D, E and G.
Figure 4. DA Responses after Local NMDAR or AMPAR Blockade Are Stable across SPNs.

The proportion of SPNs with stable DA responses is compared after local injection of aCSF, LY235959, or NBQX. More than 90% of SPNs with LY235959 or NBQX injection exhibited stable DA responses (activity increase or decrease), but fewer than 50% of SPNs with aCSF injection. Total SPNs, 117; stable responses, 39 of 43 in LY235959 tests, 43 of 45 in NBQX tests, and 12 of 29 in aCSF tests (see complementary data on unstable DA responses in Figure S3).
To further determine the relationship between the DA response and the activity reduction induced by the NMDAR or AMPAR antagonist, we analyzed the correlation of firing changes in each SPN. The lowered firing frequency after LY235959 or NBQX application at the recording site (post/pre) was a predictor of the amount of increased activity in response to L-DOPA at the initial behavioral change (on/post), or at the peak-dose effect (dys/post), accounting for 35%–50% of the change (see R2 values in Figures 5A and 5B; p < 0.01). In this subset of SPNs (units with DA-induced activity increase), the response to DA was thus highly dependent on the level of reduced basal activity. In contrast, in the SPN subset with DA-induced activity decrease, the amount of activity reduction in the “on” or dyskinesia state was not predicted by the level of reduced basal activity (Figures 5C and 5D; p > 0.05). In this subset of SPNs, DA-induced changes stabilized, but the strength of the response was independent of the reduced baseline activity. Therefore, data indicate different DA regulation across the distinguished SPN subsets.
Figure 5. Relationship between the Effect of NMDAR or AMPAR Antagonist and the Magnitude of the SPN Response to DA.

(A–D) Significant (blue) and non-significant (red) correlations between the activity reduction induced by LY235959 (A and C) or NBQX (B and D) and the DA response analyzed in the “on” state or dyskinesia state (top and bottom graphs, respectively in each panel).
Firing frequency reduction post LY235959 or NBQX: ratio of post-antagonist injection to baseline frequencies. Firing frequency increase or decrease in “on” or dyskinesia state: ratio of motor state to post-antagonist injection frequencies. SPNs included in all regression analyses had stable responses to DA (total SPNs, 82; in LY235959 tests, 24 SPNs with activity increase in response to DA and 15 with activity decrease; in NBQX tests, 26 SPNs with activity increase in response to DA and 17 with activity decrease).
Effect of Reduced Striatal NMDAR Signaling on Motor Responses to L-DOPA in Parkinsonian Primates
Motor responses to L-DOPA in primates with advanced parkinsonism are consistently complicated by dyskinesias that are associated with inversion of the SPN firing rate changes during the “on” state. To test whether the reduced glutamatergic transmission that stabilized the SPN response to L-DOPA had the expected behavioral correlate, LY235959 (9 mM) or aCSF was infused unilaterally into the putamen of primates (n = 3; Table S1) before the s.c. injection of L-DOPA. The infusion volume (10 μL) was intended to cover only the most posterolateral region corresponding to the sensorimotor territory (Sanftner et al., 2005). The preselected individual dose of L-DOPA induced reproducible monophasic peak-dose dyskinesias in the tested animals. Striatal infusion of the NMDAR antagonist reduced the global dyskinesia scores (Figures 6A and 6C) because of 71% lower scores in the hemibody contralateral to the infusion side after L-DOPA injection (p < 0.05; Figures 6B and 6D; Movies S1 and S2). LY235959 infusion did not affect the antiparkinsonian action of L-DOPA (unchanged motor disability scores [MDS], p > 0.05; Figure 6E). Therefore, LY235959 infusion into the putamen showed that the extended stabilization of DA responses across SPNs translates into motor changes with significantly reduced dyskinesias. This demonstrates that decreasing glutamatergic excitation in the striatum induces beneficial motor effects in the parkinsonian primate.
Figure 6. Infusion of NMDAR Antagonist over the Putamen Reduced Contralateral Dyskinesias Induced by L-DOPA in Parkinsonian Primates.

(A and B) Time course of global (A) and contralateral (CL) (B) dyskinesias induced by s.c. injection of a suboptimal dose of L-DOPA after unilateral infusion of LY235959 into the posterolateral putamen. Reduced global scores by LY235959 infusion (red) (9 mM) compared with the control vehicle infusion (black) reflect differences in scores on the contralateral side of the body (see also Movies S1 and S2). Time 0, before L-DOPA injection (“off” state before infusion). Scores after L-DOPA injection: 30 min post-injection and thereafter every 20 min interval until dyskinesias disappear and the mobility was returning to the “off” state. *p < 0.01, two-way ANOVAs for repeated-measures followed by Fisher’s PLSD test.
(C and D) Total and peak scores of global (C) and contralateral (D) L-DOPA-induced dyskinesias after infusion of LY235959 (red) and aCSF (black). Scales are adjusted for the contralateral side (D). AUC, area under the curve. Peak values, 50 min interval scores. *p < 0.01, paired t tests.
(E) Motor disability scores (MDS) showing no changes in the antiparkinsonian effects of L-DOPA after LY235959 infusion in comparison with aCSF infusion.
Each animal (n = 3; see details in Table S1) received one infusion of each dose (LY235959 0 and 9 mM). Error bars indicate SEM.
The motor effects of systemic administration of LY235959 and NBQX were also tested in a group of parkinsonian primates (n = 5; Table S1) that included three of the animals used in striatal application of the antagonists. Both LY235959 (3 mg/kg s.c.) and NBQX (2 mg/kg s.c.) significantly reduced dyskinesias (p < 0.05; Figures S4 and S5) without compromising the antiparkinsonian action of L-DOPA (Löschmann et al., 1991; Papa and Chase, 1996). The similarity of these effects (including animals used for SPN recordings) to those obtained with striatal infusion suggests that the reduction of glutamate signals on SPNs may be responsible for the effects induced by systemic administration.
DISCUSSION
The selective reduction of either NMDAR or AMPAR signals in SPNs tested here in parkinsonian primates supports our hypothesis that altered SPN responses to DA and their associated abnormal movements can be reversed by controlling the dysregulated glutamatergic drive. Because the animals used in these single-cell recordings had developed significant SPN hyperactivity, the impact of antagonist application on the spontaneous firing of these neurons was fully assessed. The most important finding from this study was that the reduced excitatory glutamate signals resulted in stable responses to DA in 93% of the recorded SPNs, and these effects were found across single recordings of SPNs regardless of their response to DA (i.e., units with activity increase or decrease). In addition, both the NMDAR and AMPAR antagonists equally stabilized DA responses in the large majority of neurons, and effects of the NMDAR antagonist on an extended putamenal area reduced dyskinesias improving the L-DOPA response.
The interpretation of the recorded SPN activity is limited by lack of specific cell-type identification with optogenetics, because transgenic modeling could not be applied to these primate studies. More important to this end, DA stimulation evokes pathological “unstable” responses that are widely distributed across neurons, with an increase or decrease of activity in the “on” state. Indeed, as L-DOPA reaches its peak effect, a high proportion of units do not maintain the DA-induced firing increase or decrease, which contrasts markedly with stable changes in other units and thereby creates a large imbalance of discharges. On the basis of evidence that the cooperative activity of both striatal pathways mediates normal movement execution (Cui et al., 2013), imbalance of discharges between and within the output pathways may lead to abnormal motor responses, releasing involuntary movements or causing motor fluctuations (Singh et al., 2015; Tecuapetla et al., 2014). In support of this view, the instability of DA-induced firing changes that causes discharge imbalance is associated with dyskinesias in the primate. Here, DA-induced responses were consistently stabilized across SPNs after glutamate inputs were blocked and the high baseline firing frequency was decreased, indicating that hyperactivity across SPNs is a key mechanism in the pathophysiology of PD “off” and “on” states. These data are consistent with findings in patch recordings of iSPNs (Fieblinger et al., 2014) showing that DA denervation induces marked homeostatic plasticity (reduced excitability and spine loss and pruning of corticostriatal synapses), but the corticostriatal synaptic strength increased as opposed to the expected scaling (Turrigiano, 2008). However, these patch recording data are not aligned with hyperactivity also present in dSPNs, should that be the case. Some distinctions when comparing data from largely different models may be relevant, even in the same species (Beck et al., 2017; Deffains et al., 2016). Among the most important are the lesion type and its time course, which may influence the development of adaptive and aberrant changes, such as spine remodeling (Villalba and Smith, 2017). Unlike unilateral and acute lesions, the primates used here had a bilateral and “slowly” induced parkinsonism that was classified as advanced with long-standing chronicity (Potts et al., 2014). Thus, it is plausible that extensive SPN hyperactivity does not fully develop in commonly used rodent and primate models. Of note, gene regulation after DA loss leads to changes in voltage-gated potassium and calcium (VDCC) channels in dSPNs that can increase excitability (Borgkvist et al., 2015; Calabresi et al., 2014; Meurers et al., 2009). Whether present in SPNs expressing D2R or D1R, our results indicate that exaggerated upregulation of firing plays a primary role in the activity changes of the “on” state, likely interfering with DA-modulated excitability, which then can only result in transient (short) firing changes. The application of glutamate antagonists that reduced SPN activity by half restored sustained (stable) firing increases or decreases in response to DA. Interestingly, the strength of the DA response correlated with the level of basal activity reduction by the antagonist only in SPNs with DA-induced firing increase. These data support differential DA modulation across SPNs after denervation that is congruent with the imbalance of responses to L-DOPA and the associated abnormal motor effects.
Reducing neurotransmission at NMDAR or AMPAR had the same effect on the firing frequency of SPNs and similarly stabilized their DA responses. It is important to note that the present data do not exclude the potential effects of manipulating other signals, such as gamma-aminobutyric acid (GABA). However, the activity of fast spiking interneurons, which provide most GABA inhibition of SPNs, is not affected by DA loss (Mallet et al., 2006). Instead, the reported changes in cortical and thalamic inputs after DA loss indicate a key role of glutamate signaling in SPN hyperactivity. Our findings indicate that both NMDAR and AMPAR signaling can contribute to SPN dysfunction and that controlling glutamate signaling has a significant effect. Perhaps the most parsimonious explanation of these results is that the SPN AMPARs and NMDARs are co-localized on the same postsynaptic site and act as canonical coincidence detectors. Robust NMDAR-mediated depolarization requires the prior release of a voltage-dependent block via the activation of AMPARs (Huganir and Nicoll, 2013). Therefore, in the presence of NBQX, block is not released, and the excitatory synapse is silenced. Conversely, in the presence of LY235959, AMPARs are fully activated, but too briefly to mediate a robust and prolonged depolarization. Therefore, block of either AMPARs or NMDARs is sufficient to attenuate activity at excitatory synapses and reduce firing rate. In addition, the impact may be more significant on receptors with slow deactivation kinetics, such as the SPN NMDARs (Logan et al., 2007), particularly after DA loss. Differences in glutamatergic signaling after DA lesion have been linked to changes in the expression, composition, trafficking, and localization of ionotropic receptors. Furthermore, some receptor changes are related to lesion extent, such as increased NMDAR/AMPAR ratio (Paillé et al., 2010), and some to chronic DA replacement and dyskinesia development, such as increased GluN2A/GluN2B ratio (Hallett et al., 2005; Mellone et al., 2015). Reorganization of NMDAR subunits following DA denervation leads not only to reduction of GluN2B but also to a newly developed contribution of GluN2D in functional receptors of SPNs (Zhang and Chergui, 2015). Changes in NMDAR distribution between synaptic and extrasynaptic location can also result in increased gain of transmission (Fieblinger et al., 2014). In addition, the AMPAR subunit composition plays a key role in synaptic strength. Notable changes in models of DA loss are the hyperphosphorylation of GluR1 and the expression of Ca2+-permeable GluR2-lacking AMPAR (Bagetta et al., 2012). Changes in Ca2+ kinetics are consistent with the pathological basal activity of SPNs and potentially their “unstable” response to DA. The NMDAR- and AMPAR-selective competitive antagonists (LY235959 and NBQX) used here were not subunit selective, and thus each effectively blocked most receptor subtypes within the targeted families, reducing the “off”-state hyperactivity to a similar extent as intended for comparison. Therefore, the ability of both NMDAR and AMPAR blocks to induce the same stabilization of DA responses supports the contribution of presynaptic (increased input) and postsynaptic (hyperexcitability) mechanisms in the SPN dysfunction.
The consistent glutamate antagonist effect on single neurons suggested that extended effects across SPNs would prevent the imbalance of discharges, and possibly improve the behavioral response to DA. Our tests support this idea. The infusion of the same concentration of the NMDAR antagonist over an extended area of the putamen reduced dyskinesias in the parkinsonian primate. Because the infusion into the putamen was unilateral and centered on the posterolateral motor territory using a discrete volume to avoid overflow to surrounding extrastriatal areas, most likely the covered area did not extend to the whole target region, but nevertheless, dyskinesias were reduced by 71%. In line with our results, the only drug in clinical use to treat dyskinesias is amantadine, an agent with actions at multiple sites, including the NMDAR (Oertel et al., 2017). On the basis of the effects of selective agents, the antidyskinetic effect of amantadine is likely mediated by NMDAR block. Regarding further effects of NMDAR antagonists on L-DOPA responses, it is possible that chronically reduced SPN activity also results in reduction of motor fluctuations and improved “on” state. In addition to chronic block, the antagonist intrinsic effect on parkinsonism remains to be tested, because in the present tests the infusion timeline was designed to assess L-DOPA-induced dyskinesias. Importantly, the present results provide proof of concept for the significance of reduced glutamatergic tone and stabilization of DA-induced firing changes in SPNs.
Because the current therapy for patients with PD remains symptomatic and based on dopaminergic stimulation for relief of motor disability, the present results have significant clinical implications. Treatment efficacy depends on consistently recovering mobility with responses free of dyskinesias, but most patients between mid- and late-stage disease suffer from disabling motor complications. Critically, the primates used in these experiments reproduced the phenotype of patients in that category, and SPN recordings in these patients have shown the same level of SPN hyperactivity as in the parkinsonian primate. Likewise, glutamate-driven SPN hyperactivity plays a central role in the abnormal DA responses developed in patients with PD. Strategies targeting specifically the striatal glutamate overactivity may thus help improve the efficacy of DA replacement. Of particular interest are the mechanisms regulating protein composition and posttranslational changes in AMPARs/NMDARs, including molecular changes that increase AMPAR activity or channel conductance (Hosokawa et al., 2015; Jenkins et al., 2014; Kristensen et al., 2011) and thereby upregulate SPN activity.
EXPERIMENTAL PROCEDURES
In Vitro NMDAR and AMPAR Blockade and Simulated Antagonist Diffusion
The experiments in vitro and simulations of drug diffusion in brain tissue were designed to determine whether LY235959 and NBQX would yield concentrations that selectively blocked NMDAR or AMPAR, respectively, using the specific space and time parameters of the microinjection used for recording experiments. Details are provided in Supplemental Experimental Procedures.
Non-human Primate Model of PD
Nine adult male and female macaques (Macaca mulatta and Macaca fascicularis; 5–11 kg body weight) were used in the studies (see Table S1). All procedures followed guidelines of the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Emory University. A model of “chronic and advanced” parkinsonism was produced in all animals by systemic administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and behavioral assessment using a standardized motor disability scale for primates. In these parkinsonian primates, the daily maintenance L-DOPA treatment led to the development of L-DOPA-induced dyskinesias. In each animal, the effective subcutaneous (s.c.) dose of L-DOPA methyl ester plus benserazide (Sigma-Aldrich) for use in recording experiments was determined as the dose eliciting a clear “on” state with peak-dose dyskinesias. A complete description is provided in the Supplemental Information.
Continuous Single-Cell Recording with Striatal NMDAR or AMPAR Blockade and L-DOPA-Induced Motor States
Five animals were surgically implanted with recording chambers and head-restraining devices, and striatal regions were identified with electrophysiological mapping of basal ganglia. Standard techniques were used for single-cell recordings with withdrawal of the oral maintenance L-DOPA treatment the day of recording. NMDAR or AMPAR antagonist was delivered at the site of recording, using “injectrodes” connected to a microinjection pump. The antagonist solution (or the same volume of aCSF alone as control test) was injected in a volume of 200 nL at a rate of 1 μL/min. LY235959 was dissolved in aCSF and NBQX in aCSF/water. The tip of the electrode was placed at a fixed distance from the tip of the cannula (400 μm; Figure 1C). The antagonist concentration (within the limits set from in vitro tests and simulations to maintain selective doses at the synapses) that effectively reduced activity of the recorded cell by ~50% was assessed using different drug concentrations and analyzing a total of 69 SPNs. After dose selection, complete experiments started by local application of antagonist followed by s.c. L-DOPA injection at the predetermined dose with data storage (≥3 min) according to the time-line (Figure 1G). If the baseline activity was held throughout the total duration of the experiment, the offline analysis yielded one or occasionally two units per experiment. A total of 117 SPNs were analyzed in complete experiments of DA responses, 88 SPNs for antagonists, and 29 SPNs for vehicle control tests. The animal’s behavior was monitored using a video camera. Details of experiments, including online unit isolation, behavioral changes during recordings indicative of motor states and histological verification of recording sites are included in the Supplemental Information.
Electrophysiology Data Analysis and Statistics
All data were analyzed offline using spike sorting and standard methods for differentiation and classification of SPNs (Figure S6). Significant activity changes in the “on” state (p < 0.05, ANOVAs for repeated-measures followed by post hoc Bonferroni test) separated units with increase or decrease response. Also, significant frequency changes in dyskinesia state determined whether the increased or decreased firing rate in the “on” state was stable or not. Activity changes in the “on” and dyskinesia states were analyzed for their relationship to the reduced firing frequency after the local application of LY235959 or NBQX using the regression equation
where FDopamine is frequency after L-DOPA injection in either “on” or dyskinesia state, FAntagonist is frequency after the local antagonist injection, and FBaseline is frequency before antagonist injection. All analyses are described in details in the Supplemental Information.
Striatal Infusion of Antagonist
LY235959 (9 mM) was dissolved in aCSF, and 10 μL of the solution distributed among five sites (2 μL/site) was infused unilaterally into the posterolateral putamen in three animals (see the criteria for selection of infusion parameters in the Supplemental Information). Ten minutes following the completion of infusion, the animal received s.c. injection of L-DOPA at the selected suboptimal dose. Because of the invasiveness of the procedure, each animal could receive only two infusions, one infusion of LY235959 and another infusion of vehicle alone (aCSF10 μL) as control. Movies S1 and S2 are accompanied by movie legends provided in the Supplemental Information. All details of the procedure and the behavioral assessment after brain infusion or systemic administration of antagonist are provided in the Supplemental Information.
Statistical Analysis of Behavioral Data
Behavioral responses were graded within wide ranges, and values included no integers, so data formed quantitative variables, which were analyzed using ANOVAs for repeated-measures (p < 0.05) followed by post hoc Fisher’s protected least significant difference (PLSD) tests. In all analyses, data distribution and variance homogeneity were examined, and appropriate corrections applied. Further details are provided in the Supplemental Information.
Supplementary Material
Highlights.
Selectively reducing AMPAR or NMDAR transmission stabilizes SPN responses to DA
Reduced glutamate signaling stabilizes DA-induced activity increases and decreases
Reduced glutamate signaling across SPNs suppresses dyskinesias in primates
Data support the mechanistic role of SPN hyperactivity in responses to DA in PD
Acknowledgments
This work was supported by NIH grants NS045962 and NS073994 (S.M.P.) and NS036654 and NS065371 (S.F.T.); SUNY Albany Research Foundation, and National Science Foundation (NSF) grant IOS1655365 (A.S.); and National Center for Research Resources (NCRR) grant RR000165 and Office of Research Infrastructure Programs (ORIP)/OD grant OD011132 (Yerkes National Primate Research Center). We thank Dr. Yoland Smith for advice on histological parameters and Bhagya Dyavar Shetty for technical assistance in the production and care of the primate model with advanced PD.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, one table, and two movies and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.12.095.
AUTHOR CONTRIBUTIONS
Design and Conceptualization, S.F.T. and S.M.P.; Investigation, A.S., M.A.J., K.J.B., and G.B.; Analysis, A.S., M.A.J, A.J., A.S., S.F.T., and S.M.P.; Writing, A.S., M.A.J., A.J., A.S., S.F.T., and S.M.P.; Review and Editing, A.S., M.A.J., K.J.B., G.B., A.J., A.S., S.F.T., and S.M.P.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Bagetta V, Picconi B, Marinucci S, Sgobio C, Pendolino V, Ghiglieri V, Fusco FR, Giampà C, Calabresi P. Dopamine-dependent long-term depression is expressed in striatal spiny neurons of both direct and indirect pathways: implications for Parkinson’s disease. J Neurosci. 2011;31:12513–12522. doi: 10.1523/JNEUROSCI.2236-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagetta V, Sgobio C, Pendolino V, Del Papa G, Tozzi A, Ghiglieri V, Giampà C, Zianni E, Gardoni F, Calabresi P, Picconi B. Rebalance of striatal NMDA/AMPA receptor ratio underlies the reduced emergence of dyskinesia during D2-like dopamine agonist treatment in experimental Parkinson’s disease. J Neurosci. 2012;32:17921–17931. doi: 10.1523/JNEUROSCI.2664-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck G, Singh A, Papa SM. Dysregulation of striatal projection neurons in Parkinson’s disease. J Neural Transm (Vienna) 2017 doi: 10.1007/s00702-017-1744-5. Published online June 15, 2017. https://doi.org/10.1007/s00702-017-1744-5. [DOI] [PMC free article] [PubMed]
- Borgkvist A, Avegno EM, Wong MY, Kheirbek MA, Sonders MS, Hen R, Sulzer D. Loss of striatonigral GABAergic presynaptic inhibition enables motor sensitization in parkinsonian mice. Neuron. 2015;87:976–988. doi: 10.1016/j.neuron.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravi D, Mouradian MM, Roberts JW, Davis TL, Sohn YH, Chase TN. Wearing-off fluctuations in Parkinson’s disease: contribution of postsynaptic mechanisms. Ann Neurol. 1994;36:27–31. doi: 10.1002/ana.410360108. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat Neurosci. 2014;17:1022–1030. doi: 10.1038/nn.3743. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, Costa RM. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494:238–242. doi: 10.1038/nature11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Deffains M, Iskhakova L, Katabi S, Haber SN, Israel Z, Bergman H. Subthalamic, not striatal, activity correlates with basal ganglia downstream activity in normal and parkinsonian monkeys. eLife. 2016;5:5. doi: 10.7554/eLife.16443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, Chan CS, Heiman M, Greengard P, Cenci MA, Surmeier DJ. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat Commun. 2014;5:5316. doi: 10.1038/ncomms6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze BS, Kravitz AV, Hammack N, Berke JD, Kreitzer AC. Control of basal ganglia output by direct and indirect pathway projection neurons. J Neurosci. 2013;33:18531–18539. doi: 10.1523/JNEUROSCI.1278-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, Bernardi G, Finazzi-Agrò A, Maccarrone M. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci. 2002;22:6900–6907. doi: 10.1523/JNEUROSCI.22-16-06900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Dunah AW, Ravenscroft P, Zhou S, Bezard E, Crossman AR, Brotchie JM, Standaert DG. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology. 2005;48:503–516. doi: 10.1016/j.neuropharm.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Hosokawa T, Mitsushima D, Kaneko R, Hayashi Y. Stoichiometry and phosphoisotypes of hippocampal AMPA-type glutamate receptor phosphorylation. Neuron. 2015;85:60–67. doi: 10.1016/j.neuron.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham CA, Hood SH, Taggart P, Arbuthnott GW. Plasticity of synapses in the rat neostriatum after unilateral lesion of the nigrostriatal dopaminergic pathway. J Neurosci. 1998;18:4732–4743. doi: 10.1523/JNEUROSCI.18-12-04732.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MA, Wells G, Bachman J, Snyder JP, Jenkins A, Huganir RL, Oswald RE, Traynelis SF. Regulation of GluA1 α-amino-3-hy-droxy-5-methyl-4-isoxazolepropionic acid receptor function by protein kinase C at serine-818 and threonine-840. Mol Pharmacol. 2014;85:618–629. doi: 10.1124/mol.113.091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir R, Traynelis SF. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci. 2011;14:727–735. doi: 10.1038/nn.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson’s disease. First of two parts. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- Liang L, DeLong MR, Papa SM. Inversion of dopamine responses in striatal medium spiny neurons and involuntary movements. J Neurosci. 2008;28:7537–7547. doi: 10.1523/JNEUROSCI.1176-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan SM, Partridge JG, Matta JA, Buonanno A, Vicini S. Long-lasting NMDA receptor-mediated EPSCs in mouse striatal medium spiny neurons. J Neurophysiol. 2007;98:2693–2704. doi: 10.1152/jn.00462.2007. [DOI] [PubMed] [Google Scholar]
- Löschmann PA, Lange KW, Kunow M, Rettig KJ, Jähnig P, Honoré T, Turski L, Wachtel H, Jenner P, Marsden CD. Synergism of the AMPA-antagonist NBQX and the NMDA-antagonist CPP with L-dopa in models of Parkinson’s disease. J Neural Transm Park Dis Dement Sect. 1991;3:203–213. doi: 10.1007/BF02259538. [DOI] [PubMed] [Google Scholar]
- Mallet N, Ballion B, Le Moine C, Gonon F. Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J Neurosci. 2006;26:3875–3884. doi: 10.1523/JNEUROSCI.4439-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellone M, Stanic J, Hernandez LF, Iglesias E, Zianni E, Longhi A, Prigent A, Picconi B, Calabresi P, Hirsch EC, et al. NMDA receptor GluN2A/GluN2B subunit ratio as synaptic trait of levodopa-induced dyskinesias: from experimental models to patients. Front Cell Neurosci. 2015;9:245. doi: 10.3389/fncel.2015.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurers BH, Dziewczapolski G, Shi T, Bittner A, Kamme F, Shults CW. Dopamine depletion induces distinct compensatory gene expression changes in DARPP-32 signal transduction cascades of striatonigral and striatopallidal neurons. J Neurosci. 2009;29:6828–6839. doi: 10.1523/JNEUROSCI.5310-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt JG, Obeso JA, Stocchi F. Continuous dopamine-receptor stimulation in advanced Parkinson’s disease. Trends Neurosci. 2000;23(10, Suppl):S109–S115. doi: 10.1016/s1471-1931(00)00029-x. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Olanow CW, Nutt JG. Levodopa motor complications in Parkinson’s disease. Trends Neurosci. 2000;23(10, Suppl):S2–S7. doi: 10.1016/s1471-1931(00)00031-8. [DOI] [PubMed] [Google Scholar]
- Oertel W, Eggert K, Pahwa R, Tanner CM, Hauser RA, Trenkwalder C, Ehret R, Azulay JP, Isaacson S, Felt L, et al. Randomized, placebo-controlled trial of ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (EASE LID 3) Mov Disord. 2017;32:1701–1709. doi: 10.1002/mds.27131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillé V, Picconi B, Bagetta V, Ghiglieri V, Sgobio C, Di Filippo M, Viscomi MT, Giampà C, Fusco FR, Gardoni F, et al. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J Neurosci. 2010;30:14182–14193. doi: 10.1523/JNEUROSCI.2149-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa SM, Chase TN. Levodopa-induced dyskinesias improved by a glutamate antagonist in Parkinsonian monkeys. Ann Neurol. 1996;39:574–578. doi: 10.1002/ana.410390505. [DOI] [PubMed] [Google Scholar]
- Parker PR, Lalive AL, Kreitzer AC. Pathway-specific remodeling of thalamostriatal synapses in parkinsonian mice. Neuron. 2016;89:734–740. doi: 10.1016/j.neuron.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Potts LF, Wu H, Singh A, Marcilla I, Luquin MR, Papa SM. Modeling Parkinson’s disease in monkeys for translational studies, a critical analysis. Exp Neurol. 2014;256:133–143. doi: 10.1016/j.expneurol.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanftner LM, Sommer JM, Suzuki BM, Smith PH, Vijay S, Vargas JA, Forsayeth JR, Cunningham J, Bankiewicz KS, Kao H, et al. AAV2-mediated gene delivery to monkey putamen: evaluation of an infusion device and delivery parameters. Exp Neurol. 2005;194:476–483. doi: 10.1016/j.expneurol.2005.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savtchenko LP, Rusakov DA. Extracellular diffusivity determines contribution of high-versus low-affinity receptors to neural signaling. Neuroimage. 2005;25:101–111. doi: 10.1016/j.neuroimage.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Liang L, Kaneoke Y, Cao X, Papa SM. Dopamine regulates distinctively the activity patterns of striatal output neurons in advanced parkinsonian primates. J Neurophysiol. 2015;113:1533–1544. doi: 10.1152/jn.00910.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Mewes K, Gross RE, DeLong MR, Obeso JA, Papa SM. Human striatal recordings reveal abnormal discharge of projection neurons in Parkinson’s disease. Proc Natl Acad Sci U S A. 2016;113:9629–9634. doi: 10.1073/pnas.1606792113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Graves SM, Shen W. Dopaminergic modulation of striatal networks in health and Parkinson’s disease. Curr Opin Neurobiol. 2014;29:109–117. doi: 10.1016/j.conb.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Matias S, Dugue GP, Mainen ZF, Costa RM. Balanced activity in basal ganglia projection pathways is critical for contraversive movements. Nat Commun. 2014;5:4315. doi: 10.1038/ncomms5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Kasanetz F, Kargieman L, Riquelme LA, Murer MG. Cortical slow oscillatory activity is reflected in the membrane potential and spike trains of striatal neurons in rats with chronic nigrostriatal lesions. J Neurosci. 2001;21:6430–6439. doi: 10.1523/JNEUROSCI.21-16-06430.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Smith Y. Loss and remodeling of striatal dendritic spines in Parkinson’s disease: from homeostasis to maladaptive plasticity? J Neural Transm (Vienna) 2017 doi: 10.1007/s00702-017-1735-6. Published online May 24, 2017. https://doi.org/10.1007/s00702-017-1735-6. [DOI] [PMC free article] [PubMed]
- Yin D, Valles FE, Fiandaca MS, Forsayeth J, Larson P, Starr P, Bankiewicz KS. Striatal volume differences between non-human and human primates. J Neurosci Methods. 2009;176:200–205. doi: 10.1016/j.jneumeth.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chergui K. Dopamine depletion of the striatum causes a cell-type specific reorganization of GluN2B- and GluN2D-containing NMDA receptors. Neuropharmacology. 2015;92:108–115. doi: 10.1016/j.neuropharm.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Zheng K, Scimemi A, Rusakov DA. Receptor actions of synaptically released glutamate: the role of transporters on the scale from nanometers to microns. Biophys J. 2008;95:4584–4596. doi: 10.1529/biophysj.108.129874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.