

Abstract
Bivalves exhibit an astonishing diversity of sexual systems and sex-determining mechanisms. They can be gonochoric, hermaphroditic or androgenetic, with both genetic and environmental factors known to determine or influence sex. One unique sex-determining system involving the mitochondrial genome has also been hypothesized to exist in bivalves with doubly uniparental inheritance (DUI) of mtDNA. However, the link between DUI and sex determination remains obscure. In this study, we performed a comparative gonad transcriptomics analysis for two DUI-possessing freshwater mussel species to better understand the mechanisms underlying sex determination and DUI in these bivalves. We used a BLAST reciprocal analysis to identify orthologs between Venustaconcha ellipsiformis and Utterbackia peninsularis and compared our results with previously published sex-specific bivalve transcriptomes to identify conserved sex-determining genes. We also compared our data with other DUI species to identify candidate genes possibly involved in the regulation of DUI. A total of ∼12,000 orthologous relationships were found, with 2,583 genes differentially expressed in both species. Among these genes, key sex-determining factors previously reported in vertebrates and in bivalves (e.g., Sry, Dmrt1, Foxl2) were identified, suggesting that some steps of the sex-determination pathway may be deeply conserved in metazoans. Our results also support the hypothesis that a modified ubiquitination mechanism could be responsible for the retention of the paternal mtDNA in male bivalves, and revealed that DNA methylation could also be involved in the regulation of DUI. Globally, our results suggest that sets of genes associated with sex determination and DUI are similar in distantly-related DUI species.
Keywords: sex determination, mitochondrial DNA, doubly uniparental inheritance, comparative transcriptomics, Bivalvia
Introduction
Bivalves show extreme diversity in their reproductive systems, ranging from functional (simultaneous) hermaphroditism, alternative sexuality (sequential hermaphroditism), to strict gonochorism (i.e., species that exist as separate males and females). Both genetic and environmental factors appear to determine sex in most bivalve species studied to date (reviewed in Coe 1943; Chávez-Villalba et al. 2011; Breton et al. 2017). However, because sex determination has been studied in great detail only in oysters, the genetic and environmental factors that control sexual diversification in bivalves are still poorly known. An important reason for studying sex determination in bivalves relates to their biology and ecology: with ∼25,000 living species (www.bivatol.org), they are sufficiently diverse to provide a rich source of material to better understand the evolution of sex and sex determination in general, and to provide unique examples of sex-determining mechanisms, including the only possible example of a sex-determining system involving the mitochondrial genome in animals (e.g., Breton et al. 2011).
A variety of approaches have been undertaken to elucidate the mode of sex determination in bivalves. Globally, these studies demonstrated an absence of heteromorphic sex chromosomes, an effect of several environmental factors on sex-ratio (i.e., temperature, food availability, and pollutants, such as heavy metals organochlorines and exogenous steroids), and a polygenic architecture of sex determination (Breton et al. 2017). Genes homologous to sex-determining pathway genes in other animals have been identified in several bivalve species. For example, homologs of Sry (sex determining region-y)-box 30 (Sox 30) and Dmrt1 (doublesex and mab-3 related transcription factor 1), known for their role in male sex determination in nematodes, fruit flies and vertebrates (Wallis et al. 2008; Kopp 2012), have been found in the marine clam Ruditapes philippinarum, and in some species of scallops and oysters (Yu et al. 2011; Ghiselli et al. 2012; Llera-Herrera et al. 2013; Teaniniuraitemoana et al. 2014; Zhang et al. 2014; Li et al. 2016). Several other genes known to act in sex determination in other animals or during early gonadal differentiation have been identified in bivalves, such as Oyvlg and vasph, that is, homologues of vasa, which is a gene involved in primordial germ cell development and early sex differentiation in eukaryotes; Cg-SoxE, a homologue of Sox9, and β-catenin, which are respectively expressed when sex is still not distinguishable (or in mature females and vitellogenic oocytes in Crassostrea gigas [Santerre et al. 2014]); and CgFoxL2, a homologue of FoxL2, a key gene involved in ovarian determination in vertebrates (Fabioux et al. 2004; Uhlenhaut and Treier 2006; Milani et al. 2011, 2015; Zhang et al. 2014).
An unconventional sex-determining mechanism involving doubly uniparental inheritance (DUI) of mitochondrial DNA has been hypothesized for bivalves (e.g., Breton et al. 2011; Milani et al. 2016). This unique system of mitochondrial transmission in the animal kingdom has been discovered in >100 species from different bivalve orders (Mytiloida, Unionoida, Veneroida, and Nuculanoida; Gusman et al. 2016). DUI involves two distinct mitochondrial genomes that are transmitted in a sex-specific way: the female-type, or F-type, transmitted through eggs and usually found in both male and female somatic tissues and in female gonadic tissues, and the male type, or M-type, transmitted through sperm and usually found in male gametes (reviewed in Breton et al. 2007; Passamonti and Ghiselli 2009; Zouros 2013). In addition to the typical set of 13 mtDNA-encoded proteins (Gissi et al. 2008; Capt et al. 2015), novel sex-associated mtDNA-encoded proteins have been found in bivalves with DUI; F-orf in the F mtDNA and M-orf in the M mtDNA, both of which are relatively conserved across species within a family (Breton et al. 2009, 2011; Milani et al. 2013; Mitchell et al. 2016). These genes were hypothesized to be key elements of a sex-determination system involving mitochondria, that is, they may play a role in the maintenance of a gonochoric reproductive system with separate male and female sexes (Breton et al. 2011). This hypothesis came from the observation that gonochorism in freshwater unionoid mussels is absolutely correlated with the presence of DUI and of these novel sex-specific proteins, whereas closely related hermaphroditic species lack the M mtDNA (i.e., possess strict maternal inheritance of mtDNA) and have macromutations in the F-orf gene in their F mtDNA (Breton et al. 2011). If true, this would make DUI the first animal sex-determination system involving the mitochondrial genome, and would explain its long-term persistence in bivalves. However, this hypothesis still has not been rigorously tested.
Ghiselli et al. (2012) published the first whole transcriptome analysis by RNA-Seq to better understand the mechanisms underlying DUI and sex determination in the Manila clam R. philippinarum. Because previous studies demonstrated that elimination of paternal mitochondria in mammal species with strict maternal inheritance of mtDNA is dependent on a mechanism involving the ubiquitin–proteasome system, the principal system for protein degradation (Sutovsky et al. 1999), it was proposed that a modification of the ubiquitination process in male bivalves with DUI would allow sperm mitochondria and their M genomes to escape this mechanism and invade germinal cells (Kenchington et al. 2002; Ghiselli et al. 2012). Furthermore, because ubiquitination is also involved in the regulation of gene expression and plays a role in sex determination of several animals (e.g., Caenorhabditis elegans and Drosophila; see Ghiselli et al. 2012 for a review of the literature), Ghiselli et al. (2012) proposed a relationship between ubiquitination, sex bias, and mitochondrial inheritance, and identified candidate ubiquitination genes, such as the ubiquitin activating enzyme 1 (uba-1) and proteasome subunit alpha 6 (psa-6), for further investigation. To date, this study remains the only transcriptomic analysis performed to identify genes involved in bivalve sex determination and DUI. Although an in-depth study of sex-specific genes has recently been conducted on male and female gonads of the gonochoric, DUI-possessing freshwater mussel Hyriopsis schlegelii (Shi et al. 2015), the link between DUI and sex determination has not been investigated. To sum up, a total of 45,422 genes differentially expressed in male and female gonads in H. schlegelii and key genes reported to govern sex-determination pathways in mammals were identified, including Sry, Dmrt1 and Sox9, upregulated in males, and Foxl2 and β-catenin, upregulated in females (Shi et al. 2015).
With the aim of unraveling the mechanisms underlying sex determination and DUI in freshwater mussels, we performed a comparative analysis of gonad transcriptome for the two DUI species Venustaconcha ellipsiformis (subfamily Ambleminae) and Utterbackia peninsularis (subfamily Anodontinae). Furthermore, we compared our results with published sex-specific transcriptomics data from the DUI freshwater mussel H. schlegelii and marine clams R. philippinarum and Ruditapes decussatus to identify conserved genes involved in the regulation of sex-specific aspects of DUI systems. Because of the many similarities found among the distantly-related DUI species (e.g., sex-ratio bias, mitochondrial behavior in early embryos, rates of evolution of the two genomes; see Zouros 2013 for a review), it was indeed expected that sets of genes associated with sex determination and DUI would be similar in distantly-related DUI species. We hypothesized that if a modification of the ubiquitination mechanism is responsible for the retention of the paternal mtDNA in male bivalves, then “molecular signatures” of this modification should be discernable in all DUI species.
Materials and Methods
Sample Collection and RNA Isolation
Adult specimens of V. ellipsiformis and U. peninsularis were collected in July 2014 from Straight River (Minnesota, USA; Lat 44.006509, Long −93.290899) and Suwannee River (Florida, USA; Lat 29.58684, Long −82.94095), respectively. Mussels were shipped alive to the Université de Montréal, where they were opened and sexed by microscopic examination of gonad smears. Mature gonads of all unambiguously sexed specimens were frozen at −80 °C until further analyses.
Total RNA was extracted from frozen gonad tissues with RNeasy Plus Universal Mini Kit (QIAGEN Inc., Valencia, CA) and treated with Turbo DNase (AMBION, Austin, TX) following the provided protocols. A total of 25 RNA extractions were performed, 14 for U. peninsularis (8 females and 6 males) and 11 for V. ellipsiformis (7 females and 4 males). The quality and quantity of extracted RNA were examined via electrophoresis on 1% agarose gel and BioDrop µLITE spectrophotometer. Sixteen samples (four males and four females for each species) were sent to the Institute for Research in Immunology and Cancer Institute (IRIC, Université de Montréal, Montréal, Canada) for Illumina library preparation and paired-end (2×100 bp) sequencing (Illumina HiSeq2000; San Diego, CA).
De Novo Transcriptome Assemblies
Prior to assembly, raw paired-end reads were trimmed with Trimmomatic (v0.32; Bolger et al. 2014), which is available as part of the Trinity transcriptome assembler (trinityrnaseq_r20140717; Grabherr et al. 2011), to remove low-quality reads (i.e., with Phred quality scores <30, or if the read contained more than five ambiguous nucleotides “N”). As part of the trimming process, reads similar to known polymerase chain reaction primers and Illumina adapter sequences were also removed by specifying the appropriate “ILLUMINACLIP” parameters. Reads quality was visualized with FastQC (v0.11.2; Andrews 2010). For each species, high quality reads were assembled de novo to provide species-specific reference transcriptomes using Trinity (Haas et al. 2013). Parameters were kept as default, except for the –min_contig_length, which was set at 300 to remove contigs shorter than 300 bp. This value was chosen for further annotation purposes, that is, according to the parameters fixed by TransDecoder that identifies ORFs with a minimum length of 100 amino acids to avoid false positives (https://transdecoder.github.io/).
In brief, Trinity combines overlapping reads to form longer fragments called contigs, which are subsequently clustered based on sequence similarity (i.e., grouping of related contigs that correspond to portions of alternatively-spliced transcripts or otherwise unique portions of paralogous genes). These data were then processed to extract full-length alternatively spliced isoforms and tease apart transcripts derived from paralogous genes (Grabherr et al. 2011) to acquire nonredundant unigenes.
The completeness of transcriptome assemblies was tested using Benchmarking Universal Single-Copy Orthologs (BUSCO) software (busco_v3, using the mode -tran) by comparing known core eukaryotic and metazoan genes with the contigs assembled by Trinity. BUSCO provides quantitative measures for the assessment of transcriptome completeness based on evolutionarily-informed expectations of gene content from near-universal single-copy orthologs selected from different downloadable data sets (Simão et al. 2015).
Annotation
Assembled contigs were searched against two databases, that is, Swiss-Prot and TrEMBL (Uniref90 clusters), of the Universal Protein Resource (UniProt) protein database (The UniProt Consortium, 2017) with BLASTp (E-value set at e−05) (Altschul et al. 1990). To do this, ORFs and associated protein-coding sequences were first predicted from the contigs using TransDecoder (r20140704; Haas and Papanicolaou 2017) and the longest ORF candidate was considered. BLASTx searches of assembled contigs against the SwissProt database were also performed to increase the proportion of annotated genes. Finally, BLASTp searches for both species were performed against the PANMDB database. Specifically, we used the reference data set constructed by Patnaik et al. (2016), which combines protein sequence data of Arthropoda, Nematoda, and Mollusca downloaded from the Taxonomy browser of the NCBI nr database and stored in the PANMDB (freely downloadable from the amino acid database BLAST web-interface of the Malacological Society of Korea; http://malacol.or.kr/blast/aminoacid.html; Kang et al. 2015).
Read Mapping, Differential Expression, and Functional Enrichment Analyses
The de novo transcriptomes assembled for each species served as reference for read mapping. Specifically, raw reads were mapped using Bowtie2 (v2.2.4; Langmead and Salzberg 2012), keeping the default parameters. Reads were sorted by name, and indexed with Samtools (v1.1; Li et al. 2009). Samtools idxstat was used to extract the numbers of reads mapped per contig and the results were then normalized according to Robinson and Oshlack (2010).
Two approaches were used to perform differential expression analyses between male and female gonads for each species, that is, one with the R package edgeR (version edgeR_3.12.0; Robinson et al. 2010) and one with DESeq2 (version DESeq2_1.10.1; Love et al. 2014). We used both approaches for comparative purposes, because multiple methods have been used to identify differentially expressed genes (DEG) between male and female gonads in bivalves [e.g., edgeR by Ghiselli et al. (2012) for the marine clam R. philippinarum; DESeq2 by Teaniniuraitemoana et al. (2014) for the oyster P. margaritifera; and Shi et al. (2015) used a statistical method developed by Audic and Claverie (1997) for the freshwater mussel H. schlegelii]. DESeq2 is more sensitive to outliers and generally gives lower true positive rates, whereas edgeR performs better to uncover true positives (Soneson and Delorenzi 2013; Zhang et al. 2014). For a complete comparison of both software programs, see Zhang et al. (2014).
Fragments per kilobase of exon per million fragment sequenced (FPKM) were obtained using the DESeq2 Bioconductor package (Love et al. 2014), specifying “robust = F” to use sum of raw counts, allowing analyses of differentially expressed unigenes (hereafter defined as DEG). With edgeR, DEG were selected based on FPKM values >1 in one or the other sex, and log2 fold change ≥1 and −1. Since DESeq2 removes low expression data when calculating False Discovery Rate (FDR) values, no filter was performed on FPKM values, and DEG were also selected based on log2-fold change ≥1 and ≤ −1. Only unigenes with P-values <0.05 after adjustment by FDR (Benjamini and Hochberg 1995) were considered as differentially expressed. The package edgeR (plotSmear) was used to visualize differential expression between gonad samples for each species. The proportion of unigenes that were differentially expressed according to both analytical methods was also measured.
To further understand these biased expressed unigenes, gene ontology (GO) enrichment analyses were performed for each species to test whether specific GO terms (biological process, molecular function, and cellular component levels) were over-represented in DEG using GOseq (v1.28.0; Young et al. 2010). An FDR corrected P-value of 0.05 was used for significance.
BLAST Reciprocal Analysis
A whole transcriptome reciprocal-best-BLAST-hits (RBH) analysis was applied to identify orthologous genes between V. ellipsiformis and U. peninsularis (e.g., to filter possible contamination- and species-specific transcripts). Specifically, each contig from V. ellipsiformis was searched against all contigs from U. peninsularis using the parameter p = blastn+ (default parameters were kept according to the proteinortho5.pl script, with an E-value set at e−05; Lechner et al. 2011), and conversely each sequence of U. peninsularis was searched against all sequences of V. ellipsiformis. This step allowed for identification of orthologous genes between the species (e.g., Zhao et al. 2014). From these orthologous gene sets, the orthologs found to be differentially expressed between males and females both in U. peninsularis and V. ellipsiformis using edgeR and DESeq2 were studied more thoroughly. Transcription level and annotation were individually verified for each DEG in each species to confirm the reliability of our approach, to identify orthologous sex-biased genes that may be involved in sex determination, molecular controls of DUI, or both.
Results and Discussion
Sequencing and Assemblies of the Transcriptomes
For U. peninsularis, ∼43 million paired end reads per individual were produced by the Illumina HiSeq2000 sequencing, generating a total of 348,652,616 raw reads of 2×100 bp. After trimming, 288,320,738 (83%) high-quality reads were retained for transcriptome assembly. The de novo assembled transcriptome using Trinity resulted in 200,961 contigs with a N50 of 1,655 bp and average contig length of 1,026 bp (table 1). These contigs were assembled into a total of 165,788 unigenes.
Table 1.
De Novo Assembly Quality Statistics of Utterbackia peninsularis and Venustaconcha ellipsiformis Transcriptomes
| U. peninsularis | V. ellipsiformis | |
|---|---|---|
| Total raws | 348,652,616 | 333,708,540 |
| Average raws | 43,581,577 | 41,713,567 |
| Total trimmed raws | 288,320,738 | 320,230,962 |
| Average trimmed raws | 36,040,092 | 40,028,870 |
| Total trinity “unigenes” | 165,788 | 221,362 |
| Total trinity contig | 200,961 | 285,260 |
| Percent GC | 33.36 | 37.58 |
| Contig N10 | 5,581 | 6,494 |
| Contig N20 | 3,895 | 4,650 |
| Contig N30 | 2,916 | 3,560 |
| Contig N40 | 2,224 | 2,731 |
| Contig N50 | 1,655 | 2,083 |
| Median contig length | 558 | 595 |
| Average contig | 1,026 | 1,171 |
| Total assembled bases | 206,277,984 | 334,013,631 |
| Unigene N10 | 4,882 | 5,792 |
| Unigene N20 | 3,298 | 3,896 |
| Unigene N30 | 2,409 | 2,789 |
| Unigene N40 | 1,750 | 2,019 |
| Unigene N50 | 1,251 | 1,433 |
| Median unigene length | 510 | 516 |
| Average unigene | 884 | 944 |
| Total assembled bases | 146,628,566 | 208,707,657 |
For V. ellipsiformis, 41 million reads per individual were obtained on average, giving a total of 333,708,540 raw reads generated by Illumina sequencing. After trimming, 320,230,962 (96%) clean reads were obtained and assembled into 285,260 contigs (N50 of 2,083 bp, average contig length of 1,171 bp), of which 221,362 were considered as unigenes and kept for further analyses (table 1).
Assembly quality (assembled contigs) assessed by the representation of 978 core eukaryotic and metazoan genes using BUSCO resulted in 96.8% completely recovered genes for U. peninsularis (with 24.1% duplicated, 3% fragmented, and 7% missing), and 97.8% completely recovered genes for V. ellipsiformis (with 34.4% duplicated, 0% fragmented, and 2% missing) (supplementary fig. S1, Supplementary Material online). In theory, the relatively high number of duplicates may represent alternatively spliced forms, gene duplication, or allelic variation (heterozygosity) in the samples used to construct the assemblies. Therefore, we ran the same BUSCO analysis using the assembled unigenes (i.e., filtered for alternatively spliced forms and paralogs) to better understand the origin of our duplicates (supplementary fig. S1, Supplementary Material online). This analysis still showed a similar number of completely recovered genes (96.6% for U. peninsularis and 97.5% for V. ellipsiformis), but a markedly lower number of duplicates (13.9% and 24.3%, respectively), suggesting that gene duplication and/or alternative splicing may contribute to the relatively high number of duplicates. Nevertheless, the high number of complete genes that was recovered provides an important validation of the completeness of both assemblies.
For both species, the raw sequence data in FASTQ format have been submitted to the National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database and are accessible under accession numbers SRR6279376-83 (U. peninsularis) and SRR6279374-75 and SRR6279384-89 (V. ellipsiformis).
Sequence Annotation
BLASTp searches against SwissProt and Uniref90, BLASTx against SwissProt, and BLASTx against PANMDB, respectively, allowed annotating 26,469 (13.1%), 32,515 (16.2%), 28,495 (14.2%), and 32,503 (16.1%) contigs for U. peninsularis. All databases together gave a total annotation success of 16.8% contigs. With SwissProt, most of the matches came from the extensively studied taxon Mammalia (supplementary fig. S2, Supplementary Material online). With Uniref90, the largest proportion of hits (46.4%) came from the oyster species C. gigas (fig. 1), for which whole-genome sequencing and annotation were recently completed (Zhang et al. 2012). The second largest proportion of hits (23.6%) came from the molluscan class Gastropoda (fig. 1). These results are comparable with those reported in other recent de novo transcriptome sequencing studies for freshwater mussels or other bivalve species (i.e., 15–22% annotated contigs/unigenes; Shi et al. 2015; Patnaik et al. 2016; Yarra et al. 2016; Wang et al. 2017) and indicate that it can be challenging to provide good annotations for nonmodel organisms for which few genomic resources are available. Also, very few contigs matched bacterial sequences (or other possible contaminating species such as trematodes), suggesting negligible contamination for U. peninsularis, and indicating that the assembly for this species comprised mainly gonadal coding RNAs.
Fig. 1.
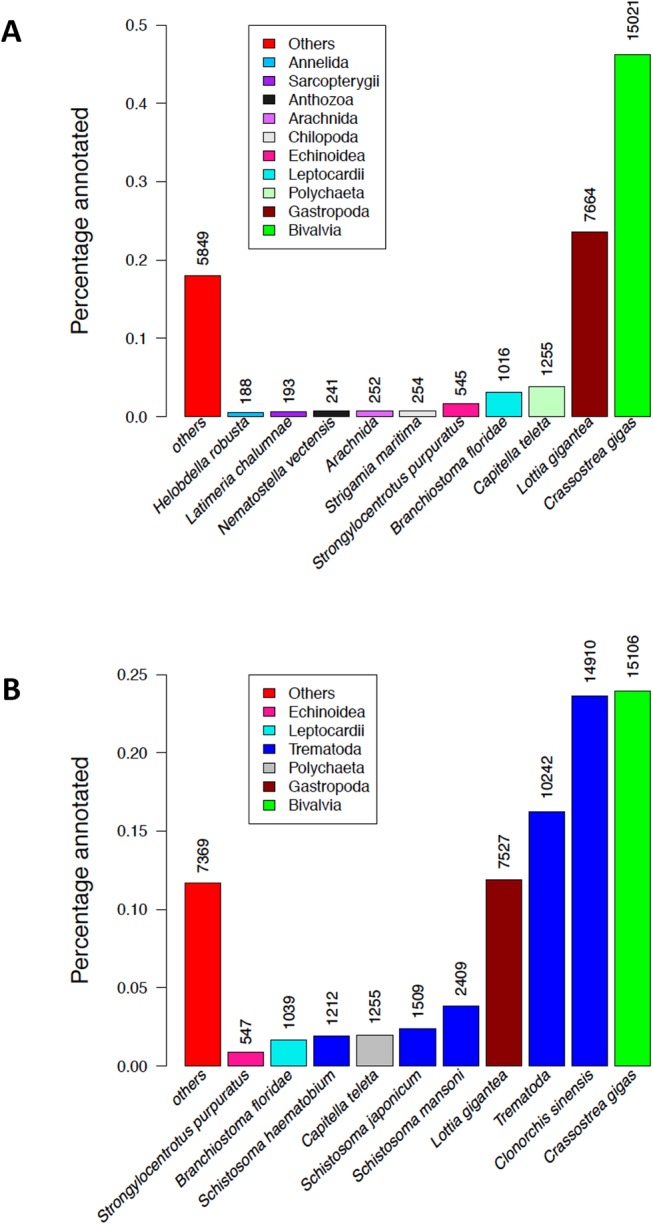
—Taxonomic distribution of top BLASTp hits in Uniref90 protein database for U. peninsularis contigs (A) and V. ellipsiformis contigs (B). Numbers of unique hits in each group are shown.
These results obtained for U. peninsularis, however, are in sharp contrast with those obtained for V. ellipsiformis, for which 63,126 (22.1%) contigs were annotated with Uniref90 but with almost half of them matching trematode species (fig. 1). The fractions of contigs annotated with SwissProt and PANMDB were also slightly larger for V. ellipsiformis, with 49,904 (17.5%) and 50,444 (17.7%) contigs annotated with BLASTp and BLASTx searches against SwissProt (see supplementary fig. S2, Supplementary Material online), and 57,215 (20%) contigs annotated with PANMDB. Again, several matches came from C. gigas with Uniref90 (fig. 1), but the discovery that ∼40% of the annotated contigs had a significant match with trematode species, which is not the case for U. peninsularis, strongly suggest a trematode infection in V. ellipsiformis, a phenomenon that is often observed in freshwater mussels, which are intermediate hosts of several trematode species (Gangloff et al. 2008). This contamination most likely explains the higher number of duplicates obtained with BUSCO as well as the higher percentage of annotated contigs for V. ellipsiformis. We performed additional analyses to verify the extent of this contamination in our samples, and found that three of four female samples, but no male samples, produced several hits when blasted against the three most expressed trematode transcripts (data not shown). This is consistent with recent report that mature female freshwater mussels are more susceptible to trematode infection (Müller et al. 2015).
We discarded contaminant trematode sequences from the transcriptome of V. ellipsiformis by relying on annotation to remove Platyhelminthes transcripts (e.g., Sayadi et al. 2016). Although this approach may result in the significant loss of mussel-specific data, we considered it to be a valid approach for retaining the maximum number of sequences related to our focal species (V. ellipsiformis). Alternatively, this approach can lead to poor decontamination, that is, with many nonannotated contigs belonging to nontarget or contaminant species. However, because the ultimate goal of this study was to identify similar sets of genes associated with sex determination and DUI in two different freshwater mussel species, we tried to minimize the contamination problem by using the best matching reciprocal BLAST between both species to filter out contamination-specific (and species-specific) contigs (see below). A total of 25,179 (40%) annotated contigs were considered as probable contaminants and discarded. In total, we retained 37,947 genes with significant blast hits in our downstream analyses.
DEG between Male and Female Gonads in Each Species
The differential gene expression analysis between sexes for U. peninsularis revealed 7,281 DEG with edgeR, and 23,107 DEG with DESeq2. The DEG identified using DESeq2 contained all the DEG identified with edgeR (fig. 2). A total of 4,315 unigenes were shown to be upregulated in males versus 2,966 in females with edgeR, whereas 14,216 genes were considered upregulated in males versus 8,891 in females with DESeq2. This is a smaller number than what was found in the DUI-possessing freshwater mussel H. schlegelii using a statistical text proposed by Audic and Claverie (1997) (45,422 DEG, with 19,511 unigenes upregulated in male gonads vs. 25,911 in female gonads; Shi et al. 2015), but a substantial number considering what has been revealed for example in the oyster species Pinctada margaritifera using DESeq2 (1,993 DEG, with 1,419 contigs upregulated in male gonads vs. 574 in female gonads; Teaniniuraitemoana et al. 2014) and in the DUI-possessing marine clam R. philippinarum using edgeR (1,575 DEG, with most of the genes being male biased; Ghiselli et al. 2012). Although different DEG analytical methods and experimental setups may explain these dissimilarities, it was suggested that a low number of DEG should be expected in bivalves considering their general lack of secondary sexual characters and sexual dimorphism, and also because sex-specific function of reproductive genes in these organisms is limited to gonad development, gametogenesis, and fertilization (Ghiselli et al. 2012). However, contrary to most bivalve species, unionoid freshwater mussels have a unique reproductive mode that may cause pronounced sexual dimorphism: 1) their larvae mature in certain areas unique to female gills called marsupia (which can also alter shell morphology) and 2) because most species require a host fish for their larvae, many gravid females, including those of V. ellipsiformis (Allen et al. 2007), display adaptations such as mantle-derived lures to actively attract a prospective fish for their parasitic larvae (Haag and Warren 1999; Barnhart et al. 2008; Zieritz and Aldridge 2011; Haag 2012). Interestingly, sexual dimorphism in burrowing behavior has also been reported in the freshwater mussel DUI species Elliptio complanata (Flynn et al. 2013). To our knowledge, however, no sexual dimorphism has been reported in U. peninsularis. Most of the DEG found in this species are male-biased, which is consistent with what has been found in most bivalve species, as well as in many other animals (Ghiselli et al. 2012; Teaniniuraitemoana et al. 2014; Peng et al. 2015; Li et al. 2016), and may be explained by a higher proportion of essential functions for female-biased genes (those shared by the two sexes) than for male-biased genes (see Ghiselli et al. 2012). This explanation is also consistent with the previous assumption that male development is associated with an activation of several testis-specific genes and/or a repression of several genes vital for ovarian development (see Peng et al. 2015).
Fig. 2.

—Smear plots of DEG between male and female gonads in U. peninsularis (A) and V. ellipsiformis (B). Black dots represent genes normally expressed in both tissues. Green dots, DEG revealed by edgeR software; orange dot, DEG revealed by DESeq2. Green dots surrounded by orange represent genes considered as DEG by both software packages. Positive fold-changes represent genes upregulated in males, while negative ones represent genes upregulated in females.
For V. ellipsiformis, 11,408 DEG were revealed with edgeR and 52,257 DEG with DESeq2 (fig. 2). Only 60 unigenes were upregulated in males versus 11,348 in females with edgeR, whereas DESeq2 revealed 3,134 unigenes upregulated in males and 49,123 in females. This important female-biased gene expression in V. ellipsiformis is most likely explained by trematode contamination found in three out of four female gonad samples (see the annotation section above). After having filtered out 25,179 Platyhelminthes contigs from our data, the number of DEG did not vary much with 9,769 DEG revealed by edgeR (765 upregulated in males and 9,004 in females) and 52,652 revealed by DESeq2 (3,317 upregulated in males and 49,335 in females). These numbers, although not surprising since the percentage of annotation (< 22%), was very low, either suggest 1) that the contamination could not be completely removed from our data using the approach described above, or 2) that changes in female host gene expression occurred in response to parasite infection (e.g., Wijayawardena et al. 2016). Fortunately, because our U. peninsularis samples were not infected by trematodes, our BLAST reciprocal analysis discussed below allowed us to mitigate this problem and to accomplish our main objective, that is, identify similar sets of genes associated with sex determination and DUI in two different freshwater mussel species.
BLAST Reciprocal Analysis and Enriched GO Terms and Pathways in Male and Female Gonads
To identify the main genes possibly involved in sex determination and/or DUI in U. peninsularis and V. ellipsiformis, we performed a whole transcriptome BLAST analysis. From the reciprocal BLAST results, we selected the genes found to be differentially expressed in U. peninsularis in our previous analyses using edgeR and DESeq2, and verified their sex-bias in V. ellipsiformis DEG filtered for “flatworm contigs” (i.e., we verified that every male-biased or female-biased gene in one species was also male-biased of female-biased in the other).
A total of ∼12,000 orthologous relationships was found between both species, 16,740 when isoforms were included. From these orthologous genes, 2,342 were found to be differentially expressed in U. peninsularis by edgeR and 2,583 (including the 2,342 from edgeR) by DESeq2. Specific attention was given to these 2,583 genes (1,568 male-biased and 1,015 female-biased) to increase the probability of identifying candidate genes that could have a role in sex determination and/or DUI. Of the 1,568 male-biased orthologous genes, 1,432 (91.3%) had a significant BLASTp (or BLASTx) match in the SwissProt and/or UniRef90 protein databases (E-value set at e−05), whereas of the 1,015 female-biased orthologous genes, 903 (89%) had a significant BLASTp (or BLASTx) match in the SwissProt and/or UniRef90 protein databases (supplementary table S1, Supplementary Material online). The same genes were also found to be differentially expressed in V. ellipsiformis.
Using all annotated and differentially expressed orthologous genes, GOSeq analyses revealed 57 ontology categories significantly enriched in both species (supplementary table S2, Supplementary Material online). Top GO terms enriched in female and male gonads of both species are shown in figure 3 (for complete GOSeq results for U. peninsularis and V. ellipsiformis after trimming for trematode contigs, see supplementary tables S3 and S4, respectively, Supplementary Material online). In transcriptomes of both species, GO terms show higher counts for male-biased genes. At the biological process level, single-organism cellular process, cellular component organization or biogenesis, and organelle organization had the top counts. The intracellular and organelle components represented the majority of terms at the cellular component level, whereas at the molecular function level, protein binding was the most represented term (fig. 3). Similar results were also obtained for other bivalve species, including the oyster Crassostrea hongkongensis (Tong et al. 2015), the scallop Patinopecten yessoensis (Li et al. 2016), as well as the DUI-containing marine clam R. philippinarum (Ghiselli et al. 2012) and freshwater mussel Cristaria plicata (Patnaik et al. 2016). Unexpectedly, “reproduction” and “ubiquitination,” the two categories on which Ghiselli et al. (2012) previously focused because of their direct relationship with sex determination and DUI, were not significantly overrepresented in our results. We thus concentrated our effort on the reciprocal BLAST/DEG analyses.
Fig. 3.

—Significantly enriched-GO terms involved in molecular function (top), biological process (middle), and cellular component (bottom) in ovary (pink) and testis (blue) of both species. Analyses were performed on DEGs from blast reciprocal analyses between V. ellipsiformis and U. peninsularis. X-axis shows the number of DEGs contained in each GO category.
Identification of Candidate Genes Involved in Sex Determination
To identify candidate genes that contribute to sex determination/differentiation, or maintenance in mature gonads, we first searched in our reciprocal BLAST/DEG results for sex-specific expression patterns (i.e., transcribed only in one sex)—as expected for sex-determining genes (table 2). In addition, we identified the top ten upregulated (and annotated) genes in male and female gonads of both freshwater mussel species (table 3). Among these, six genes were exclusively expressed in male gonads and three in female gonads. Not surprisingly, these sex-specifically expressed and upregulated genes appear to play roles in spermatogenesis and oogenesis, which are the ultimate biological processes in the male and female gonads, and in reproduction in general. In males, for example Kelch-like protein 10 and Proteasome activator complex are members of the E3 ubiquitin–protein ligase complex process, which mediates the ubiquitination and subsequent proteasomal degradation of target proteins during spermatogenesis (Arama et al. 2007). In addition, sperm motility kinase 2B and WD repeat-containing protein on Y chromosome are involved in sperm fertility and motility, whereas testis-specific serine/threonine-protein kinases 3 and 5 are involved in germ cell survival during meiosis (e.g., Peng et al. 2015). Among other male-biased sequences, we also identified genes homologous to spermatogenesis-associated protein (SPATA 4, 17, 22, and 24), sperm flagellar protein 1/2, sperm motility kinase, sperm-associated antigen and surface protein, and spermine oxidase potentially expressed in the male reproductive tissues (supplementary table S4, Supplementary Material online).
Table 2.
Sex-Specifically Expressed Genes in Male and Female Gonads of Utterbackia peninsularis and Venustaconcha ellipsiformis
| Unigene ID | Gene Symbol | Description | Sex (fold change) |
|---|---|---|---|
| c43183_g1 | – | Uncharacterized protein | M (10) |
| c63770_g1 | – | Uncharacterized protein | M (10) |
| c106662_g8 | TSSK3_MOUSE | Testis-specific serine/threonine-protein kinase 3 | M (11) |
| c69491_g1 | RDS2_CHICK | Photoreceptor outer segment membrane glycoprotein 2 | M (12) |
| c106233_g1 | EDR1_ARATH | Serine/threonine-protein kinase EDR1 | M (12) |
| c55198_g1 | CE049_MOUSE | Uncharacterized protein C5orf49 homolog | M (12) |
| c85464_g1 | CALL3_MOUSE | Calmodulin-like protein 3 | F (9) |
| c66486_g1 | SMCO3_MOUSE | Single-pass membrane and coiled-coil domain-containing protein 3 | F (13) |
| c237103_g1 | – | Uncharacterized protein | F (13) |
Note.—Unigene ID indicates the names of the unigene given by Trinity to the assembly files. Gene symbol indicates the annotation format provided by the Uniprot database followed by a quick short description. Sex indicates in which sex the unigene is specifically expressed, fold change values (obtained from DESEq2 software) are based on the results obtained for the noncontaminated U. peninsularis.
Table 3.
Top Ten Genes Showing the Greatest Difference in Expression in Male and Female Gonads of Utterbackia peninsularis and Venustaconcha ellipsiformis
| Unigene ID | Gene Symbol | Description | Sex (fold change) |
|---|---|---|---|
| c1056_g1 | K1S6Q8_CRAGI | Serine/threonine-protein phosphatase 6 regulatory ankyrin repeat subunit B | M (12) |
| c235886_g1 | KLH10_HUMAN | Kelch-like protein 10 | M (11) |
| c104796_g5 | K1RU43_CRAGI | WD repeat-containing protein on Y chromosome | M (11) |
| c47982_g1 | SAM15_MACFA | Sterile alpha motif domain-containing protein 15 | M (11) |
| c847_g1 | K1Q7E2_CRAGI | Sperm motility kinase X | M (11) |
| c46585_g1 | K1RFU6_CRAGI | Proteasome activator complex subunit 3 | M (11) |
| c173681_g1 | K1RFD4_CRAGI | Probable 4-coumarate–CoA ligase 3 | M (11) |
| c44943_g1 | PYG_DROME | Glycogen phosphorylase | M (11) |
| c36459_g1 | CC151_BOVIN | Coiled-coil domain-containing protein 151 | M (11) |
| c110789_g1 | K1R8H1_CRAGI | Testis-specific serine/threonine-protein kinase 5 | M (11) |
| c69487_g1 | LECG_THANI | Galactose-specific lectin nattectin; | F (14) |
| c92390_g1 | LECG_THANI | Galactose-specific lectin nattectin; | F (14) |
| c82290_g1 | LECM2_ERYPO | C-type lectin lectoxin-Lio2; | F (14) |
| c83693_g1 | PSM_MYTCA | Shell matrix protein; | F (14) |
| c79356_g1 | LECM1_PHIOL | C-type lectin lectoxin-Phi1; | F (14) |
| c99224_g1 | PLC_HALLA | Perlucin; | F (13) |
| c173670_g1 | LPSBP_PERAM | Hemolymph lipopolysaccharide-binding protein; | F (13) |
| c96153_g1 | LECG_THANI | Galactose-specific lectin nattectin; | F (13) |
| c105339_g1 | LECM2_ERYPO | C-type lectin lectoxin-Lio2; | F (13) |
| c55454_g1 | CL17A_HUMAN | C-type lectin domain family 17, member A; | F (13) |
Note.—Unigene ID indicates the names of the unigene given by Trinity to the assembly files. Gene symbol indicates the annotation format provided by the SwissProt or Uniprot database followed by a short description. Sex indicates in which sex the unigene is upregulated, fold change values (obtained from DESEq2 software) are based on the results obtained for the noncontaminated U. peninsularis
Specifically expressed and top upregulated genes in females were mostly lectin-type genes, which are often highly represented in ovary transcriptomes in animal species, including bivalves. These genes have a number of biological functions such as immune defense, block to polyspermy at fertilization, and sperm–egg recognition (Luckenbach et al. 2008; Moy et al. 2008; Patnaik et al. 2016). Among other female-biased genes, we identified vitellogenin, putative vitellogenin receptor, and Hsp90 organizing protein, which are involved in vitellogenesis, a central process in oogenesis. We also found cyclin-dependent kinase 2-associated protein 2, which is involved in another vital process in oogenesis, that is, oocyte maturation, and superoxide dismutase [Cu–Zn], which could neutralize reactive oxygen species and ensure oocyte quality (Peng et al. 2015 and above).
To better identify genes related to sex-determining pathways, we also searched in our reciprocal BLAST/DEG results for candidate genes previously identified in the literature in 1) the model organisms Ca. elegans, Drosophila melanogaster, Mus musculus (see Zhang et al. 2014 for the list); 2) four bivalve species without DUI, that is, the oysters C. gigas and C. honkongensis, the scallop P. yessoensis and the clam R. decussatus (Zhang et al. 2014; Tong et al. 2015; Li et al. 2016; Ghiselli F, Iannello M, Puccio G, Chang PL, Plazzi F, Nuzhdin SV, Passamonti M. unpublished data); and 3) two bivalve species with DUI, that is, the marine clams R. philippinarum and the freshwater mussel H. schlegelii (Ghiselli et al. 2012; Shi et al. 2015). Of the 26 key sex-determining pathway genes examined, putative homologs were found for 8 genes or gene families in our two freshwater mussel species (table 4; supplementary table S5, Supplementary Material online). Of these, four genes (Sry, Fem-1, MAB-3, and FoxL2) were differentially expressed but did not show sex-specific expression in our samples, although we cannot rule out the possibility that they may have sex-specific expression at an earlier developmental stage. For example, Sry/Sox30 was found to be expressed only in male oysters and clams (table 4).
Table 4.
Key Sex-Determining Pathway Genes in Model Organisms and Bivalve Species
| Gene | Caenorhabditis elegans | Drosophila melanogaster | Mus musculus | Crassostrea gigas | Crassostrea hongkongensis | Patinopecten yessoensis | Ruditapes decussatus | Ruditapes philippinarum | Hyriopsis schlegelii | Utterbackia peninsularis/Venustaconcha ellipsiformis |
|---|---|---|---|---|---|---|---|---|---|---|
| Amh (Mmu) | ms | |||||||||
| Amhr2 (Mmu), | ||||||||||
| Bar-1 (Cel), Arm (Dme), Ctnnb1 (Mmu) | fs | fb | ||||||||
| Cbx2 (Mmu) | ms | |||||||||
| Doa (Dme) | fs | |||||||||
| Dsx (Dme), Dmrt1 (Mmu), Mab-3 (Cel) | ms | ms | ms | ms | ms | mb | mb | mb | ||
| Fog-1,-2,-3 (Cel, Dme, Mmu) | ms | ms | ||||||||
| Fem-1 (Cel, Dme), Fem1b (Mmu) | ms | mb | mb | mb | ||||||
| FoxL2 (Mmu) | fs | fs | fs | fb | fb | fb | ||||
| Fru (Dme) | ms | |||||||||
| Zglp1 (Mmu) | ||||||||||
| Her-1 (Cel), Her (Dme) | fs | |||||||||
| Nr0b1 (Mmu) | ms | |||||||||
| Rspo1 (Mmu) | fs | fb | ||||||||
| Rnt-1 (Cel), Run (Dme) | fs | |||||||||
| Sdc (Cel, Dme, Mmu) | fs | |||||||||
| Sxl (Dme) | fs | |||||||||
| Sry/Sox30/SoxH (Mmu) | ms | ms | ms | mb | ms | ms | mb | mb | ||
| Sis-a (Dme) | fs | |||||||||
| Sf1 (Mmu) | ms | |||||||||
| Sox9 (Mmu), Sox100B (Dme) | ms | mb | ||||||||
| Gata4 (Mmu) | mb | mb | ||||||||
| Tra-1 -2 -3 (Cel), Tra2 (Dme) | fs | fs | fb | |||||||
| Wt1 (Mmu) | ms | mb | ||||||||
| Wnt4 (Dme, Mmu) | fs | fb | ||||||||
| Xol-1 (Cel) | ms |
Note.—Names in parentheses indicate the species in which each gene has been identified; shaded box indicates presence of the gene in the genome/transcriptome; fs, female-specific expression (i.e., transcribed only in females); ms, male-specific expression; fb, female-biased expression; mb, male-biased expression. See supplementary table S5, Supplementary Material online, for a short description of each gene
As previously mentioned, both Sry (sex determining region-y)-box 30 (Sox 30) and Dmrt1 (MAB-3 and dsx related transcription factor 1) are known to be involved in male sex determination in nematodes, fruit flies, and vertebrates (Wallis et al. 2008; Kopp 2012), whereas FoxL2 is known for its role in ovarian determination in vertebrates (Uhlenhaut and Treier 2006). Fem-1 encodes an ankyrin repeat-containing protein orthologous to human FEM1A and is involved in male sex-determination in Ca. elegans, and also in the protein ubiquitination pathway (Doniach and Hodgkin 1984). Two other genes that seem well represented in bivalves, and more generally in mollusks (Kim et al. 2017), are Bar-1/Arm/Ctnnb1 (Beta-catenin) and Tra-1 (table 4). Tra-1 controls female somatic sex differentiation in Drosophila, whereas Drosophila Beta-catenin homolog is named Armadillo and is known to be involved (among other processes) in oogenesis and ovarian follicle cell development. The Rspo1/Wnt/Beta-catenin pathway is also involved in bird sex determination (Beukeboom and Perrin 2014).
Collectively, the results presented in table 4 suggest shared sex-determining pathway genes in distantly-related, as well as in SMI and DUI bivalves. Zhang et al. (2014) suggested a deeply conserved role in sex determination for Dmrt1/Dsx/MAB-3 in both invertebrates and vertebrates, but also for Sry and FoxL2, even if they were thought to be new recruits to sex-determining pathways in vertebrates or placental mammals (Gamble and Zarkower 2012; Matson and Zarkower 2012). Indeed, some steps of the sex determination pathway may be deeply conserved in metazoans, despite rapid evolution of the regulatory pathways that in bivalves, for example, may involve both genetic and environmental factors (Zhang et al. 2014; Breton et al. 2017).
Zhang et al. (2014) proposed a working model for sex determination in the oyster C. gigas, in which CgSoxH, a Sry-like gene that is strictly expressed in testis, would play a leading role in the sex-determining pathway by directly or indirectly activating CgDsx, a DM domain gene like those (e.g., Dmrt1) that have been identified as master switches for testis development in animal species studied so far. Both CgSoxH and CgDsx would interact with, or inhibit, CgFoxL2, which is usually specifically expressed in ovaries (this scenario is consistent with the reported interaction among Sry, Dmrt1, and FoxL2 in mammals). Even if the number of species studied is still limited, we suggest that this general model probably applies to most bivalve species. We also propose that major interspecific differences in bivalves will be found in cis/trans genetic elements and/or environmental factors that may control these three “sex-determining key genes.” For example, a mito-nuclear sex determination system in which mitochondrially encoded elements distorting sex ratios has been hypothesized to occur in species with DUI (e.g., Breton et al. 2011; Milani et al. 2016; Pozzi et al. 2017). In both plants and animals, cases of mitochondrial genomes acting on germ line development and/or with sex ratio distortion properties have been documented (e.g., Chase 2007; Perlman et al. 2015). In bivalves with DUI, the existence of two sex-specific mitochondrial genomes on which selection can operate represents a possible source of novel sex determination mechanisms.
Identification of Candidate Genes Involved in DUI
As mentioned earlier, a modification of the ubiquitin–proteasome system has been hypothesized as responsible for the retention of sperm mitochondria in male embryos of species with DUI (Kenchington et al. 2002; Ghiselli et al. 2012). To identify putative factors that could be involved in the degradation of sperm mitochondria through ubiquitination in DUI bivalves, Milani et al. (2013) analyzed structure and localization of three transcripts—that is, baculoviral IAP repeat-containing 4 (birc), proteasome subunit alpha 6 (psa), and AN1 zinc finger ubiquitin-like domain (anubl1)—previously shown to present sex-specific and family biases (i.e., family producing predominantly females or males) in the Manila clam R. philippinarum. In situ hybridization confirmed the localization of these transcripts in gametogenic cells (Milani et al. 2013). Also, homologs of these genes were shown to be involved in reproduction and ubiquitination in other animals: PSA is a subunit of the proteasome, a complex known to be involved in male sexual differentiation, whereas ANUBL1 possesses an ubiquitin domain, and BIRC acts as an inhibitor of apoptosis (Milani et al. 2013). For these reasons, the authors hypothesized that these genes could have a role in sex determination and could also be responsible for the maintenance/degradation of spermatozoon mitochondria during embryo development of the DUI species R. philippinarum. It is worth mentioning that PSA has also been reported in male-biased families in the DUI species Mytilus edulis (Diz et al. 2013), supporting the hypothesis of an involvement of the ubiquitination system (and of the proteasome subunit alpha) in sex determination in bivalves.
To identify genes related to the DUI mechanism, we searched our reciprocal BLAST results for genes previously suggested to be involved in this process in the DUI species R. philippinarum and M. edulis, that is, for genes involved in ubiquitination that show male-biased transcription, indicating their potential role in spermatogenesis (Ghiselli et al. 2012; Diz et al. 2013; Milani et al. 2013). Annotation, function, and expression ratio of male-biased (and female-biased) ubiquitination genes are reported in supplementary table S1, Supplementary Material online. Compared to females in which 18 ubiquitination genes were differentially expressed, 53 ubiquitination genes were male-biased. Among them, psa6 and anubl1, two male-biased ubiquitination genes were absent from female-biased genes, as were birc5 and birc2. These results, consistent with the hypothesis of Milani et al. (2013), suggest that psa6 might be important in sex determination and/or differentiation in DUI species, that anubl1 could be a factor tagging sperm mitochondria that differentiate them from egg mitochondria, and that birc could be an additional mitochondrial tag that would protect the midpiece mitochondria from degradation. Our results are also consistent with our hypothesis that sets of genes associated with sex determination and DUI are similar in distantly-related DUI species, as exemplified here with freshwater mussels versus a marine clam and a marine mussel. However, additional RNA- and protein-based studies such as those conducted by Milani et al. (2013) and Diz et al. (2013) will be needed to confirm our results.
Lastly, although speculative, one of the factors that could also influence inheritance of mitochondrial genes is DNA methylation. For example, in the green alga Chlamydomonas, maternal inheritance of chloroplast DNA is thought to be influenced by DNA methylation (Sager and Grabowy, 1983; Umen and Goodenough 2001). A growing body of literature has also shown that epigenetic modifications, such as DNA methylation, can also regulate sex determination and differentiation (see Liu et al. 2015). Strikingly, in the leafhopper Zyginidia pullula, the symbiont Wolbachia pipientis disrupts male imprinting by modifying methylation patterns in gonads; as a result, Wolbachia feminizes genetic males of Z. pullula (Negri et al. 2009). In our results, transcripts associated with DNA methyltransferases, histone deacetylases, and histone acetyltransferases were identified, and most showed male-biased expression (supplementary table S1, Supplementary Material online). Among these male-biased transcripts, DNMT1, a DNA methyltransferase associated with mitochondria, was detected. However, because the epigenetic mechanisms underlying organellar DNA inheritance and/or sex determination/differentiation are still poorly understood, we cannot infer any detailed functions of these genes from their expression patterns. Future studies, currently underway in our laboratory, are clearly needed to reveal the complex interplay between methylation, sex bias, and mitochondrial inheritance in bivalves with DUI.
Conclusion
In this study, we present the first comparative gonadal transcriptomic analysis of two freshwater mussel species aimed at assessing the link between DUI of mtDNA and sex determination in Unionoida. Using a BLAST reciprocal analysis to identify orthologs between V. ellipsiformis and U. peninsularis and comparing our results with previously published sex-specific transcriptomics data from distantly-related DUI species, we were able to identify elements possibly involved in the regulation of sex-specific aspects of DUI systems. Our search for candidate genes involved in sex determination suggests that some steps of the sex determination pathway may be deeply conserved in metazoans, whereas our search for candidate genes involved in DUI supports previous hypotheses that a modification of the ubiquitination mechanism could be responsible for the retention of the paternal mtDNA in male bivalves. Our results also suggest that DNA methylation could be involved in the maintenance of DUI in bivalves.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This study was supported by Natural Sciences and Engineering Research Council Discovery Grants awarded to S.B. (RGPIN/435656-2013) and D.T.S. (RGPIN/217175-2013). Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the US Government.
Literature Cited
- Allen DC, et al. 2007. Early life-history and conservation status of Venustaconcha ellipsiformis (Bivalvia, Unionidae) in Minnesota. Am Midl Nat. 157:74–91.http://dx.doi.org/10.1674/0003-0031(2007)157[74:ELACSO]2.0.CO;2 [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. Babraham Bioinform. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- Arama E, Bader M, Rieckhof GE, Steller H.. 2007. A ubiquitin ligase complex regulates caspase activation during sperm differentiation in Drosophila. PLoS Biol. 5(10):e251.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic S, Claverie J-M.. 1997. The significance of digital gene expression profiles. Genome Res. 7(10):986–995.http://dx.doi.org/10.1101/gr.7.10.986 [DOI] [PubMed] [Google Scholar]
- Barnhart CM, Haag WR, Roston WN.. 2008. Adaptations to host infection and larval parasitism in Unionoida. J N Am Benthol Soc. 27(2):370–374.http://dx.doi.org/10.1899/07-093.1 [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 57:289–300. [Google Scholar]
- Beukeboom LW, Perrin N.. 2014. The evolution of sex determination. Oxford: University Press. [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120.http://dx.doi.org/10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, et al. 2009. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: Unionoida) with doubly uniparental inheritance of mtDNA: gender-specific open reading frames and putative origins of replication. Genetics 183(4):1575–1589.http://dx.doi.org/10.1534/genetics.109.110700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, et al. 2011. Novel protein genes in animal mtDNA: a new sex determination system in freshwater mussels (Bivalvia: Unionoida)? Mol Biol Evol. 28(5):1645–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, Beaupré HD, Stewart DT, Hoeh WR, Blier PU.. 2007. The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends Genet. 23(9):465–474. [DOI] [PubMed] [Google Scholar]
- Breton S, Capt C, Guerra D, Stewart D.. 2017. Sex determining mechanisms in bivalves. Preprints 2017:2017060127. [Google Scholar]
- Capt C, Passamonti M, Breton S.. 2015. The human mitochondrial genome may code for more than 13 proteins. Mitochondrial DNA 27:3098–3101. [DOI] [PubMed] [Google Scholar]
- Chase CD. 2007. Cytoplasmic male sterility: a window to the world of plant mitochondrial–nuclear interactions. Trends Genet. 23(2):81–90.http://dx.doi.org/10.1016/j.tig.2006.12.004 [DOI] [PubMed] [Google Scholar]
- Chávez-Villalba J, et al. 2011. Determination of gender in the pearl oyster Pinctada margaritifera. J Shellfish Res. 30(2):231–240. [Google Scholar]
- Coe WR. 1943. Sexual differentiation in Mollusks. I. Pelecypods. Q Rev Biol. 18(2):154–164.http://dx.doi.org/10.1086/394673 [Google Scholar]
- Diz AP, et al. 2013. Proteomic analysis of eggs from Mytilus edulis females differing in mitochondrial DNA transmission mode. Mol Cell Proteomics 12(11):3068–3080.http://dx.doi.org/10.1074/mcp.M113.031401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doniach T, Hodgkin J.. 1984. A sex-determining gene, fem-1, required for both male and hermaphrodite development in Caenorhabditis elegans. Dev Biol. 106(1):223–235. [DOI] [PubMed] [Google Scholar]
- Fabioux C, et al. 2004. Oyster vasa-like gene as a marker of the germline cell development in Crassostrea gigas. Biochem Biophys Res Commun. 320(2):592–598. [DOI] [PubMed] [Google Scholar]
- Flynn K, et al. 2013. Burrowing in the freshwater mussel Elliptio complanata is sexually dimorphic and feminized by low levels of atrazine. J Toxicol Environ Health A 76(20):1168–1181.http://dx.doi.org/10.1080/15287394.2013.845865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble T, Zarkower D.. 2012. Sex determination. Curr Biol. 22(8):R257–R262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff MM, Lenertz KK, Feminella JW.. 2008. Parasitic mite and trematode abundance are associated with reduced reproductive output and physiological condition of freshwater mussels. Hydrobiologia 610(1):25..http://dx.doi.org/10.1007/s10750-008-9419-8 [Google Scholar]
- Ghiselli F, et al. 2012. De novo assembly of the manila clam Ruditapes philippinarum transcriptome provides new insights into expression bias, mitochondrial doubly uniparental inheritance and sex determination. Mol Biol Evol. 29(2):771–786.http://dx.doi.org/10.1093/molbev/msr248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissi C, Iannelli F, Pesole G.. 2008. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101(4):301–320.http://dx.doi.org/10.1038/hdy.2008.62 [DOI] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644–652.http://dx.doi.org/10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusman A, Lecomte S, Stewart DT, Passamonti M, Breton S.. 2016. Pursuing the quest for better understanding the taxonomic distribution of the system of doubly uniparental inheritance of mtDNA. PeerJ 4:e2760.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag WR. 2012. North American freshwater mussels: natural history, ecology, and conservationHost use and host infection strategies. Cambridge University Press; p. 140–179. [Google Scholar]
- Haag WR, Warren ML.. 1999. Mantle displays of freshwater mussels elicit attacks from fish. Fresh Biol. 42(1):35–40.http://dx.doi.org/10.1046/j.1365-2427.1999.00454.x [Google Scholar]
- Haas BJ, Papanicolaou A. 2017. TransDecoder. Retrieved from http://transdecoder.github.io [Google Scholar]
- Kang SW, et al. 2015. Construction of PANM database (Protostome DB) for rapid annotation of NGS data in Mollusks. Korean J Malacol. 31(3):243–247.http://dx.doi.org/10.9710/kjm.2015.31.3.243 [Google Scholar]
- Kenchington E, MacDonald B, Cao L, Tsagkarakis D, Zouros E.. 2002. Genetics of mother-dependent sex ratio in blue mussels (Mytilus spp.) and implications for doubly uniparental inheritance of mitochondrial DNA. Genetics 161(4):1579–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MA, et al. 2017. Alternative splicing profile and sex-preferential gene expression in the female and male pacific abalone Haliotis discus hannai. Genes (Basel) 8(3):99.http://dx.doi.org/10.3390/genes8030099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp A. 2012. Dmrt genes in the development and evolution of sexual dimorphism. Trends Genet. 28(4):175–184.http://dx.doi.org/10.1016/j.tig.2012.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Rev Clin Oncol. 9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner M, et al. 2011. Proteinortho: detection of (Co-)orthologs in large-scale analysis. BMC Bioinformatics 12:124.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079.http://dx.doi.org/10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. 2016. Transcriptome sequencing and comparative analysis of ovary and testis identifies potential key sex-related genes and pathways in scallop Patinopecten yessoensis. Mar Biotechnol. 18(4):453–465.http://dx.doi.org/10.1007/s10126-016-9706-8 [DOI] [PubMed] [Google Scholar]
- Liu H, et al. 2015. Large-scale transcriptome sequencing reveals novel expression patterns for key sex-related genes in a sex-changing fish. Biol Sex Differ. 6(1):26.http://dx.doi.org/10.1186/s13293-015-0044-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llera-Herrera R, García-Gasca A, Abreu-Goodger C, Huvet A, Ibarra AM.. 2013. Identification of male gametogenesis expressed genes from the scallop Nodipecten subnodosus by suppressive subtraction hybridization and pyrosequencing. PLoS One 8(9):e73176.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S.. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12):550..http://dx.doi.org/10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckenbach JA, Iliev DB, Goetz FW, Swanson P.. 2008. Identification of differentially expressed ovarian genes during primary and early secondary oocyte growth in coho salmon, Oncorhynchus kisutch. Reprod Biol Endocrinol. 6:2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson CK, Zarkower D.. 2012. Sex and the singular DM domain: insights into sexual regulation, evolution and plasticity. Nat Rev Genet. 13(3):163–174.http://dx.doi.org/10.1038/nrg3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani L, Ghiselli F, Guerra D, Breton S, Passamonti M.. 2013. A comparative analysis of mitochondrial ORFans: new clues on their origin and role in species with doubly uniparental inheritance of mitochondria. Genome Biol Evol. 5(7):1408–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani L, Ghiselli F, Maurizii MG, Passamonti M.. 2011. Doubly uniparental inheritance of mitochondria as a model system for studying germ line formation. PLoS One 6(11):e28194.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani L, Ghiselli F, Passamonti M.. 2016. Mitochondrial selfish elements and the evolution of biological novelties. Curr Zool. 62(6):687–697.http://dx.doi.org/10.1093/cz/zow044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani L, Ghiselli F, Pecci A, Maurizii MG, Passamonti M.. 2015. The expression of a novel mitochondrially-encoded gene in gonadic precursors may drive paternal inheritance of mitochondria. PLoS One 10(9):e0137468.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A, Guerra D, Stewart D, Breton S.. 2016. In silico analyses of mitochondrial ORFans in freshwater mussels (Bivalvia: Unionoida) provide a framework for future studies of their origin and function. BMC Genomics 17:597.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy GW, Springer SA, Adams SL, Swanson WJ, Vacquier VD.. 2008. Extraordinary intraspecific diversity in oyster sperm bindin. Proc Natl Acad Sci U S A. 105:1993–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, et al. 2015. Factors affecting trematode infection rates in freshwater mussels. Hydrobiologia 742(1):59–70. [Google Scholar]
- Negri I, et al. 2009. Unravelling the Wolbachia evolutionary role: the reprogramming of the host genomic imprinting. Proc R Soc B Biol Sci. 276(1666):2485–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passamonti M, Ghiselli F.. 2009. Doubly uniparental inheritance: two mitochondrial genomes, one precious model for organelle DNA inheritance and evolution. DNA Cell Biol. 28(2):79–89.http://dx.doi.org/10.1089/dna.2008.0807 [DOI] [PubMed] [Google Scholar]
- Patnaik BB, et al. 2016. Sequencing, de novo assembly, and annotation of the transcriptome of the endangered freshwater pearl bivalve, Cristaria plicata, provides novel insights into functional genes and marker discovery. PLoS One 11(2):e0148622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, et al. 2015. Gonadal transcriptomic analysis and differentially expressed genes in the testis and ovary of the Pacific white shrimp (Litopenaeus vannamei). BMC Genomics 16:1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman SJ, Hodson CN, Hamilton PT, Opit GP, Gowen BE.. 2015. Maternal transmission, sex ratio distortion, and mitochondria. Proc Natl Acad Sci U S A. 112(33):10162–10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi A, Plazzi F, Milani L, Ghiselli F, Passamonti M.. 2017. SmithRNAs: could mitochondria “bend” nuclear regulation? Mol Biol Evol. 34(8):1960–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140.http://dx.doi.org/10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A.. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11(3):R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager R, Grabowy C.. 1983. Differential methylation of chloroplast DNA regulates maternal inheritance in a methylated mutant of Chlamydomonas. Proc Natl Acad Sci U S A. 80(10):3025–3029.http://dx.doi.org/10.1073/pnas.80.10.3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santerre C, Sourdaine P, Adeline B, Martinez A-S.. 2014. Cg-SoxE and Cg-β-catenin, two new potential actors of the sex-determining pathway in a hermaphrodite lophotrochozoan, the Pacific oyster Crassostrea gigas. Comp Biochem Physiol Part A Mol Integr Physiol. 167:68–76. [DOI] [PubMed] [Google Scholar]
- Sayadi A, Immonen E, Bayram H, Arnqvist G, Falabella P.. 2016. The de novo transcriptome and its functional annotation in the seed beetle Callosobruchus maculatus. PLoS One 11(7):e0158565.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Hong Y, Sheng J, Peng K, Wang J.. 2015. De novo transcriptome sequencing to identify the sex-determination genes in Hyriopsis schlegelii. Biosci Biotechnol Biochem. 0:1–9. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212. [DOI] [PubMed] [Google Scholar]
- Soneson C, Delorenzi M.. 2013. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics 14:91.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutovsky P, et al. 1999. Ubiquitin tag for sperm mitochondria. Nature 402(6760):371–372.http://dx.doi.org/10.1038/46466 [DOI] [PubMed] [Google Scholar]
- Teaniniuraitemoana V, et al. 2014. Gonad transcriptome analysis of pearl oyster Pinctada margaritifera: identification of potential sex differentiation and sex determining genes. BMC Genomics 15:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium 2017. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45(D1):D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, et al. 2015. Transcriptomics analysis of Crassostrea hongkongensis for the discovery of reproduction-related genes. PLoS One 10(8):e0134280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlenhaut NH, Treier M.. 2006. Foxl2 function in ovarian development. Mol Genet Metab. 88(3):225–234.http://dx.doi.org/10.1016/j.ymgme.2006.03.005 [DOI] [PubMed] [Google Scholar]
- Umen JG, Goodenough UW.. 2001. Chloroplast DNA methylation and inheritance in Chlamydomonas. Genes Dev. 15(19):2585–2597.http://dx.doi.org/10.1101/gad.906701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis MC, Waters PD, Graves JAM.. 2008. Sex determination in mammals—before and after the evolution of SRY. Cell Mol Life Sci. 65(20):3182–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. 2017. Transcriptome analysis of the freshwater pearl mussel (Cristaria plicata) mantle unravels genes involved in the formation of shell and pearl. Mol Genet Genomics 292(2):343–352.http://dx.doi.org/10.1007/s00438-016-1278-9 [DOI] [PubMed] [Google Scholar]
- Wijayawardena BK, Minchella DJ, DeWoody JA.. 2016. The influence of trematode parasite burden on gene expression in a mammalian host. BMC Genomics 17(1):600..http://dx.doi.org/10.1186/s12864-016-2950-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarra T, Gharbi K, Blaxter M, Peck LS, Clark MS.. 2016. Characterization of the mantle transcriptome in bivalves: Pecten maximus, Mytilus edulis and Crassostrea gigas. Mar Genomics 27:9–15.http://dx.doi.org/10.1016/j.margen.2016.04.003 [DOI] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, Oshlack A.. 2010. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11(2):R14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F-F, Wang M-F, Zhou L, Gui J-F, Yu X-Y.. 2011. Molecular cloning and expression characterization of Dmrt2 in akoya pearl oysters, Pinctada martensii. J Shellfish Res. 30(2):247–254.http://dx.doi.org/10.2983/035.030.0208 [Google Scholar]
- Zhang G, et al. 2012. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490(7418):49–54.http://dx.doi.org/10.1038/nature11413 [DOI] [PubMed] [Google Scholar]
- Zhang ZH, et al. 2014. A comparative study of techniques for differential expression analysis of RNA-seq data. PLoS One 9:e1360 doi: 10.1371/journal.pone.0103207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Xu F, Guo X.. 2014. Genomic analysis of the Pacific oyster (Crassostrea gigas) reveals possible conservation of vertebrate sex determination in a mollusc. G3 (Bethesda) 4:2207–2217.http://dx.doi.org/10.1534/g3.114.013904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Yu H, Kong L, Liu S, Li Q.. 2014. Comparative transcriptome analysis of two oysters, Crassostrea gigas and Crassostrea hongkongensis provides insights into adaptation to hypo-osmotic conditions. PLoS One 9(11):e111915.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieritz A, Aldridge DC.. 2011. Sexual, habitat-constrained and parasite-induced dimorphism in the shell of a freshwater mussel (Anodonta anatina, Unionidae). J Morphol. 272(11):1365–1375. [DOI] [PubMed] [Google Scholar]
- Zouros E. 2013. biparental inheritance through uniparental transmission: the doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol Biol. 40(1):1–31.http://dx.doi.org/10.1007/s11692-012-9195-2 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.