

Abstract
Interactions of T cell receptors (TCR) to peptides in complex with MHC (p:MHC) are key features that mediate cellular immune responses. While MHC binding is required for a peptide to be presented to T cells, not all MHC binders are immunogenic. The interaction of a TCR to the p:MHC complex holds a key, but currently poorly comprehended, component for our understanding of this variation in the immunogenicity of MHC binding peptides. Here, we demonstrate that identification of the cognate target of a TCR from a set of p:MHC complexes to a high degree is achievable using simple force-field energy terms. Building a benchmark of TCR:p:MHC complexes where epitopes and non-epitopes are modelled using state-of-the-art molecular modelling tools, scoring p:MHC to a given TCR using force-fields, optimized in a cross-validation setup to evaluate TCR inter atomic interactions involved with each p:MHC, we demonstrate that this approach can successfully be used to distinguish between epitopes and non-epitopes. A detailed analysis of the performance of this force-field-based approach demonstrate that its predictive performance depend on the ability to both accurately predict the binding of the peptide to the MHC and model the TCR:p:MHC complex structure. In summary, we conclude that it is possible to identify the TCR cognate target among different candidate peptides by using a force-field based model, and believe this works could lay the foundation for future work within prediction of TCR:p:MHC interactions.
Keywords: MHC, T cell receptor, Antigens/Peptides/Epitopes, modelling, pipeline
1. Introduction
Binding to MHC (Major Histocompatibility Complex) is a prerequisite for peptide T cell immunogenicity. Given this, large efforts have been dedicated to the development of methods capable of accurately predict this event (some of the most accurate and publicly available at the IEDB are described in: Andreatta and Nielsen 2016; Nielsen and Andreatta 2016; Andreatta et al. 2015; Karosiene et al. 2013; Kim et al. 2009). The accuracy of the state-of-the-art methods has proven to be very high (in particularly for MHC class I), and most projects will in one way or another apply such prediction tools to guide the process of rational T cell epitope discovery (a few examples include Braendstrup et al. 2014; Pérez et al. 2008; Paul et al. 2015). However, not all peptides processed along the MHC pathways and bound by MHC turn out immunogenic. The main reason for this is the unavailability of T cells reactive to the given peptide-MHC (p:MHC) complex due to tolerance. The general rules underlying tolerance are well defined and deal with negative selection of T cells expressing a T cell receptor (TCR) with binding specificity towards p:MHC complexes of self-peptides. However, the details of these rules remain poorly described, and our understanding of the rules that define which p:MHCs are the targets of a given TCR remains highly limited.
In the last years, many efforts have been made modelling TCR:p:MHC systems. These efforts include simulation methods that have evolved from simulating the peptide in the MHC binding pocket for 1 nanosecond to simulating the entire TCR:p:MHC complex for more than 1 microsecond (Kass et al. 2014). Also, as more TCR:p:MHC complexes have been resolved by crystallography, template-based modelling techniques have achieved considerable accuracy, either using a single template or multiple templates (Liu et al. 2011). In other studies, force fields have been adapted in order to estimate changes in binding affinities, proving that structure-based methods are useful tools to design and engineer TCR and pMHC (mainly class I) interactions modulating both affinity and specificity (Pierce et al. 2014; Laugel et al. 2005). Also, docking approaches have shown that interactions between TCRs and pMHC complexes can be modelled when “good” scoring functions are used (Riley et al. 2016; Pierce and Weng 2013). Focusing on peptide immunogenicity, TCR interactions with pMHC class I complexes, in particular for HLA-A*02:01, have shown that CDR loops interactions to unknown epitopes can be predicted using a very simple rule-based model learning from known complexes of the same allele (Roomp and Domingues 2011). Also, a particular case (LC13 TCR and HLA-B*08:01) was characterized using 100ns molecular dynamics simulations. Here, however no strong difference was found regarding the binding behaviour between more and less immunogenic peptides (Knapp et al. 2014).
Given this background, we seek to answer, given TCR:p:MHC modelled complexes of different peptides interacting with the same MHC and TCR molecules, which structural properties can be used in order to predict the cognate target (i.e. the p:MHC complex) of the TCR. To address this question, first we built a benchmark set based on solved TCR:p:MHC of class II and generated homology models for both the bound epitope and a set of natural MHC-binding non-epitopes. Next, we used two well-known force fields, FoldX (Guerois et al. 2002) and Rosetta’s Talaris2013 (Leaver-Fay et al. 2013; O’Meara et al. 2015), to mimic the molecular interactions and chemical properties between the TCR and the p:MHC complex. FoldX (Schymkowitz et al. 2005) has in earlier studies demonstrated high performance predicting the impact of a mutation in the context of a given biological assembly. Rosetta (Bradley et al. 2005) has been extensively used in a large range of applications, from de novo protein design to understanding the folding process. These two force fields are described as weighted sums of terms modelling interactions in a given molecular assembly. Here, we investigate how these force fields could be used to identify the target of a given TCR. The weights of the two force fields were adjusted in a cross-validation setup in order to detect correlations between each force field term and the peptide immunogenicity. This approach allowed us to define a robust model, that given the sequences of the MHC alpha and beta subunits, TCR alpha and beta subunits and a set of peptides, could discriminate between epitopes and non-epitopes, and thus correctly predict the cognate target for the given TCR.
2. Material and Methods
2.1 The TCR:p:MHCII data set
A data set of 43 TCR:p:MHCII was downloaded from the PDB (Berman et al. 2003). Entries presenting extreme TCR orientations compared with all other entries in the dataset were excluded (4Y1A, 4Y19, 4C56, 3PL6, 2WBJ and 1YMM) (see Supplementary Figure 1). Finally, entries found to be single mutants of other cases in the data set were also excluded (4P23, 3T0E, 2IAM, 3QIW, 4E41 and 4P46) leaving a final benchmark data set of 31 TCR:p:MHCII complexes.
2.2 Similarity measures between TCR:p:MHCII complexes
To calculate the structural similarity between two TCR:p:MHCII structures, the two MHC beta-chain subunits are aligned using the TMalign software (Zhang and Skolnick 2014) and a transformation matrix is obtained. The corresponding alpha (TCRA) and beta (TCRB) TCR subunits in the complex are next translated and rotated using this matrix, and RMSD values (RMSD-TCRA, RMSD-TCRB) for the alpha and beta chains, respectively, are calculated for corresponding alpha carbons according to a pairwise alignment computed using CLUSTALW. Finally, we define RMSD-TCR as the average between RMSD-TCRA and RMSD-TCRB.
Similarity at the sequence level is calculated using TCR sequence identity from a BLASTP local alignment between a pair of structural complexes. Using identities between TCR alpha chains (TCRA_ID%) and TCR beta chains (TCRB_ID%), we define TDR_ID% as the average between TCRA_ID% and TCRB_ID%.
2.3 Benchmark of MHC class II epitopes and non-epitopes
We defined epitopes as the peptides in the TCR:p:MHCII structures as shown in Table 1. For each epitope, 4 peptides of the same length, extracted from the epitope source protein sequence, and presenting a similar predicted binding score as the epitope to the corresponding MHC, were selected as non-binding epitopes. The binding scores were obtained using the predicted percentile rank score obtained from NetMHCIIpan (version 3.1), and the non-epitopes were selected, with predicted percentile rank values in the range +/-5% of the percentile rank of the epitope so that the epitopes fall, on average, in the middle rank between corresponding non-epitopes. In 2 cases (3qib and 3qiu) no other peptide in the sequence had a score in the 5% interval, and therefore the range was extended to +/-17%. In situations where the epitope had no source protein (non-natural peptides), the non-epitopes were selected from UniRef50 as described above.
Table 1. Epitopes in TCR:p:MHCII complexes.
First column corresponds to the PDB code, underlined entries are excluded from the benchmark (see section 2.1). Second column is for MHC family (DR, DP, DQ are human alleles and IE, IA are mouse alleles). TCR is specified in third column and the peptide or protein name is in fourth column. Fifth column corresponds to the Epitope sequence (cores in bold) and sixth column is for the UniprotID that was used to extract other peptides as Non-Epitopes (* is for cases that have no sequence, so random peptides were chosen from UniRef50).
| PDB ID | MHCII | TCR | Peptide Name | Epitope sequence | Pept Full Seq |
|---|---|---|---|---|---|
| 1D9K | IAk | D10 | ConAlb | HRGAIEWEGIESG | P02789 |
| 1FYT | DR1 | HA1.7 | HA | PKYVKQNTLKLAT | Q38SR9 |
| 1J8H | DR4 | HA1.7 | HA | PKYVKQNTLKLAT | Q38SR9 |
| 1U3H | IAu | 172.1 | MBP1-11 | SRGGASQYRPSQ | P04370 |
| 1YMM | DR2 | OB.1A12 | MBP85-99 | ENPVVHFFKNIVTPR | P02686 |
| 1ZGL | DR2a | 3A6 | MBP89-101 | VHFFKNIVTPRTP | P02686 |
| 2IAM | DR1 | E8 | mutTPI | GELIGTLNAAKVPAD | P60174 |
| 2IAN | DR1 | E8 | TPI | GELIGTLNAAKVPAD | P60174 |
| 2PXY | IAu | 1934 | MBP1-11 | SRGGASQYRPSQ | P04370 |
| 2WBJ | DR2 | OB.1A12 | ENGA | FARVHFISALHG | A7ZPV4 |
| 2Z31 | IAu | Cl19 | MBP1-11 | SRGGASQYRPSQ | P04370 |
| 3C5Z | IAb | B3K506 | p3K | FEAQKAKANKA | * |
| 3C60 | IAb | YAE62 | p3K | FEAQKAKANKA | * |
| 3C6L | IAb | 2W20 | P3K | FEAQKAKANKA | * |
| 3MBE | IAg | 21.3 | HEL | AMKRHGLDNYRGYSLGN | P00698 |
| 3O6F | DR4 | MS2-3C8 | MBP114-126 | FSWGAEGQRPGFG | P02686 |
| 3PL6 | DQ1 | Hy.1B11 | MBP85-99 | ENPVVHFFKNIVTPR | P02686 |
| 3QIB | IEk | 2B4 | MCC88-104 | ADLIAYLKQATK | P00039 |
| 3QIU | IEk | 226 | MCC88-104 | ADLIAYLKQATK | P00039 |
| 3QIW | IEk | 226 | MCC88-104p5E | ADLIAYLKQATK | P00039 |
| 3RDT | IAb | J809.B5 | P3K | FEAQKAKANKA | * |
| 3T0E | DR4 | MS2-3C8 | MBP114-126 | FSWGAEGQRPGFG | P02686 |
| 4C56 | DR1 | AV22/BV19 | HA | PKYVKQNTLKLAT | Q38SR9 |
| 4E41 | DRA1 | G4 | mutTPI | GELIGTLNAAKVPAD | P60174 |
| 4GG6 | DQ8 | SP3.4 | Glia-alpha1 | SGEGSFQPSQENP | X2KVI4 |
| 4GRL | DQ1 | Hy.1B11 | pMM | DRLLMLFAKDVVSRN | P26276 |
| 4H1L | DR52c | Ani2.3 | pHIR(Ni2+) | HIRCNIPKRI | * |
| 4MAY | DQ1 | Hy.1B11 | UL15 | FRQLVHFVRDFAQLL | P04295 |
| 4OZF | DQ2 | JR5.1 | Glia-alpha2 | PFPQPELPYPQPQ | X2KWL1 |
| 4OZG | DQ2 | D2 | Glia-alpha2 | PFPQPELPYPQPQ | X2KWL1 |
| 4OZH | DQ2 | S16 | Glia-alpha2 | PFPQPELPYPQPQ | X2KWL1 |
| 4OZI | DQ2.5 | S2 | Glia-alpha1a | LQPFPQPELPYPQ | X2KWL1 |
| 4P23 | IAb | J809.B5 | p3K | FEAQKAKANKA | * |
| 4P2Q | IEk | 5cc7 | 5c2 | ADGLAYFRSSFK | * |
| 4P2R | IEk | 5cc7 | 5c1 | ANGVAFFLTPFKA | * |
| 4P46 | IAb | J809.B5 Y31A | p3K | FEAQKAKANKA | * |
| 4P4K | DP2 | AV22 | M2(Be2+) | FWIDLFETIG | * |
| 4P5T | IAb | 14.C6 | p3K | FEAQKAKANKA | * |
| 4Y19 | DR4 | FS18 | Insulin | GSLQPLALEGSLQKRGIV | P01308 |
| 4Y1A | DR4 | FS19 | Insulin | GSLQPLALEGSLQKRGIV | P01308 |
| 4Z7U | DQ8 | S13 | Glia-alpha1 | SGEGSFQPSQENP | X2KVI4 |
| 4Z7V | DQ8 | L3 | Glia-alpha1 | SGEGSFQPSQENP | X2KVI4 |
| 4Z7W | DQ8 | T316 | Glia-alpha1 | SGEGSFQPSQENP | X2KVI4 |
We clustered the complexes using sequence identity between TCRA, TCRB and peptide to eliminate sequence biases. First, clusters were formed using exact peptides match (see Supplementary Figure 2A). Next, as similarities were found between some TCRs (Supplementary Figure 2B), clusters were joined if TCR_ID% was above 90%.
2.4 TCR:p:MHCII modeling pipeline
We developed a pipeline to model TCR:p:MHCII complexes as shown in Figure 1A, which takes as input 5 sequences, MHC alpha and beta subunits, the peptide and TCR alpha and beta subunits. We first created a template library for homology modelling based on the set of available TCR:p:MHCII structures described in subsection 2.3. From each TCR:p:MHCII structure, the first complex, following the chain order in the original PDB file, was taken in the cases where more than one appeared in a given PDB entry. Next, the MHC binding core was identified for each epitope as the nine consecutive residues after P1, that was in turn defined as the residue with the largest number of contacts (residues with less than 8.0Å between alpha carbons) with the MHC molecule. Note that epitope cores were defined only to align target and template peptides. Full-length peptides were used for force field predictions (see later).
Figure 1. Flow diagram of the TCR:p:MHCII modelling pipeline and the cross-validation.
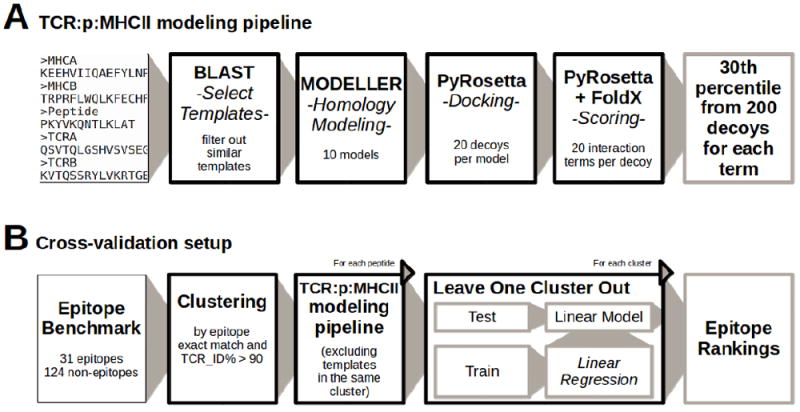
A) The pipeline input consists in a set of 5 sequences corresponding to MHC alpha and Beta subunits, the peptide sequence and TCR alpha and beta chains, and outputs a set of percentile values that describe the interaction energies between TCR and p:MHC using 200 models. B) The setup validates the pipeline splitting the set of epitopes in clusters and leaving one cluster out at a time. It models each peptide excluding the templates in the same cluster during the modelling step and also the percentile values when the model is trained. Finally, each cluster is tested separately avoiding the effect of overfitting.
For template selection, a BLASTP search was performed against the template library (excluding entries in the same cluster as the query), and templates were sorted according to TCR_ID%. Next, the top template was included and, iterating over templates from the sorted list, additional templates were added if no other templates in the same sequence cluster or with similar conformation (RMSD-TCR<3.0Å) were previously included. This procedure was repeated until no more templates were left.
Once the templates have been selected, alignments between MHC and TCR chains were computed using CLUSTALW. Target peptides and template peptides were aligned on the binding core. In the case of non-epitopes, the cores were predicted using NetMHCIIpan (version 3.1). Next, 10 models were built with MODELLER (Sali et al. Blundell 1993) using the multiple template approach on selected templates. For each model, decoys were produced using a docking protocol based on the Monte Carlo Minimization implemented within PyRosetta (Chaudhury et al. 2010). First, the position of the TCR dimer was perturbed by random translations in 1Å and rotations in 1°. Second, the distance between the TCR and p:MHC was adjusted using the SlideIntoContact procedure. Third, high resolution MCM optimized the complex orientation with respect to the PyRosetta Talaris2013 full-atom scoring function. This was repeated 20 times for each model producing a total of 200 decoys for each entry in the benchmark.
2.5 Scoring of TCR:p:MHCII complexes
To score each decoy, two force fields were tested: Talaris2013 (hereafter referred to as PyRosetta) and FoldX. Both force fields are linear compositions of terms that evaluate several types of properties related to protein interactions. Interaction energies between TCR and p:MHC were calculated and each energy term was saved separately after this calculation. In the case of PyRosetta, the energy was calculated computing the energy of the entire complex and subtracting the energy of the TCR and p:MHC in isolation. In the case of FoldX, the energy was calculated using the analyzecomplex method on each complex. For each term, 30th percentile was calculated from the distribution of 200 decoys, obtaining a single value. This approach is similar to what has been suggested earlier for interaction energy landscapes characterizations in similar systems (Yanover and Bradley 2011). Here however, we calculate the distributions per energy term and next evaluate the percentile score independently of the overall score. Finally, for each input the pipeline produces 20 values (6 from PyRosetta and 14 from FoldX) corresponding to the force field terms with a non-zero values when calculating the interaction energy between TCR and p:MHC.
2.6 Cross-validation setup
As depicted in Figure 1B, for the sake of validation, we clustered the benchmark data as mentioned previously and each entry was modelled using the previously explained pipeline excluding all templates in the cluster of the query molecule in order to reduce the effect of redundancy and overfitting.
Leave-one-cluster-out experiments were performed searching for optimal weights on the different force filed terms using as training set the benchmark data excluding a given cluster and next evaluating the predictive performance on the left out cluster. This was performed over all clusters. In this setup, a model was thus developed leaving out all data belonging to one cluster, and the model development was performed without ever referring to the left out data for model refinement. Optimization of the force field weights was performed on the training data using linear regression models computed to fit the weighted features to a binary target value Epitope=1 and Non-epitope=0. Models were built for each force field separately (PyRosetta and FoldX) and merging all terms together. For the three models, all values were normalized before fitting, subtracting the minimum and dividing by the max-min difference. AUC values were calculated from predictions over the complete benchmark data set measuring a global prediction performance. Also, for each epitope and its corresponding non-epitopes, a per-structure AUC value was calculated. To compare the performance of different models, a binomial test was used comparing those entries with better predictions excluding ties.
3. Results
3.1 Benchmark for epitope model training and validation
A total of 31 PDBs with TCR:p:MHCII complexes were included in the study; 17 human and 14 mouse complexes. The number of structures is not equally spread across the different species and loci since there is only one entry for DP, 10 DQ and 6 for DR, respectively. For mouse, only one entry is included for IAk and IAg, whereas 5 entries are included for IAb, and 3 and 4 for IAu and IEk respectively. For each of these cases, 4 non-epitopes were generated as described in material and methods, resulting in a total number of 155 entries (epitopes and non-epitopes). 13 clusters were obtained grouping the benchmark by sequence identities (Supplementary Table 1). As shown in Supplementary Table 2, for all cases, non-epitopes are separated on average by only 1.9% from their corresponding epitope in terms of predicted percentile rank binding value (maximal difference is less than 17%).
3.2 TCR:p:MHCII modeling pipeline
Structures of TCR:p:MHCII complexes were constructed using a multiple template homology modelling pipeline in addition to a docking protocol (for details materials and methods). A critical issue when constructing a modelling pipeline is to access its ability to generate accurate models. To validate this, we built 10 models for both multiple and single template approaches and analysed the best model (in terms of RMSD-TCR) obtained for each epitope with its native structure. Comparing the non-redundant multiple template approach to an approach using only single templates revealed that the former produced more accurate models (binomial p-value<0.05, Supplementary Figure 3A). Also, the non-redundant multiple template approach achieved better results when comparing against models built using all templates (binomial p-value<0.2, Supplementary Figure 3B). This latter finding suggests that using all templates is sub-optimal because the underlying templates is biased in terms of sequence diversity and docking orientations. After the initial model construction, decoys were produced using the PyRosetta docking protocol. To access if this docking step increased the ability to generate accurate models, we built 200 models using MODELLER and 200 models using the PyRosetta docking protocol, and next compared the lowest RMSD-TCR conformations between both approaches. As shown in Supplementary Figure 3C, this docking procedure resulted in significantly improved model accuracy compared to the MODELLER procedure (binomial p-value=5.5e-04). In conclusion, the TCR:p:MHCII modelling pipeline is found to be robust and contain good models with minimum RMSD-TCR values (among 200 decoys) to the native structure in the range 2-5Å. Note, that we here only have evaluated the ability of the modelling pipeline to produce ensembles containing one or more accurate models. We have not evaluated our ability to identify these accurate models within the ensembles. We do not address this otherwise critical issue here, since we in the final modelling pipeline use all the models in the ensemble, and hence at no point include or in other ways use the native structure to make predictions.
3.3 Global prediction performance
For each peptide (epitope and non-epitope), the TCR:p:MHCII force field scoring computes 20 interaction terms (6 from PyRosetta and 14 from FoldX). We first assessed the prediction capabilities of PyRosetta and FoldX to distinguish between epitopes and non-epitopes in terms of the AUC. For both cases, the original function with default weights showed random performance (data not shown). Next, we fitted models performing leave-one-cluster-out (LOCO) validation experiments, where the weight for each force field term was refitted using data from all clusters but one, and evaluating performance on the epitopes and non-epitopes from the left-out clusterv (for details see material and method). Here, we evaluated the performance either “globally”, merging predictions from epitopes and non-epitopes from all structures, or on per-structure level, comparing the rank of the epitope to the 4 non-epitope peptides in each structure. As shown in Figure 2, these LOCO experiments resulted in better than random predictions for both PyRosetta (AUC=0.62) and FoldX (AUC=0.68). Performing the LOCO experiments on the combined set of 6 PyRosetta and 14 FoldX terms, resulted in a further improve in global performance with an AUC of 0.72. In Supplementary Figure 4, we display a heatmap of the weights for each force field term obtained in each LOCO experiments. From this figure, it is apparent that the different terms consistently have either positive or negative weights in the different LOCO models, confirming that force field refitting is highly robust and reproducible. Analysing each term individually, several were found to have substantial predictive performance (Supplementary Figure 5A). In particular, 2 PyRosetta and 5 FoldX terms where found to have an average predictive performance (measured in terms of Pearson correlation) of more than +/-0.05. However, as expected, no single term outperforms the combined linear model (Supplementary Figure 5B).
Figure 2. Global prediction performance.
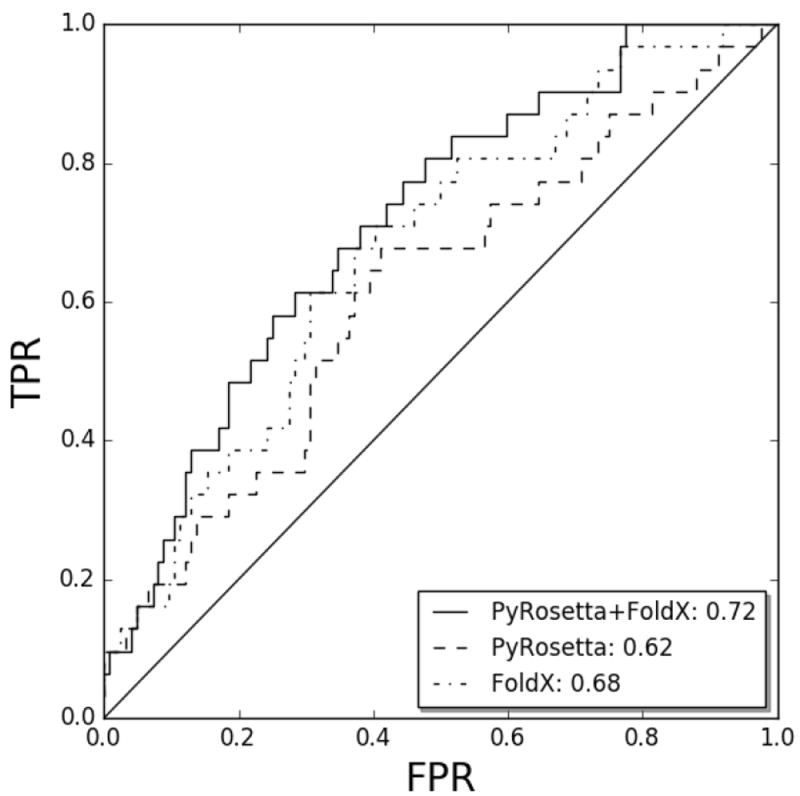
ROC curves for the prediction performance using different sets of terms to fit linear models. All terms together are depicted in solid line, PyRosetta terms in dashed line and FoldX terms in dotted line. FPR: False positive proportion, TPR: True positive proportion. The x=y diagonal is included as a reference corresponding to random predictions.
3.4 Pipeline accuracy on epitope rankings
To evaluate predictions at per-structure level, we calculated the AUC for each epitope and its corresponding 4 non-epitopes. First of all, as shown in Figure 3, the predictive performance of the model is clearly different from random. 24 cases had AUC above 0.5 and only 5 cases obtained an AUC less than 0.5, corresponding to a p-value=0.0005 (binomial test excluding ties). We analysed how the pipeline accuracy depended on two parameters that can be deduced from its input. First, MHC binding, which is required for a peptide to be presented to the T cell. Second, the availability of TCR:p:MHC template structures used to build the models, which was analysed in terms of maximum TCR beta sequence identity between the query and all the templates used. Similar analyses were performed for the MHC alpha, MHC beta and TCR alpha sequences, in all cases showing lower predictive performance (data not shown). Of the 15 entries with predicted percentile rank binding to the MHC of less than 25% and with the maximum sequence identity (over all templates) to the TCR beta subunit above 75%, 13 (87%) had AUC>0.5 and only 2 (13%) had AUC<=0.5 (p-value=0.0074). For the remaining 16 entries (predicted rank MHC binding above 25% or TCR beta sequence identity less than 75%), only 68% (11) had AUC>0.5 and 5 had AUC<=0.5 (p-value=0.21). These values suggest as expected that the accuracy of the proposed pipeline is dependent on both the binding strength of the peptide-MHC interaction and the availability of suitable templates.
Figure 3. Prediction performance refitting all terms together.
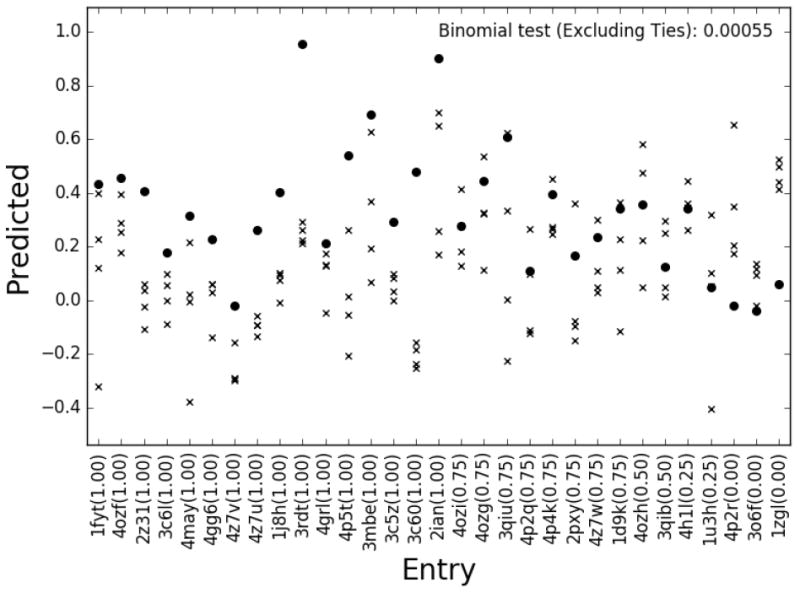
Peptide rankings tested on each benchmark entry individually (epitopes shown in circles, non-epitopes in crosses). The x-axis gives the entry evaluated and epitope ranking (AUCs given in parenthesis).
3.5 TCR:p:MHC Pipeline in a real application
Focusing on the subset of 19 epitopes with predicted binding to the restrictive MHC (percentile rank <=25%), we turned to construct a real-life benchmark of the TCR:p:MHCII scoring pipeline. To do this, we selected from the benchmark the source protein sequence for each epitope, and predicted for all overlapping peptides of the length of epitopes MHC binding and TCR:p:MHC interaction. Next, all peptides were scored using a combined model α*(100-MHCrank/100) + (1-α)*TCR:p:MHC, where MHCrank is the percentile rank score reported by NetMHCIIpan (version 3.1), and TCR:p:MHC is the interaction score obtained using the model proposed here. Based on these prediction scores, an AUC value was calculated taking the known epitope as positive and all other peptides as negatives. The optimal value of α was found using a simple grid search in a leave one out experiment (see Supplementary Figure 6), and was consistently found to be 0.9. The result of this benchmark is summarized in Table 2 and shows that the combined model outperform the MHC binding method alone in 12 cases (p-value 0.075), and the TCR:p:MHC interaction method alone in 14 cases (p-value 0.03) confirming that also in such a real-life experiment does the proposed model achieve improved predictive performance values.
Table 2. Epitope rankings using TCR:p:MHC pipeline in addition to MHC binding affinity predictions.
The predicted entry is specified from first to fourth column. The fifth column is a rank% computed as the entry percentile in the context of a set of random peptides of the same length taken from UniRef50. From the sixth to the eighth columns, are shown the position in the rankings obtained using NetMHCIIpan, TCR:p:MHC and a combined model using 0.9*(100-MHCRank/100)+0.1*TCR:p:MHC
| PDB ID | MHCII | TCR | Epitope sequence | Numbe of Peptides | NetMHCIIpan Rank% | NetMHCIIpan ranking | TCRpMHC ranking | Combined ranking |
|---|---|---|---|---|---|---|---|---|
| 4MAY | DQ1 | Hy.1B11 | FRQLVHFVRDFAQLL | 734 | 0.44 | 8 | 2 | 1 |
| 4P4K | DP2 | AV22 | FWIDLFETIG | 315 | 0.46 | 4 | 58 | 9 |
| 3C5Z | IAb | B3K506 | FEAQKAKANKA | 133 | 0.76 | 4 | 21 | 2 |
| 3C6L | IAb | 2W20 | FEAQKAKANKA | 133 | 0.76 | 4 | 2 | 1 |
| 3C60 | IAb | YAE62 | FEAQKAKANKA | 133 | 0.76 | 4 | 15 | 1 |
| 3RDT | IAb | J809.B5 | FEAQKAKANKA | 133 | 0.76 | 4 | 4 | 1 |
| 4P5T | IAb | 14.C6 | FEAQKAKANKA | 133 | 0.76 | 4 | 10 | 1 |
| 1J8H | DR4 | HA1.7 | PKYVKQNTLKLAT | 565 | 0.82 | 4 | 14 | 3 |
| 1FYT | DR1 | HA1.7 | PKYVKQNTLKLAT | 565 | 1.58 | 11 | 25 | 15 |
| 1ZGL | DR2a | 3A6 | VHFFKNIVTPRTP | 303 | 1.58 | 2 | 46 | 6 |
| 4H1L | DR52c | Ani2.3 | HIRCNIPKRI | 120 | 1.62 | 13 | 32 | 8 |
| 3QIB | IEk | 2B4 | ADLIAYLKQATK | 107 | 2.10 | 1 | 3 | 1 |
| 3QIU | IEk | 226 | ADLIAYLKQATK | 107 | 2.10 | 1 | 1 | 1 |
| 2IAN | DR1 | E8 | GELIGTLNAAKVPAD | 285 | 4.28 | 4 | 16 | 3 |
| 4GRL | DQ1 | Hy.1B11 | DRLLMLFAKDVVSRN | 462 | 8.72 | 13 | 28 | 9 |
| 4GG6 | DQ8 | SP3.4 | SGEGSFQPSQENP | 282 | 24.48 | 58 | 54 | 59 |
| 4Z7U | DQ8 | S13 | SGEGSFQPSQENP | 282 | 24.48 | 58 | 20 | 51 |
| 4Z7V | DQ8 | L3 | SGEGSFQPSQENP | 282 | 24.48 | 58 | 24 | 54 |
| 4Z7W | DQ8 | T316 | SGEGSFQPSQENP | 282 | 24.48 | 58 | 50 | 58 |
4. Discussion
In this work, we have demonstrated that reliable predictions of the cognate target of a TCR can be achieved using refined version of the conventional FoldX and Rosetta force fields. We built a benchmark of TCR:p:MHCII complexes, structures of epitopes and non-epitopes in complex with MHC and TCR were constructed using homology modelling techniques. Peptide immunogenicity was evaluated calculating interaction energies between TCR and each p:MHC complex. Weights assigned to each force field term were refitted, observing that some terms were not correlated with peptide immunogenicity. Leave-one-cluster-out experiments showed that TCR cognate identification can be predicted at a significant level (p-value<0.0005). We found a higher predictive performance of our approach for complexes where we could accurately predict the peptide binding to the restrictive MHCII element. Given this, we conclude that prediction of the TCR cognate target on basis of interaction energies is possible when MHC binding can be predicted. Likewise, we found that our TCR:p:MHCII scoring pipeline achieved superior performance in situations were TCR templates with high sequence identity (TCR-beta > 75%) to the query TCR were available.
Generally, epitopes are ranked using MHC binding information as the main prioritization factor. Here, investigated if our proposed TCR:p:MHC pipeline combined with MHC binding affinity predictions could result in improved predictions. In a simulated real life application, we took as input the MHC class II sequences, the TCR sequence, and a target protein sequence. Next, all overlapping peptides of length corresponding to the epitope were sorted according to a score combining predicted MHC binding and TCR recognition aiming to identify the most immunogenic peptides in the top of the ranking. In this benchmark, we found a consistent increase in predictive performance of the combined model compared to each of the two models (MHC binding, and TCR interaction) individually.
As stipulated for the MHC class II antigen presenting pathway, MHC binding is required for a given peptide to be presented to the T cell. Ideally, MHC binding predictors used in addition to TCR cognate identification methods are required to fully understand peptide immunogenic behaviour. However, some of epitopes used in this work showed extreme low predicted MHC binding potential. At this point, we do not fully descern the source of this seemingly inconsistency but it could suggest that we need to learn more about the underlying principles that govern MHC binding since these epitopes can not be explained using state-of-the-art MHC binding predictions.
In our analysis, we have found that, in order to achieve accurate predictive performance, refined versions of the conventional FoldX and Rosetta force fields, with optimized relative weights on the different energy terms, was needed. In the last few years there have been several works performing such optimisation on different classes of proteins (Leaver-Fay et al. 2013; O’Meara et al. 2015; Park et al. 2016; Alford et al. 2017). In most of the cases, and particularly for the force fields used in this work, such optimisation was performed on both intra- and extra-cellular proteins, that are subject to completely different environments. Given the very special nature of TCR:p:MHC interactions, and the relatively small difference in energy between immunogenic and non-immunogenic peptides (Knapp et al. 2014; Harndahl et al. 2012), we believe that it is fundamental to perform such optimisation in a targeted way, only using TCR:p:MHC complexes.
Another issue to address is the energy relaxation of the structures. In this work we did not relax structures after the modelling and docking procedures. Despite of this, we obtained robust linear models and performance values significantly better than random. We believe that by sampling the single-term energy distributions over 200 models provides enough samples to stabilize the distribution of each term so that relaxation is unneeded. We also tried to adjust the weights using 30th percentiles with fewer than 200 models and we saw a drop in the accuracy of the predictions. A more profound analysis of the effect of relaxation and the number of models sampled is suggested to address these issues and their impact on the predictive performance of the proposed pipeline. Such analyses could also include other force fields, beyond FoldX and PyRosetta to investigate if other terms could be included to obtain even better predictions.
In conclusion, the work presented demonstrates that prediction of the cognate TCR target is possible given accurate predicted MHC binding of the target peptides and high sequence identity of the TCR beta subunit to the template database. When these conditions are satisfied, the suggested force-field based model performs significantly better than random when it comes to identification of the cognate TCR target. We believe this work lays the foundation for future works within prediction of TCR:p:MHCII interactions, and expect that refined models will be developed along the lines outlined here as more structural and sequence data describing TCR:p:MHCII interactions becomes available.
Supplementary Material
Highlights.
Identification the TCR cognate target is achieved using force-field based models.
Performance depends on the ability to both accurately predict the binding of the peptide to the MHC and modeling the TCR:p:MHC complex structure
A modeling pipeline for TCR:p:MHC structure prediction is proposed and evaluated.
Cross-validation experiments confirms model robustness.
Acknowledgments
EL is a posdoc student at the Argentinean national research council (CONICET). MN is a researcher at the Argentinean national research council (CONICET). This work was funded partially by National Institutes of Health [HHSN272201200010C].
Abbreviations
- TCR
T cel receptors
- MHC
Major Histocompatibility Complex
- IEDB
Immune Epitope Database
- HLA
Human Leukocyte Antigen
- CDR
Complementarity Determining Region
- RMSD
Root Mean Square Deviation
- PDB
Protein Data Bank
- MCM
Monte Carlo Method
- AUC
Area Under the Curve
- LOCO
Leave One Cluster Out
Footnotes
Conflict of interests
Authors declare that there is no conflict of interest regarding the publication of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K, Labonte JW, Pacella MS, Bonneau R, Bradley P, Dunbrack RL, Jr, Das R, Baker D, Kuhlman B, Kortemme T, Gray JJ. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory Comput. 2017;13(6):3031–3048. doi: 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511–7. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, Nielsen M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics. 2015;67(11-12):641–50. doi: 10.1007/s00251-015-0873-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10(12):980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 5.Bradley P, Misura K, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309(5742):1868–71. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 6.Braendstrup P, Mortensen B, Justesen S, Osterby T, Rasmussen M, Hansen A, Christiansen C, Hansen M, Nielsen M, Vindeløv L, Buus S, Stryhn A. Identification and HLA-tetramer-validation of human CD4+ and CD8+ T cell responses against HCMV proteins IE1 and IE2. PLoS One. 2014;9(4):e94892. doi: 10.1371/journal.pone.0094892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaudhury S, Lyskov S, Gray JJ. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26(5):689–91. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320(2):369–87. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 9.Harndahl M, Rasmussen M, Roder G, Dalgaard Pedersen I, Sørensen M, Nielsen M, Buus S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur J Immunol. 2012;42(6):1405–16. doi: 10.1002/eji.201141774. [DOI] [PubMed] [Google Scholar]
- 10.Karosiene E, Rasmussen M, Blicher T, Lund O, Buus S, Nielsen M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics. 2013;65(10):711–24. doi: 10.1007/s00251-013-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kass I, Buckle A, Borg N. Understanding the structural dynamics of TCR-pMHC complex interactions. Trends Immunol. 2014;35(12):604–612. doi: 10.1016/j.it.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics. 2009;10:394. doi: 10.1186/1471-2105-10-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knapp B, Dunbar J, Deane CM. Large scale characterization of the LC13 TCR and HLA-B8 structural landscape in reaction to 172 altered peptide ligands: a molecular dynamics simulation study. PLoS Comput Biol. 2014;10(8):e1003748. doi: 10.1371/journal.pcbi.1003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laugel B, Boulter J, Lissin N, Vuidepot A, Li Y, Gostick E, Crotty L, Douek D, Hemelaar J, Price D, Jakobsen B, Sewell A. Design of soluble recombinant T cell receptors for antigen targeting and T cell inhibition. J Biol Chem. 2005;280(3):1882–92. doi: 10.1074/jbc.M409427200. [DOI] [PubMed] [Google Scholar]
- 15.Leaver-Fay A, O’Meara MJ, Tyka M, Jacak R, Song Y, Kellogg EH, Thompson J, Davis IW, Pache RA, Lyskov S, Gray JJ, Kortemme T, Richardson JS, Havranek JJ, Snoeyink J, Baker D, Kuhlman B. Scientific benchmarks for guiding macromolecular energy function improvement. Methods Enzymol. 2013;523:109–43. doi: 10.1016/B978-0-12-394292-0.00006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu I, Lo Y, Yang J. PAComplex: a web server to infer peptide antigen families and binding models from TCR-pMHC complexes. Nucleic Acids Res. 2011;39:W254–60. doi: 10.1093/nar/gkr434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8(1):33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Meara MJ, Leaver-Fay A, Tyka MD, Stein A, Houlihan K, DiMaio F, Bradley P, Kortemme T, Baker D, Snoeyink J, Kuhlman B. Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J Chem Theory Comput. 2015;11(2):609–22. doi: 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park H, Bradley P, Greisen P, Jr, Liu Y, Mulligan VK, Kim DE, Baker D, DiMaio F. Simultaneous Optimization of Biomolecular Energy Functions on Features from Small Molecules and Macromolecules. J Chem Theory Comput. 2016;12(12):6201–6212. doi: 10.1021/acs.jctc.6b00819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paul S, Lindestam Arlehamn C, Scriba T, Dillon M, Oseroff C, Hinz D, McKinney D, Carrasco Pro S, Sidney J, Peters B, Sette A. Development and validation of a broad scheme for prediction of HLA class II restricted T cell epitopes. J Immunol Methods. 2015;422:28–34. doi: 10.1016/j.jim.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pérez C, Larsen M, Gustafsson R, Norström M, Atlas A, Nixon D, Nielsen M, Lund O, Karlsson A. Broadly immunogenic HLA class I supertype-restricted elite CTL epitopes recognized in a diverse population infected with different HIV-1 subtypes. J Immunol. 2008;180(7):5092–100. doi: 10.4049/jimmunol.180.7.5092. [DOI] [PubMed] [Google Scholar]
- 22.Pierce B, Hellman L, Hossain M, Singh N, Vander Kooi C, Weng Z, Baker B. Computational design of the affinity and specificity of a therapeutic T cell receptor. PLoS Comput Biol. 2014;10(2):e1003478. doi: 10.1371/journal.pcbi.1003478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierce B, Weng Z. A flexible docking approach for prediction of T cell receptor-peptide-MHC complexes. Protein Sci. 2013;22(1):35–46. doi: 10.1002/pro.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riley T, Singh N, Pierce B, Weng Z, Baker B. Computational Modeling of T Cell Receptor Complexes. Methods Mol Biol. 2016;1414:319–40. doi: 10.1007/978-1-4939-3569-7_19. [DOI] [PubMed] [Google Scholar]
- 25.Roomp K, Domingues FS. Predicting interactions between T cell receptors and MHC-peptide complexes. Mol Immunol. 2011;48(4):553–62. doi: 10.1016/j.molimm.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 26.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 27.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(Web Server issue):W382–8. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yanover C, Bradley P. Large-scale characterization of peptide-MHC binding landscapes with structural simulations. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(17):6981–6986. doi: 10.1073/pnas.1018165108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2014;33(7):2302–9. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.