

Abstract
It is not well understood how ribosome antibiotics affect a wide range of biochemical pathways; changes in RNA-mediated protein quinary interactions and consequent activity inside the crowded cytosol may provide one possible mechanism. We developed real-time (RT) in-cell NMR spectroscopy to monitor temporal changes in protein quinary structure, for 24 hours and longer, in response to external and internal stimuli. RT in-cell NMR consists of a bioreactor containing gel encapsulated cells inside a 5 mm NMR tube, a gravity siphon for continuous exchange of medium, and a horizontal drip irrigation system to supply nutrients to the cells during the experiment. We showed that adding antibiotics that bind to the small ribosomal subunit result in more extensive quinary interactions between thioredoxin and mRNA. The results substantiate the idea that RNA-mediated modulation of quinary protein interactions may provide the physical basis for ribosome inhibition and other regulatory pathways.
Keywords: in cell NMR spectroscopy, flow cell, ribosome, thioredoxin, tetracycline, chloramphenicol, streptomycin, RNA interactome
Graphical abstract

Introduction
As the singular protein synthesizing machine, the bacterial ribosome is a main target of various classes of antibiotics.1 Ribosome inhibition depends on where the antibiotic is bound: Those that bind to the small (30S) ribosomal subunit, such as tetracycline and streptomycin, alter the ribosome interaction with mRNA or tRNA, and those that bind to the large (50S) ribosomal subunit, such as chloramphenicol, interfere with the peptydyl transferase activity of the ribosome. Ribosome inhibition affects many biochemical pathways, only some of which ultimately determine the lethality of a particular antibiotic.2 The physical mechanisms of these effects are not well understood.3
Recently, we and others showed that weak, quinary protein interactions4, 5 inside the crowded cytosol are mostly mediated by ribosomes, which represent up to 90% of the total RNA in the cell6 and mRNA.7–10 Quinary interactions also affect protein stability, substrate binding and activity.7, 9, 11–14 We tested the idea that antibiotic ribosome inhibitors act along specific biochemical pathways by directly or indirectly altering the quinary interactions of a target protein. As a first step toward addressing this idea, we developed real-time, RT, in-cell NMR spectroscopy, a flow technique that extends the lifetime of cells in the NMR tube and facilitates the introduction of various stimuli that affect quinary interactions.
Maintaining cell viability is critical because intracellular changes induced by stimuli can take hours to fully manifest into detectable signals. We showed that the continuous flow of media sustains intracellular phosphate-containing metabolites at steady-state levels for up to 24 hours in both bacterial and HeLa cells. We then tested a model of ribosomal inhibition in which the addition of bacteriostatic ribosome antibiotics altered the quinary structure of a target protein. The results are consistent with a physical mechanism in which ribosome inhibitors affect ribosome-mRNA interactions both directly by competing with mRNA for ribosome binding and indirectly by reducing the ribosome concentration in the cell.
Materials and Methods
Expression of [U-15 N, 2H] Trx
We followed the procedure previously reported to over-express Trx.7 Five mL of Lurie Broth (LB) medium supplemented with 75 μg/mL of kanamycin was inoculated with a single colony of pRSF-Trx and grown overnight at 37 °C. The overnight culture was transferred into 200 mL of fresh LB medium containing 75 μg/mL of kanamycin and grown at 37 °C to an OD600 of 0.7–1.0. The culture was centrifuged at 200g for 20 min, the cell pellet washed twice with minimal medium (M9), and re-suspended in 100 mL of deuterated M9 medium containing 1.0 g/L 15N-ammonium chloride as the sole nitrogen source and 0.2 % glucose as the sole carbon source. Over-expression of Trx was induced by adding 1 mM of isopropyl β-D-1-thiogalactopyranoside and allowed to proceed for 18 hrs. Following over-expression the cells were pelleted and washed twice with M9 medium before being cast into agarose. All experiments were repeated in duplicate.
Preparation of HeLa cells
We followed the procedure previously reported to grow HeLa cells.7 HeLa cells (Sigma-Aldrich) were seeded flask into four 150 cm2 Corning flasks at ~5 × 106 cells/flask and cultured for 3 days in complete medium, low-glucose Dulbecco’s modified Eagle medium (DMEM, Gibco), supplemented with 10% fetal bovine serum (FBS, Gibco), until 80% confluence was reached (2 × 107 cells/flask). Cells were harvested by exposure to 0.25% trypsin/EDTA (Sigma-Aldrich) for 5 min at 37 °C. Trypsin was neutralized by a 5-fold dilution with complete medium. Cells were pelleted at 200g for 10 min at 25 °C and washed with 15 mL of prewarmed (37 °C) PBS.
Preparation of Ribosomes
Functionally active ribosomes were purified based on the protocol15 with slight modifications. E. coli cells were grown in LB medium to an OD550 of 0.5 to 0.7 and resuspended in lysis buffer, 20 mM Tris-HCl, pH 7.2, 100 mM ammonium chloride, 10 mM magnesium chloride, 0.5 mM EDTA, and 6 mM βME, at a density of 1 g cells per mL before sonicating with a Model 250 Digital Sonifier (Branson). The lysate was centrifuged at 30,000g for 45 minutes and the supernatant layered onto lysis buffer containing 37.7% sucrose prior to centrifuging at 280,000g for 18 h at 4 °C in an Optima LE-90K Ultracentrifuge (Beckman Coulter) using a SW41 Ti rotor. The clear pellet was washed four times with wash buffer, 10 mM Tris-HCl, pH 7.4, 1 M ammonium chloride, 10 mM magnesium acetate, and 2.5 mM DTT, to remove residual ATPase activity15. The ribosome pellet was re-suspended in 10 mM potassium phosphate, pH 6.5, 10 mM magnesium acetate, and 1 mM DTT. Concentration was determined by absorbance at 260 nm, using an ε0.1% = 15 mg/(mL×cm). Only ribosome solutions with a 260/280 nm ratio of 1.97 to 1.98 were used.
Cell casting
This procedure was modified from a previously reported method16. Pelleted E. coli cells, ~350 μL, containing over-expressed protein were mixed 1:1 (v/v) with 3.0% Cambrex Seaprep Agarose in M9 medium at 37 °C so that the final concentration of the gel was 1.5%. Pelleted HeLa cells, 8 × 107 cells, were mixed 1:1 with 3.0% Cambrex Seaprep Agarose in DMEM at 37 °C so that the final concentration of the gel was 1.5%. The resulting mixtures were injected into 2 m of polytetrafluoroethylene (PTFE) tubing with an inner diameter of 0.5 mm, wrapped around a 2.5 cm diameter cylinder and placed into an ice bath for 15 min to allow the agarose gel to set. Cast cells were extruded into a 5 mm, 600 MHz screw cap NMR tube with a PTFE/silicone septum (New Era) containing a 135 μL plug of 3.0% Cambrex Seaprep Agarose in D2O in the bottom by injecting sterile M9 medium into the tubing.
The perfusion system
A reservoir of fresh M9 medium was connected to the NMR tube using 2 m of PTFE tubing, 0.5 mm I.D., to supply fresh medium and antibiotics to the cells. The end of the tubing was thermally sealed. Eight 50 μm orifices were made over a 3 cm length at the end of the supply tube using a stainless steel dissection needle with 10 μm tip (Roboz Surgical Instrument Co.) held in a microdissection needle holder (Roboz Surgical Instrument Co.). A second, 3 m length of PTFE tubing led to a floor reservoir to receive the spent medium. A gravity siphon was started using a syringe filled with fresh medium. The syringe was attached to a low-pressure tee (Idex Peek) to ensure that no bubbles were present in the system. To maintain a constant flow rate of 100 μL/min, a height difference of 0.86 m was maintained between the two reservoirs. Once the flow of medium was established the sample was equilibrated for 30 min to ensure that no bubbles remained in the system.
Total RNA Extraction
E. coli (~1 ×108 cells) were incubated for 4 hrs in LB with either 15 μg/mL tetracycline (Tet), 50 μg/mL streptomycin (Sm), or 33 μg/mL chloramphenicol (Cm). Total RNA was prepared as described.17 The concentration of RNA preparations was measured by absorbance at 260 nm. The amount of total RNA loaded onto 1 % agarose gels was 500 ng. Ethidium bromide was used to stain the nucleic acids.
Cell Extract
Cellular metabolites were extracted based on a previously reported protocol.18 5 mL of H2O was added to a 3 g cell pellet of E. coli with 0.5 mL of 35% (v/v) perchloric acid. Following sonication the supernatant was neutralized with 2 M KOH and the potassium perchlorate salt was removed. The solution was lyophilized and reconstituted in 1 mL of 90% H2O/10% D2O with 5 mM ethylenediaminetetraacetic acid.
Cell Viability
HeLa cells (8 ×107 cells) were packaged into agarose threads and divided into two aliquots in DMEM. The first aliquot was treated with Trypan blue immediately after casting to selectively stain dead cells. The second aliquot was placed in a 5% CO2 incubator at 37 °C for 24 h before Trypan blue staining. All images were obtained using a Nikon Eclipse TS100 inverted microscope equipped with a Canon EOS digital camera.
NMR experiments
All NMR spectra were recorded at 298K using a 600 MHz Avance III NMR spectrometer equipped with a QCI-P cryoprobe (Bruker). To prepare in vitro NMR samples, 10 μM of ribosome were added to 50 μM of [U- 15N] Trx dissolved in NMR buffer (80 mM potassium phosphate, pH 6.5, 10% D20/90% H20). 1H- 15N-HSQC with Watergate water suppression19 was used to monitor protein chemical shift changes due to ribosome binding. 1024 × 128 points in the proton and nitrogen dimensions, respectively, were acquired with 512 transients. [U- 15N] Trx in E. coli strain BL21(DE3) in the presence of antibiotics was monitored by using cross-correlated relaxation-enhanced polarization transfer transverse relaxation optimized heteronuclear multiple quantum coherence, 1H–15N CRINEPT–HMQC–TROSY, with a CRIPT transfer delay of 1.4 ms and a recycle time of 300 ms. The in-cell [U- 15N] Trx spectrum was first collected in the absence of tetracycline to establish a reference spectrum for each trial. Tetracycline (15 μg/mL) was added to the reservoir of fresh medium and 1H-15N CRINEPT–HMQC–TROSY spectra were collected 5 times in succession over ~18 hours. Chloramphenicol (33 μg/mL) and streptomycin (50 μg/mL) were used in ensuing experiments following the same procedure. Spectral widths in the 1H and 15N dimensions were 12 and 30 ppm, respectively, and 1024 and 128 points were collected in the 1H and 15N dimensions, respectively. Proton decoupled 31P spectra were collected for E.coli and HeLa cells cast in agarose with and without the perfusion system. Spectra were acquired at 3.5 h time s. The 31P peak intensity at −11.5 ppm that contains contributions from the alpha phosphate of both ATP and ADP and diphosphate of NAD+ and NAD(H) was integrated with three different integration ranges to estimate a standard error of the mean. All spectra were processed with Topspin version 3.2 (Bruker) and analyzed by using CARA software.
Data analysis
NMR spectral data was analyzed as previously reported.7, 20 To reassign the in-cell [U- 15N] thioredoxin peaks that changed position from in vitro assignments,21 minimum chemical shift changes were assumed to occur in both the proton and nitrogen dimensions.22 93 out of 102 residues were used in the analysis. Chemical shift changes were calculated as Δ = (δH2 +(δN/42)1/2, where δH(N) represents the change in hydrogen and nitrogen chemical shifts in the absence and presence of antibiotic.7 Changes in intensity were calculated by using ΔI = (I/Iref)bound/(I/Iref)free, where (I/Iref)free is the scaled intensity of an individual peak in the in-cell spectrum of Trx in the absence of antibiotic, (I/Iref)bound is the scaled intensity of individual peaks in the in-cell spectrum of Trx in the presence of antibiotic, and Iref is a glutamine amide side chain at 7.49 ppm and 112.4 ppm in the proton and nitrogen dimensions, respectively, that does not shift in the presence of antibiotic. In our analyses, the chemical shift changes were too small to resolve a principal binding mode, therefore only peak intensities were used. The data were compiled into matrix M (Tables S1–S4). Matrix M was assembled in Excel (Microsoft, Inc), exported as an ASCII text file, and read into MATLAB (Mathworks, Inc.). SVD of matrix M was accomplished by using the [U, S, V] = svd[M] command. The output matrices generated, U, S, V, are the left singular vectors, the singular value matrix, and right singular vectors, respectively. Scree plots of singular values were used to visualize the contribution of each binding mode to M. The Scree plot was further assessed by fitting the singular values with linear regression to determine the coefficient of determination, r2,23. Experimental noise is described by the magnitudes of the second and higher order binding modes. The threshold to determine the amino acids involved in the changes in quinary interactions was set to one and a half times the maximum contribution of the second binding mode.
Results
RT in-cell NMR spectroscopy
Intracellular changes induced by stimuli, such as cellular adaptation to metabolic24, 25 or antibiotic26 stress, can take hours to fully manifest into detectable signals. To achieve at least a 3:1 signal to noise ratio, in-cell NMR experiments often require 2–3 hours or more.27–31 During this time it is important to ensure that the cells remain in a metabolically active state. RT in-cell NMR significantly simplifies and standardizes previously described flow in-cell NMR methods32–34 to guarantee that the experiments are conducted with stable, metabolically active cells.
A schematic of the experimental set-up is shown in Figure 1A. Flow is passively sustained by using a gravity siphon. The continuous exchange of medium eliminates problems associated with cell leakage of labeled proteins, which plagued early applications of in-cell NMR.35 Inlet and outlet tubing are sealed in a standard screw-cap NMR tube with a PTFE/silicone septum. A low-pressure three-way valve attached to a syringe is used to prime the system and to eliminate bubbles that may obstruct the flow. To remove the possibility of potentially costly damage to the NMR spectrometer by flooding the bore, the inlet tubing reservoir is placed below the bottom of the NMR tube so that if vacuum is lost the flow will stop.
Figure 1.
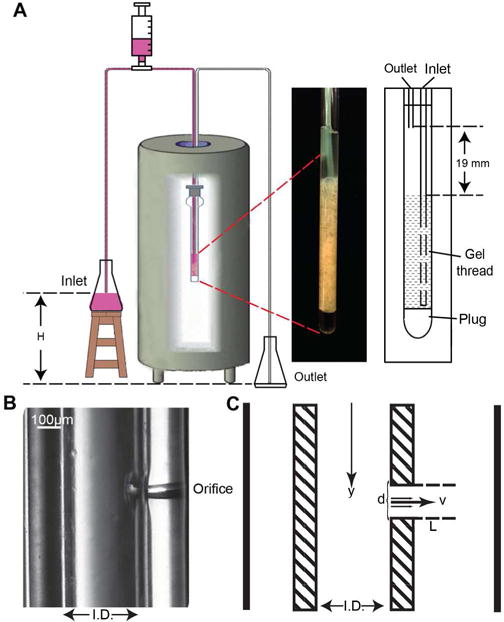
RT in-cell NMR. A gravity siphon drives the continuous flow of fresh medium through the cells for the duration of the experiment. Antibiotics are introduced through the inlet reservoir. B Magnified image of an inlet tubing horizontal drip irrigation orifice. C Schematic of a horizontal drip irrigation orifice showing the formation of the irrigation jet.
Cells containing the labeled target protein are cast in 1.5% low melting agarose to prevent the sample from floating to the top of the NMR tube. Agarose has been shown to maintain cell viability for a variety of cell lines and primary cells over an extended period of time in cell culture studies.36 To facilitate locking on solvent and to eliminate the need for deuterated solvent in the medium, a 3% agarose plug in 100% D2O is placed in the bottom of the NMR tube. A horizontal “drip irrigation” system was developed in which the flow is delivered through orifices in the sides of the inlet tubing (Figure 1B, 1C). This maximizes exposure of the cells to fresh medium and minimizes the upward drag that often dislodges cells from their positions in the NMR tube, which can significantly reduce the quality of the in-cell NMR spectrum.
RT in-cell NMR maintains cell physiology for up to 24 hours
The horizontal drip irrigation design ensures that cell physiology remains stable for the duration of long-term in-cell NMR experiments. Without a continuous supply of fresh medium during in-cell NMR experiments that last for more than 2 hours, cells are significantly affected by changes in the cellular energy state. Cellular levels of phosphate-containing metabolites, ATP, ADP, NAD+, and NAD(H), were monitored during in-cell NMR experiments performed with and without flow by using 31P NMR (Figures 2A, 2B, S1). Fresh M9 medium or HEPES buffered DMEM were used to sustain E. coli and HeLa cells, respectively. Under these conditions, both cell types are maintained in a metabolically active state for up to 24 hours without significant, more than 40%, loss in metabolite concentration. Cell viability was confirmed by assessing cell death within the gel threads. HeLa cells stained with trypan blue were 99 ± 1% viable after 24 hours (Figure 2C and 2D). Visible cell growth after 24 hours confirms the metabolically active status of the cells inside the threads (Figure 2C and 2D).
Figure 2.
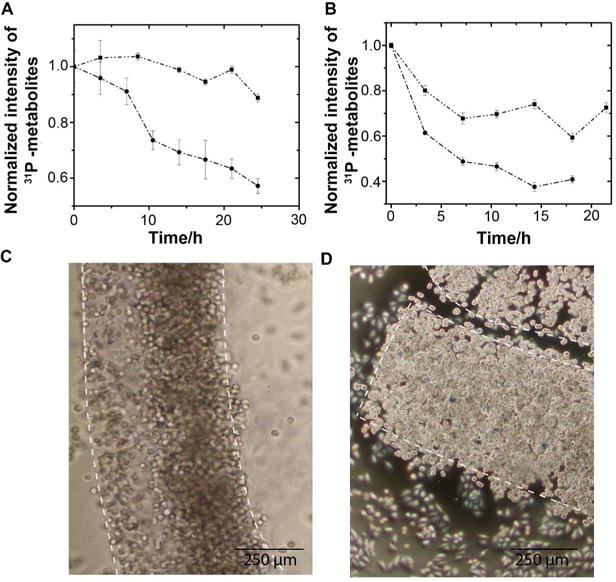
RT in-cell NMR maintains E.coli and HeLa cells in a physiologically stable state for up to 24 hours. A. Signal intensity of cellular 31P-containing metabolites, including ATP, ADP, NAD+ and NAD(H), over time for HeLa cells with (square) and without (circle) flow of DMEM. B. Signal intensity of cellular 31P-containing metabolites over time for E.coli cells with (square) and without (circle) flow of minimal medium. Error bars are the standard deviation of each data point. C. HeLa cells are 99 ± 1% viable immediately after casting. D. HeLa cells are 99 ± 1% viable 24 h after casting; the increase in cell density is consistent with on-going metabolic activity. White dashed lines outline the threads. HeLa cell samples contained 8 × 107 cells. E. coli samples contained cells from 100 mL of culture suspended in ~0.7 mL.
The number of orifices and the length of the irrigation jets were optimized to deliver the flow required to support normal cell physiology (Figure 1). A flow rate of 100 μL/min was required to avoid a rapid drop in the phosphate containing metabolite levels of both E. coli and HeLa cells. To support this flow, eight 50 μm diameter orifices were provided. The length of the submerged jets from the orifices can be estimated by assuming that for a laminary flow of viscous fluids37 the hydrostatic force, ρgH × πd2/4, is equal to the frictional force, μ(dv/dy) × πdL, where ρ is the fluid density; g is the acceleration of gravity; H is the distance between the inlet and outlet reservoirs, 0.86 m; d is the diameter of the orifice, 50 μm; μ is the coefficient of water viscosity, ≈10−3 kg/(mxs); (dv/dy) is the gradient of the flow velocity across the orifice opening,; and L is the length of the jet (Figure 1C). For a 50 μm diameter orifice L ~1 mm, which is smaller than the 5 mm internal diameter of the NMR tube, and permits effective mixing of the medium without disturbing the cell packing.
Analysis of RT in-cell NMR spectra
The chemostatic nature of RT in-cell NMR ensures that the cell culture is maintained in a steady state, allowing changes in quinary interactions to be monitored at atomic resolution and revealing the ability of the cell to adapt to internal and external stimuli. Temporal changes in cellular metabolism resulting from normal cell cycle processes can lead to changes in NMR spectra38. To unambiguously differentiate between signals that result from stimulus specific and non-specific processes, in-cell NMR spectra collected over time are analyzed by using Singular Value Decomposition, SVD20. SVD is a mathematical technique used to identify the principal components of an arbitrary matrix that contribute maximally to the variance of its elements.39 This method has been successfully used to identify protein-protein and protein-drug interactions in live cells by using in-cell NMR.20, 23
By analyzing a temporal series of intrinsically noisy in-cell NMR spectra of target proteins, SVD identifies the residues that exhibit the most prominent changes in both chemical shifts and intensities due to external or internal stimuli. These residues constitute binding modes due to quinary interactions. A Scree plot shows the distribution of singular values that defines the relative contribution of each binding mode to the change in chemical shift or intensity. An abrupt drop in the singular values following the first binding mode indicates a change in quinary interactions due to stimuli; a gradual decrease in singular values indicates no change in quinary interactions and corresponds to noise in the in-cell NMR spectra.20, 23 In our analyses, the chemical shift changes were too small to resolve a principal binding mode, therefore only peak intensities were used. The analysis objectively identifies the amino acid residues involved in the principal binding mode of a target protein with its interactor.
Antibiotic binding to ribosomes alters Trx quinary interactions
To test the influence of antibiotic ribosome inhibitors on quinary interactions in live cells a target protein that does not directly interact with the ribosome (Figure S2) or the antibiotics (Figure S3) was chosen. E. coli thioredoxin, Trx, is a 12-kD oxidoreductase enzyme containing a dithiol-disulfide active site that engages in extensive quinary interactions mediated primarily by mRNA7. We expected that the quinary interactions of Trx will not vary over the time course of the experiment in the absence of antibiotics. On the other hand, since ribosome inhibitors affect ribosome-mRNA interactions both directly by competing with mRNA for ribosome binding (Figure 3A) and indirectly by reducing the ribosome concentration in the cell (Figure 3B & 3C), Trx quinary interactions could be strongly affected by ribosomal inhibitors.
Figure 3.
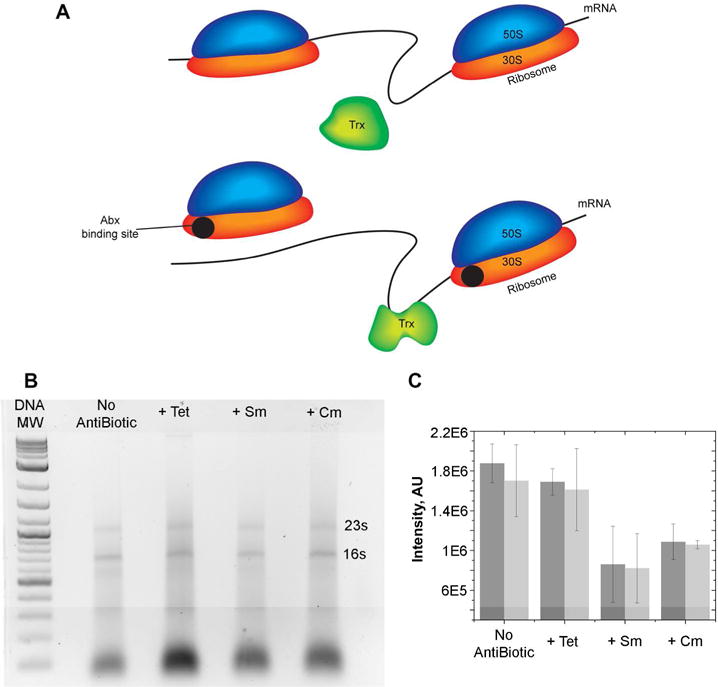
Possible mechanism of ribosome inhibition-dependent changes in biochemical pathways. A. Upper: Trx-mRNA quinary interactions in E.coli. Lower: Antibiotic binding to the small, 30S, ribosome particle alters Trx-mRNA quinary interactions. B. Profile of total E. Coli RNA after adding tetracycline (Tet), streptomycin (Sm), or chloramphenicol (Cm). DNA MW are molecular weight markers. C. Adding Sm or Cm reduces the intracellular concentration of ribosomal RNA bands, 16S (dark gray) and 23S (light gray). Each experiment was performed in duplicate.
Five RT in-cell NMR spectra, each requiring 3 hours, were collected back to back over a 15 hour period. As expected, SVD analysis of the spectra of Trx without antibiotics showed no change in quinary interactions. The Scree plot of singular values showed a monotonic decrease and a linear fit with r2 = 0.84, indicating that there is no change in the quinary interactions20, 23 (Figures S4A and S4B).
After collecting a Trx in-cell spectrum, antibiotic was added to the medium in the inlet reservoir and another series of spectra were collected. Adding tetracycline (Figure 4) or streptomycin, antibiotics that bind to the 30S subunit, resulted in a sharp drop in the Scree plot of singular values (Figure 5) and correspondingly poor linear fits with r2 equal to 0.67 and 0.66, respectively, indicating specific changes in quinary interactions. Extensive broadening of the in-cell NMR spectra provided additional evidence that adding 30S ribosomal antibiotics altered Trx quinary interactions (Figure 4). Adding chloramphenicol, which binds to the 50S ribosomal subunit,40 resulted in a linear decrease in singular values with r2 = 0.94, suggesting that quinary interactions of Trx are not perturbed (Figures S4C and S4D). This result was expected since chloramphenicol does not affect mRNA binding to the ribosome and thus is unlikely to change mRNA-mediated Trx quinary interactions.
Figure 4.
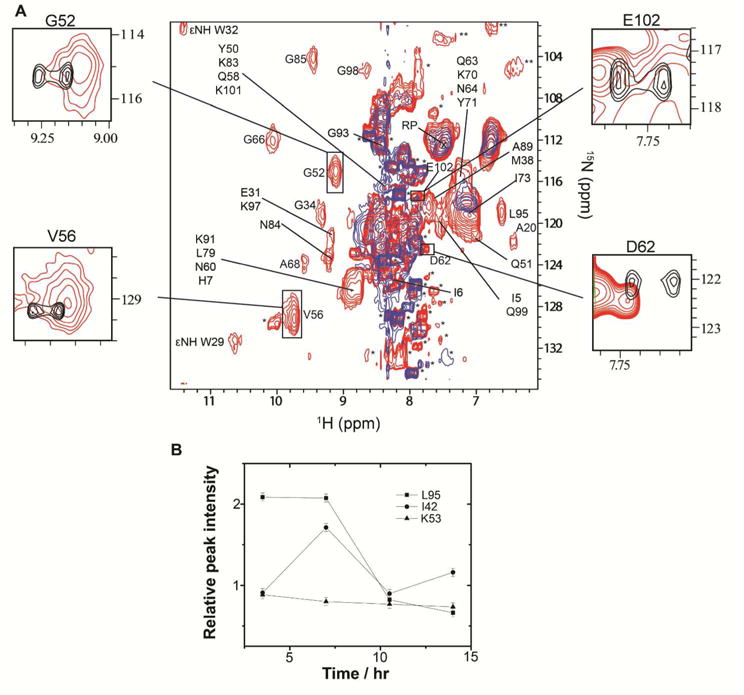
Tetracycline binding to ribosomes changes the quinary structure of Trx in E. coli. A. Overlay of the in-cell 1H–15N CRINEPT–HMQC–TROSY spectra of Trx without (red) and with (blue) tetracycline. The insets show overlays of the boxed regions of the in-cell spectrum (red) and the in vitro 1H–15N CRINEPT–HMQC–TROSY spectrum of purified Trx (black). The intensities of the G52, V56, D62 and E102 peaks, residues involved in quinary interactions, are broadened out in the presence of tetracycline. Single and double asterisks indicate peaks from metabolites and unassigned side chain protons, respectively. The overlaid spectra are at the same contour levels. Reference peak used for peak intensities normalization is indicated by RP. B. Changes in peak intensities of L95, I42 and K53 with time. K53 shows no change, L95 exhibits a drop, and I42 exhibits an early rise then drop in peak intensity. Errors were determined from S/N of the in-cell NMR spectra.
Figure 5.
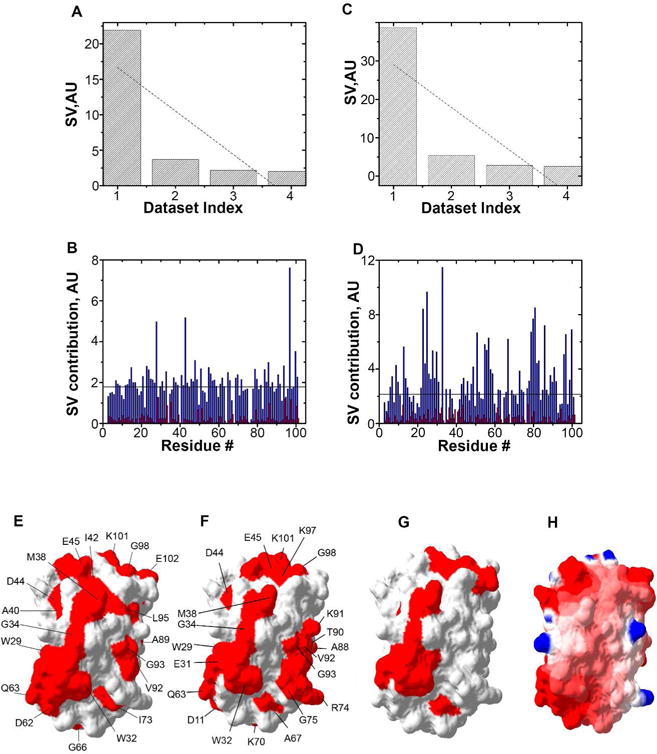
Antibiotic binding to ribosomes affects residues that match the Trx quinary interaction surface. A. Distribution of singular values, SV, of each dataset index (binding mode) for Trx residues in the presence of tetracycline. B. The contribution of each amino acid in response to adding tetracycline is shown for the first (blue) and second (red) binding modes. C. Distribution of SV of each dataset index (binding mode) for Trx residues in the presence of streptomycin. D. The contribution of each amino in response to adding streptomycin is shown for the first (blue) and second (red) binding modes. E. Residues involved in quinary interactions (red) due to the presence of tetracycline are mapped onto the molecular surface of Trx (PDB entry 1X0B). F. Residues involved in quinary interactions (red) due to the presence of streptomycin. G. Quinary interaction surface (red) of Trx in the absence of antibiotics. H. Electrostatic surface map of Trx showing regions of positive, 4 kT/e, (blue) and negative, −4 kT/e (red) potential, where k is the Boltzmann constant, T is the temperature and e is the electron charge. In A and C, linear fits of SVs are shown by dotted lines. In B and D, the horizontal line represents the threshold used to highlight the amino acids with the strongest effect on quinary structure. The threshold value was set at one and a half times the largest contribution from the second binding mode, which represents the average noise of the NMR spectra.
The changes in the quinary interaction surface due to adding streptomycin and tetracycline are similar to each other and involve three contiguous patches located on one face of Trx (Figures 5E & 5F). These patches reflect a changing Trx interactome due to antibiotic stimulus. The first patch, consisting of W29, E31, W32, G34, M38, I42, E45, D62, Q63, and L95 for tetracycline-induced changes, and D11, W29, E31, W32, G34, M38, and Q63 for streptomycin-induced changes, contains hydrophobic and negatively charged residues. This patch is similar to the quinary interaction surface of Trx (Figure 5G) identified by comparing in-cell and lysate Trx spectra7, further validating the role of RNA in forming Trx quinary interactions.
The second and third patches consist of G98, K101, E102 and V92, G93 for tetracycline-induced changes, and E45, K97, G98, K101 and R74, G75 A88, T90, K91, V92, G93 for streptomycin-induced changes. Both contain positively charged and hydrophobic residues consistent with functional groups that mediate RNA binding (Figure 5H). Several other stretches of contiguous amino acids contributed to the first binding mode of both tetracycline-and streptomycin-induced changes (Figures 5B & 5D). These regions consist primarily of buried hydrophobic residues and likely reflect tertiary structural rearrangements of Trx. The effect is more pronounced for streptomycin.
Discussion
The results of RT in-cell NMR spectroscopy are consistent with a model in which ribosomes and Trx compete for mRNA binding (Figure 3A): Weakened ribosome-mRNA interactions result in more extensive mRNA-Trx interactions as indicated by broadening of the Trx in-cell NMR spectra (Figure 4). The changes in quinary interactions due to ribosomal inhibition are almost certainly not unique to Trx. A combination of chemical cross-linking and mass spectrometry resulted in the positive identification of more than 800 proteins that can potentially bind to mRNA or ribosome in various eukaryotic cells11, 12, 41. Among these RNA binding proteins are enzymes involved in intermediary metabolism and important drug targets.42 The ubiquity of RNA-mediated changes in quinary protein interactions may therefore provide a physical basis for ribosome inhibition and other regulatory pathways.
Developed in the course of the present studies, RT in-cell NMR spectroscopy maintains cells in a metabolically active state for more than 24 hours in the NMR tube. This is a critical step towards monitoring intracellular responses to stimuli, in particular those induced by drugs, which can take hours to maturate into detectable signals. The advent of this technology will allow temporal changes in target protein structural interactions to be monitored in real time and will help reveal the inner workings of the cell with unprecedented accuracy and resolution.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant R01GM085006 to AS.
Footnotes
Supporting Information
31P NMR spectra of E. coli and HeLa, overlay of Trx 15N-HSQC spectra without and with ribosomes, SVD analysis of in-cell NMR spectra of Trx in the absence and presence of chloramphenicol, Trx does not interact with antibiotics, in-cell thioredoxin peak intensities in the presence of antibiotics.
References
- 1.Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol. 2014;12:35–48. doi: 10.1038/nrmicro3155. [DOI] [PubMed] [Google Scholar]
- 2.Belenky P, Ye JD, Porter CB, Cohen NR, Lobritz MA, Ferrante T, Jain S, Korry BJ, Schwarz EG, Walker GC, Collins JJ. Bactericidal Antibiotics Induce Toxic Metabolic Perturbations that Lead to Cellular Damage. Cell Rep. 2015;13:968–980. doi: 10.1016/j.celrep.2015.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imlay JA. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol. 2013;11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McConkey EH. Molecular evolution, intracellular organization, and the quinary structure of proteins. Proc Natl Acad Sci U S A. 1982;79:3236–3240. doi: 10.1073/pnas.79.10.3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen RD, Pielak GJ. A cell is more than the sum of its (dilute) parts: A brief history of quinary structure. Protein Sci. 2017;26:403–413. doi: 10.1002/pro.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milo R, Philips R. Cell Biology by the Numbers. 1st. Garland Science; New York, USA: 2015. [Google Scholar]
- 7.Majumder S, Xue J, DeMott CM, Reverdatto S, Burz DS, Shekhtman A. Probing protein quinary interactions by in-cell nuclear magnetic resonance spectroscopy. Biochemistry. 2015;54:2727–2738. doi: 10.1021/acs.biochem.5b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kyne C, Ruhle B, Gautier VW, Crowley PB. Specific ion effects on macromolecular interactions in Escherichia coli extracts. Protein Sci. 2015;24:310–318. doi: 10.1002/pro.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majumder S, DeMott CM, Reverdatto S, Burz DS, Shekhtman A. Total Cellular RNA Modulates Protein Activity. Biochemistry. 2016;55:4568–4573. doi: 10.1021/acs.biochem.6b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barbieri L, Luchinat E, Banci L. Protein interaction patterns in different cellular environments are revealed by in-cell NMR. Sci Rep. 2015;5:14456. doi: 10.1038/srep14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simsek D, Tiu GC, Flynn RA, Byeon GW, Leppek K, Xu AF, Chang HY, Barna M. The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell. 2017;169:1051–1065 e1018. doi: 10.1016/j.cell.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMott CM, Majumder S, Burz DS, Reverdatto S, Shekhtman A. Ribosome mediated quinary interactions modulate in-cell protein activities. Biochemistry. 2017;56:4117–4126. doi: 10.1021/acs.biochem.7b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danielsson J, Mu X, Lang L, Wang H, Binolfi A, Theillet FX, Bekei B, Logan DT, Selenko P, Wennerstrom H, Oliveberg M. Thermodynamics of protein destabilization in live cells. Proc Natl Acad Sci U S A. 2015;112:12402–12407. doi: 10.1073/pnas.1511308112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monteith WB, Cohen RD, Smith AE, Guzman-Cisneros E, Pielak GJ. Quinary structure modulates protein stability in cells. Proc Natl Acad Sci U S A. 2015;112:1739–1742. doi: 10.1073/pnas.1417415112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiel MC, Aoki H, Ganoza MC. Identification of a ribosomal ATPase in Escherichia coli cells. Biochimie. 1999;81:1097–1108. doi: 10.1016/s0300-9084(99)00352-1. [DOI] [PubMed] [Google Scholar]
- 16.Foxall DL, Cohen JS, Mitchell JB. Continuous perfusion of mammalian cells embedded in agarose gel threads. Exp Cell Res. 1984;154:521–529. doi: 10.1016/0014-4827(84)90176-9. [DOI] [PubMed] [Google Scholar]
- 17.Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 1993;15:532–534. 536–537. [PubMed] [Google Scholar]
- 18.Evans FE, Kaplan NO. 31P nuclear magnetic resonance studies of HeLa cells. Proc Natl Acad Sci U S A. 1977;74:4909–4913. doi: 10.1073/pnas.74.11.4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 20.Majumder S, DeMott CM, Burz DS, Shekhtman A. Using singular value decomposition to characterize protein-protein interactions by in-cell NMR spectroscopy. Chembiochem. 2014;15:929–933. doi: 10.1002/cbic.201400030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reverdatto S, Rai V, Xue J, Burz DS, Schmidt AM, Shekhtman A. Combinatorial Library of Improved Peptide Aptamers (CLIPs) to inhibit RAGE signal transduction in mammalian cells. PLoS One. 2013;8:e65180. doi: 10.1371/journal.pone.0065180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farmer BT, 2nd, Constantine KL, Goldfarb V, Friedrichs MS, Wittekind M, Yanchunas J, Jr, Robertson JG, Mueller L. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat Struct Biol. 1996;3:995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- 23.Cobbert JD, DeMott C, Majumder S, Smith EA, Reverdatto S, Burz DS, McDonough KA, Shekhtman A. Caught in action: selecting peptide aptamers against intrinsically disordered proteins in live cells. Sci Rep. 2015;5:9402. doi: 10.1038/srep09402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masters JR, Stacey GN. Changing medium and passaging cell lines. Nat Protoc. 2007;2:2276–2284. doi: 10.1038/nprot.2007.319. [DOI] [PubMed] [Google Scholar]
- 25.Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40:323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol. 2007;42:399–435. doi: 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maldonado AY, Burz DS, Shekhtman A. In-cell NMR spectroscopy. Prog Nucl Magn Reson Spectrosc. 2011;59:197–212. doi: 10.1016/j.pnmrs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature. 2016;530:45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 29.Banci L, Barbieri L, Bertini I, Luchinat E, Secci E, Zhao Y, Aricescu AR. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat Chem Biol. 2013;9:297–299. doi: 10.1038/nchembio.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serber Z, Corsini L, Durst F, Dotsch V. In-cell NMR spectroscopy. Methods Enzymol. 2005;394:17–41. doi: 10.1016/S0076-6879(05)94002-0. [DOI] [PubMed] [Google Scholar]
- 31.Sakakibara D, Sasaki A, Ikeya T, Hamatsu J, Hanashima T, Mishima M, Yoshimasu M, Hayashi N, Mikawa T, Walchli M, Smith BO, Shirakawa M, Guntert P, Ito Y. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature. 2009;458:102–105. doi: 10.1038/nature07814. [DOI] [PubMed] [Google Scholar]
- 32.Sharaf NG, Barnes CO, Charlton LM, Young GB, Pielak GJ. A bioreactor for in-cell protein NMR. J Magn Reson. 2010;202:140–146. doi: 10.1016/j.jmr.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kubo S, Nishida N, Udagawa Y, Takarada O, Ogino S, Shimada I. A gel-encapsulated bioreactor system for NMR studies of protein-protein interactions in living mammalian cells. Angew Chem Int Ed Engl. 2013;52:1208–1211. doi: 10.1002/anie.201207243. [DOI] [PubMed] [Google Scholar]
- 34.Inomata K, Kamoshida H, Ikari M, Ito Y, Kigawa T. Impact of cellular health conditions on the protein folding state in mammalian cells. Chem Commun (Camb) 2017;53:11245–11248. doi: 10.1039/c7cc06004a. [DOI] [PubMed] [Google Scholar]
- 35.Li C, Wang GF, Wang Y, Creager-Allen R, Lutz EA, Scronce H, Slade KM, Ruf RA, Mehl RA, Pielak GJ. Protein (19)F NMR in Escherichia coli. J Am Chem Soc. 2010;132:321–327. doi: 10.1021/ja907966n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang L, Li C, Chen L, Li Z. An agarose-gel based method for transporting cell lines. Curr Chem Genomics. 2009;3:50–53. doi: 10.2174/1875397300903010050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landau LD, Lifshitz EM. Fluid Mechanics. 2d. Pergamon Press; New York, USA: 1987. [Google Scholar]
- 38.Bertrand K, Reverdatto S, Burz DS, Zitomer R, Shekhtman A. Structure of proteins in eukaryotic compartments. J Am Chem Soc. 2012;134:12798–12806. doi: 10.1021/ja304809s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Golub GH, Van Loan CF. Matrix Computations. 4. The Johns Hopkins University Press; Baltimore, USA: 2012. [Google Scholar]
- 40.Dunkle JA, Xiong L, Mankin AS, Cate JH. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc Natl Acad Sci U S A. 2010;107:17152–17157. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, Krijgsveld J, Hentze MW. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 42.Castello A, Hentze MW, Preiss T. Metabolic Enzymes Enjoying New Partnerships as RNA-Binding Proteins. Trends Endocrinol Metab. 2015;26:746–757. doi: 10.1016/j.tem.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.