

Summary
Experimental models of Graves' hyperthyroid disease accompanied by Graves' orbitopathy (GO) can be induced efficiently in susceptible inbred strains of mice by immunization by electroporation of heterologous human TSH receptor (TSHR) A‐subunit plasmid. In this study, we report on the development of a bona fide murine model of autoimmune Graves' disease induced with homologous mouse TSHR A‐subunit plasmid. Autoimmune thyroid disease in the self‐antigen model was accompanied by GO and characterized by histopathology of hyperplastic glands with large thyroid follicular cells. Examination of orbital tissues showed significant inflammation in extra‐ocular muscle with accumulation of T cells and macrophages together with substantial deposition of adipose tissue. Notably, increased levels of brown adipose tissue were present in the orbital tissue of animals undergoing experimental GO. Further analysis of inflammatory loci by 19F‐magnetic resonance imaging showed inflammation to be confined to orbital muscle and optic nerve, but orbital fat showed no difference in inflammatory signs in comparison to control β‐Gal‐immunized animals. Pathogenic antibodies induced to mouse TSHR were specific for the self‐antigen, with minimal cross‐reactivity to human TSHR. Moreover, compared to other self‐antigen models of murine Graves' disease induced in TSHR knock‐out mice, the repertoire of autoantibodies to mouse TSHR generated following the breakdown of thymic self‐tolerance is different to those that arise when tolerance is not breached immunologically, as in the knock‐out models. Overall, we show that mouse TSHR A‐subunit plasmid immunization by electroporation overcomes tolerance to self‐antigen to provide a faithful model of Graves' disease and GO.
Keywords: autoimmunity, Graves' disease, Graves' orbitopathy, self‐antigen mouse model, thyroid disease
Introduction
Graves' hyperthyroid disease is a common autoimmune condition that results from pathogenic autoantibodies to the thyrotrophin hormone receptor (TSHR), which act as agonists for the receptor to mimic the hormone TSH 1. The disease is accompanied frequently by extrathyroidal manifestations such as Graves' orbitopathy (GO) 2. Naturally spontaneous models of Graves' hyperthyroid disease are not available as the illness is uniquely human, although recently a transgenic model in NOD.H2h4 mice over‐expressing human TSHR A‐subunit in the thyroid that develops spontaneously thyroid‐stimulating antibodies (TSAbs) specific for the human receptor has been described 3. Animal models of autoimmune conditions have proved useful to study the immunological basis of autoimmunity and evaluation of novel therapeutic approaches for the treatment of disease 4, 5. We have reported recently the development of a robust and reproducible model of experimental Graves' disease with GO in female BALB/c mice by immunization using electroporation with plasmid‐encoding heterologous human TSHR ectodomain (human TSHR A‐subunit) 6, 7, 8. Another efficient murine model of Graves' disease uses recombinant adenovirus encoding either the human TSHR holoreceptor or the A‐subunit for the induction of autoimmune hyperthyroidism 9, 10. In this model, prolonged regular immunization throughout several months also leads to the development of GO 11. In the models of autoimmune hyperthyroidism induced by delivery of human TSHR A‐subunit cDNA, the induced pathogenic stimulatory antibodies to human TSHR cross‐react with endogenous mouse TSHR in vivo to induce disease. There is a high degree of amino acid homology (> 87%) between mouse and human TSHRs, which explains the cross‐reactivity of induced anti‐human TSHR antibodies with the endogenous mouse receptor for induction of disease. However, despite the high sequence homology in TSHR between these two species, immunization of mice with the homologous mouse TSHR A‐subunit with adenovirus does not lead to induction of autoimmunity to the self‐antigen 12. These results suggest that immune tolerance to the mouse TSHR is robust in wild‐type mice and not easily breached to promote autoimmune disease. One early study has reported the use of cellular delivery of mouse TSHR or purified mouse recombinant receptor protein to induce self‐autoimmunity with features of Graves'‐like disease, but the model remains to be capitalized 13.
Other studies have used genetic models to overcome self‐tolerance to mouse TSHR to develop an experimental self‐antigen model of Graves' hyperthyroid disease. In these studies, TSHR knock‐out (KO) animals, where the TSHR has been deleted genetically, do not develop self‐tolerance to the receptor. Immunization with adenovirus expressing mouse TSHR A‐subunit leads readily to induction of anti‐mouse TSHR antibodies but, naturally, the immune animals cannot respond to the pathogenic antibody as they lack endogenous receptor. For disease induction, adoptive transfer of spleen cells from immune KO animals to T cell‐deficient mice expressing endogenous TSHR was sufficient for the onset of self‐autoimmunity with features of Graves' disease and orbital inflammation 14. Another study has reported a self‐antigen TSHR model of Graves'‐like disease using a variant of mouse TSHR where exon 5, coding for 25 amino acids (mouse TSHR739), is deleted, but the onset of GO was not examined in the study 15.
In the present study, we show that immunization by electroporation of mouse TSHR A‐subunit plasmid in female BALB/c mice is sufficient to break self‐tolerance and leads to the successful development of a faithful model of Graves' thyroid disease and accompanying GO.
Materials and methods
Construction of pTRiEx1.1 neo‐mouse TSHR A‐subunit plasmid
Mouse TSHR A‐subunit gene coding for amino acids 1–289 (including the signal sequence) was synthesized as a synthetic gene with mouse codon optimization with added Kozak sequence and RNA instability motif to improve translation, with BamH1 and Not1 cloning sites for in‐frame cloning into the multi‐system expression plasmid, pTriEx‐1.1‐neo 7. For plasmid cloning of the synthetic gene, the human TSHR A‐subunit gene was excised from pTRiEx1.1 neo‐human TSHR A‐subunit plasmid 7 by BamH1 and Not1 digestion and replaced with the mouse TSHR A‐subunit gene. The cloned mouse TSHR A‐subunit gene in the recombinant plasmid was sequenced fully to verify faithful sequence. The construction of the synthetic mouse TSHR A‐subunit gene, plasmid subcloning steps and DNA sequencing was undertaken by GenScript USA, Inc. (Piscataway, NJ, USA).
The control pTRiEx1.1 neo‐β‐Gal plasmid was available from our previous study 7. All plasmids were grown by transformation of Escherichia coli XL‐1 blue competent cells. Plasmids were purified with Qiagen Giga Prep kits (Qiagen, Valencia, CA, USA) to yield mg quantities. The plasmid constructs used in this study are referred to as mouse TSHR A‐subunit plasmid and control β‐Gal plasmid.
Mice
BALB/c mice were bred and housed in a specific pathogen‐free environment in animal care facilities at University Hospital Essen. Investigations involving animals were approved by the institutional ethics animal committee of North Rhine Westphalia State Agency for Nature, Environment and Consumer Protection, Germany. At the age of 6–8 weeks, mice were immunized as described 7, 8. Briefly, female BALB/c mice were immunized by intramuscular (i.m.) injection and electroporation of 50 µg (1 mg/ml) plasmid into each biceps femoris muscle four times, 3 weeks apart. A total of 15 animals were immunized with pTRiEx1.1 neo‐mouse TSHR A‐subunit plasmid and 17 animals with control β‐Gal plasmid. Mice were monitored for disease development by almost daily examination (5–6 days a week) of eye signs during immunizations for 6 weeks after the last immunization. The disease was divided into three stages: initiation/early onset (red eyes and lids, proptosis, swelling of the lid) acute (upcoming pus in one or both eyes) and chronic (persistent symptoms for at least 5 days). Animals were weighed every 3 weeks. After killing, blood was collected from inferior vena cava puncture and serum stored aliquoted at −80°C.
Transfection of plasmid and Western blotting
For transfection of plasmid wild‐type Chinese hamster ovary (CHO) cells were transfected with pTRiEx1.1 neo‐mouse TSHR A‐subunit plasmid (1 µg) with FuGene and grown for 72 h. Transfected cells or stably transfected mouse TSHR CHO cell cultures were lysed as described previously 16 and 70 µg total cell lysate was subjected to 7·5% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and blotted to nitrocellulose. Western blotting was performed with goat anti‐TSHR polyclonal antibody (1 : 200; Bioss GmbH, Düsseldorf, Germany) overnight at 4°C, detection with horseradish peroxidase (HRP)‐labelled anti‐goat immunoglobulin IgG (1 : 5000; Sigma‐Aldrich, St Louis, MO, USA) and enhanced chemiluminescence (ECL; GE Healthcare, Chicago, IL, USA).
Assessment of thyroid disease and GO
Total thyroxine (T4) was measured using 50 µl serum by enzyme‐linked immunosorbent assay (ELISA) (DRG Diagnostics GmbH, Marburg, Germany). Serum antibodies to TSHR were measured using human TRAK assay, which uses luminescence‐labelled (acridium derivative) bTSH as competitor (Thermo Fisher BRAHMS TRAK Human LIA; Thermo Fisher, Waltham, MA, USA) with 100 µl diluted serum (diluted 1 : 3 with normal human serum). TSAbs were determined from 3 µl serum in stably transfected mouse TSHR CHO cells 17, 18 and cyclic adenosine monophosphate (cAMP) in the supernatants measured by the ELISA kit (Enzo Laboratories, Farmingdale, NY, USA) 7.
Thyroid and orbital pathology
Microsurgical excision of thyroid and one orbital tissue was performed and tissue fixed in buffered formalin and paraffin‐embedded. The second orbital tissue was used for isolating and growing in vitro low passage cultures of orbital fibroblasts and stored frozen in liquid nitrogen. Sections of 1 µm were then cut and stained using haematoxylin and eosin (H&E) (thyroid glands and orbits) and PicroSirius red for collagen deposition (orbits only). Sections were deparaffinized and stained for 10 min in Weigert's iron haematoxylin solution for 10 min, followed by PicroSirius Red solution for 60 min. PicroSirius red staining of slices was analysed quantitatively with CellProfiler. All orbits were examined at the anterior, middle and posterior areas. All thyroid and orbital sections were analysed blindly.
Immunohistochemistry for detection of orbital infiltrating leucocytes and cytokines
Consecutive slices of orbital tissue from the middle area were stained for F4/80, CD3 and uncoupling protein‐1 (UCP‐1) with the HRP‐conjugated polymer system (Zytomed, Berlin, Germany), as described previously 8. Total fat area and UCP1‐positive stained adipose tissue were determined with ImageJ software (National Institutes of Health, Rockville, MD, USA). F4/80 and CD3‐positive cells were counted in muscle, perineural connective tissue and fat tissue of one entire slide of the middle area. Images were generated using an Olympus BX51 microscope (Olympus, Düsseldorf, Germany). Numbers of cells were normalized per mm2.
Magnetic resonance imaging (MRI)
From the group of immunized animals, five animals were selected randomly from each group (mouse TSHR A‐subunit plasmid and control β‐Gal) for MRI. At least 48 h before MRI, animals were injected intravenously (i.v.) with 300 µl perfluorocarbon (PFC). PFC is ingested avidly by macrophages and monocytes to visualize areas of inflammation by 19F MRI 19, 20. Final MRI in immune animals was performed 6 weeks after the last immunization, after which they were killed. Data were recorded on a Bruker AVANCEIII® 9.4T wide‐bore NMR spectrometer driven by ParaVision® 5.1 (Bruker, Rheinstetten, Germany). Mice were anaesthetized with 1·5% isoflurane and maintained at 37°C. Vital functions were supervised with a pneumatic pillow and monitored by a M1025 system (SA Instruments, Stony Brook, NY, USA). Data acquisition and analysis were carried out in a non‐blinded fashion by an experienced investigator (with more than 25 years’ practical knowledge in experimental MRI). For acquisition of anatomical reference images, a multi‐slice T2‐weighted turbo spin echo (TSE) sequence was used with an in‐plane resolution of 50 × 50 µm2 and a slice thickness (ST) of 0·5 mm. The slice package covered the entire orbit for analysis of different tissue types (muscle, adipogenesis, nerve and all anatomical structures). After the acquisition of all 1H data sets, the resonator was tuned to 19F, and morphologically matching 19F MR images were recorded. Inflammatory foci were determined from 19F MR images by planimetric analysis of PFC signals using the return on investment (ROI) tool of ParaVision® (Billerica, MA, USA). The full experimental protocol took 60–90 min and was well tolerated by all mice, which recovered from anaesthesia within a few minutes with all animals surviving the procedure.
Statistical analyses
Statistical analyses were performed using GraphPad (Prism 7). The level of significance was set at P < 0·05. Statistical analyses carried out with two‐tailed Student's t‐tests with a confidence level greater than 95%. Data are presented as arithmetic mean ± standard error of the mean (s.e.m.). P‐values are marked with stars representing *P < 0·05; **P < 0·01; ***P < 0·001 and ****P < 0·0001.
Results
In‐vitro expression of pTRiEx1.1 neo‐mouse TSHR A‐subunit plasmid
We first confirmed that the polyclonal anti‐mouse TSHR antibody recognizes the mouse receptor by Western blotting by testing with an extract from stably transfected mouse TSHR CHO cells, which shows a band co‐migrating at approximately 125 kD, representing the intact TSHR (Supporting information, Fig. S1a). Most probably, the high glycosylation (40%) of TSHR protein accounted for irregular migration on SDS‐PAGE gels 21. Examination of mouse TSHR A‐subunit protein expression by transfection of the plasmid into CHO cells and Western blotting showed a specific band co‐migrating at approximately 41 kD, which was absent in lysates from control wild‐type CHO cells (Supporting information, Fig. S1b, lanes 1 and 2, respectively). The 41 kD band represents mouse TSHR A‐subunit most probably with all four N‐linked sites glycosylated 21. The data show that the pTRiEx1.1 neo‐mouse TSHR A‐subunit plasmid results in high protein expression of the mouse receptor in in‐vitro transfection studies and suitable for use as an immunogen for in‐vivo expression of the receptor.
Antibody in mouse TSHR‐immunized animals
Female BALB/c mice were immunized by electroporation with pTRiEx1.1 neo‐mouse TSHR A‐subunit plasmid, as described in our earlier studies with the heterologous human TSHR A‐subunit plasmid that provides a robust model of experimental GO 7, 8. As controls, animals were immunized with β‐Gal plasmid. Serum was assessed for anti‐mouse TSHR responses when animals were killed 6 weeks after the end of immunization. Measurement of TSAbs using transfected mouse TSHR CHO cells showed significant positivity in serum from the mouse TSHR A‐subunit plasmid‐immunized group compared to the control β‐Gal group (P < 0·0001) (Fig. 1a). Next, we determined if anti‐mouse TSHR antibodies induced in mouse TSHR plasmid immunizations cross‐react with human TSHR. In the first experiment, we assessed TSH binding inhibiting immunoglobulins (TBII) using commercial TRAK kits, where captured human TSHR serves as an antigen for binding luminescence‐labelled bTSH as tracer. The results show that antibodies generated to mouse TSHR fail to inhibit the binding of labelled tracer to human TSHR and hence fail to recognize the human receptor. In contrast, serum samples from human TSHR A‐subunit plasmid‐immunized animals (from a different study) used as controls in the same experiment show > 80% inhibition of tracer binding in the assay (Fig. 1b). To confirm these results further, we tested the anti‐mouse TSHR antibodies for TSAb activity in stably transfected human TSHR CHO cells and showed no measurable cross‐reactivity to human TSHR (Fig. 1c). However, when the anti‐mouse TSHR antibodies were evaluated for TSAbs in stably transfected mouse TSHR CHO cells, 11 of 14 immune animals showed significant stimulating antibodies (Fig. 1d). Nevertheless, TSAb activity was weak, consistent with no changes in serum total thyroxine 4 (TT4) levels and indicative of the absence of endocrine biofunction, despite the pathological effects observed in the thyroid glands. Overall, these findings indicate that anti‐mouse TSHR antibodies induced by mouse TSHR A‐subunit plasmid immunizations are highly specific for the self‐antigen with minimal cross‐reactivity. Conversely, anti‐human TSHR antibodies induced by human TSHR A‐subunit plasmid immunizations are cross‐reactive with mouse TSHR.
Figure 1.
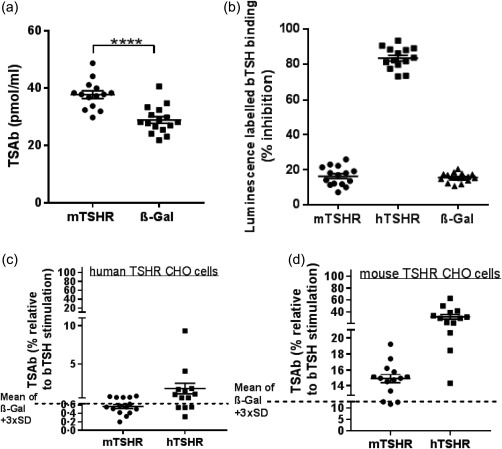
Anti‐thyroid stimulating hormone receptor (TSHR) antibodies induced in mouse TSHR A‐subunit‐immunized female BALB/c mice. (a) Thyroid stimulating antibodies (TSAbs) measured in bioassay for cyclic adenosine monophosphate (cAMP) production using stably transfected mouse TSHR expressing Chinese hamster ovary (CHO) cells. TSAb activity (pmol/ml cAMP) in individual mouse serum is shown, together with serum from control β‐Gal mice. The mouse TSHR A‐subunit‐immunized group (labelled mTSHR) was significantly different in TSAb activity from the control group (labelled β‐Gal); ****P ≤ 0.0001 (t‐test). (b) Serum antibodies to human TSHR were measured using human TRAK assay, which uses luminescence‐labelled (acridium derivative) bovine thyroid‐stimulating hormone (bTSH) as competitor (Thermo Fisher BRAHMS TRAK human LIA) with 100 µl diluted serum (diluted 1 : 3 with normal human serum). The y‐axis shows % inhibition of tracer binding to immobilized human TSHR in the test. Individual sera from the mouse TSHR A‐subunit‐immunized group showed low levels (< 25%) of % inhibition of tracer binding considered to be in the ‘grey’ zone, which was in the same range as control β‐Gal animal serum. As a positive control, serum from human TSHR A‐subunit mice (labelled hTSHR) (from a different experimental group) showed high levels (> 80%) of inhibition of tracer binding to immobilized human TSHR in the assay. (c,d) Assessment of cross‐reactivity by measurement of relative TSAb activity in individual serum samples in mouse TSHR A‐subunit‐immunized animals, assayed in stably transfected human TSHR‐expressing Chinese hamster ovary (CHO) cells and in mouse TSHR‐expressing CHO cells. Serum from human TSHR A‐subunit‐immunized animals was also measured for cross‐reactive antibodies. The y‐axis shows the stimulation obtained with 0·9 mU/ml bTSH shown as 100% value, where the stimulatory response of immune serum samples was recorded as a % of the bTSH response. The mean response obtained with control β‐Gal serum +3 standard deviation (s.d.) is marked (broken line); values above scored as significantly positive. The results in (c) in human TSHR CHO cells show serum from mouse TSHR A‐subunit animals to poorly stimulate human TSHR and hence to fail to be cross‐reactive to human receptor. The results in (d) measured in mouse TSHR CHO cells show serum from mouse TSHR A‐subunit animals to stimulate mouse TSHR (like panel a data); similarly, serum from human TSHR A‐subunit mice cross‐react strongly with mouse TSHR.
Thyroid histology in mouse TSHR A‐subunit‐immunized mice
Thyroid gland sections were examined blindly by a reader unaware of the immunization scheme. Mouse TSHR‐immunized animals showed predominantly hyperplastic thyroids characterized by cuboid cylindrical follicular cells with a small amount of colloid (seven of 15 animals), with only one gland with hypothyroidism features characterized by thin follicles (one of 15 animals). The remaining thyroid glands from immune animals showed a histologically normal appearance, similar to control β‐Gal‐immunized animals (Fig. 2a). There was no evidence of follicular destruction, fibrosis or lymphocytic infiltration in any of the thyroid glands (Fig. 2b–d). Most probably as a consequence of ‘hyperthyroidism’, the mTSHR‐immunized mice showed an increase in heart size (Supporting information, Fig. S2a), as reported earlier for the adenovirus model 11, while body weights were stable (Supporting information, Fig. S2b). However, despite the activated thyroid follicular cells and increased size of the heart in mouse TSHR A‐subunit plasmid‐immunized animals, the mean TT4 levels were not elevated and not different from control β‐Gal plasmid‐immunized mice (Supporting information, Fig. S3).
Figure 2.
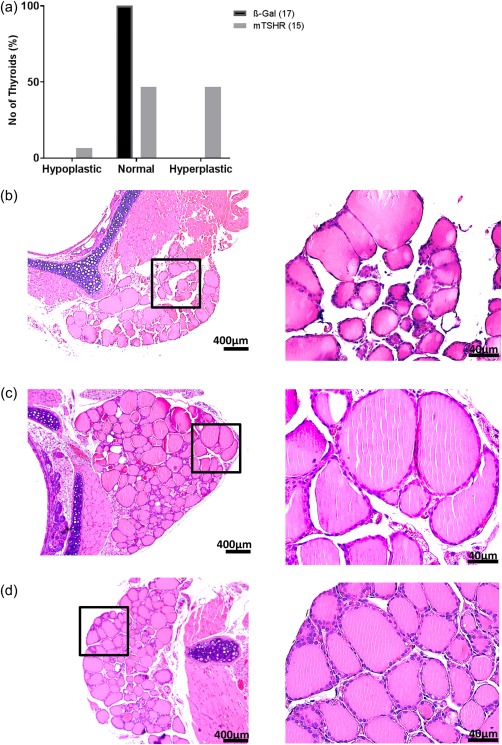
Thyroid histology of mouse thyroid stimulating hormone receptor (TSHR) A‐subunit‐immunized mice. (a) Thyroid slices of the animals were haematoxylin and eosin (H&E)‐stained, and scored for thyroid status as described in the Methods and Results. Histologically scores of β‐Gal and mouse TSHR‐immunized mice were performed blindly by a reader unaware of the immunization scheme. A number of hyper‐, hypo‐ and euthyroid individual mice are shown. All control β‐Gal mice were scored as euthyroid. One mouse TSHR A‐subunit animal was scored as hypothyroid, seven of 15 immune animals as euthyroid and the last seven as hyperthyroid. (b) Thyroid histology of mouse TSHR A‐subunit‐immunized mouse which showed signs of hypothyroidism with characteristically thin follicular epithelium. (c) Example of thyroid of control β‐Gal scored as euthyroid. (d) Thyroid of mouse TSHR A‐subunit‐immunized mouse which demonstrates signs of hyperthyroidism‐like empty follicles and thicker thyrocytes (magnification ×100 and ×400). [Colour figure can be viewed at wileyonlinelibrary.com]
Orbital histopathology and MRI of mouse TSHR A‐subunit‐immunized mice
Orbital inflammation examination by MRI
We first examined for signs of orbital inflammation in live animals by 1H/19F MRI in five randomly selected immune animals undergoing experimental GO at −3 days before killing at 6 weeks after the end of immunization (Supporting information, Fig. S4a,b). 19F MRI showed increased orbital inflammation in mouse TSHR‐immunized mice compared to control animals (Fig. 3a, Supporting information, Fig. S4c,d) and 1H MRI showed increased deposition of adipose tissue around the optic nerve (Supporting information, Fig. S4e,f).
Figure 3.

Inflammation of orbital tissue from mouse thyroid stimulating hormone receptor (TSHR) A‐subunit‐immunized mice evaluated by magnetic resonance imaging (MRI) and F4/80 staining. Immune animals 6 weeks after the last immunization; five mice from each group were selected randomly for MRI. At least 48 h before imaging, animals were injected intravenously (i.v.) with perfluorcarbon (PFC), which is taken up avidly by circulating monocytes and macrophages and inflammatory tissue detected with 19F M RI. (a) Quantification of 19F signal from MRI expressed as 19F integral in arbitrary units (a.u.). Significantly increased orbital inflammation in mouse TSHR A‐subunit immunized animals compared to controls, *P < 0.05. B: F4/80 positive cells were counted in the periorbital tissue (in No/mm2). Significant increased localized inflammation in animals undergoing experimental GO in comparison to control β‐Gal was found in muscle and perineural tissue (****P < 0.001). (c) CD3‐positive cells were counted in the periorbital tissue (in No/mm2). Again, most of the CD3‐positive cells in mouse TSHR‐immunized mice were localized in muscle and periorbital tissue in comparison to control β‐Gal orbits. (d,e) Section in the middle region of the orbital perineural region from mouse TSHR A‐subunit immune animal (×400). The optic nerve is marked as ‘nerve’ in the photomicrographs, adipose tissue as ‘AT’. The arrows in photomicrographs indicate areas of F4/80 active accumulation of inflammatory cells in the perineural tissue (d) and muscle tissue (e) of animals undergoing experimental GO. (f,g) Section in the middle region of orbital muscle from mouse TSHR A‐subunit immune animal undergoing CD3 IHC (×400). The arrows in photomicrographs indicate areas of CD3‐positive cell accumulation in the perineural tissue (f) and in the orbital muscle (g) of animals undergoing experimental GO. Overall, the MRI studies and immunohistochemistry (IHC) studies both provide independent evidence for increased inflammation in the orbital tissue of animals undergoing experimental GO. [Colour figure can be viewed at wileyonlinelibrary.com]
Examination by IHC
Additional studies on inflammation in diseased orbital tissues was performed in H&E‐stained sections. Compared to orbits from control β‐Gal animals which show a normal appearance, all orbital tissues from animals undergoing GO showed histological abnormalities. Monocytes/macrophage infiltrate in orbital adipose tissue and skeletal muscle was also confirmed by F4/80 immunostaining (Fig. 3b,d,e). Quantitation of F4/80 infiltrating cells in the entire orbits of mouse TSHR A‐subunit‐immunized orbital tissue gave a significant increase in the number of inflammatory cells (Fig. 3b), located mainly in orbital muscle and optic nerve tissue (Fig. 3d,e). In contrast, adipose tissue in the orbits showed a low abundance of F4/80 cells in animals undergoing GO and similar in numbers to control β‐Gal animals (Fig. 3d). We also quantified the abundance of inflammatory CD3‐positive T cells by immunohistochemistry (IHC), showing a significant increase in orbital tissues in GO animals compared to control β‐Gal animals (Fig. 3c,f,g). The data provide evidence of substantial inflammatory infiltrate in orbital muscle and optic nerve tissue in animals undergoing GO, while orbital fat tissue showed no difference of inflammatory signs in comparison to control β‐Gal animals.
Orbital adipogenesis
The extent of orbital adipogenesis in diseased animals was measured by MR imaging, without measurably significant differences in the content of orbital fat tissue in diseased orbits compared to orbital tissue from control animals (Fig. 4a, Supporting information, Fig. S4e,f).
Figure 4.
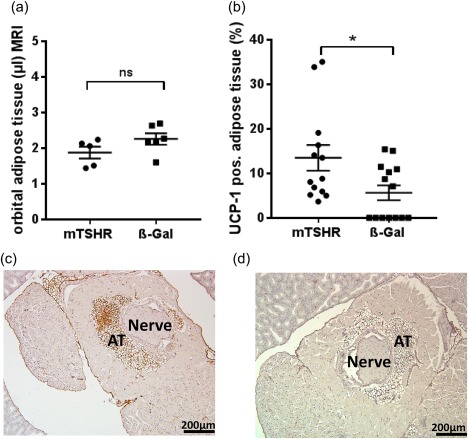
Analysis of adipose tissue (AT) in orbital region from mouse thyroid stimulating hormone receptor (TSHR)‐immunized animals evaluated by magnetic resonance imaging (MRI) and uncoupling protein‐1 (UCP‐1) staining. (a) The orbital regions of the animals (in Fig. 2) were imaged additionally by T2‐weighted 1H magnetic resonance imaging (MRI). The volume (µl) of the AT in the periorbital and retro‐orbital area was measured. No statistically significant (n.s.) expansion of total adipose tissue was found in the orbital tissue of animals undergoing experimental Graves' orbitopathy (GO) compared to controls. (b) Slices of the middle orbital area were stained by immunohistochemistry (IHC) for UCP‐1 as a marker for brown/beige fat. The total area of AT and area of UCP‐1‐positive tissue was measured. The mouse TSHR A‐subunit‐immunized mice show a significant increase in brown/beige AT in comparison to control β‐Gal‐immunized mice, *P ≤ 0·05. (c,d) Representative images of orbital tissue stained by IHC for UCP‐1 of mouse TSHR A‐subunit‐immunized mice (c) and control β‐Gal (d). UCP‐1‐positive stained AT (brown‐coloured) was quantified and is expressed as UCP‐1‐positive AT % of total adipose tissue. [Colour figure can be viewed at wileyonlinelibrary.com]
Further examination of orbital adipogenesis was evaluated by UCP‐1 expression by IHC for quantitating brown/beige adipose tissue (BAT) (Fig. 4b–d). The significantly elevated UCP‐1 positive BAT was shown to be present in the total orbital adipose tissue of mice undergoing GO compared to control β‐Gal mice (Fig. 4b). Notably, the total area of adipose tissue in orbit remained unchanged, suggesting that white orbital tissue (WAT) was being replaced by BAT during orbital pathogenesis. It is also important to highlight that the two different techniques of MR imaging and UCP‐1 staining of orbital tissue used in this study provide different information on quantitative aspects of adipogenesis in animals undergoing GO. Thus, quantitation of MR imaging indicates total fat expansion in the orbit without distinguishing between WAT and BAT, but UCP‐1‐stained sections provide quantitation of percentage of BAT and WAT in the orbital tissue. Overall, quantitation of total adipogenesis in the orbit by both methods give comparable results (Fig. 4a).
Changes in orbital muscle
We further examined for evidence of disease orbital muscles in H&E sections by studying inferior rectus and medial rectus muscle in all immune animals undergoing GO due to their appropriate anatomical position. Pictures were taken and prepared with Image J® for quantitative analysis with CellProfiler®. Area of muscle fibre and total muscle area were measured quantitatively in µm2. The whole area of the muscle did not show any statistically significant difference (data not shown). Measuring the single muscle fibres from all animals in the study group, there were statistically significantly increased mean areas of muscle fibres in mouse TSHR‐immunized mice in comparison to control mice (Fig. 5a–c). Moreover, analysis of the 75th percentile (218 µm2) of orbital muscle fibres revealed that mouse TSHR‐immunized mice have a higher number of thicker muscle fibres (> 218 µm2) than control animals as a sign of inflammation (Fig. 5d).
Figure 5.
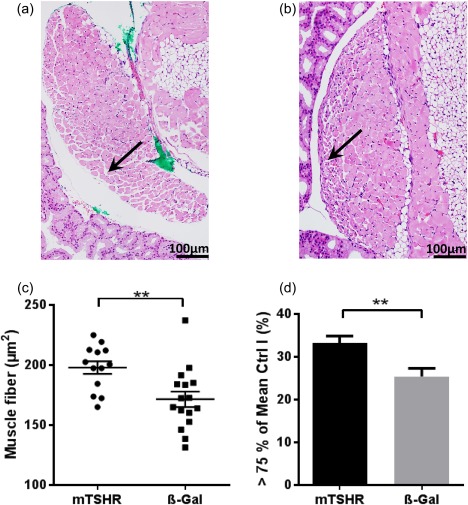
Muscle changes in the orbital tissue of mouse thyroid stimulating hormone receptor (TSHR) A‐subunit‐immunized mice. Slices of the middle orbital area of one orbit were haematoxylin and eosin (H&E)‐stained to analyse the morphology of muscle. Rectus inferior muscle and obliquus inferior muscle of every individual mouse were analysed quantitatively with Image J to analyse muscle changes. (a,b) representative muscle from mouse TSHR A‐subunit‐immunized animal (magnification ×200). The green colour in A was used as a marker for orientation after preparation of the section. Arrow indicates larger muscle fibres at the edge. (b) An example of a muscle of β‐Gal mouse with an arrow marking smaller muscle fibres at the edge. (c) Further analyses showed that the individual muscle fibres in each muscle are increased in animals undergoing experimental Graves' orbitopathy (GO) (**P ≤ 0·01), but orbital muscles in both groups (mouse TSHR A‐subunit and control β‐Gal) have the same total size (data not shown). (d) After determining the 75th percentile of all control β‐Gal mice muscle fibres (218 µm2) of rectus inferior muscle and obliquus inferior muscle, animals undergoing experimental GO have a statistically significant increased number of larger muscle fibres (**P ≤ 0·01). [Colour figure can be viewed at wileyonlinelibrary.com]
Clinical evidence of disease
Eye signs examined daily for 5‐6 days every week during the immunization period and 6 weeks after the end of immunization did not show any GO typical changes in the eyes of β‐Gal control mice, while all mouse TSHR‐immunized mice showed eye signs (acute inflammation, proptosis or chronic disease) 10 times or more often during the whole time (Supporting information, Fig. S5a–d). The earliest inflammation was shown after the second immunization step.
Summary of orbital disease
In summary, analyses of orbital inflammation and adipogenesis in mouse TSHR immune animals by IHC and MR imaging studies show increased inflammation and adipogenesis in the orbital tissue of BAT compared to control β‐Gal animals. Importantly, orbital inflammation was restricted to orbital muscle and optic nerve tissue, while orbital adipose tissue showed no difference in inflammatory signs in comparison to control β‐Gal animals. Orbital muscle showed thicker muscle fibres as a sign of inflammation, and clinical signs of inflammation shown as eye signs in mouse TSHR‐immunized mice could also be evaluated.
Discussion
In this study, we show that immunization by electroporation of homologous mouse TSHR A‐subunit plasmid provides sufficiently strong stimulus to break self‐tolerance for induction of autoimmune disease. Graves' disease was confirmed by characteristic histopathology of hyperplastic glands without follicular destruction or fibrosis, which was absent in control β‐Gal‐immunized animals. Moreover, the induced ‘hyperthyroidism’ was accompanied by tissue remodelling by orbital muscle inflammation and changes in adipose tissue. With some exceptions, attempts to induce autoimmune Graves'‐like hyperthyroidism with mouse TSHR cDNA constructs have proved difficult. The most efficient mouse model for Graves' disease is dependent upon immunization with the adenovirus‐expressing human TSHR A‐subunit 10. In this model, the antibodies induced to human TSHR cross‐react with endogenous mouse TSHR, leading to stimulation of the thyroid gland with resulting autoimmune hyperthyroidism. In contrast, immunization with adenovirus expressing the corresponding region in mouse TSHR A‐subunit failed to induce disease in different inbred strains of mice 12. Additional manipulations such as depletion of the regulatory T cell (Treg) population of CD4 T cells had negligible effects on disease induction 12.
Another novel approach has used genetic knock‐out (KO) mice deficient in mouse TSHR in conjunction with adoptive transfer to develop an autoimmune model of Graves' hyperthyroidism and GO, together with the presence of infiltrating macrophages in the orbital adipose tissue and muscle in a small number of immune animals 14. Our data on antibodies induced to mouse TSHR in our mouse TSHR A‐subunit plasmid electroporation model show similarities and differences from those arising in genetic KO models 14. In the latter study, the induced anti‐mouse TSAbs were detectable more readily by bioassay with mouse TSHR‐transfected CHO cells rather than with human TSHR‐transfected CHO cells, although there was an apparently wide variability in the detection of the TSAbs in each of these transfected CHO cells 12. These data are in marked contrast to the anti‐mouse TSHR antibodies induced in our model, as they fail to cross‐react with human TSHR using two different assay systems. The data suggest that the antibody repertoire of autoantibodies to mouse TSHR generated by inducing breakdown of self‐tolerance in our model may differ from those autoantibodies that arise when tolerance is not breached immunologically, as occurs in KO animal models.
Why genetic delivery of mouse TSHR A‐subunit plasmid by electroporation is more efficient in the induction of self‐autoimmunity than delivery by adenovirus can only be speculated. Our understanding of the outcome of genetic delivery by intramuscular injection suggests that migrating skin and mucosal dendritic cells (DCs) endocytose plasmid to express processed peptides on major histocompatibility complex (MHC) classes I and II molecules for presentation to T cells in germinal centres of local lymph nodes to initiate an immune response 22, 23. It is possible that the use of a highly efficient eukaryotic expression plasmid (pTriEx1.1neo), together with a synthetic gene coding for mouse TSHR A‐subunit that has been optimized specially for mouse codon usage with added Kozak sequence and RNA instability motif to improve translation, results in vivo in higher levels of expression of mouse TSHR. This, in a combination of efficient electroporation delivery of the plasmid, leads to increased levels of processed mouse TSHR A‐subunit peptides displayed on MHC molecules in DCs which may overcome peripheral tolerance to activate autoreactive T and B cells leading to self‐autoimmunity in the model 24.
A variant of mouse TSHR with a 25 amino acid deletion (mouse TSHR739) representing exon 5 has been described. Interestingly, similar shorter variants of TSHR have been described in human thyroid tissue 25. Plasmid immunization of the shorter mouse TSHR variant results in the induction of self‐autoimmunity 15. This remarkable result suggests that immune tolerance to mouse TSHR may be focused predominantly on a minimal region of 25 amino acids encoded by exon 5 of the receptor to prevent autoimmunity. Until the model with mouse TSHR739 plasmid immunization is verified independently, this searching question remains open.
There were some other notable parameters in the self‐antigen mouse TSHR A‐subunit model of GO that were observed in this study. Earlier, we reported on a large proportion of UCP‐1‐positive BAT in the retrobulbar tissue of several mice strains 26. Notably, the BAT co‐expressed high levels of TSHR, suggesting the BAT to be prone to the TSHR‐directed autoimmune attack 27. We observed in quantitative studies that orbital adipogenesis in the model was accompanied by an increase in BAT, but the total amount of adipose tissue in the orbit was shown to remain at steady and comparable levels during disease progression by different techniques of MR imaging and UCP‐1 staining by IHC. This finding provides compelling evidence that disease progression in the GO model resulted in a progressive replacement of white by brown adipose tissue. The basis for white–brown replacement in the orbital tissue in the model may be dependent upon pathogenic TSAbs, T3 hormone or local inflammatory stimuli from infiltrating cells during disease progression 28, 29, 30. Although it has been argued as an expression of white–brown transdifferentiation, generation of brown adipose tissue from orbital stem cells cannot be ruled out. Another important observation in the mouse TSHR A‐subunit model was the increase in size and thickening of muscle fibres in the orbital tissue of GO mice. This unique finding suggests that the mouse TSHR A‐subunit model, where during disease progression the orbital adipose content remains at steady levels, the muscle fibre hypertrophy is reminiscent of human orbital disease subset, manifested by an increase in orbital muscle, rather than an increase in orbital adipose tissue, for pathogenesis of GO.
Studies in human and mouse models indicate maintenance of self‐tolerance to TSHR to prevent Graves' autoimmune disease to be mediated primarily by central tolerance operating in the thymus 25, 31, 32, 33, 34. In humans, loss of immune tolerance to TSHR occurs in genetically susceptible individuals in association with environmental factors resulting in the onset of Graves' disease and extra‐thyroidal complications of Graves' orbitopathy 35. When central tolerance is compromised to TSHR in Graves' disease, this can lead to activation of the adaptive immune system of T and B cells to an endogenous receptor expressed in the thyroid gland and other tissues in genetically susceptible individuals. We have a detailed understanding of T and B cell tolerance mechanisms operating in thymus and bone marrow, respectively, as well in the periphery, that results collectively in the prevention of autoimmune disease 36, 37, 38, 39. Central tolerance in the thymus is regulated principally by a transcription factor termed ‘autoimmune regulator’ and encoded by the autoimmune regulator (AIRE) gene, which allows expression of thousands of self‐antigen peptides in medullary thymic epithelial cells (mTECs) that specialize in promiscuous gene expression 40. The display of tissue‐specific antigens (TSAs) on mTECs allows developing T cells with potential self‐reactivity to recognize the MHC‐associated self‐antigen peptides to undergo deletion by a process known as negative selection. Low‐affinity, self‐reactive T cells that escape negative deletion and emigrate to the periphery are then restrained by a variety of peripheral tolerance mechanisms including active suppression by Treg cells. The mechanisms for maintaining central tolerance to TSHR are not understood completely, but recent studies show that AIRE may not be a critical factor for tolerance to the receptor to prevent Graves' hyperthyroid disease 41, 42. With TSHR acting as an AIRE‐independent TSA, it is possible that non‐AIRE transcription factors in the thymus 39, 43, 44 or factors regulating B cell tolerance in the bone marrow 45 are involved in regulating central tolerance to TSHR to prevent autoimmune disease.
With limited knowledge on thymic central tolerance regulation of TSHR to prevent autoimmunity, it is unclear how immune tolerance may be breached leading to hyperthyroidism in Graves' disease. One consensus model deals with an inciting infectious agent displaying cross‐reactive determinants to TSHR which, in combination with other underlying factors, increases the risk of autoimmunity in Graves' disease 46. Another recent model developed by Pujol Borrell and colleagues has focused attention on the developing thymocytes as the critical mediators for loss of tolerance to TSHR 47. Immature thymocytes have been shown to express high levels of TSHR, which is functional with the capacity to respond to stimulation by TSH or TSAbs by initiating intracellular signalling events. This has led to the proposal that continuous stimulation of thymocyte TSHR during T cell maturation results in strong ‘help’ from a recently identified population of CD4 helper T cells, called T follicular helper cells (Tfh cells) to low‐affinity TSHR cross‐reactive B cells in germinal centres of draining lymph nodes. The gradual increase in affinity of TSAbs for TSHR in the right genetic background and environmental exposure is sufficient for the onset of Graves' hyperthyroid disease 48. This exciting model is plausible, as it neatly explains the unique origins and maturation of high‐affinity pathogenic TSAbs in Graves' disease 46.
In conclusion, we show that immunization by electroporation of mouse TSHR A‐subunit plasmid in female BALB/c mice provides a bona fide autoimmune model for experimental Graves' thyroid disease accompanied by orbital pathology resembling the human condition. The availability of a self‐antigen mouse TSHR A‐subunit model for autoimmune Graves' disease will allow studies to investigate mechanisms of tolerance to TSHR and evaluation of novel therapeutic options for curing hyperthyroidism and orbital manifestations in a faithful autoimmune model.
Disclosure
The authors declare no competing interests.
Author contributions
Conception and design: J . P. B., U. B. P., A. E. and A. S. Carrying out o the experiments: A. S., M H. and U. F. Provision of study materials: J. P. B., U. B. P., A. E., S. L., S. M., K. S. and M. O. Collection and assembly of data: A. S., M. H., U. F., U. B. P. and S. P. Data analysis and interpretation: A. S., J. P. B., U. B. P., A. E., S. D. C. and U. F. Manuscript writing: A. S., J. P. B., U. B. P. and A. E. Final approval of manuscript: all authors.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. In‐vitro expression of pTRiEx1.1 neo‐mouse thyroid stimulating hormone receptor (TSHR) A‐subunit plasmid. (a) Western blot with stably transfected mouse TSHR Chinese hamster ovary (CHO) cells. It reacted faithfully with the mouse receptor and showed a correctly co‐migrating band at approximately 12 5kD. The high glycosylation (40%) of TSHR protein gives irregular migration on sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS PAGE) gels of the receptor rather than the actual molecular size of ∼115 kDa. (b) We examined mouse TSHR A‐subunit protein expression by pTRiEx1.1 neo‐mouse TSHR A‐subunit plasmid by transfection into wild‐type CHO cells. Western blotting showed a specific band co‐migrating at approximately 41 kD (lane 1), which was absent in lysates from control wild‐type CHO cells (lane 2). The 41 kD band represents the mouse TSHR A‐subunit with all four N‐linked sites glycosylated. The molecular weight of marker proteins is given in kDa.
Fig. S2. Clinical signs of thyroid disease of mouse thyroid stimulating hormone receptor (TSHR) A‐subunit‐immunized mice. (a) Mouse TSHR A‐subunit immunized mice showed statistically significant increased heart size measured in mm (***P ≤ 0·001). (b) Animals undergoing disease and control mice showed no difference in their weights.
Fig. S3. Thyroid hormone status of mouse 1A‐subunit‐immunized mice. (a) Total thyroxine 4 (TT4) concentrations were measured in 50 µl serum by enzyme‐linked immunosorbent assay (ELISA). Values were expressed in µg/dl (y‐axis). Student's t‐test revealed no statistically significant difference between mouse TSHR mice A‐subunit animals and control β‐Gal animals.
Fig. S4. Magnetic resonance imaging (MRI) images of mouse thyroid stimulating hormone receptor (TSHR) A‐subunit‐immunized mice. Supporting information Fig. S2 shows the result of MRI analysis. From the group of immunized animals, five animals were selected from each group (mouse TSHR A‐subunit plasmid and control β‐Gal) for MRI. At least 48 h before MRI, animals were injected intravenously (i.v.) with 300 µl perfluorocarbon (PFC). PFC is ingested avidly by macrophages and monocytes to visualize areas of inflammation by 19F MRI. Data were recorded on a Bruker AVANCEIII® 9.4T wide‐bore nuclear magnetic resonance (NMR) spectrometer driven by ParaVision® 5.1 (Bruker, Rheinstetten, Germany). (a,b) Anatomical reference overview of mouse TSHR‐immunized mice (a) and β‐Gal control mice by 1H MRI. (c,d) After the acquisition of all 1H data sets, the resonator was tuned to 19F. Inflammatory foci were determined from 19F MR images presented with a higher signal in mouse TSHR‐immunized mice (c) than in β‐Gal control mice (d). (e,f) Presentation of sagittal MRI analysis of mice orbit. It shows an increased area of adipose tissue in mouse TSHR‐immunized mice. The result was statistically not significant, but showed an overall increase in deposition of adipose tissue (Fig. 3).
Fig. S5. Clinical data of mouse thyroid stimulating hormone receptor (TSHR) A‐subunit‐immunized mice. (a) Eye signs did not show any Graves' orbitopathy (GO) typical changes in the eyes of control β‐Gal mice, while all mouse TSHR‐immunized animals showed typical ocular signs of GO (acute inflammation, proptosis or chronic disease) 10 times or more often when examined during the entire time of the experiment. (b) Representative pictures of a control β‐Gal mouse without any eye signs as normal control. (c) Example of one mouse immunized with mouse TSHR plasmid revealed an acute inflammation in the left eye with swelling of the lid, redness of the lid and pus inside the eye. (d) Picture shows one of the mice with proptosis.
Acknowledgements
We gratefully acknowledge Drs Sandra McLachlan and Basil Rapoport for providing stably transfected mouse TSHR CHO cells. We thank Alexandra Brenzel (Imaging Center Essen IMCES, University Duisburg‐Essen) and Christoph Jesenek (Molecular Ophthalmology, University Duisburg‐Essen) for technical assistance. This project was funded by Interne Forschungsförderung Essen (IFORES) (AS), Stiftung Universitätsmedizin Essen (AE) and DFG/GRK 2098 (AE, UBP).
References
- 1. Weetman AP. Graves' disease. N Engl J Med 2000; 343:1236–48. [DOI] [PubMed] [Google Scholar]
- 2. Bahn RS. Graves' ophthalmopathy. N Engl J Med 2010; 362:726–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rapoport B, Aliesky HA, Banuelos B, Chen CR, McLachlan SM. A unique mouse strain that develops spontaneous, iodine‐accelerated, pathogenic antibodies to the human thyrotrophin receptor. J Immunol 2015; 194:4154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagayama Y, Nakahara M, Abiru N. Animal models of Graves' disease and Graves' orbitopathy. Curr Opin Endocrinol Diabetes Obes 2015; 22:381–6. [DOI] [PubMed] [Google Scholar]
- 5. Banga JP, Moshkelgosha S, Berchner‐Pfannschmidt U, Eckstein A. Modeling Graves' orbitopathy in experimental Graves' disease. Horm Metab Res 2015; 47:797–803. [DOI] [PubMed] [Google Scholar]
- 6. Bahn RS. News and views: at long last, an animal model of Graves' orbitopathy. Endocrinology 2013; 154:2989–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moshkelgosha S, So PW, Deasy N, Diaz‐Cano S, Banga JP. Cutting edge: retrobulbar inflammation, adipogenesis, and acute orbital congestion in a preclinical female mouse model of Graves' orbitopathy induced by thyrotropin receptor plasmid‐in vivo electroporation. Endocrinology 2013; 154:3008–15. [DOI] [PubMed] [Google Scholar]
- 8. Berchner‐Pfannschmidt U, Moshkelgosha S, Diaz‐Cano S, Edelmann B, Gortz GE, Horstmann M. Comparative assessment of female mouse model of Graves' orbitopathy under different environments, accompanied by proinflammatory cytokine and T‐cell responses to thyrotropin hormone receptor antigen. Endocrinology 2016; 157:1673–82. [DOI] [PubMed] [Google Scholar]
- 9. Nagayama Y, Kita‐Furuyama M, Ando T et al A novel murine model of Graves' hyperthyroidism with intramuscular injection of adenovirus expressing the thyrotropin receptor. J Immunol 2002; 168:2789–94. [DOI] [PubMed] [Google Scholar]
- 10. Chen CR, Pichurin P, Nagayama Y, Latrofa F, Rapoport B, McLachlan SM. The thyrotropin receptor autoantigen in Graves disease is the culprit as well as the victim. J Clin Invest 2003; 111:1897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ungerer M, Faßbender J, Li Z, Münch G, Holthoff H‐P. Review of mouse models of Graves' disease and orbitopathy‐novel treatment by induction of tolerance. Clin Rev Allergy Immunol 2017; 52:182–93. [CrossRef’] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakahara M, Mitsutake N, Sakamoto H et al Enhanced response to mouse thyroid‐stimulating hormone (TSH) receptor immunization in TSH receptor‐knockout mice. Endocrinology 2010; 151:4047–54. [DOI] [PubMed] [Google Scholar]
- 13. Kaithamana S, Fan J, Osuga Y, Liang SG, Prabhakar BS. Induction of experimental autoimmune Graves' disease in BALB/c mice. J Immunol 1999; 163:5157–64. [PubMed] [Google Scholar]
- 14. Nakahara M, Johnson K, Eckstein A et al Adoptive transfer of antithyrotropin receptor (TSHR) autoimmunity from TSHR knockout mice to athymic nude mice. Endocrinology 2012; 153:2034–42. [DOI] [PubMed] [Google Scholar]
- 15. Endo T, Kobayashi T. Immunization of mice with a newly identified thyroid‐stimulating hormone receptor splice variant induces Graves'‐like disease. J Autoimmun 2013; 43:18–25. [DOI] [PubMed] [Google Scholar]
- 16. Gortz GE, Moshkelgosha S, Jesenek C et al Pathogenic phenotype of adipogenesis and hyaluronan in orbital fibroblasts from female Graves' orbitopathy mouse model. Endocrinology 2016; 157:3771–8. [DOI] [PubMed] [Google Scholar]
- 17. Chen CR, McLachlan SM, Rapoport B. Thyrotropin (TSH) receptor residue E251 in the extracellular leucine‐rich repeat domain is critical for linking TSH binding to receptor activation. Endocrinology 2010; 151:1940–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Misharin A, Hewison M, Chen CR et al Vitamin D deficiency modulates Graves' hyperthyroidism induced in BALB/c mice by thyrotropin receptor immunization. Endocrinology 2009; 150:1051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flogel U, Ding Z, Hardung H et al In vivo monitoring of inflammation after cardiac and cerebral ischemia by fluorine magnetic resonance imaging. Circulation 2008; 118:140–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Temme S, Bonner F, Schrader J, Flogel U. 19F magnetic resonance imaging of endogenous macrophages in inflammation. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2012; 4:329–43. [DOI] [PubMed] [Google Scholar]
- 21. Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH) receptor: interaction with TSH and autoantibodies. Endocr Rev 1998; 19:673–716. [DOI] [PubMed] [Google Scholar]
- 22. Wolff JA, Malone RW, Williams P et al Direct gene transfer into mouse muscle in vivo . Science 1990; 247:1465–8. [DOI] [PubMed] [Google Scholar]
- 23. Sardesai NY, Weiner DB. Electroporation delivery of DNA vaccines: prospects for success. Curr Opin Immunol 2011; 23:421–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grodeland G, Fredriksen AB, Loset GA, Vikse E, Fugger L, Bogen B. Antigen targeting to human HLA class II molecules increases efficacy of DNA vaccination. J Immunol 2016; 197:3575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brand OJ, Barrett JC, Simmonds MJ et al Association of the thyroid stimulating hormone receptor gene (TSHR) with Graves' disease. Hum Mol Genet 2009; 18:1704–13. [DOI] [PubMed] [Google Scholar]
- 26. Johnson KT, Wiesweg B, Schott M et al Examination of orbital tissues in murine models of Graves' disease reveals expression of UCP‐1 and the TSHR in retrobulbar adipose tissues. Horm Metab Res 2013; 45:401–7. [DOI] [PubMed] [Google Scholar]
- 27. Wiesweg B, Johnson KT, Eckstein AK, Berchner‐Pfannschmidt U. Current insights into animal models of Graves' disease and orbitopathy. Horm Metab Res 2013; 45:549–55. [DOI] [PubMed] [Google Scholar]
- 28. Smorlesi A, Frontini A, Giordano A, Cinti S. The adipose organ: white‐brown adipocyte plasticity and metabolic inflammation. Obes Rev 2012; 13: 83–96. [DOI] [PubMed] [Google Scholar]
- 29. Whittle A. Searching for ways to switch on brown fat: are we getting warmer? J Mol Endocrinol 2012; 49:R79–87. [DOI] [PubMed] [Google Scholar]
- 30. Rosell M, Kaforou M, Frontini A et al Brown and white adipose tissues: intrinsic differences in gene expression and response to cold exposure in mice. Am J Physiol Endocrinol Metab 2014; 306:E945–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Colobran R, Armengol Mdel P, Faner R et al Association of an SNP with intrathymic transcription of TSHR and Graves' disease: a role for defective thymic tolerance. Hum Mol Genet 2011; 20:3415–23. [DOI] [PubMed] [Google Scholar]
- 32. McLachlan SM, Aliesky HA, Chen C‐R, Chong G, Rapoport B, Ludgate M. Breaking tolerance in transgenic mice expressing the human TSH receptor A‐subunit: thyroiditis, epitope spreading and adjuvant as a ‘double edged sword’. PLOS ONE 2012; 7:e43517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stefan M, Wei C, Lombardi A et al Genetic‐epigenetic dysregulation of thymic TSH receptor gene expression triggers thyroid autoimmunity. Proc Natl Acad Sci USA 2014; 111:12562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McLachlan SM, Aliesky HA, Banuelos B, Lesage S, Collin R, Rapoport B. High‐level intrathymic thyrotrophin receptor expression in thyroiditis‐prone mice protects against the spontaneous generation of pathogenic thyrotrophin receptor autoantibodies. Clin Exp Immunol 2017; 188:243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tomer Y. Mechanisms of autoimmune thyroid diseases: from genetics to epigenetics. Annu Rev Pathol 2014; 9:147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hogquist KA, Baldwin TA, Jameson SC. Central tolerance: learning self‐control in the thymus. Nat Rev Immunol 2005; 5:772–82. [DOI] [PubMed] [Google Scholar]
- 37. Gauld SB, Merrell KT, Cambier JC. Silencing of autoreactive B cells by anergy: a fresh perspective. Curr Opin Immunol 2006; 18:292–7. [DOI] [PubMed] [Google Scholar]
- 38. Melchers F. Checkpoints that control B cell development. J Clin Invest 2015; 125:2203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takaba H, Takayanagi H. The mechanisms of T cell selection in the thymus. Trends Immunol 2017; 11:805–16. [DOI] [PubMed] [Google Scholar]
- 40. Anderson MS, Venanzi ES, Klein L et al Projection of an immunological self shadow within the thymus by the Aire protein. Science 2002; 298:1395–401. [DOI] [PubMed] [Google Scholar]
- 41. McLachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocr Rev 2014; 35:59–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Misharin AV, Nagayama Y, Aliesky HA, Rapoport B, McLachlan SM. Studies in mice deficient for the autoimmune regulator (Aire) and transgenic for the thyrotropin receptor reveal a role for Aire in tolerance for thyroid autoantigens. Endocrinology 2009; 150:2948–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takaba H, Morishita Y, Tomofuji Y et al Fezf2 orchestrates a thymic program of self‐antigen expression for immune tolerance. Cell 2015; 163:975–87. [DOI] [PubMed] [Google Scholar]
- 44. Roberts NA, Adams BD, McCarthy NI et al Prdm1 regulates thymic epithelial function to prevent autoimmunity. J Immunol 2017; 199:1250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Russell L, John S, Cullen J, Luo W, Shlomchik MJ, Garrett‐Sinha LA. Requirement for transcription factor Ets1 in B cell tolerance to self‐antigens. J Immunol 2015; 195:3574–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hargreaves CE, Grasso M, Hampe CS et al Yersinia enterocolitica provides the link between thyroid‐stimulating antibodies and their germline counterparts in Graves' disease. J Immunol 2013; 190:5373–81. [DOI] [PubMed] [Google Scholar]
- 47. Pujol‐Borrell R, Gimenez‐Barcons M, Marin‐Sanchez A, Colobran R. Genetics of Graves' disease: special focus on the role of TSHR gene. Horm Metab Res 2015; 47:753–66. [DOI] [PubMed] [Google Scholar]
- 48. Gimenez‐Barcons M, Colobran R, Gomez‐Pau A et al Graves' disease TSHR‐stimulating antibodies (TSAbs) induce the activation of immature thymocytes: a clue to the riddle of TSAbs generation? J Immunol 2015; 194:4199–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. In‐vitro expression of pTRiEx1.1 neo‐mouse thyroid stimulating hormone receptor (TSHR) A‐subunit plasmid. (a) Western blot with stably transfected mouse TSHR Chinese hamster ovary (CHO) cells. It reacted faithfully with the mouse receptor and showed a correctly co‐migrating band at approximately 12 5kD. The high glycosylation (40%) of TSHR protein gives irregular migration on sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS PAGE) gels of the receptor rather than the actual molecular size of ∼115 kDa. (b) We examined mouse TSHR A‐subunit protein expression by pTRiEx1.1 neo‐mouse TSHR A‐subunit plasmid by transfection into wild‐type CHO cells. Western blotting showed a specific band co‐migrating at approximately 41 kD (lane 1), which was absent in lysates from control wild‐type CHO cells (lane 2). The 41 kD band represents the mouse TSHR A‐subunit with all four N‐linked sites glycosylated. The molecular weight of marker proteins is given in kDa.
Fig. S2. Clinical signs of thyroid disease of mouse thyroid stimulating hormone receptor (TSHR) A‐subunit‐immunized mice. (a) Mouse TSHR A‐subunit immunized mice showed statistically significant increased heart size measured in mm (***P ≤ 0·001). (b) Animals undergoing disease and control mice showed no difference in their weights.
Fig. S3. Thyroid hormone status of mouse 1A‐subunit‐immunized mice. (a) Total thyroxine 4 (TT4) concentrations were measured in 50 µl serum by enzyme‐linked immunosorbent assay (ELISA). Values were expressed in µg/dl (y‐axis). Student's t‐test revealed no statistically significant difference between mouse TSHR mice A‐subunit animals and control β‐Gal animals.
Fig. S4. Magnetic resonance imaging (MRI) images of mouse thyroid stimulating hormone receptor (TSHR) A‐subunit‐immunized mice. Supporting information Fig. S2 shows the result of MRI analysis. From the group of immunized animals, five animals were selected from each group (mouse TSHR A‐subunit plasmid and control β‐Gal) for MRI. At least 48 h before MRI, animals were injected intravenously (i.v.) with 300 µl perfluorocarbon (PFC). PFC is ingested avidly by macrophages and monocytes to visualize areas of inflammation by 19F MRI. Data were recorded on a Bruker AVANCEIII® 9.4T wide‐bore nuclear magnetic resonance (NMR) spectrometer driven by ParaVision® 5.1 (Bruker, Rheinstetten, Germany). (a,b) Anatomical reference overview of mouse TSHR‐immunized mice (a) and β‐Gal control mice by 1H MRI. (c,d) After the acquisition of all 1H data sets, the resonator was tuned to 19F. Inflammatory foci were determined from 19F MR images presented with a higher signal in mouse TSHR‐immunized mice (c) than in β‐Gal control mice (d). (e,f) Presentation of sagittal MRI analysis of mice orbit. It shows an increased area of adipose tissue in mouse TSHR‐immunized mice. The result was statistically not significant, but showed an overall increase in deposition of adipose tissue (Fig. 3).
Fig. S5. Clinical data of mouse thyroid stimulating hormone receptor (TSHR) A‐subunit‐immunized mice. (a) Eye signs did not show any Graves' orbitopathy (GO) typical changes in the eyes of control β‐Gal mice, while all mouse TSHR‐immunized animals showed typical ocular signs of GO (acute inflammation, proptosis or chronic disease) 10 times or more often when examined during the entire time of the experiment. (b) Representative pictures of a control β‐Gal mouse without any eye signs as normal control. (c) Example of one mouse immunized with mouse TSHR plasmid revealed an acute inflammation in the left eye with swelling of the lid, redness of the lid and pus inside the eye. (d) Picture shows one of the mice with proptosis.