

Abstract
Antibody-mediated injury is a major cause of allograft dysfunction and loss. Antibodies to ABH(O) blood group antigens are classic mediators of ABO-incompatible graft rejection, whereas donor-specific anti-HLA antibodies and, more recently, autoantibodies are appreciated as important contributors to allograft inflammation and dysfunction. In August 2016, the International Summit of the Canadian National Transplant Research Program focused on recent advances in the field of antibody-mediated rejection. Here, we describe work presented and discussed at the meeting, with a focus on 3 major themes: the importance of (1) natural antibodies and autoantibodies, (2) tissue injury–derived exosomes and autoimmunity, (3) inflammasome activation and innate immune responses in regulating allograft inflammation and dysfunction. Finally, we explore novel areas of therapeutic intervention that have recently emerged from these 3 major and overlapping fields of transplantation research.
A summary report on these cutting-edge topics, from basic to translational, that impact allograft outcomes as discussed at the recent Canadian National Transplant Research Program (CNTRP) International Summit.
Transplantation is often considered by patients and transplant physicians as a unique opportunity to start a new life. However, it is important to remember that before transplantation, both recipients and allografts sustain various insults that shape both the adaptive and innate immune repertoires. In solid organ transplantation, recognition is emerging that signals from tissue injury due to various chronic diseases in patients awaiting transplant, the organ donor before donation and the organ at the time of transplantation are major contributors to allograft inflammation, rejection, and dysfunction. Recent discoveries in the molecular pathways linking cell death, autoimmunity, and allograft inflammation have uncovered novel mechanisms and therapeutic options to target these pathways and reduce allograft dysfunction. The Canadian National Transplant Research Program is actively contributing to this emerging field through its work in the areas of cell death, acute organ dysfunction, ex vivo organ perfusion, graft-versus-host disease (GvHD), and antibody-mediated rejection (AMR).1 The Canadian National Transplant Research Program International Summit united a group of internationally renowned scientists from both the solid organ and bone marrow/stem cell transplantation fields to focus on the interplay between natural antibodies (Nabs), autoantibodies, tissue injury–derived exosomes, innate immune responses, and alloimmunity (Figure 1). This review summarizes key concepts that emerged from the work and discussions of the attendees.
FIGURE 1.
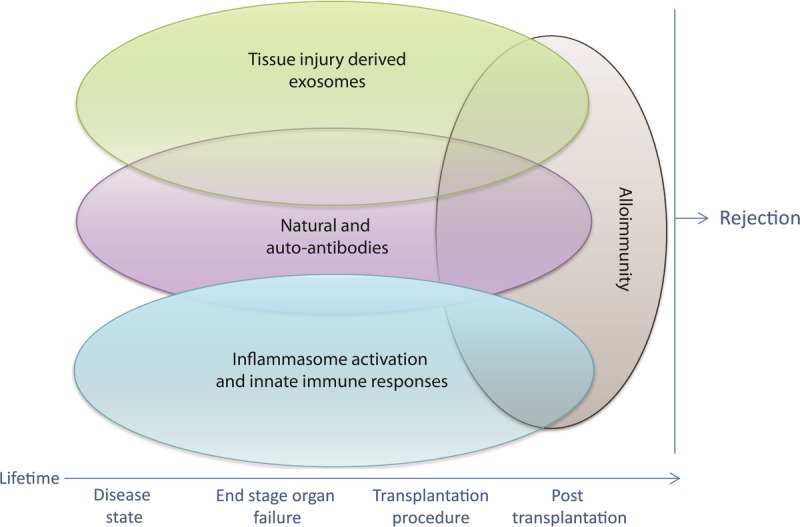
Organ transplantation can be considered a multistep process that starts in the recipient and donor well before the transplant operation itself is performed. Recent discoveries are shedding light on the interplay between tissue injury-derived exosomes, natural and autoantibodies, inflammasome activation, and innate immune responses and alloimmunity that regulates allograft inflammation and dysfunction.
Nabs and Autoantibodies
Antibody-Mediated Rejection of Organ Transplants: Lessons From Animal Models
Antibodies are reported to be the cause of up to 50% of acute rejection episodes and more than 60% of late graft failures in kidney transplants.2-4 The approaches currently used to investigate antidonor antibody–mediated allograft injury in animal models include passive transfer of antibody and presensitization of recipients. Each approach has its limitations in providing clinically relevant insights to the problem. Fairchild's group5 developed a unique mouse model of AMR in which they observed acute humoral rejection of renal allografts in CCR5-deficient recipients. Rejected allografts showed margination of neutrophils and macrophages, diffused C3d deposition within peritubular capillaries, interstitial hemorrhage accompanied by edema, and glomerular fibrin deposition—all features classically observed during AMR of human kidney allografts. Use of CD8−/−CCR5−/− mice demonstrated that CD8+ T cells were dispensable during the development of AMR, whereas NK cells seemed to play a critical role in allograft inflammation.6 In the absence of NK cells, donor-specific antibodies (DSA) did not cause acute allograft inflammation and injury; instead, they slowly induced development of chronic glomerular injury and renal failure. Furthermore, the authors found that depletion of recipient B cells promoted long-term graft survival in this model. Collectively, these results suggest a major role for NK cells and antibody-mediated injury in long-term microvascular damage and allograft dysfunction. Monitoring leukocyte-endothelium interactions in real time will be essential to understand the role of cell-mediated injury during AMR.
New Understanding of ABH Glycans and ABO Antibodies
Antibodies to ABH(O) blood group antigens were the first type of antibody implicated in allograft rejection, manifested as hyperacute rejection of ABO-incompatible (ABOi) kidney transplants. Natural anti-A and anti-B antibodies are produced as a presumed immunologic cross-reaction to similar epitopes found on the gut flora.7 The first protocol for intentional ABOi heart transplantation was developed 20 years ago by West’s group,7,8 with the rationale that the ontogeny of natural isohemagglutinins begins after 4 to 6 months of age, providing a window of opportunity to transplant infants across the ABO barrier safely in the absence of preformed anti-A and anti-B antibodies.9 Since then, multiple teams worldwide have confirmed that ABOi heart transplantation is a safe procedure in infants and young patients with absent or low circulating isohemagglutinins, saving thousands of lives in the last 2 decades by increasing the donor pool.10,11 In this setting, a unique form of donor-specific B cell tolerance was observed, induced by the ABOi heart graft. This was characterized by minimal or no production of antibodies to the donor ABH antigens, together with normal production of third-party antibodies, measured both in vivo in patient serum and in vitro by cultured patients’ cells, and deletion of peripheral B cells specific for donor ABH structures.7,8 These observations have raised many questions about the precise mechanisms of tolerance to foreign ABH structures carried on graft vascular endothelium in the setting of immunologic immaturity and how this differs from resistance of vascular endothelium to antibody-mediated injury (or “accommodation”) typically observed in ABOi organ transplants in older individuals.
The standard technique for measuring ABO antibodies in clinical laboratories is the hemagglutination assay using reagent erythrocytes. The application of this assay to management of ABOi organ transplantation is based on the presumption that ABH antigens expressed by erythrocytes are identical to those in the graft vasculature and in other graft structures.7,8 However, ABH carbohydrate antigens are carried by precursor structures called type I-IV chains,12 creating unique antigen epitopes that may differ in expression between reagent erythrocytes and graft vascular cells. West’s group generated monoclonal antibodies that differentiate between types I, II, III, and IV precursor structures of ABH antigens and found that ABH carbohydrate-subtype chains expressed on erythrocytes were not identical to subtype chains expressed on vascular endothelium.13 This observation has a major consequence on the interpretation of erythrocyte agglutination assays, because agglutination titres may not reflect the antibodies that can actually bind to and affect the graft vasculature. Recently, a collaborative team of carbohydrate chemists and transplant researchers led by Dr. West developed an ABH-glycan microarray that characterizes and quantifies serum antibodies to antigen subtypes, identifying those targeting the ABH antigens actually expressed in the graft.14 This assay will provide precise understanding of the actual ABH compatibility that cannot be captured by erythrocyte-based agglutination assays alone, which will help mitigate unnecessary exclusion of patients for ABOi transplantation or invasive antibody-removal interventions. In addition, this and related new glyconanotechnology tools15 will allow for a more detailed investigation of mechanisms of tolerance induced by nonself graft antigens,14 as well as endothelial accommodation in the presence of ABH antibodies.
Antigen-Specific Autoantibodies
Autoantibodies are increasingly recognized as important contributors to allograft rejection and decreased long-term survival.16 Nearly a decade ago, Bharat and Mohanakumar17 provided evidence of the importance of antibodies in chronic rejection of lung allografts presenting as bronchiolitis obliterans syndrome (BOS) Interestingly, the vast majority of BOS patients did not exhibit anti-HLA antibodies, but rather autoantibodies against the autoantigens K-alpha 1 tubulin, a filamentous protein and collagen V expressed in airway epithelial cells.17 A T-cell response to collagen V was also described in patients developing BOS independently of DSA.17
Autoantibodies to vimentin, angiotensin II type 1 receptor, tubulin, fibronectin, and perlecan/LG3 have been associated with increased rejection rates and reduced survival of heart, kidney, and lung allografts.16,18 Antiperlecan/LG3 antibodies were also found to increase the risk of delayed graft function in renal transplant patients and reduce long-term allograft function in patients with delayed graft function. Indeed, passively transferred anti-LG3 antibodies to mice during renal ischemia-reperfusion injury (IRI) led to complement activation, microvascular injury, and aggravated renal dysfunction.
Autoimmunity has also been observed after both autologous and allogeneic stem cell transplantation. Many of the clinical and laboratory features of GvHD, especially in its chronic form, resemble those of autoimmune diseases, such as systemic sclerosis. However, low levels of antiperlecan antibodies are significantly associated with the onset of chronic GvHD,19 suggesting that the manifestations of autoimmunity in solid organ and bone marrow transplant patients are at least partly different or that the antiperlecan response occurred early as part of an inflammatory phase of GvHD20 with a significant decrease by the onset of clinical disease. Chronic GvHD appears to be caused by an imbalance between regulatory mechanisms mediated by T regulatory, B regulatory, and NK regulatory cells and effector mechanisms mediated by effector B cells and T cells.21-23 Yet, the exact mechanistic interactions between these regulatory and effector mechanisms are still poorly understood and the stem cell transplantation fields have yet to explore activation of autoimmune pathways before transplantation. Because myeloablative chemotherapy induces major increases in cell death, with immune reconstitution very similar to infants, the impact of tissue injury in this setting could prove dramatically different from that of solid organ transplantation.
Autoantibodies Reactive to Apoptotic Cells
Autoantibodies reactive to apoptotic cells appear to display promiscuous reactivity. Zorn and colleagues tested up to 8000 proteins by protein microarray for autoantibody profiling, comparing sera from renal allograft recipients with either chronic rejection or stable function. Chronic humoral rejection of human kidney allografts associates with broad autoantibody responses. The specificity of various monoclonal antibodies was characterized using immortalized B cells purified from graft infiltrates or the blood of chronically rejecting patients. Intriguingly, a significant fraction of monoclonal antibodies were found to be reactive to multiple antigens, and to apoptotic, but not viable, cells.24,25 Subsequently Zorn's group demonstrated that the presence of polyreactive autoantibodies with reactivity to apoptotic cells pretransplant correlated with late kidney allograft loss.24 This reactivity to apoptotic but not to viable cells could be explained by profound cell membrane changes, potentially permitting the occurrence of autoantigen-autoantibody interactions. These polyreactive autoantibodies can also bind oxidized epitopes such as malondialdehyde, a byproduct of lipid peroxidation that is produced during apoptosis.26
The polyreactive nature of these autoantibodies is compatible with Nabs that bind multiple antigens with relatively low affinity. It is suggested, at least in murine studies, that Nabs are produced by innate B cells, such as B-1 B cells, and likely represent the first line of defense against pathogens, as well as a clearing mechanism for dying cells and cellular debris.24 Nabs can also display anti-HLA reactivity, as exemplified by their reactivity to HLA proteins coated on luminex beads.25,27 The implantation of a left ventricular assisted device in patients awaiting heart transplantation is associated with elevated levels of polyreactive Nabs (personal communication from E. Zorn). In this setting, the presence of Nabs with reactivity to malondialdehyde and apoptotic cells is associated with an increased risk of primary graft dysfunction. Furthermore, in human heart transplant patients with cardiac allograft vasculopathy, B-cell clones derived from immune infiltrates present around coronary arteries are predominantly polyreactive (personal communication from E. Zorn).
Tissue Injury–Derived Exosomes and Autoimmunity
Antibody reactivity to apoptotic cells or antigens expressed or released by apoptotic cells, such as vimentin and perlecan/LG3, suggests that apoptosis somehow contributes to autoantibody production.16,18 This finding was surprising given the classic notion that apoptosis is a nonimmunogenic, if not tolerogenic, type of regulated cell death.28,29 This idea has recently been revisited with the discovery of a novel type of membrane vesicle called the apoptotic exosome (ApoExo)-like vesicle, which is released through a caspase-3–dependent pathway, but is strikingly different from apoptotic bodies in size, ultrastructure, and enzymatic activity.30 The ApoExo is characterized by the presence of an active 20S proteasome, which plays a key role in triggering autoantibody production and acceleration of rejection. Inhibition of proteasome activity within ApoExo by bortezomib largely reduces their autoimmune activity.30 Vascular injury in mice, whether in the form of hindlimb ischemia or renal IRI, increases circulating levels of ApoExo and prompts the production of antiperlecan/LG3 and antinuclear antibodies.30 These observations provide novel insights into the mechanisms controlling the production of autoantibodies. They suggest that vascular and tissue injury, whether present before, at the time of, or after transplantation, prompt the production of exosome-like vesicles that can, in turn, foster the production of autoantibodies.
A recent study by Mohanakumar's group31 evaluated the importance of circulating exosomes in prompting autoimmunity to lung allografts. Exosomes expressing collagen V were isolated from the sera of human lung transplant recipients diagnosed with both acute and chronic rejection. As expected, their presence in the circulation preceded the occurrence of acute rejection and BOS.31 In another study, exosomes isolated from the sera of cardiac transplant patients with coronary artery vasculopathy were found to express self-antigens, such as myosin and vimentin.32 Administration of exosomes isolated from transplant recipients with an inflamed syngeneic graft induced antivimentin and antimyosin antibody production, whereas exosomes purified from recipients with quiescent grafts did not induce autoantibody production (personal communication from T. Mohanakumar). Interestingly, in recipients of syngeneic heart transplants, exosomes were found to aggravate inflammation, but only when administered at the time of transplantation (personal communication from T. Mohanakumar). Collectively, these results suggest that tissue injury and cell death play 3 important and potentially additive functions: first, triggering the production of immunogenic exosomes that can in turn induce autoantibody production; second, potentiating the proinflammatory activity of exosomes; and third, facilitating autoantibody/autoantigen interactions and complement activation when autoantibodies are already present in circulation at the time of tissue injury.
Inflammasome Activation and Innate Immune Responses
Tissue damage mediated by IRI is an unfortunate but integral part of solid organ transplantation. However, it is important to remember that enhanced cell death can also manifest in both the donor and the recipient before transplantation. Progressive organ failure leading to transplantation is associated with programmed death of parenchymal cells in the failing organ. Organ donors also experience increased loads of dying cells within various tissues, especially at the time of brain death or in association with hemodynamic disturbances. Cell death prompts the release of damage-associated molecular patterns that are important in shaping innate and adaptive immune responses to injury. The inflammasome is a multiprotein caspase-activating platform that regulates a variety of pathways in response to damage-associated molecular patterns.33 The inflammasome complex has been recognized as a critical mediator in various autoimmune diseases, with its role in chronic kidney disease and acute kidney injury becoming more widely accepted.34 The canonical NLRP3-inflammasome platform controls caspase-1 activation, which in turn regulates the maturation and secretion of proinflammatory cytokines, such as IL-1β and IL-18. Muruve’s group35 recently described that high NLRP3 expression levels in human kidney biopsies were correlated with disease outcomes in patients with IgA nephropathy.
The canonical inflammasone was initially characterized in macrophages and other leukocytes. Growing evidence is uncovering inflammasome-independent or noncanonical roles for NLRP3 in renal tubular epithelial injury and fibrosis.36-40 Noncanonical activation of the NLRP3 platform has recently been described by Muruve's team in renal epithelial cells, and the platform has been shown to regulate ROS and TGFβ production, as well as caspase 8 activation.41 Intriguingly, NLRP platforms can be secreted upon activation and may represent a biomarker of tissue injury–triggered inflammation. These results also suggest that the inflammasome is a pivotal pathway connecting tissue injury with inflammation and autoimmunity.
Collectively, these observations highlight the possibility of interfering with inflammasome activation during organ preservation or transplantation to prevent the onset of inflammatory pathways that contribute to maladaptive organ healing and organ dysfunction. Whether inflammasome inhibition could prove useful in preventing autoantibody production is an intriguing question requiring further investigation.
Importance of Phagocytosis in Tissue Repair (the Kidney Example)
Recent reports suggest that accelerating the clearance of apoptotic and necrotic cells after IRI could represent a novel therapeutic strategy to mitigate inflammation at the time of transplantation.42 An important step in phagocytic clearance is the presentation of “eat me” signals, such as phosphatidylserine, on the apoptotic cell surface, which promotes specific recognition by phagocytes and subsequent internalization of dying cells.43 Kidney injury molecule-1 (KIM-1) is a type-I transmembrane glycoprotein that is highly upregulated on the apical side of renal proximal tubule epithelial cells after IRI.44 As a phosphatidylserine receptor, KIM-1 binds to PdtSer and confers a phagocytic phenotype on epithelial cells, enabling their phagocytosis of apoptotic cells within the tubule lumen.44 KIM-1 also functions as a scavenger receptor, mediating the uptake of necrotic cells (via apoptosis inhibitor of macrophage [AIM]), oxidized lipids, albumin.42,45 Importantly, the extracellular portion of KIM-1 can be cleaved and detected in the urine.44 Because high levels have been shown to correlate with kidney tissue damage, KIM-1 may be a sensitive and specific biomarker of kidney injury.46 Interestingly, Dr. Gunaratnam's group47 demonstrated that the shedding of Kim-1 mediated by the metalloproteinase, a disintegrin and metalloproteinase 17, may serve to regulate its phagocytic function. The central importance of KIM-1 in the clearance of dying cells was reported by a number of groups including that of Gunaratnam, who reported the occurrence of aggravated acute and long-term tissue damage after renal IRI in KIM-defective mice.42,48,49 It has recently been demonstrated that the AIM protein, which is freely filtered by the kidney during AKI, binds intraluminal debris and interacts with KIM-1.42 This in turn enhances the capacity of renal epithelial cells to phagocytose necrotic debris, thereby contributing to kidney repair. Importantly, exogenous AIM administration promotes the rapid removal of cellular debris, ameliorating acute kidney injury in both AIM-deficient and wild-type mice, but not KIM-1–deficient mice. These results open new avenues of exploration for accelerating renal repair after IRI. It is also well known that the rapid clearance of intraluminal debris prevents inflammation and potentially immune responses against tissue antigens.42 The scope of future investigation will be to determine whether AIM/KIM-1–dependent phagocytic clearance of dying cells can prevent delayed graft function and moreover the development of autoimmune responses that are detrimental to allograft function.
Limiting Tissue Injury With Ex Vivo Organ Perfusion Techniques
High-mobility group box 1 (HMGB1), a classic DAMP, is released in association with brain death. HMGB1 activates inflammatory responses primarily through its binding to receptor for advanced glycation end (RAGE) products and/or Toll-like receptors, leading to distal organ inflammation. In a rodent model of traumatic brain injury followed by lung transplantation, RAGE-KO mice were protected from acute lung injury. This observation in rodents confirms the associations seen between high circulating HMGB1 in human organ donors and greater acute lung injury posttransplantation. The HMGB1-RAGE axis mediates traumatic brain injury-induced pulmonary dysfunction in lung transplantation.50,51 These results raise the possibility of preconditioning or rehabilitating donor lungs by targeting the HMGB1/RAGE axis. Indeed, normothermic ex vivo lung perfusion provides a unique opportunity for targeted pharmacological intervention before transplantation. This approach allows for a comprehensive anatomical and functional assessment of the donor organ. It also opens the possibility of therapeutic intervention in diseased or damaged organs, as well as immunologic modulation to improve long-term function of the donor organ. However, if performed inappropriately, ex vivo lung perfusion and other ex vivo organ perfusion methods could induce mechanical stress in the organ, leading to an inflammatory response characterized by cytokine release.52 Continued focus on device refinement, proper parameters to assess organ function and well-being, and methods to monitor cell death in real time are needed to harvest the full potential of this new technology.
Masking the Signals: Tolerance Through Synthetic Glycoconjugates
Another option to prevent the formation of harmful antibodies during organ rejection may be to suppress B-cell activation. CD22 is an inhibitory B-cell coreceptor and is a member of the sialic acid-binding immunoglobulin-like lectin family. CD22 can inhibit signalling of the B-cell antigen receptor when engaged with a sialylated conjugate or antigen.53 Because sialylated glycans are often absent on microbes but abundant in higher vertebrates, these residues may provide an important signal for immune tolerance. Antigens covered with sialic acid can recruit CD22, which in turn prevents B-cell activation through B-cell antigen receptor inhibition.54 Thus, glycosylation of self-antigens may affect CD22 activation and immune response. Targeting CD22, or other inhibitory receptors, on B cells directly may be a mode of tolerance induction. Multivalent conjugates displaying sialic acid together with other carbohydrate antigens, such as ABH structures on persistent scaffolds, are currently under investigation as an in vivo tolerogenic strategy.55
CONCLUSIONS
Although organ transplantation can be viewed as an isolated surgical procedure, it can also be considered as a multistep process that starts in the recipient and donor well before the transplant operation itself is performed. Tissue damage often accompanies solid organ transplantation, but may also be present at other times, such as during end-stage organ failure in patients awaiting transplant, or in donors during brainstem death. Tissue injury triggers innate and adaptive immune and autoimmune responses that can impact long-term patient outcomes by contributing to acute and chronic rejection events. This summit highlighted the interplay between Nabs, innate immune responses, tissue injury, and autoimmunity, and identified exciting emerging areas of biomedical research in transplantation (Figure 1).
Footnotes
The authors declare no funding or conflicts of interest.
All authors significantly contributed to the writing and revision of the article. M.D. and M.J.H. consolidated the article and designed Figure 1. All authors gave final approval of the version to be published.
REFERENCES
- 1.Hebert MJ, Hartell D, West L. Transdisciplinary tour-de-force: The Canadian National Transplant Research Program. Transplantation. 2016;100:466–470. [DOI] [PubMed] [Google Scholar]
- 2.Gosset C, Lefaucheur C, Glotz D. New insights in antibody-mediated rejection. Curr Opin Nephrol Hypertens. 2014;23:597–604. [DOI] [PubMed] [Google Scholar]
- 3.Sellares J, de Freitas DG, Mengel M, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant. 2012;12:388–399. [DOI] [PubMed] [Google Scholar]
- 4.Lefaucheur C, Loupy A, Vernerey D, et al. Antibody-mediated vascular rejection of kidney allografts: a population-based study. Lancet. 2013;381:313–319. [DOI] [PubMed] [Google Scholar]
- 5.Bickerstaff A, Nozaki T, Wang JJ, et al. Acute humoral rejection of renal allografts in CCR5(−/−) recipients. Am J Transplant. 2008;8:557–566. [DOI] [PubMed] [Google Scholar]
- 6.Kohei N, Tanaka T, Tanabe N, et al. Natural killer cells play a critical role in mediating inflammation and graft failure during antibody-mediated rejection of kidney allografts. Kidney Int. 2016;89:1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urschel S, West LJ. ABO-incompatible heart transplantation. Curr Opin Pediatr. 2016;28:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.West LJ. Neonatal tolerance: applicability to solid organ transplantation. Curr Opin Organ Transplant. 2016;21:66–73. [DOI] [PubMed] [Google Scholar]
- 9.West LJ, Pollock-Barziv SM, Dipchand AI, et al. ABO-incompatible heart transplantation in infants. N Engl J Med. 2001;344:793–800. [DOI] [PubMed] [Google Scholar]
- 10.Urschel S, Larsen IM, Kirk R, et al. ABO-incompatible heart transplantation in early childhood: an international multicenter study of clinical experiences and limits. J Heart Lung Transplant. 2013;32:285–292. [DOI] [PubMed] [Google Scholar]
- 11.West LJ. ABO-incompatible heart transplantation: an alternative to improve the donor shortage in infants. Curr Opin Organ Transplant. 2005;10:364–368. [Google Scholar]
- 12.Meloncelli PJ, West LJ, Lowary TL. Synthesis and NMR studies on the ABO histo-blood group antigens: synthesis of type III and IV structures and NMR characterization of type I-VI antigens. Carbohydr Res. 2011;346:1406–1426. [DOI] [PubMed] [Google Scholar]
- 13.Jeyakanthan M, Tao K, Zou L, et al. Chemical basis for qualitative and quantitative differences between ABO blood groups and subgroups: implications for organ transplantation. Am J Transplant. 2015;15:2602–2615. [DOI] [PubMed] [Google Scholar]
- 14.Jeyakanthan M, Meloncelli PJ, Zou L, et al. ABH-glycan microarray characterizes ABO subtype antibodies: fine specificity of immune tolerance after ABO-incompatible transplantation. Am J Transplant. 2016;16:1548–1558, (2016). [DOI] [PubMed] [Google Scholar]
- 15.Slaney AM, Dijke IE, Jeyakanthan M, et al. Conjugation of A and B blood group structures to silica microparticles for the detection of antigen-specific B cells. Bioconjug Chem. 2016;27:705–715. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Q, Reed EF. The importance of non-HLA antibodies in transplantation. Nat Rev Nephrol. 2016;12:484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bharat A, Mohanakumar T. Autoimmunity and lung transplantation. Front Biosci (Elite Ed). 2012;4:2378–2388. [DOI] [PubMed] [Google Scholar]
- 18.Cardinal H, Dieude M, Hebert MJ. The emerging importance of non-HLA autoantibodies in kidney transplant complications. J Am Soc Nephrol. 2017;28:400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kariminia A, Holtan SG, Ivison S, et al. Heterogeneity of chronic graft-versus-host disease biomarkers: association with CXCL10 and CXCR3+ NK cells. Blood. 2016;127:3082–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooke KR, Luznik L, Sarantopoulos S, et al. The biology of chronic graft-versus-host disease: a task force report from the National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2017;23:211–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarantopoulos S, Ritz J. Aberrant B-cell homeostasis in chronic GVHD. Blood. 2015;125:1703–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Masson A, Socie G, Bagot M, et al. Deficient regulatory B cells in human chronic graft-versus-host disease. Oncoimmunology. 2015;4:e1016707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alho AC, Kim HT, Chammas MJ, et al. Unbalanced recovery of regulatory and effector T cells after allogeneic stem cell transplantation contributes to chronic GVHD. Blood. 2016;127:646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao B, Moore C, Porcheray F, et al. Pretransplant IgG reactivity to apoptotic cells correlates with late kidney allograft loss. Am J Transplant. 2014;14:1581–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porcheray F, DeVito J, Helou Y, et al. Expansion of polyreactive B cells cross-reactive to HLA and self in the blood of a patient with kidney graft rejection. Am J Transplant. 2012;12:2088–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zorn E, See SB. Polyreactive natural antibodies in transplantation. Curr Opin Organ Transplant. 2017;22:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao B, Rong C, Porcheray F, et al. Evidence to support a contribution of polyreactive antibodies to HLA serum reactivity. Transplantation. 2016;100:217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poon IK, Lucas CD, Rossi AG, et al. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Getts DR, McCarthy DP, Miller SD. Exploiting apoptosis for therapeutic tolerance induction. J Immunol. 2013;191:5341–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dieudé M, Bell C, Turgeon J, et al. The 20S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci Transl Med. 2015;7:318ra200. [DOI] [PubMed] [Google Scholar]
- 31.Gunasekaran M, Xu Z, Nayak DK, et al. Donor-derived exosomes with lung self-antigens in human lung allograft rejection. Am J Transplant. 2016;5:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nath DS, Ilias Basha H, Tiriveedhi V, et al. Characterization of immune responses to cardiac self-antigens myosin and vimentin in human cardiac allograft recipients with antibody-mediated rejection and cardiac allograft vasculopathy. J Heart Lung Transplant. 2010;29:1277–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. [DOI] [PubMed] [Google Scholar]
- 34.Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011;22:1007–1018. [DOI] [PubMed] [Google Scholar]
- 35.Chun J, Chung H, Wang X, et al. NLRP3 localizes to the tubular epithelium in human kidney and correlates with outcome in IgA nephropathy. Sci Rep. 2016;6:24667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bakker PJ, Butter LM, Claessen N, et al. A tissue-specific role for Nlrp3 in tubular epithelial repair after renal ischemia/reperfusion. Am J Pathol. 2014;184:2013–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lech M, Lorenz G, Kulkarni OP, et al. NLRP3 and ASC suppress lupus-like autoimmunity by driving the immunosuppressive effects of TGF-β receptor signalling. Ann Rheum Dis. 2015;74:2224–2235. [DOI] [PubMed] [Google Scholar]
- 38.Shigeoka AA, Mueller JL, Kambo A, et al. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol. 2010;185:6277–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vilaysane A, Chun J, Seamone ME, et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol. 2010;21:1732–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W, Wang X, Chun J, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol. 2013;190:1239–1249. [DOI] [PubMed] [Google Scholar]
- 41.Chung H, Vilaysane A, Lau A, et al. NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ. 2016;23:1331–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arai S, Kitada K, Yamazaki T, et al. Apoptosis inhibitor of macrophage protein enhances intraluminal debris clearance and ameliorates acute kidney injury in mice. Nat Med. 2016;22:183–193. [DOI] [PubMed] [Google Scholar]
- 43.Penberthy KK, Ravichandran KS. Apoptotic cell recognition receptors and scavenger receptors. Immunol Rev. 2016;269:44–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonventre JV. Kidney injury molecule-1: a translational journey. Trans Am Clin Climatol Assoc. 2014;125:293–299, discussion 299. [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao X, Jiang C, Olufade R, et al. Kidney injury molecule-1 enhances endocytosis of albumin in renal proximal tubular cells. J Cell Physiol. 2016;231:896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaidya VS, Ozer JS, Dieterle F, et al. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol. 2010;28:478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gandhi R, Yi J, Ha J, et al. Accelerated receptor shedding inhibits kidney injury molecule-1 (KIM-1)-mediated efferocytosis. Am J Physiol Renal Physiol. 2014;307:F205–F221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ismail OZ, Zhang X, Wei J, et al. Kidney injury molecule-1 protects against Gα12 activation and tissue damage in renal ischemia-reperfusion injury. Am J Pathol. 2015;185:1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L, Brooks CR, Xiao S, et al. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest. 2015;125:1620–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weber DJ, Gracon AS, Ripsch MS, et al. The HMGB1-RAGE axis mediates traumatic brain injury-induced pulmonary dysfunction in lung transplantation. Sci Transl Med. 2014;6:252ra124. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Nicolls MR, Laubach VE. Traumatic brain injury: lungs in a RAGE. Sci Transl Med. 2014;6:252fs234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stone JP, Critchley WR, Major T, et al. Altered immunogenicity of donor lungs via removal of passenger leukocytes using ex vivo lung perfusion. Am J Transplant. 2016;16:33–43. [DOI] [PubMed] [Google Scholar]
- 53.Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 2014;14:653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nitschke L. CD22 and Siglec-G regulate inhibition of B-cell signaling by sialic acid ligand binding and control B-cell tolerance. Glycobiology. 2014;24:807–817. [DOI] [PubMed] [Google Scholar]
- 55.Mahajan VS, Pillai S. Sialic acids and autoimmune disease. Immunol Rev. 2016;269:145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]