

Abstract
We describe a new library generation method, Machine-based Identification of Molecules Inside Characterized Space (MIMICS), that generates sets of molecules inspired by a text-based input. MIMICS-generated libraries were found to preserve distributions of properties while simultaneously increasing structural diversity. Newly identified MIMICS-generated compounds were found to be bioactive as inhibitors of specific components of the unfolded protein response (UPR) and the VEGFR2 pathway in cell-based assays, thus confirming the applicability of this methodology toward drug design applications. Wider application of MIMICS could facilitate the efficient utilization of chemical space.
Graphical abstract
Effective enumeration of unknown and novel compounds has the potential to change the way discovery of new molecular entities is pursued. In the regime of drug design, these types of compounds can be used to populate libraries, providing an effective starting point for the identification of new leads and motifs. In particular, Vishrup and Rupakheti1,2 described an iterative method to enumerate compounds over all of chemical space in a way that maximizes structural diversity and demonstrated the potential of this approach toward drug design applications.
We show that novel compounds can be generated in a facile manner with minimal a priori information and that compounds generated in this way can function in a bioactive manner. Our approach, called Machine-based Identification of Molecules Inside Characterized Space (MIMICS), considers the properties of a set of molecules rather than an individual molecule and generates an inspired set with both increased structural diversity and chemical novelty. The structures of the reference set are not needed for molecule generation, and instead only a partial text-based representation is used for reference. Additionally, the particular physical property for optimization does not need to be known: MIMICS can preserve multiple descriptors despite limited initial information.
GENERATION OF MOLECULAR LIBRARIES
The Simplified Molecular Input Line Entry System (SMILES) is used to encode molecules in a linear, text-based format for use in MIMICS. SMILES lacks implicit hydrogens, and interpretation of SMILES strings as complete structures requires the use of outside algorithms.3 Stereochemical information present in SMILES is retained, but not the information needed to interpret it. The starting input information available to MIMICS is thus necessarily incomplete.
The creation of a set of molecules requires only two steps: character generation and filtration. First, SMILES strings from an enumerated input set of molecules, whose physical properties inform the resultant properties of the MIMICS molecules generated, are used to generate a section of text. A randomly selected set of bioactive molecules from ChemBank4 was used for this. This is done using the character-level Recurrent Neural Network5 (char-RNN), freely available software that generates context-independent text based on analysis of character sequences from an input. Recurrent neural networks identify patterns from both the state of each input provided and the order in which it is provided. While the output produced is more dynamic than would be expected from an algorithmic approach, the method is inherently probabilistic, and the rationale behind a given output cannot be elucidated. The characters from the generated text take the form of SMILES-encoded molecules. Through identifying patterns both within and between sequences of characters that corresponded to molecules, we hypothesized that this method could produce chemically meaningful output.
Second, filtration of generated characters allows the population of a library of molecules. Strings filtered out include those with syntax errors, complete strings copied from the input set, identical strings generated more than once, and strings representing invalid molecules (as a result of invalid valences, aromaticity, or ring-strain errors).6,7 The threshold for chemical correctness was set to avoid manual curation of structures. There is no property- or structure-based filtration; all valid and unique SMILES strings are retained. The populated library represents the final output of MIMICS.
MIMICS-GENERATED LIBRARIES ARE DESCRIPTIVELY CONSERVATIVE BUT INTERNALLY DIVERSE
An input set was created using 880 000 molecules from the ChemBank4 database. Molecules were randomly selected from a set that adhered to Lipinski’s rule of five, with the additional restriction that no input molecules would have a molecular weight greater than 500 Da. From these molecules, 7.0 × 108 characters were generated and processed into a library of 1.09 × 106 molecules using MIMICS that was then compared with the input set. From the set of initially generated strings, 9.2% were filtered out as unusable because of repetition, syntax errors, or invalidity and removed during processing. However, the percentage removed for chemical invalidity was only 0.5%.
Generated molecules were first compared to the input set using Bemis–Murcko (BM)8 and nearest-neighbor analyses. We hypothesized that in order to be chemically and medicinally useful, the generated set of compounds must contain both novelty and structural diversity. The 880 000 molecule input set required 158 000 BM clusters for a complete description, while the generated set required more than 340 000 (Figure 1A). An additional 3 × 106 MIMICS molecules were generated, and the required number of clusters was not observed to converge. MIMICS coverage of the input scaffolds was found to scale with molecule count, beginning at 14.1% with 10 000 molecules analyzed and rising to 31.5% with the entire 880 000 molecule set considered. Nearest-neighbor analysis (Figure 1B–D) shows much higher density for input molecules on the higher-scoring end of the histogram. This implies that clusters that enumerate MIMICS molecules contain more structural diversity than input molecule clusters.
Figure 1.
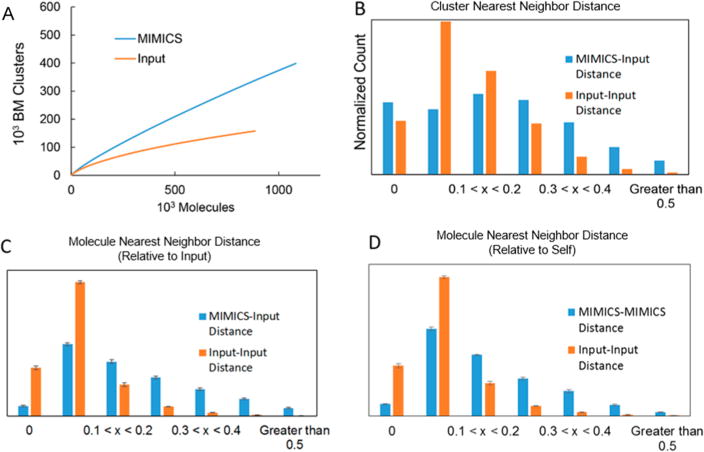
Structural novelty comparison. (A) Bemis–Murcko clustering8 was conducted on the MIMICS and input molecule sets to assess the diversity and novelty of central structural motifs. The number of unique scaffolds produced as a function of MIMICS molecules generated is displayed. (B) The Tanimoto distance between a particular structure and its nearest neighbor in the input set was computed using the Open Babel7 FP2 fingerprint for samples of MIMICS and input molecules. (C) Nearest-neighbor distance histogram for MIMICS molecules and input molecules relative to the input. (D) Nearest-neighbor distance histogram for MIMICS molecules and input molecules relative to themselves.
Nearest-neighbor analysis on samples of the molecules themselves (Figure 1C) supports this and reinforces the lack of a one-to-one correlation between the generated and input molecules. There were more than 19 times more MIMICS molecules than input molecules with nearest-neighbor distances higher than 0.50; 81% of the input molecules had distances below 0.10, compared with only 36% of MIMICS molecules. Overall, the generated set contains both novel structures and more structural diversity than the parent input set.
Generated molecules were compared to the input set both descriptively and structurally. It should be noted that character generation and thus molecule generation were informed only by the SMILES strings of the input molecules. No other information was available to the neural network, including atomic masses and identities, bond lengths, implicit hydrogen positions, ground-state three-dimensional conformations, or the metrics and descriptors that would later be used to generate the molecules in question. Out of the 1.09 × 106 compounds generated, only 37 000 independently generated input compounds (that is, a new SMILES that corresponded to an input molecule) were present (3.4%). Because MIMICS had no information regarding the existence or structure of compounds outside its input, the remainder of the generated molecules represent novel, independent creations.
Figure 2 compares the distributions of properties of the MIMICS and input sets. Filtering of the generated molecules on the basis of chemical properties was not conducted, and therefore, the property distributions reflect creations of MIMICS rather than an artificial subset of molecules. A principal moment of inertia (PMI) ratio plot9 of each set (Figure 2A) shows that even having only the input SMILES to work with, MIMICS was able to generate sets of molecules exhibiting similar distributions of overall molecular shapes as their input (Figures 2A and S2). Distributions of descriptor properties (Figure 2B–I) show that the two sets are comparable. Distortion on the heavier side of the molecular weight histogram (Figure 2I) is attributed to the fact that no compounds with weight greater than 500 Da were present in the input set. The relative lack of compounds between 400 and 500 Da is offset by the population of compounds heavier than 500 Da.
Figure 2.
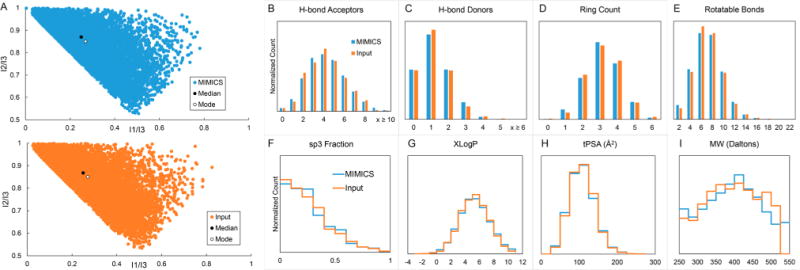
Comparison with input. MIMICS (blue) and input (orange) molecules are compared structurally and descriptively. (A) Normalized PMI ratio plots for each set of compounds were computed. The points labeled Median and Mode correspond to the median and mode coordinates of all points. Descriptive properties computed using PaDEL-descriptor10 include (B, C) numbers of hydrogen-bond acceptors and donors, (D) ring count, (E) rotatable bond count, (F) fraction of sp3-hybridized carbons, (G) XLogP, (H) topological polar surface area, and (I) molecular weight (MW). For all of the computed descriptors, both the average values and overall distributions were preserved in going from the input set to the generated MIMICS set.
MIMICS NEURONS DISPLAY ORGANIZATIONAL STRUCTURE
The ability of MIMICS to construct SMILES strings with high fidelity prompted an examination of the molecule generation process. Neuron activations, decimal values between −1 and 1, were recorded as a function of the component letters of a SMILES string. The resultant output formed a map of neuron activation patterns across a string (Figure 3).
Figure 3.
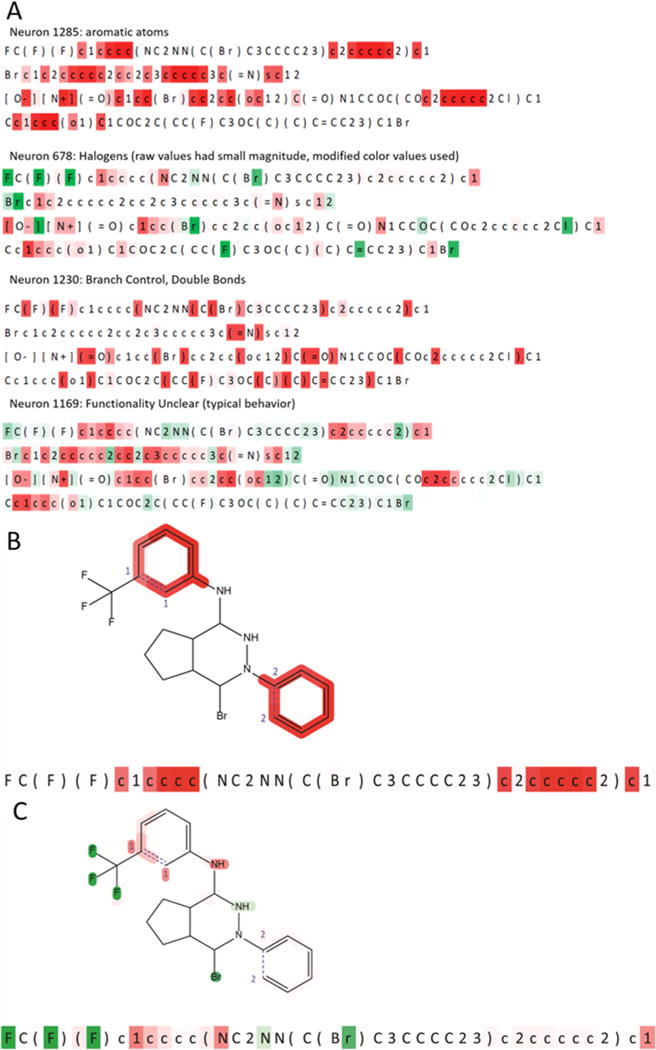
(A) Neuron activations for four different neurons. Letter colors indicate neuron activation at those particular letters, with green corresponding to positive activation and red corresponding to negative activation. Activations of three neurons with well-defined behavior and one without (out of 1538 neurons total) are displayed. Neuron 678 has been recolored because of the low magnitude of raw activations. (B, C) Mapping of neuron activations from SMILES to the molecular structure for neurons 1285 (B) and 678 (C).
Neuron 1285 was found to consistently activate negatively for letters corresponding to aromatic atoms, over four different molecules. Similarly, neuron 678 was found to consistently activate for letters corresponding to halogens, and neuron 1230 was found to consistently activate negatively on parentheses and equals signs, corresponding to control of branching and double bonds. Neuron 1285 appears to have erred on the fourth string, negatively activating on an aliphatic atom (capital letter “C”). This could alternately be conceptualized as the neuron expecting to see a six-membered aromat rather than the furan that is present. The second letter of two-letter halogens such as Br or Cl was found to activate neuron 678 much more strongly than the first letter. This suggests that the neuron in question has interpreted the defining feature of these atoms to be the second letter, which allows for the distinction between bromine and boron or chlorine and carbon.
A majority of neurons had no easily identifiable functionality (neuron 1169). The easily identified neurons are most likely counting or simply keeping track of useful information and feeding it along to other neurons. The typical neuron is likely a single intermediate step in a much larger computation.
MIMICS-GENERATED MOLECULES CAN ACT IN A BIOACTIVE MANNER
To demonstrate that MIMICS molecules have the capability of acting in a bioactive manner, we compared the MIMICS molecules with a group of small molecules that were previously identified as potential unfolded protein response (UPR) inhibitors from a previously completed high-throughput screen.11,12 From this subset of overlapping compounds, 23 were commercially available. The UPR was chosen as an example of a previously validated screen based upon biological activity. In total, of the 23 MIMICS-generated molecules tested, 12 novel molecules with potent and specific inhibitory activity (as measured by biologically significant EC50 values and other off-target activity assays) against the UPR were identified (Figure 4, two examples). None of the identified molecules were present in the input set. Furthermore, within the MIMICS set, there were 19 independently enumerated FDA-approved drugs.
Figure 4.
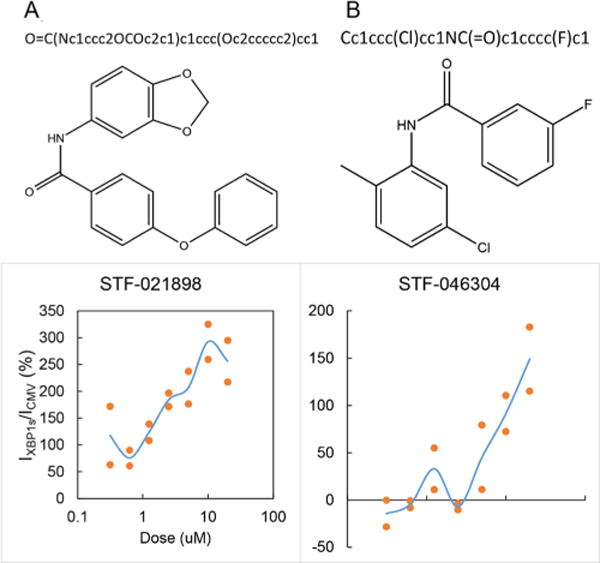
Confirmation of bioactivity against the IRE1α/XBP1 pathway, a branch of the UPR. The HT1080 (human fibrosarcoma) cell line was stably transduced with an XBP1-luciferase reporter construct. (top) Generated SMILES expressions, (middle) structures, and (bottom) dose–response curves showing inhibitory action relative to CMV control toward IRE1α/XBP1 are presented for the two identified inhibitors, (A) STF-021898 and (B) STF-046304.
MIMICS GENERATES NOVEL VEGFR-2 INHIBITORS
A more targeted application of MIMICS involves the generation of a screening library for a single disease target and the identification of novel molecules against the target. VEGFR-2, a mediator of the VEGF angiogenesis pathway, was chosen as a model because of the number of training ligands available. A set of 25 000 SMILES strings representing various VEGFR-2 ligands from BindingDB13 formed the basis for the MIMICS input. Three sets of ligands were virtually docked against the VEGFR-2 protein: the MIMICS-generated set, an existing screening library (the Stanford High-Throughput Bioscience Center library), and a set of randomly selected bioactive molecules (Figure 5).
Figure 5.
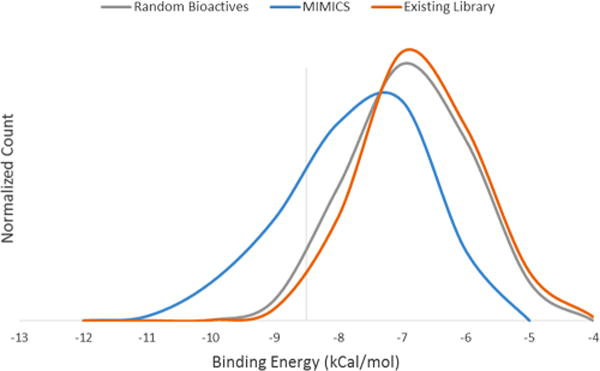
Frequency distributions of binding energies (in kCal/mol) for MIMICS and an existing screening library (Stanford High-Throughput Bioscience Center (HTBC) library). More negative values indicate more stable ligand–protein complexes and higher binding affinities.
In terms of ligand affinity, the MIMICS-generated library was found to significantly outperform both the existing screening library and the set of randomly selected bioactive molecules. The 10 000 member MIMICS library contained 40 compounds with higher binding affinity than the single best performer in the existing 110 000-member screening library. Furthermore, the high-affinity compound density (< −9 kCal/mol) was 11 times higher in the MIMICS set compared with the existing set. A properly targeted MIMICS-generated library thus provides a quantifiable benefit over existing screening libraries that is greater than what would be expected by chance.
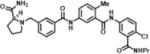
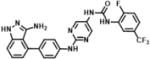
Five molecules with extremely high affinity (< −11.5 kCal/mol) were chosen for synthesis on the basis of synthetic accessibility/stability, solubility, and similarity to known VEGFR-2 inhibitors (Table 1). To ensure that the biological activity against VEGFR-2 was retained in these MIMICS-generated compounds, we chose to perform human umbilical vein endothelial cell (HUVEC) tube formation, a standard and widely used assay to assess the effect of drugs on angiogenesis in vitro.14,15 This assay was chosen to focus on the functional performance of the evaluated compounds. As shown in Figure 6, of the five compounds selected, three showed significant potency in inhibiting HUVEC tube formation. A known VEGFR2 inhibitor, vatalanib, was used as a positive control for comparison. Two compounds, SN38488 and SN38676, displayed greatly improved potency in inhibiting in vitro angiogenesis compared with vatalanib over two different dose ranges. Furthermore, neither compound displayed significant cytotoxicity on a normal human mammary epithelial cell line (MCF10A) within the dose range where tube formation inhibition occurred, suggesting that the inhibitory effect on tube formation was not due to nonspecific cytotoxicity of the compounds. These results demonstrate the ability of MIMICS to generate useful novelty when combined with a scoring method.
Table 1.
Structures and Properties of the Novel VEGFR-2 Inhibitors Synthesized
| Cpd. | ID | Structure | Name | MW (Da) | Purity (%) |
|---|---|---|---|---|---|
| 1 | SN38501 |
|
(S)-1-(3-((3-((4-chloro-3-(propylcarbamoyl)phenyl)carbamoyl)-4-methylphenyl)carbamoyl)benzyl)pyrrolidine-2-carboxamide | 576.09 | 97.3 |
| 2 | SN38502 |
|
1-(2-((4-(3-amino-1H-indazol-4-yl)phenyl)amino)pyrimidin-5-yl)-3-(2-fluoro-5-(trifluoromethyl)phenyl)urea | 522.47 | 95.2 |
| 3 | SN38488 |
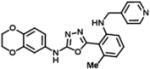
|
N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-5-(2-methyl-6-((pyridin-4-ylmethyl)amino)phenyl)-1,3,4-oxadiazol-2-amine | 415.45 | 97.7 |
| 4 | SN38676 |
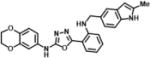
|
N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-5-(2-(((2-methyl-1H-indol-5-yl)methyl)amino)phenyl)-1,3,4-oxadiazol-2-amine | 453.5 | 99.4 |
| 5 | SN38677 |
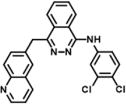
|
N-(3,4-dichlorophenyl)-4-(quinolin-6-ylmethyl)phthalazin-1-amine | 431.32 | 96.1 |
| Ref. | Vatalanib |
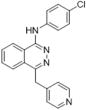
|
N-(4-chlorophenyl)-4-(pyridin-4-ylmethyl)phthalazin-1-amine | 346.81 | N/A |
Figure 6.
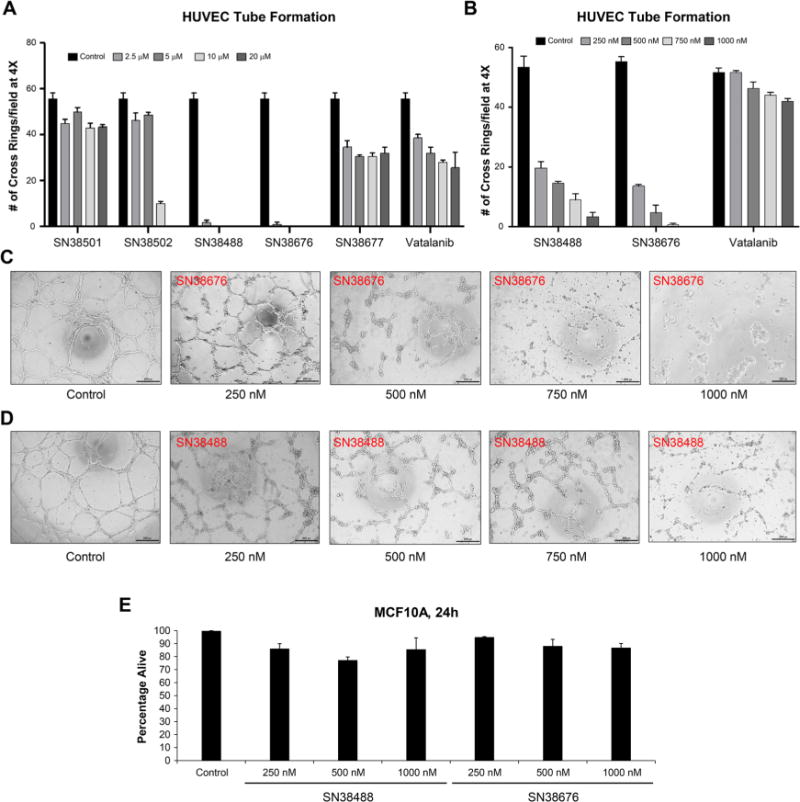
Novel VEGFR-2 inhibitors inhibit HUVEC tube formation with improved potency compared with a known VEGFR-2 inhibitor and minimal nonspecific cytotoxicity to normal cells. (A) Five novel VEGFR-2 inhibitors were tested on HUVEC tube formation at a higher dose range of 0–20 μM. DMSO (solvent) was used as the control treatment. A known VEGFR-2 inhibitor, vatalanib, was used as a positive control and for potency comparison. (B) Two inhibitors that displayed the highest potencies in inhibiting tube formation at the higher dose range were tested at a lower dose range (1–1000 nM). (C, D) Bright-field images of the effects of the two most potent compounds on HUVEC tube formation at the lower dose range. (E) Normal human mammary epithelial cell line (MCF10A) was treated with the two most potent compounds at the lower dose range, and cell viability was assessed by trypan blue staining after 24 h. Data represent means of triplicate experiments. Error bars represent standard errors of the mean.
DISCUSSION
MIMICS thus represents a unique methodology for identifying drug-like molecules, particularly in terms of the way in which compounds are generated. Rather than manipulating input molecules directly, MIMICS generates molecules informed by the properties of the whole input set. This results in the generation of similar sets of molecules rather than molecules informed by a single parent. While current approaches to enumerate “maximally diverse” libraries from a parent currently exist, doing so invariably changes the physical properties of the resultant library. Daughter libraries generated in such a manner are necessarily smaller than their parents: MIMICS allows the generation of much larger libraries. The ability to direct library generation toward a set of properties allows a middle ground between randomly screening whatever libraries may be available or committing to a set of scaffolds in a combinatorial approach. After a chemical space has been defined, MIMICS can be directed to generate novel, structurally diverse libraries within that space.
Comparisons of MIMICS-generated sets to others represents an open question. MIMICS output does not have a defined end point: strings can continue to be generated for as long the user wishes. As a result, computation of metrics such as percent overlap will scale and shrink depending on output size. Computation of overlap with the set of synthesizable molecules represents an interesting challenge, particularly given the challenge of defining “synthesizable”, but part of MIMICS’s intended purpose is that, when used with an appropriate scoring function, that resultant output molecules serve as targets for future synthetic advances.
A further contribution of MIMICS to the chemical regime of drug discovery is its ease of use. The core component, char-RNN, is freely available and was popularized by its ability to replicate Shakespearian prose and political speeches. Although the specific rationale behind the inclusion or construction of any given molecule cannot be enumerated, the ability to generate meaningful, chemically useful sets with minimal a priori information makes MIMICS a unique method for generating novel molecules.
The implementation of MIMICS allows it to be used as a general purpose analytics tool. If given a sample of a “chemical universe” and sufficient computing power, it could be used to populate areas around compounds of interest, analogues created not by the substitution of an R group or heteroatom but rather informed by the universe of bioactivity around it. Alternatively, gaps in chemical space that have already been shown to exist could be filled with MIMICS compounds that not only occupy the same space but also have desired physical or structural properties. The ability of MIMICS to mimic sets of molecules in an efficient, facile manner can make practical utilization of the vastness of chemical space possible.
Supplementary Material
Acknowledgments
The authors acknowledge grant support from P01 CA67166 (Q.-T.L., A.C.K.) and the award of a Sarah and Nadine Pole Scholarship (W.Y.) as well as support from ChemAxon for providing an academic license.
ABBREVIATIONS
- MIMICS
Machine-based Identification of Molecules Inside Controlled Space
- UPR
unfolded protein response
- SMILES
Simplified Molecular Input Line Entry System
- char-RNN
character-level Recurrent Neural Network
- BM
Bemis–Murcko
- PMI
principal moment of inertia
- IRE1α/XBP1
inositol-requiring enzyme 1/X-box binding protein 1
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.6b00754.
Methods, synthesis protocol for VEGFR-2 inhibitors, and structures of the 12 identified UPR inhibitors and MIMICS-generated FDA-approved drugs (PDF)
ORCID
William Yuan: 0000-0002-2682-0416
Notes
The authors declare no competing financial interest.
References
- 1.Virshup AM, Contreras-García J, Wipf P, Yang W, Beratan DN. Stochastic Voyages into Uncharted Chemical Space Produce a Representative Library of All Possible Drug-Like Compounds. J Am Chem Soc. 2013;135(19):7296–7303. doi: 10.1021/ja401184g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rupakheti C, Virshup A, Yang W, Beratan DNJ. Strategy To Discover Diverse Optimal Molecules in the Small Molecule Universe. J Chem Inf Model. 2015;55(3):529–537. doi: 10.1021/ci500749q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson E, Veith GD, Weininger D. SMILES: A Line Notation and Computerized Interpreter for Chemical Structures. Environmental Research Laboratory, U.S. Environmental Protection Agency; Duluth, MN: 1987. (Report EPA/600/M-87/021). [Google Scholar]
- 4.Seiler KP, George GA, Happ MP, Bodycombe NE, Carrinski HA, Norton S, Brudz S, Sullivan JP, Muhlich J, Serrano M, Ferraiolo P, Tolliday NJ, Schreiber SL, Clemons PA. ChemBank: a Small-Molecule Screening and Cheminformatics Resource Database. Nucleic Acids Res. 2007;36:D351–9. doi: 10.1093/nar/gkm843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karpathy A. Multi-layer Recurrent Neural Networks for character-level language models in Torch. 2015 https://github.com/karpathy/char-rnn (accessed September 15, 2016)
- 6. (version 15.10.5.0).Chemical validity filtration was computed using Marvin. ( http://www.chemaxon.com)
- 7.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An Open Chemical Toolbox. J Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. (version 15.10.5.0).Bemis–Murcko clustering was computed using JKlustor. ( http://www.chemaxon.com)
- 9.Sauer WHB, Schwarz MK. Molecular Shape Diversity of Combinatorial Libraries: a Prerequisite for Broad Bioactivity. J Chem Inf Comput Sci. 2003;43:987–1003. doi: 10.1021/ci025599w. [DOI] [PubMed] [Google Scholar]
- 10.Yap CW. PaDEL-descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J Comput Chem. 2011;32:1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- 11.Jiang D, Niwa M, Koong AC. Targeting the IRE1–XBP1 Branch of the Unfolded Protein Response in Human Diseases. Semin Cancer Biol. 2015;33:48–56. doi: 10.1016/j.semcancer.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117(4):1311–4. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: A Web-accessible Database of Experimentally Determined Protein-ligand Binding Affinities. Nucleic Acids Res. 2007;35:D198–D201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan KC, Ko JM, Lung HL, Sedlacek R, Zhang ZF, Luo DZ, Feng ZB, Chen S, Chen H, Chan KW, Tsao SW, Chua DT, Zabarovsky ER, Stanbridge EJ, Lung ML. Catalytic Activity of Matrix Metalloproteinase-19 is Essential for Tumor Suppressor and Anti-angiogenic Activities in Nasopharyngeal Carcinoma. Int J Cancer. 2011;129(8):1826–1837. doi: 10.1002/ijc.25855. [DOI] [PubMed] [Google Scholar]
- 15.Kong D, Li Y, Wang Z, Banerjee S, Sarkar FH. Inhibition of Angiogenesis and Invasion by 3,3′-diindolylmethane is Mediated by the Nuclear Factor-kappaB Downstream Target Genes MMP-9 and uPA That Regulated Bioavailability of Vascular Endothelial Growth Factor in Prostate Cancer. Cancer Res. 2007;67(7):3310–3319. doi: 10.1158/0008-5472.CAN-06-4277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.