

Abstract
Exposure to trichloroethylene (TCE) is linked to formation of congenital heart defects in humans and animals. Prior interactome analysis identified the transcription factor, Hepatocyte Nuclear Factor 4 alpha (HNF4a), as a potential target of TCE exposure. As a role for HNF4a is unknown in the heart, we examined developing avian hearts for HNF4a expression and for sensitivity to TCE and the HNF4a agonist, Benfluorex. In vitro analysis using a HNF4a reporter construct showed both TCE and HFN4a to be antagonists of HNF4a-mediated transcription at the concentrations tested. HNF4a mRNA is expressed transiently in the embryonic heart during valve formation and cardiac development. Embryos were examined for altered gene expression in the presence of TCE or Benfluorex. TCE altered expression of selected mRNAs including HNF4a, TRAF6 and CYP2C45. There was a transition between inhibition and induction of marker gene expression in embryos as TCE concentration increased. Benfluorex was largely inhibitory to selected markers. Echocardiography of exposed embryos showed reduced cardiac function with both TCE and Benfluorex. Cardiac contraction was reduced by 29% and 23%, respectively at 10 ppb. The effects of TCE and Benfluorex on autocrine regulation of HNF4a, selected markers and cardiac function argue for a functional interaction of TCE and HNF4a. Further, the dose-sensitive shift between inhibition and induction of marker expression may explain the nonmonotonic-like dose response observed with TCE exposure in the heart.
Keywords: TCE, Benfluorex, heart defects, CYP2C45, CYP2H1, TRAF6, NFKBIE, nonmonotonic dose response
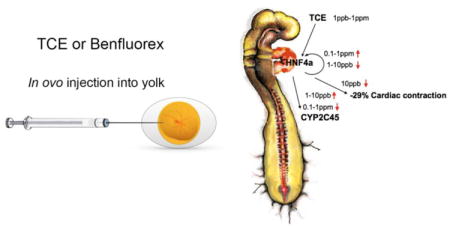
Graphical abstract
Introduction
Trichloroethylene (TCE) is an industrial degreasing solvent and a substrate used in chemical manufacturing. It is identified as a cardiac teratogen and carcinogen by the EPA and has a maximum contaminant level (MCL) of 5 ppb in water (Environmental Protection Agency, (Chiu et al., 2006). The volatile nature of TCE allows it to easily be dispersed into the environment at large, often through the air and the groundwater supply. TCE was first associated with congenital heart defects (CHDs) following a water system exposure that occurred in Tucson, AZ where exposure at levels up to 270 ppb (parts per billion) produced an odds ratio of CHDs between 2.5 and 3 (Bove et al., 2002; Goldberg et al., 1990). Other studies in Madison, WI and Endicott, NY related to vapor exposure supported the results found in Tucson, AZ but the overall, epidemiological data are limited (Forand et al., 2012; Yauck et al., 2004).
Chick, mouse and rat models were used to explore the mechanistic effects of TCE on fetal cardiac formation and gene expression. Studies showed that TCE inhibits epithelial mesenchymal cell transformation in embryonic heart tissue in an in vitro assay at 50-250 ppm (parts per million) (Boyer et al., 2000). Abnormal cardiac gene expression, including molecules involved in calcium homeostasis and several cytochrome P450s, was observed by exposures with a rat cell line in vitro, in mouse maternal drinking water or injection in ovo of chick embryos (Caldwell et al., 2008; Collier et al., 2003; Makwana et al., 2010; Selmin et al., 2008). TCE exposure is associated with significantly increased incidence of ventricular septal defects in chick embryos (Drake et al., 2006a; Drake et al., 2006b; Rufer et al., 2010). An EPA IRIS assessment stated that “weight-of-evidence analysis of epidemiological, toxicological, in vitro, in ovo, and mechanistic/AOP data concluded that TCE has the potential to cause cardiac defects in humans when exposure occurs at sufficient doses during a sensitive window of fetal development” (Makris et al., 2016).
TCE has a poorly understood but nonmonotonic-like dose curve. Low doses of TCE can produce greater changes in gene expression and function in cells and embryos compared to higher doses (Caldwell et al., 2008; Drake et al., 2006b; Makwana et al., 2013; Makwana et al., 2010; Mishima et al., 2006). H9C2 myoblasts show a greater decrease in calcium homoeostasis at 10 ppb than they do at 100 ppb and 10 ppm doses (Caldwell et al., 2008). Epithelial-mesenchymal transition mediates the formation of cardiac valve tissues in the embryonic heart (Person et al., 2005). Mesenchymal cell formation is inhibited at higher doses of TCE (50–250 ppm) while mesenchymal cell proliferation is stimulated at 80 ppb (Mishima et al., 2006). Other work has shown that markers of blood flow show more change at 8 ppb than 800 ppb (Drake et al., 2006b; Makwana et al., 2010). It was suggested that low levels of TCE might be insufficient to induce detoxification, producing a greater concentration of TCE or an active metabolite in the tissue. However, CYP 2H1 expression was higher at the lower dose while CYP 1A4 showed higher expression at the greater dose (Makwana et al., 2013). While it is unclear which cytochromes in the chick metabolize TCE, there appears to be some cytochrome P450 enzyme induction at the lowest tested exposure.
Microarray analysis of chick embryos treated with 8 ppb TCE, in ovo, identified approximately 4,000 genes significantly altered by TCE exposure. Among these genes, approximately 1,400 were linked to genes identifiable in an interactome database (Selmin et al., 2014). Within this TCE interactome, Hepatocyte Nuclear Factor 4 alpha (HNF4a) was the most interconnected molecule as it was linked directly or indirectly to approximately 75% of the nodes (Selmin et al., 2014). HNF4a is a master transcription factor associated with the liver and not identified with the developing heart, but Western Blot and PCR data show that it can be found in the early chick heart and in cultured rat cardiomyocytes (Selmin et al., 2014).
We hypothesize that HNF4a is an early and significant target of TCE-mediated cardiac teratogenicity. To explore the hypothesis, we first tested the HNF4a-expressing HepG2 cell line to examine regulation of a transcriptional reporter by TCE and its reported agonist, Benfluorex (Lee et al., 2013). The data show that both TCE and Benfluorex inhibit HNF4a-mediated transcription in vitro at the doses examined. We then explored HNF4a expression during chick heart development. Quantitative PCR analysis showed that HNF4a is expressed in the developing heart during the developmental window that coincides with the published interval of sensitivity to TCE exposure (Drake et al., 2006b). Chick embryos were exposed, in ovo, to examine effects of TCE and Benfluorex on expression of identified targets of HNF4a and/or TCE in the heart. The data identify HNF4a, TRAF6 and CYP2C45 as markers with altered gene expression when exposed to both Benfluorex and TCE. Consistent with a nonmonotonic-like response, TCE is an inhibitor of gene expression at doses of 10 ppb or lower and an inducer of expression at doses of 100 ppb or greater. Finally, as myocyte contraction is perturbed by TCE exposure (Makwana et al., 2010), we compared embryonic heart function after low-dose TCE and Benfluorex exposure. Echocardiographic analysis showed similar decreases in cardiac contraction with either Benfluorex or TCE at 10 ppb. These data support the temporal teratogenicity of TCE in the heart during the period of cardiac myocyte and valve formation. The dose response data suggest that a basis for the nonmonotonic-like response in the developing heart may lie in the shift between inhibition or induction of gene expression by TCE.
Materials and Methods
Reporter Analysis with HepG2 Cells
Human HepG2 cells (ATCC, HB-8065) were grown in DMEM media with 10% bovine serum and 1% pen/strep. Cells were incubated with either Benfluorex Hydrochloride (5μM) (sc-291931, Santa Cruz Biotechnology) or Trichloroethylene (Cas # 70-01-6, Sigma, Aldrich). For measurement of HNF4a-mediated transcription, HepG2 cells were transfected using the Cignal HNF4a reporter kit (CCS-3039L, Qiagen) with Attractene transfection reagent (Qiagen). This kit quantifies up or down regulation of an HNF4a-responsive luciferase construct encoding firefly luciferase under control of a minimal CMV promoter and tandem repeats of the HNF4a transcriptional response element. Expression is normalized to a co-transfected constitutively active Renilla luciferase reporter. Cells were exposed to the indicated final concentrations in multiwall plates for 18 hr before collection in Cell Culture Lysis Reagent (Promega, #E1531). Equal aliquots of cell lysate were mixed with Luciferase Assay Reagent (Promega, #E1500) and measured in a luminometer.
Embryos for gene expression analysis
Fertilized chick eggs were obtained from MacEntire Eggs, San Diego, California and were incubated to Hamilton and Hamburger stages (Hamburger and Hamilton, 1992) 15-26 at 37.5°C as needed. After incubation to appropriate stages, hearts were dissected from the embryos and pooled by stages for extraction. Each pool was from 2-25 hearts (larger numbers were used for stages 15-18) and mean levels were calculated from 3 independent pools for each stage. RNA was collected for analysis using E.Z.N.A. Total RNA Kit 1 (R6834-02, Omega Bio-Tek). Chicken embryos, at the stages used here, are exempt from institutional, NIH and AALAC oversight.
In ovo injection
Chick embryos were incubated to Hamilton and Hamburger stage 13 before being exposed to either Trichloroethylene (Cas # 70-01-6, Sigma, Aldrich) or Benfluorex Hydrochloride (sc-291931B, Santa Cruz Biotechnology). 1× Tyrode's solution was used as the control and to dilute the chemicals. The method of exposure was a single in ovo injection with a 250 μL syringe (370166, Hamilton).
For injection, the eggs were turned on their side and a small hole was made with an 18 G needle (305196, BD Biosciences). 50 μL of solution was injected into the yolk of the egg. The exposure dose was either: 1 ppb, 5 ppb, 10 ppb, 100 ppb, 1 ppm of Benfluorex Hydrochloride or Trichloroethylene. The hole was covered with tape and the egg was reoriented and incubated until it reached stage 17. At HH stage 17, the heart was dissected from the chick embryo and RNA was collected for analysis.
As environmental exposures and standards are often described in ppm for TCE, we use that nomenclature here for both TCE and Benfluorex. Table 1 shows the conversion of ppm or ppb levels used here to molar concentrations and the final concentration of both reagents in the egg for each of the injected concentrations.
Table 1. Conversion of ppm to molarity.
| TCE ppm/ppb | 1 ppb | 10 ppb | 100 ppb | 270 ppb | 1 ppm | 10ppm | |||
| TCE μM | 7.6 nM | 76 nM | 0.76 μM | 2.05 μM | 7.6 μM | 76 μM | |||
| Total TCE/egg | 0.5 nmol | 5 nmol | 50 nmol | 500 nmol | |||||
| Benfluorex ppm/ppb | 1 ppb | 10 ppb | 100 ppb | 440 ppb | 880 ppb | 1 ppm | 1.75 ppm | 3.5 ppm | 10 ppm |
| Benfluorex μM | 2.85 nM | 28.5 nM | 0.285 μM | 1.25 μM | 2.5 μM | 2.85 μM | 5.0 μM | 10.0 μM | 28.5 μM |
| Total Benfluorex/egg | 0.19 nmol | 1.9 nmol | 19 nmol | 190 nmol |
Echocardiographic analysis
Chicken eggs were injected 10 ppb of either Benfluorex Hydrochloride or Trichloroethylene at embryonic stage 13 as above. Twenty four hours later (approx. stage 17), embryos were transferred to a shell-less culture method (Dunn et al., 1981). Utilization of shell-less culture provided an enhancement of sensitivity and access to the heart compared to the usual windowed egg approach. Briefly, a plastic film (Anchor All Purpose Food Film, PW-18) was draped in a disposable coffee cup to produce a supporting sling. Treated eggs were cracked and the egg contents were gently transferred into the plastic sling, covered with a plastic petri dish top and incubated in a humidified incubator at 37°C. Only embryos that remained viable with an intact vasculature after transfer were retained for analysis. For echocardiography, petri dish covers were removed and a plastic ring, larger in diameter than the length of the embryo was placed over the embryo and flooded with warmed Tyrode's solution. Echocardiographic images were collected using a Vevo 2100 system (Visual Sonics) with a Micro-scan 700 transducer. The transducer was placed near the heart parallel to the head and tail of the embryo. The fractional shortening of the heart was determined using B-mode recordings and Vevo LAB software (Visual Sonics). It was determined that stage 19-20 embryos were the earliest stage that we could reliably measure ventricular contraction and the data shown are from these stages. Three to six embryos were measured for control, Benfluorex and TCE treatments during each experiment to produce mean measures of contraction. The experiment was repeated 3 times and the data show the mean and S.E.M. of the three independent experiments.
Quantitative real-time PCR
Total RNA from the heart of the chick embryo was isolated using the E.Z.N.A. Total RNA Kit 1 (R6834-02, Omega Bio-Tek). This RNA was treated with DNase I (E1091-02, Omega Bio-Tek) prior to cDNA synthesis. cDNA was created using the qScript cDNA synthesis kit (Cat# 95047-100, Quanta Biosciences). cDNA was quantitated using the Quanti-iT Oligreen ssDNA assay kit (011492, Invitrogen) and a fluorescent plate reader relative to a set of ssDNA standards. 10 ng of cDNA was added to each PCR tube for measurement. Quantitative PCR was accomplished using a Rotorgene Instrument (Qiagen) using a SensiFAST SYBR buffer (Bio-98002, Bioline). Cycling parameters used a 60°C annealing temperature and 72°C temperature for elongation and data collection. Primers were designed for chick marker genes using NCBI Primer-Blast and selected for pairs that gave melting curves with a single peak. Effective primer sequences are shown in Table 2.
Table 2. Primers used for qPCR (Gallus gallus).
| Symbol | ID | Genbank ID | Forward Primer | Reverse Primer |
|---|---|---|---|---|
| HNF4a | hepatocyte nuclear factor 4, alpha | NM 001030855.1 | GAGAAGACCGTGTGGGAGAA | CCGATAAGCAAGTCCGTGTG |
| TRAF6 | TNF receptor-associated factor 6, E3 ubiquitin protein ligase | XM 421089.3 | TCAGGGGAGAAAGGCAGAAG | ACTCTTAGAGCACAGGCAGG |
| NFKBIE | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, epsilon | XM 419490.5 | CGGAGAAGAGTTTTGTGGGC | CACGGGGCACAAATCTGAAA |
| CYP2H1 | Cytochrome p450 2H1 | NM_001001616.1 | TGGCTTGAAAGGCAACCTACG | TTGTCTGCTCAGTATGGAGGAAGG |
| CYP2C45 | Cytochrome p450 2C45 | NM_001001752.1 | GGTTTGTGTTGCTTGCCTGC | TTCACCTCCAGTATGTTCCCTACG |
Results
TCE and Benfluorex inhibit transcription of an HNF4a reporter
To explore the direct effect of TCE on HNF4a-mediated transcription, experiments were performed using the HNF4a-expressing cell line, HepG2, and a luciferase reporter with a concatamerized HNF4a binding site. Cells transfected with the reporter construct were incubated for 18 hr with the reagents. As shown in Figure 1, both TCE and Benfluorex inhibited luciferase activity. A nonmonotonic-like response was observed for TCE showing greater inhibition at 10 and 10 ppb than at intermediate doses of 1 ppm and 100 ppb. These data show that both reagents inhibit reporter transcription at the doses achieved in culture. Though Benfluorex is not volatile, note that TCE is volatile and the indicated dose was the starting concentration.
Figure 1.

TCE and Benfluorex are inhibitory of HNF4a-mediated transcription. A luciferase reporter assay was performed with HepG2 cells and an HNF4a regulated reporter plasmid with varied doses of Benfluorex or TCE. HepG2 cells were chosen for their endogenous expression of HNF4a (Liu et al., 2014). Both TCE and Benfluorex were significant inhibitors of luciferase expression in vitro at the doses tested. Values are the average of 4-10 independent measurements for each exposure. Error bars indicate S.E.M. All treatments are significant, P<.001.
Analysis of stage based differential expression of TCE linked genes
The developmental window of sensitivity to TCE exposure in the chick embryo is between HH stages 15-24 (Drake et al., 2006b). To explore the relationship between the three genes with the highest identified linkage in the TCE interactome, pooled and staged hearts were extracted for total RNA from stages 15 to 26 and examined by qPCR for HNF4a, NFKBIE and TRAF6 (Selmin et al., 2014). Measurement of expression at each stage was compared using equal aliquots of cDNA and the data were normalized to the level observed from HH stage 19 hearts (set to 1.0). Data shown for each stage represent the average and SEM of three independent pools collected and measured on different days. As shown in Fig. 1, HNF4a expression begins to increase in HH stage 18 hearts, peaks between HH stages 20 and 23 and drops at stage 26 to a level similar to stage 18. The patterns of Traf6 and NFKBIE expression are slightly delayed and peak at stage 24. HNF4a is a known regulator of cytochrome P450 expression (Jover et al., 2009). Though the specific regulation of avian cytochromes by HNF4a is unknown, expression of both CYP2C45 and CYP2H1 was observed in the embryonic heart (Makwana et al., 2013). Figure 2 shows that both CYPs are observed in the heart and that CYP2C45 peaks at stages 22-23 similar to HNF4a. CYP2H1 expression is elevated throughout Stages 22-25 and that there is an early peak at stage 15-16 when HNF4a expression is low. These data show that the three most highly linked genes in the TCE interactome are expressed in the embryonic chick heart at a time consistent with the window of sensitivity to TCE. Normal cardiac expression of the two cytochromes examined peaks late in this period as well but CYP 2C45 expression coincides with HNF4a expression to a greater extent.
Figure 2.

Normal expression of the HNF4a, TRAF6 and NFKBIE in the embryonic chick heart during the window of sensitivity to TCE. Embryonic chick hearts were collected from staged embryos and pooled for mRNA collection. Quantitative PCR measurement was performed with equal aliquots of cDNA from each stage and compared to the stage 19 pool which was normalized to a value of 1. The data show a slight bimodal distribution for HNF4a gene expression in the developing chick heart with peaks at stage 20 and again at stage 22-23. TRAF6 and NFKBIE both rise from stage 19 and peak at stage 24. The interval from stage 19-24 is one of rapid growth and differentiation of the ventricular myocardium. Measurements shown are the mean and S.E.M. of at least 3 independent pools samples at each stage.
Analysis of in ovo dose response to TCE and HNF4a reagent exposure
Previous exploration of TCE and cardiac gene expression and function found that the dose sensitivity in the developing heart was described as nonmonotonic-like and that low doses could be more toxic than higher doses (Selmin et al., 2014). To explore the relationship of expression of TCE-linked markers to the dose, chicks were injected in ovo at HH stage 13 with varied doses of TCE. Experimental doses were calculated to produce final concentrations of 1 ppb, 5 ppb, 10 ppb, 100 ppb or 1 ppm as calculated with an egg volume of 66.7 ml (Table 1) with a single injection (Drake et al., 2006b). After 24 hr of exposure, at a stage equivalent to HH 17, hearts were collected by dissection and mRNA was isolated. For each treatment, a total of 30 exposed hearts were pooled. Quantitative PCR was performed for each dose and the averages were calculated. Data were normalized to the average expression and standard error of the mean of pooled untreated stage 17 hearts. qPCR measurements were made for HNF4a and TRAF6 as both molecules were identified in the interactome and are known to be regulated by HNF4a transcription (Selmin et al., 2014). CYP2C45 was selected for its apparent link to HNF4a regulation (above), Figure 3 shows that transcription of HNF4a, TRAF6 and CYP2C45 are significantly inhibited at doses of 10 ppb and lower. At 100 ppb and 1 ppm, the markers were either significantly upregulated by TCE or unaffected. The data show that low dose TCE is inhibitory to marker expression and HNF4a auto-regulation. At higher doses, the toxin is inductive of marker expression. The pattern of transcriptional regulation suggests that an interaction between TCE and the transcriptional complex varies somewhat for each marker.
Figure 3.

Expression of Cytochrome P450 genes in the developing heart. The staged comparison of CYP gene expression shows both an early peak of expression by CYP2H1 and again between stages 22 and 25. In contrast, CYP2C45 peaks at stage 22-23. While neither pattern matches expression of HNF4a, the pattern of CYP2C45 is closest of the two. Measurements are made from the same sample pools as shown in Fig. 2 and are normalized to expression observed at stage 19 for each gene. Error bars reflect the S.E.M.
A dose response analysis was also performed on HNF4a agonist, Benfluorex. Figure 4 shows a significant down regulation of CYP2C45 at the lowest and highest doses (1 ppb, 100 ppb, 1 ppm) while the intermediate doses of 5 and 10 ppb were not significant from controls. Benfluorex showed some indication of inhibition of HNF4a expression but did not reach significance at any dose tested. In contrast to TCE, Benflorex showed an opposing concentration-dependent shift between up and down-regulation of TRAF6. Benfluorex significantly inhibited CYP2C45 expression only at the 100 ppb and 1 ppm levels. In the publication that identified Benfluorex as an agonist of HNF4a, the effective concentration was higher than tested here (700-2800 ppm) (Lee et al., 2013).
Figure 4.

TCE effects switch from inhibition to induction of selected markers in chick hearts. The two highest linked genes in the TCE interactome, HNF4a and TRAF6, were measured in the heart after in ovo injection of varied doses. Comparison was made with CYP2C45 as a potential target of HNF4a. TCE exposure significantly reduced mRNA expression of these markers at 10 ppb. Exposure at 1 ppm TCE significantly increases expression of HNF4a and TRAF6. Expression of CYP2C45 is increased at exposure levels of 100 ppb and 1 ppm. Significance calculated by Student's T-test for values obtained (unpaired, two-tailed).*p< 0.05, **p<0.005.
Analysis of cardiac contraction in the heart
It was previously shown that TCE exposure alters calcium homeostasis and reduces cardiac contraction in vitro (Caldwell et al., 2008; Makwana et al., 2010). Echocardiographic analysis was used to compare the effects of TCE and Benfluorex on function in the developing heart. An exposure level of 10 ppb for both TCE and Benfluorex was selected based upon marker expression. Embryos were injected in ovo at HH stage 13, moved to shell-less culture at HH stage 17, and incubated overnight prior to analysis. Consistent analysis of early chick embryos required incubation until the embryos reached HH stages 19 or 20. Fractional shortening was measured based upon wall movement in the primitive ventricle. At these stages, the common ventricle produced a C-shaped image and the shortest diameter across the C was selected for capture. Figure 5 shows a significant decrease in the percentage of fractional shortening in the heart for both TCE and Benfluorex exposure. Cardiac contraction decreased by 29% in embryos exposed to TCE and 23% in embryos exposed to Benfluorex.
Figure 5.

Benfluorex inhibits marker expression in chick hearts. Benfluorex was injected M at the same concentrations used for TCE and the markers shown in Fig. 4 were compared. Benfluorex exposure in the heart significantly reduced mRNA expression of CYP2C45 at the lowest dose (1ppb) and the highest doses (100 ppb and 1 ppm). TRAF6 showed significantly increased expression at 1ppb and decreased expression at all other doses but 1 ppm. Benfluorex showed no significant effect on the regulation of HNF4a expression at any dose tested although it was inhibitory at the higher doses tested in cell culture in Figure 1. The data show that TCE and Benfluorex are not acting identically on the HNF4a transcription complex. Significance calculated by Student's T-test for values obtained (unpaired, two-tailed).*p< 0.05, **p<0.005.
Discussion
Embryonic exposure to TCE is associated with congenital heart defects but not without a level of controversy (Bukowski, 2014; DeSesso and Risotto, 2017; Hardin et al., 2005). A combination of epidemiological, animal model and in vitro data was recently reevaluated by the EPA and it was concluded that TCE is likely to cause congenital heart defects (Makris et al., 2016). The defects associated with exposures in humans include atrial septal defects, ventricular septal defects, pulmonary stenosis and membranous ventricular septal defects (Goldberg et al., 1990; Yauck et al., 2004).
Data obtained in a series of studies of embryonic rat, mouse and chick hearts as well as cultured myoblasts show large numbers of genes as having altered transcription (Caldwell et al., 2010; Collier et al., 2003; Selmin et al., 2014; Selmin et al., 2008). Among these genes, there was a consistent perturbation of molecules associated with calcium homeostasis (Selmin et al., 2008). It was hypothesized that perturbation of calcium homeostasis would reduce cardiac contraction and this was confirmed by in vitro studies with a muscle cell line and in ovo studies (Caldwell et al. 2008, Rufer et al. 2010; Makwana et al., 2010).
Functional effects of TCE exposure include impaired myocyte contraction, reduced calcium homeostasis and altered cytochrome expression (Makwana et al., 2010; 2013). However, there is a lack of understanding of the transcriptional regulation that mediates these effects. Microarray data identified approximately 4,000 elements from chick embryos as significantly altered by 8 ppb TCE injected in ovo (Selmin et al. 2014). The data were compared with a chick interactome database and approximately 1300 genes were found in both datasets. These 1300 genes were explored within the chicken interactome to identify links between them. The highest linked node within the set of genes was the transcription factor HNF4a (Selmin et al., 2014). This data suggested that HNF4a could be a target of TCE toxicity despite a lack of data concerning this molecule in the heart. As shown here, HNF4a, and the next two most highly linked molecules in the interactome, TRAF6 and NFKBIE are expressed in the embryonic heart during the period of TCE-sensitivity. TRAF6 and NFKBIE appear to be early downstream targets of HNF4a and their expression as well as that of cardiac CYP2C45 is consistent with the temporal expression of HNF4a as a central transcription factor. HNF4a is not well recognized in the developing heart as the HNF4a null mouse is embryonic lethal prior to heart development and there is little or no expression in fetal or adult hearts (Chen et al., 1994). Its cardiac expression is consistent with the idea that it may be uniquely involved in the organogenesis stage of this organ. Consistent with the data shown here, a stage-specific examination of transcription factor clusters during cardiomyocyte differentiation by embryonic stem cells in vitro identified a temporally restricted co-expression of HNF4a with AP1 during a stage corresponding to early cardiomyocyte differentiation (Zeidler et al., 2016).
Benfluorex is a small molecule inhibitor that was used to treat hyperlipidemia and type II diabetes until its withdrawal from the market for cardiovascular side effects. Its side effects appear to be due to its agonist activity towards a serotonin receptor (Rothman and Baumann, 2009). It was identified as an agonist of HNF4a subsequent to a high throughput screen showed that it could reverse the inhibitory effect of palmitate on the insulin promoter (Lee et al., 2013). It is utilized here to provide a comparison between a known ligand of HNF4a and TCE. Benfluorex has a strong overlap with TCE in regulation of specific targets but does not act identically in regulation of gene expression. TCE appears to produce a more consistent dose-sensitive shift in gene transcription while Benfluorex is largely an antagonist at the levels used here. However, Benfluorex exposure in ovo is less than the exposure that lead to its identification as an agonist of HNF4a by 2 or 3 orders of magnitude (Lee et al., 2013). Previously identified TCE-sensitive targets are also associated with HNF4a as shown by their altered expression in the presence of this identified HNF4a agonist (Fig. 4). HNF4a is regulatory of its own transcription and it is variably sensitive to both inhibition and induction with increasing doses of TCE (Fig. 3).
Previous analyses of TCE exposure in relation to heart development showed perturbed gene expression in both cell and animal models at low doses (Caldwell et al., 2010; Caldwell et al., 2008; Makwana et al., 2010; Selmin et al., 2014). An ongoing issue with TCE exposure in embryos is the nonmonotonic-like dose effect. Low doses of TCE appear to have a more severe effect on gene expression than higher doses (Drake et al., 2006b; Makwana et al., 2013; Makwana et al., 2010; Rufer et al., 2010). It was speculated that toxicity was related to the relative induction of Cytochrome p450 enzymes in the heart, a metabolic explanation of the nonmonotonic-like response (Lagarde et al., 2015). Examination of Cytochrome P450 expression in early chick embryos revealed that cytochromes were largely expressed in the heart prior to the development of the liver (Makwana et al., 2013). Though specific roles for cytochrome P450s in the developing heart are unknown, avian CYP2C45 is linked to the chicken xenobiotic receptor (Baader et al., 2002).
In the present study, there was a decrease in cytochrome CYP2C45 expression at a low dose (10 ppb) and an increase in expression at a high dose (1 ppm). This contrasts to a higher expression of another CYP2C molecule, CYP2H1, at 8 ppb than at 800 ppb, shown previously (Makwana et al., 2013). The data suggest that the interaction of TCE (or a metabolite) with HNF4a is dose-dependent and that there is a switch between agonist and antagonist activity at different concentrations in the regulation of specific gene targets. The inhibition of gene expression at low level exposures may be more critical than upregulation at greater exposures in the myocyte. CYP2C45 is used here as a marker of altered HNF4a transcription. Its relationship to calcium homeostasis and myocyte contraction is unknown. However, the data is consistent with transcriptional misregulation observed in this tissue that alters myocyte contraction, cell proliferation and valve formation seen at different exposures. Disruption of HNF4a was shown to impair calcium homeostasis in pancreatic beta cells and may regulate critical gene expression similarly in the heart (Moore et al., 2016).
There are reports of nonmonotonic dose responses to toxins such as Bisphenol A and DEHP as endocrine disruptors acting on nuclear steroid receptors (Andrade et al., 2006; Vandenberg et al., 2012). Though HNF4a is not a steroid receptor, it is a nuclear receptor. The data suggest that nonmonotonic-like regulation of transcription of this receptor by TCE may be critical to its cardiac teratogenicity.
Previous data suggest that HNF4a is more commonly expressed in the embryonic liver and kidney than the heart. Which begs the question, why does TCE have cardiac specific effects? We argue that the transient expression of HNF4a in combination with the morphogenetic role of calcium-mediated contraction is critical. If metabolism of TCE to an active metabolite is involved, the expression of phase I and II enzymes may also be important. During the window of TCE toxicity in the chick heart, the heart is the principle organ in the embryo that expresses HNF4a and cytochrome p450 enzymes (Makwana et al., 2013). Direct interaction of HNF4a at this time by TCE or a metabolite of TCE leads to altered gene regulation. HNF4a is a highly linked molecule and one aspect of its altered activity appears to result in altered calcium homeostasis and impaired contraction (Moore et al., 2016). It is clear that altered myocyte contraction is sufficient to produce cardiac malformation (Hogers et al., 1997). Chick embryos exposed to 8 ppb TCE had a high incidence of ventricular septal defects (Rufer et al., 2010). The reduction in fractional shortening observed here with 10 ppb TCE (29%), is consistent with modeling that observed ventricular septal defects chick embryos with 10-35% restricted outflow (Midgett et al., 2017).
Additional effects of altered HNF4a regulation may also contribute to valve cell proliferation or a loss of valve cell formation at the same or different exposure levels, leading to other types of cardiac defects. The subsequent development of the liver and a concurrent loss of HNF4a expression in the heart in the embryo would shift metabolism of TCE to the liver as the window of cardiac sensitivity to TCE closes. Though TCE likely alters regulation of HNF4a in other embryonic or fetal tissues, misregulation of HNF4a in other organs may not lead to teratological defects in differentiation.
Though the chick embryo is an unconventional target for toxicity studies it has been a useful model for cardiac development for many decades and a substantial portion of our knowledge of heart development has been derived from this model (Kain et al., 2014). In the present context, in ovo injection provides useful control of exposure levels in the embryo and likely reduces the loss of volatile TCE during exposure. The ability to produce multiple, independently exposed, embryos enables a cost-effective exploration of dose responses compared to rodent models. One limitation of the chick embryo model is that there is no placental communication with the mother and a maternal contribution to metabolism of TCE could be missed. Thus, it would be useful to extend this data to an investigation of HNF4a in a mammalian model in the future.
Delivery to pregnant rodents in drinking water has been useful to deliver low dose exposures to entire litters of embryos but total exposure is dependent on the thirst of the pregnant dam and could vary (Caldwell et al., 2010). While a defined exposure was also produced by gavage in rats (Fisher et al., 2001), this method of exposure involves a bolus of TCE and, as shown here, higher exposures could produce induction of gene expression rather than inhibition. The epidemiological studies of Yauck et al. and Forand et el. focus on heart defects and the inhalation of TCE with relevance to vapor intrusion (Forand et al., 2012; Yauck et al., 2004). An experimental study of TCE inhalation in pregnant rats showed no cardiac defects in the embryos but the dose range varied from 50-600 ppm (Carney et al., 2006). If the minimum exposure in this study produced an exposure in cardiac tissue of at least 100ppb, our data would predict an induction rather than inhibition of HNF4a with significant differences in transcriptional targets. Thus, exposure to TCE in thus study may have been too high to observe cardiac teratogenesis via mechanisms described here. This result raises a point that both laboratory and epidemiological studies require some novelty in design to accommodate non-monotonic-like dose responses. The identification of dose-sensitive molecular markers of TCE exposure in the heart provides an avenue for future studies designed to directly compare drinking water and inhalation exposure routes for TCE.
In summary, there is an abundance of literature that shows TCE can alter cardiac formation in several animal models, consistent with reported effects in exposed human populations. Controversy exists largely due to an inconsistency in effects and a nonmonotonic-like dose curve (Bukowski, 2014; DeSesso and Risotto, 2017; Hardin et al., 2005). The data displayed here show that exposure, in ovo in embryonic chick hearts, to a defined agonist of HNF4a produces a perturbation of marker expression. TCE shows a strong overlap perturbation of the same markers. Perturbed expression of these marker genes is dose specific and TCE exposure shifts between inhibition and induction of gene expression at very low levels. Together, these data support the previous bioinformatic analysis and verify a new target of TCE exposure. As TCE exposure has been implicated in adult pathologies, it may be appropriate to investigate the contribution of HNF4a misregulation to these processes as well.
Figure 6.

Fractional shortening in the heart after TCE exposure. Chick embryos exposed to TCE or Benfluorex at stage 13 were evaluated at stage 19 or 20 for cardiac contraction percentage in the common ventricle. The data show a significant decrease in cardiac function in developing hearts exposed to TCE or Benfluorex compared to the control. The changes were significant as calculated by the Student's T-test (unpaired, two-tailed). *p<0.05, **p<0.005.
Highlights.
HNF4a-mediated transcription is inhibited in vitro by Trichloroethylene (TCE)
HNF4a is expressed in the chick embryo heart during the window of TCE sensitivity
TCE and Benfluorex (HNF4a agonist) perturb marker expression in a non-linear manner
Both TCE and Benfluorex inhibit cardiac contraction
Data suggest that HNF4a is a significant component of TCE-mediated teratogenicity
Acknowledgments
Research support was by Pilot Project Funding from P30 ES006694 (NIH). AH was supported by R25 ES025494 (NIH). KI and IM were visiting trainees partially supported by a Science Education Award from the Howard Hughes Medical Institute (HHMI) to Macalester College. We thank a former member of the laboratory, Shannon Shoemaker, for the chick embryo artwork in the graphical abstract.
Footnotes
No Conflicts of Interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrade AJ, Grande SW, Talsness CE, Gericke C, Grote K, Golombiewski A, Sterner-Kock A, Chahoud I. A dose response study following in utero and lactational exposure to di-(2-ethylhexyl) phthalate (DEHP): reproductive effects on adult male offspring rats. Toxicology. 2006;228:85–97. doi: 10.1016/j.tox.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Baader M, Gnerre C, Stegeman JJ, Meyer UA. Transcriptional activation of cytochrome P450 CYP2C45 by drugs is mediated by the chicken xenobiotic receptor (CXR) interacting with a phenobarbital response enhancer unit. J Biol Chem. 2002;277:15647–15653. doi: 10.1074/jbc.M109882200. [DOI] [PubMed] [Google Scholar]
- Bove F, Shim Y, Zeitz P. Drinking water contaminants and adverse pregnancy outcomes: A review. Environmental Health Perspectives. 2002;110:61–74. doi: 10.1289/ehp.02110s161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer AS, Finch WT, Runyan RB. Trichloroethylene inhibits development of embryonic heart valve precursors in vitro [see comments] Toxicological Sciences. 2000;53:109–117. doi: 10.1093/toxsci/53.1.109. [DOI] [PubMed] [Google Scholar]
- Bukowski J. Critical review of the epidemiologic literature regarding the association between congenital heart defects and exposure to trichloroethylene. Critical reviews in toxicology. 2014;44:581–589. doi: 10.3109/10408444.2014.910755. [DOI] [PubMed] [Google Scholar]
- Caldwell PT, Manziello A, Howard J, Palbykin B, Runyan RB, Selmin O. Gene Expression Profiling in the Fetal Cardiac Tissue after Folate and Low-Dose Trichloroethylene Exposure. Birth Defects Research Part a-Clinical and Molecular Teratology. 2010;88:111–127. doi: 10.1002/bdra.20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell PT, Thorne PA, Johnson PD, Boitano S, Runyan RB, Selmin O. Trichloroethylene disrupts cardiac gene expression and calcium homeostasis in rat myocytes. Toxicological Sciences. 2008;104:135–143. doi: 10.1093/toxsci/kfn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney EW, Thorsrud BA, Dugard PH, Zablotny CL. Developmental toxicity studies in Crl:CD (SD) rats following inhalation exposure to trichloroethylene and perchloroethylene. Birth Defects Res B Dev Reprod Toxicol. 2006;77:405–412. doi: 10.1002/bdrb.20091. [DOI] [PubMed] [Google Scholar]
- Chen WS, Manova K, Weinstein DC, Duncan SA, Plump AS, Prezioso VR, Bachvarova RF, Darnell JE. Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes & Development. 1994;8:2466–2477. doi: 10.1101/gad.8.20.2466. [DOI] [PubMed] [Google Scholar]
- Chiu WA, Caldwell JC, Keshava N, Scott CS. Key scientific issues in the health risk assessment of trichloroethylene. Environ Health Perspect. 2006;114:1445–1449. doi: 10.1289/ehp.8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier J, Selmin O, Johnson P, Runyan R. Trichloroethylene effects on gene expression during cardiac development. Birth Defects Research Part a-Clinical and Molecular Teratology. 2003;67:488–495. doi: 10.1002/bdra.10073. [DOI] [PubMed] [Google Scholar]
- DeSesso JM, Risotto SP. Review of TCE cardiac defects data by Makris et al. is not systematic. Reproductive Toxicology. 2017;71:134. doi: 10.1016/j.reprotox.2017.05.012. [DOI] [PubMed] [Google Scholar]
- Drake VJ, Koprowski SL, Hu N, Smith SM, Lough J. Cardiogenic effects of trichloroethylene and trichloroacetic acid following exposure during heart specification of avian development. Toxicological Sciences. 2006a;94:153–162. doi: 10.1093/toxsci/kfl083. [DOI] [PubMed] [Google Scholar]
- Drake VJ, Koprowski SL, Lough J, Hu N, Smith SM. Trichloroethylene exposure during cardiac valvuloseptal morphogenesis alters cushion formation and cardiac hemodynamics in the avian embryo. Environmental Health Perspectives. 2006b;114:842–847. doi: 10.1289/ehp.8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn BE, Fitzharris TP, Barnett BD. Effects of varying chamber construction and embryo pre-incubation age on survival and growth of chick embryos in shell-less culture. The Anatomical Record. 1981;199:33–43. doi: 10.1002/ar.1091990105. [DOI] [PubMed] [Google Scholar]
- Fisher JW, Channel SR, Eggers JS, Johnson PD, MacMahon KL, Goodyear CD, Sudberry GL, Warren DA, Latendresse JR, Graeter LJ. Trichloroethylene, trichloroacetic acid, and dichloroacetic acid: do they affect fetal rat heart development? Int J Toxicol. 2001;20:257–267. doi: 10.1080/109158101753252992. [DOI] [PubMed] [Google Scholar]
- Forand SP, Lewis-Michl EL, Gomez MI. Adverse Birth Outcomes and Maternal Exposure to Trichloroethylene and Tetrachloroethylene through Soil Vapor Intrusion in New York State. Environmental Health Perspectives. 2012;120:616–621. doi: 10.1289/ehp.1103884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg SJ, Lebowitz MD, Graver E. Association of human congential cardiac malformations and drinking water contaminants. J Am Coll Cardiol. 1990;16:155–164. doi: 10.1016/0735-1097(90)90473-3. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stagesin the development of the chick embryo. Develompmental Dynamics. 1992;195:231–272. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- Hardin BD, Kelman BJ, Brent RL. Trichloroethylene and dichloroethylene: a critical review of teratogenicity. Birth defects research Part A, Clinical and molecular teratology. 2005;73:931–955. doi: 10.1002/bdra.20192. [DOI] [PubMed] [Google Scholar]
- Hogers B, DeRuiter MC, Gittenberger de Groot AC, Poelmam RE. Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circulation Research. 1997;80:473–481. doi: 10.1161/01.res.80.4.473. [DOI] [PubMed] [Google Scholar]
- Jover R, Moya M, Gomez-Lechon MJ. Transcriptional Regulation of Cytochrome P450 Genes by the Nuclear Receptor Hepatocyte Nuclear Factor 4-Alpha. Curr Drug Metab. 2009;10:508–519. doi: 10.2174/138920009788898000. [DOI] [PubMed] [Google Scholar]
- Kain KH, Miller JW, Jones-Paris CR, Thomason RT, Lewis JD, Bader DM, Barnett JV, Zijlstra A. The chick embryo as an expanding experimental model for cancer and cardiovascular research. Dev Dyn. 2014;243:216–228. doi: 10.1002/dvdy.24093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarde F, Beausoleil C, Belcher SM, Belzunces LP, Emond C, Guerbet M, Rousselle C. Non-monotonic dose-response relationships and endocrine disruptors: a qualitative method of assessment. Environmental Health. 2015;14:13. doi: 10.1186/1476-069X-14-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Athavankar S, Cohen T, Piran R, Kiselyuk A, Levine F. Identification of alverine and benfluorex as HNF4alpha activators. ACS Chem Biol. 2013;8:1730–1736. doi: 10.1021/cb4000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris SL, Scott CS, Fox J, Knudsen TB, Hotchkiss AK, Arzuaga X, Euling SY, Powers CM, Jinot J, Hogan KA, Abbott BD, Hunter ES, Narotsky MG. A systematic evaluation of the potential effects of trichloroethylene exposure on cardiac development. Reproductive Toxicology. 2016;65:321–358. doi: 10.1016/j.reprotox.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makwana O, Ahles L, Lencinas A, Selmin OI, Runyan RB. Low-Dose Trichloroethylene Alters Cytochrome P450-2C Subfamily Expression in the Developing Chick Heart. Cardiovascular Toxicology. 2013;13:77–84. doi: 10.1007/s12012-012-9180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makwana O, King NM, Ahles L, Selmin O, Granzier HL, Runyan RB. Exposure to low-dose trichloroethylene alters shear stress gene expression and function in the developing chick heart. Cardiovascular toxicology. 2010;10:100–107. doi: 10.1007/s12012-010-9066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgett M, Thornburg K, Rugonyi S. Blood flow patterns underlie developmental heart defects. Am J Physiol Heart Circ Physiol. 2017;312:H632–H642. doi: 10.1152/ajpheart.00641.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima N, Hoffman S, Hill EG, Krug EL. Chick embryos exposed to trichloroethylene in an ex ovo culture model show selective defects in early endocardial cushion tissue formation. Birth Defects Research Part a-Clinical and Molecular Teratology. 2006;76:517–527. doi: 10.1002/bdra.20283. [DOI] [PubMed] [Google Scholar]
- Moore BD, Jin RU, Lo H, Jung M, Wang H, Battle MA, Wollheim CB, Urano F, Mills JC. Transcriptional Regulation of X-Box-binding Protein One (XBP1) by Hepatocyte Nuclear Factor 4alpha (HNF4Alpha) Is Vital to Beta-cell Function. J Biol Chem. 2016;291:6146–6157. doi: 10.1074/jbc.M115.685750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person AD, Klewer SE, Runyan RB. Cell Biology of Cardiac Cushion Development. International Review of Cytology. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH. Serotonergic Drugs and Valvular Heart Disease. Expert opinion on drug safety. 2009;8:317–329. doi: 10.1517/14740330902931524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufer ES, Hacker TA, Flentke GR, Drake VJ, Brody MJ, Lough J, Smith SM. Altered cardiac function and ventricular septal defect in avian embryos exposed to low-dose trichloroethylene. Toxicol Sci. 2010;113:444–452. doi: 10.1093/toxsci/kfp269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmin O, Makwana O, Runyan R. Environmental Sensitivity to Trichloroethylene (TCE) in the Developing Heart. In: Gilbert KM, Blossom SJ, editors. Trichloroethylene: Toxicity and Health Risks. Springer; London: 2014. pp. 153–169. [Google Scholar]
- Selmin OI, Thorne PA, Caldwell PT, Taylor MR. Trichloroethylene and trichloroacetic acid regulate calcium signaling pathways in murine embryonal carcinoma cells p19. Cardiovascular toxicology. 2008;8:47–56. doi: 10.1007/s12012-008-9014-2. [DOI] [PubMed] [Google Scholar]
- Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Lee DH. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose-responses. Endocr Rev. 2012;33 doi: 10.1210/er.2011-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauck JS, Malloy ME, Blair K, Simpson PM, McCarver DG. Proximity of residence to trichloroethylene-emitting sites and increased risk of offspring congenital heart defects among older women. Birth Defects Research Part a-Clinical and Molecular Teratology. 2004;70:808–814. doi: 10.1002/bdra.20060. [DOI] [PubMed] [Google Scholar]
- Zeidler S, Meckbach C, Tacke R, Raad FS, Roa A, Uchida S, Zimmermann WH, Wingender E, Gultas M. Computational Detection of Stage-Specific Transcription Factor Clusters during Heart Development. Front Genet. 2016;7 doi: 10.3389/fgene.2016.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]