

Abstract
Renewable commodity chemicals can be generated from plant materials. Often abundant materials such as sugars are used for this purpose. However, these lack appropriate functionalities and, therefore, they require extensive chemical modifications before they can be used as commodity chemicals. The plant kingdom is capable of producing an almost endless variety of compounds, including compounds with highly appropriate functionalities, but these are often not available in high quantities. It has been demonstrated that it is possible to produce functionalized plant compounds on a large scale by fermentation in microorganisms. This opens up the potential to exploit plant compounds that are less abundant, but functionally resemble commodity chemicals more closely. To elaborate this concept, we demonstrate the suitability of a highly functionalized plant compound, methyl perillate, as a precursor for the commodity chemical terephthalic acid.
Keywords: bio-based commodity chemicals, methyl perillate, monoterpene, natural functionalization, terephthalic acid
Global material demands inspire research towards bio‐based building blocks. For instance, terephthalic acid (TA) is currently produced from petrochemical sources by oxidation of para‐xylene (pX).1 Global TA demand is expected to reach 65 million tons in 2018, predominantly for the production of polyethylene terephthalate (PET).2 One approach for the bio‐based supply of TA is to use sugars or polysaccharides as starting materials, which are abundantly available from biomass.3 However, the structural similarity between sugars and TA is limited and, therefore, a considerable number of harsh synthesis steps are needed for TA production (Table 1).
Table 1.
Comparison of sugars and methyl perillate as precursors for TA.
| Compound | Functional group(s) | Reactions needed | Natural source |
|---|---|---|---|
| sugars | – oxygenated ring | – solubilization – hydrodeoxygenation – oxidation (100–600 °C, 0.1–83 bar)3 |
– biomass (sugars) |
| methyl perillate | – oxygenated ring – acid group – functionalized p‐position |
– dehydrogenation – oxidation |
– essential oil (Perilla, Salvia) or fermentation |
These steps include solubilizing sugars, hydrodeoxygenation of the sugar ring, dehydrogenation to produce a mixture of aromatics, separation of pX, and oxidation, with temperatures up to 600 °C and high pressure.2 Methyl perillate (MPA), on the other hand, carries an unsaturated six‐membered ring, is functionalized at the para position, and has an acid group at the C7 position. MPA is a monoterpenoid, which occurs in the plant species Salvia dorisiana (see Figure S1 and Table S1 in the Supporting Information). Terpenoids such as artemisinic acid and farnesene are produced with high yields through microbial fermentation,4 using metabolic engineering of microorganisms upon the introduction of plant metabolic pathways. Though this has not yet been achieved for MPA, its precursors limonene and perillic acid (PA) can be obtained by using microbial systems.5 Therefore, when compared to glucose (Table 1) and other bio‐based precursors (Table S2), MPA could be considered as a highly functionalized precursor for TA synthesis.
A mild conversion of MPA to TA was designed (Scheme 1), using dehydrogenation and oxidation reactions, and subsequently tested.
Scheme 1.
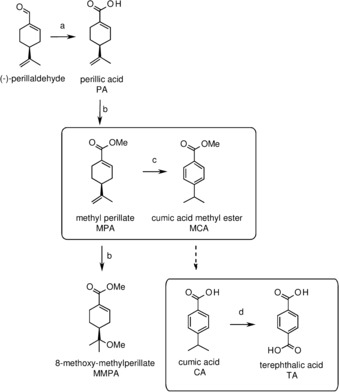
Synthesis of terephthalic acid from methylperillate, as developed in this study.
First, MPA was synthesized from commercially available (−)‐perillaldehyde as the starting material, using silver oxide as a catalyst, providing PA in reasonable isolated yield (Scheme 1, reaction a) (66 % yield, 94 % pure) (see Figure S2 and Table S3).6 The resulting PA was methylated to MPA by esterification in excess methanol with catalytic p‐toluene sulfonic acid (Scheme 1, reaction b). During methylation, a by‐product was formed, 8‐methoxy‐methyl perillate (MMPA). Short reaction times ensured minimal MMPA formation, and it could be readily separated from MPA (see Figures S3 and S4). Nearly pure MPA (3.0 g, 48 % yield, 96 % pure) was used as the starting material for the synthesis of TA.
Three procedures were considered for the dehydrogenation of MPA to methyl cumic acid (MCA). A dehydrogenation procedure using ethylene diamine and metallic sodium has been reported for the dehydrogenation of limonene.7 We could reproduce these results; however, this procedure consumes metallic sodium and was, therefore, considered not fully sustainable. Another procedure, using zeolite NaY as a catalyst, yielded a mixture of isomers, with only 25 % of MCA; this procedure was, therefore, not considered.8 The third procedure, a heterogenic catalytic procedure using a solid‐supported Pd catalyst for hydrogenation was followed,9 involving sequential catalytic double‐bond isomerization, hydrogenation, and dehydrogenation. Acetone was used as a hydrogen acceptor10 to steer the reaction towards complete ring dehydrogenation. Initially, limonene was used as a testing substrate. When a Pd/C catalyst was used, formation of dehydrogenated product p‐cymene was observed only when the reaction was performed at 150 °C (20 h); whereas, at 100 °C (20 h), no conversion of limonene could be observed. Changing the support material of the catalyst to alumina (5 % Pd/Al2O3) resulted in a somewhat reduced yield at 150 °C, but it had a strongly improved reactivity and selectivity towards the desired product (80 %, Table S4) at 125 °C and 100 °C for 20 h. At 125 °C, the reaction time could be reduced to 1 h for full conversion of limonene and comparable, even superior, yields of p‐cymene (see Figure S5 and Table S5). Also, these shorter reaction times significantly reduced the formation of acetone‐related by‐products diacetone alcohol and mesityl oxide. Thus, a short dehydrogenation procedure for limonene‐like substances was developed, which operates efficiently at mild temperature.
Using the developed procedure (125 °C, 1 h, 5 % Pd/Al2O3), MPA was efficiently converted to MCA (see Figures 1 and S6). A plausible reaction mechanism involves sequential isomerization and dehydrogenation (Scheme S1),9 in which transfer hydrogenation to acetone pulls the equilibria towards the fully dehydrogenated product MCA. The ratio of MCA to MPA was 1:0.17 (85 % conversion). The end product contained similar acetone aldol addition products to those detected in the limonene dehydrogenation. Isopropanol (iPrOH) and water are formed by transfer hydrogenation and aldol–addition reactions, respectively. However, no hydrolyzed or trans‐esterified forms of MPA or MCA were detected in the end product.
Figure 1.
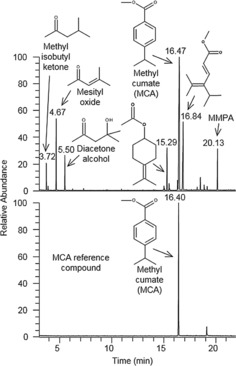
Methyl cumate, the product of dehydrogenation from MP. GC‐MS chromatograms of dehydrogenation product and MCA reference compound. Side products related to acetone are detected; the side product of PA methylation (MMPA) is still present, and some other side products are visible; a library‐hit of their identity is indicated in the chromatogram.
Use of highly functionalized starting materials such as MPA provide benefits by reducing the number of synthetic steps. This is the case for the oxidation to form TA. Previous studies have shown that oxidation of the isopropyl group at position 4 can be readily achieved, but oxidation of the methyl group at position 7 of limonene and its dehydrogenation product p‐cymene is more difficult and needs an extra step.11 In MPA and MCA, the 7 position is already occupied by a carboxyl group. Therefore, oxidation to form the end product TA can be achieved by an efficient single oxidation step (Scheme 1, reaction d).
Two oxidation methods were tested to convert intermediate MCA to TA. First, oxidation with KMnO4 was tested,12 but yielded only 12 % TA. Second, a procedure for the oxidation with nitric acid was tested. Aromatic isopropyl groups can be efficiently oxidized to the corresponding carboxylic acid group by nitric acid.7, 13 Under aqueous nitric acid conditions, the methyl ester group of MCA is rapidly hydrolyzed, yielding the free cumic acid (CA) in situ. Oxidation of CA using 65 % nitric acid resulted in full conversion and 89 % isolated yield (24 h reflux, non‐optimized; Figure S7). The product contained 70 % TA (Figure S7) and a single side‐product was identified as 1,1‐dinitroethyl benzoic acid by using NMR, LC‐MS, and IR analyses (Figure S7).14 It is known that 1,1‐dinitroethyl benzoic acid is also converted to TA by heating in 30 % nitric acid at 180 °C.14b This protocol improves earlier described nitric acid oxidation yields starting from benzene (42 % yield of TA)15 and p‐cymene (51 % yield of p‐toluic acid).16 These results show that it is possible to oxidize MCA to TA in a single step with good yield.
In conclusion, we have demonstrated the applicability of two mild catalytic steps to convert the natural monoterpenoid MPA to the commodity chemical TA. By employing palladium‐catalyzed dehydrogenation, short and mild conditions can be deployed, which offer advantages in terms of sustainability and yield. Subsequently, oxidation using nitric acid is efficient in producing TA with high yield and high purity. Our work clearly outlines the advantages of selecting highly functionalized molecules as starting materials to produce commodity materials such as TA (Figure 2). This approach anticipates the ability of the fermentation industry to produce functionalized terpenoid compounds at affordable prices. The fermentative production of complex functionalized molecules such as farnesene, which is positioned as a jet‐fuel, indicates that this is a realistic scenario. Therefore, the identification of compounds that carry appropriate functionalization and the development of sustainable procedures to convert them into bio‐based building blocks may have high potential for future applications.
Figure 2.
Naturally highly functionalized starting materials from fermentative production allow mild chemistry to produce commodity chemicals.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was funded by the Dutch Ministry of Economic Affairs [Grant KB‐13‐006‐042]. Source of the pictures in Figure 2 and the graphical abstract: Wageningen Food & Biobased Research
E. Jongedijk, F. van der Klis, R. de Zwart, D. S. van Es, J. Beekwilder, ChemistryOpen 2018, 7, 201.
References
- 1.
- 1a. Rezaei V., Sajadi S. A. A., Russ J Appl Chem. 2015, 88, 1201–1206; [Google Scholar]
- 1b. Schwartz T. J., O'Neill B. J., Shanks B. H., Dumesic J. A., ACS Catal. 2014, 4, 2060–2069; [Google Scholar]
- 1c. Schwartz T. J., Shanks B. H., Dumesic J. A., Curr Opin Biotechnol 2016, 38, 54–62. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Collias D. I., Harris A. M., Nagpal V., Cottrell I. W., Schultheis M. W., Ind. Biotechnol. 2014, 10, 91–105; [Google Scholar]
- 2b. Williams C. L., Vinter K. P., Patet R. E., R. E., Chang C. C., Nikbin N., Feng S. T., Wiatrowski M. R., Caratzoulas S., Fan W., Vlachos D. G., Dauenhauer P. J., ACS Catal. 2016, 6, 2076–2088. [Google Scholar]
- 3.
- 3a. Lu R., Lu F., Chen J., Yu W., Huang Q., Zhang J., Xu J., Angew. Chem. Int. Ed. 2016, 55, 249–253; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 257–261; [Google Scholar]
- 3b. Wang F., Tong Z., RSC Adv. 2014, 4, 6314; [Google Scholar]
- 3c. Lee J. J., Kraus G. A., Green Chem. 2014, 16, 2111; [Google Scholar]
- 3d.M. W. T. Peters, J. D. Jenni, M. Manzer, L. E. Henton, D. E. US patent 2011/0087000 A1 2011.
- 4.
- 4a. Meadows A. L., Hawkins K. M., Tsegaye Y., Antipov E., Kim Y., Raetz L., Dahl R. H., Tai A., Mahatdejkul-Meadows T., Xu L., Zhao L., Dasika M. S., Murarka A., Lenihan J., Eng D., Leng J. S., Liu C. L., Wenger J. W., Jiang H., Chao L., Westfall P., Lai J., Ganesan S., Jackson P., Mans R., Platt D., Reeves C. D., Saija P. R., Wichmann G., Holmes V. F., Benjamin K., Hill P. W., Gardner T. S., Tsong A. E., Nature 2016, 537, 694–697; [DOI] [PubMed] [Google Scholar]
- 4b. Paddon C. J., Westfall P. J., Pitera D. J., Benjamin K., Fisher K., McPhee D., Leavell M. D., Tai A., Main A., Eng D., Polichuk D. R., Teoh K. H., Reed D. W., Treynor T., Lenihan J., Fleck M., Bajad S., Dang G., Dengrove D., Diola D., Dorin G., Ellens K. W., Fickes S., Galazzo J., Gaucher S. P., Geistlinger T., Henry R., Hepp M., Horning T., Iqbal T., Jiang H., Kizer L., Lieu B., Melis D., Moss N., Regentin R., Secrest S., Tsuruta H., Vazquez R., Westblade L. F., Xu L., Yu M., Zhang Y., Zhao L., Lievense J., Covello P. S., Keasling J. D., Reiling K. K., Renninger N. S., Newman J. D., Nature 2013, 496, 528–532. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Jongedijk E., Cankar K., Buchhaupt M., Schrader J., Bouwmeester H., Beekwilder J., Appl. Microbiol. Biotechnol. 2016, 100, 2927–2938; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Mars A. E., Gorissen J. P., van den Beld I., Eggink G., Appl. Microbiol. Biotechnol. 2001, 56, 101–107. [DOI] [PubMed] [Google Scholar]
- 6. Wang Q., Fan S. Y., Wong H. N. C., Li Z., Fung B. M., Twieg R. J., Nguyen H. T., Tetrahedron 1993, 49, 619–638. [Google Scholar]
- 7. Colonna M., Berti C., Fiorini M., Binassi E., Mazzacurati M., Vannini M., Karanam S., Green Chem. 2011, 13, 2543–2548. [Google Scholar]
- 8. Hatzakis E., Opsenica I., Solaja B. A., Stratakis M., ARKIVOC 2007, 124–135. [Google Scholar]
- 9. Grau R. J., Zgolicz P. D., Gutierrez C., Taher H. A., J Mol Catal A-Chem. 1999, 148, 203–214. [Google Scholar]
- 10. Wang L., Xiao J., Top. Curr. Chem. 2016, 374, 17. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Neaţu F., Culica G., Florea M., Parvulescu V. I., Cavani F., ChemSusChem 2016, 9, 3102–3112; [DOI] [PubMed] [Google Scholar]
- 11b.S. Lundmark, M. Kangas, B. Häggman, WO Patent 2014/133433 A1, Sweden, 2014, p. 14;
- 11c. Hronec M., Holotík S., Ilavsky J., Collect Czech Chem. C 1980, 45, 880–887. [Google Scholar]
- 12. Meyer R., Ber. Dtsch. Chem. Ges. 1878, 11, 1283–1287. [Google Scholar]
- 13.
- 13a. Ferguson L. N., Wims A. I., J. Org. Chem. 1960, 25, 668–678; [Google Scholar]
- 13b. Newton A., J. Am. Chem. Soc. 1943, 65, 2444–2445. [Google Scholar]
- 14.
- 14a. Gasco A. M., Distilo A., Sorba G., Gasco A., Ferioli R., Folco G., Civelli M., Caruso P., Eur. J. Med. Chem. 1993, 28, 433–438; [Google Scholar]
- 14b. Mcintyre J. E., J. Chem. Soc. 1964, 3540. [Google Scholar]
- 15. Newton A., J. Am. Chem. Soc. 1943, 65, 320–323. [Google Scholar]
- 16. Tuley W. F., Marvel C. S., Org. Synth. 1947, 27, 86–88. [Google Scholar]
- 17. Lamsen E. N., Atsumi S., Front. Microbiol. 2012, 3, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.J. W. Frost, K. M. Draths, US patent 5616496, 1997.
- 19. Koopman F., Wierckx N., de Winde J. H., Ruijssenaars H. J., Bioresour. Technol. 2010, 101, 6291–6296. [DOI] [PubMed] [Google Scholar]
- 20. Jongedijk E., Cankar K., Ranzijn J., van der Krol S., Bouwmeester H., Beekwilder J., Yeast 2015, 32, 159–171. [DOI] [PubMed] [Google Scholar]
- 21. West T., Fermentation 2017, 3, 14. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary