

Abstract
Marine invertebrates and fish are well known for their remarkable genetic diversity, which is commonly explained by large population size and the characteristic dispersive nature of their early, planktonic life history. Other potential sources of diversity in marine animals, such as a higher mutation rate, have been much less considered, though evidence for a high genetic load in marine bivalves has been accumulating for nearly half a century. In this review, I examine evidence for a higher genetic load in marine animals from studies of molecular marker segregation and linkage over the last 40 years, and survey recent work examining mutational load with molecular evolution approaches. Overall, marine animals appear to have higher genetic load than terrestrial animals (higher dn/ds ratios, inbreeding load, and segregation dis`tortion), though results are mixed for marine fish and data are lacking for many marine animal groups. Bivalves (oysters) have the highest loads observed among marine animals, comparable only to long-lived plants; however, more data is needed from other bivalves and more marine invertebrate taxa generally. For oysters, a higher load may be related to a chronically lower effective population size that, in concert with a higher mutational rate, elevate the number of deleterious mutations observed. I suggest that future studies use high-throughput sequencing approaches to examine (1) polymorphism in genome-scale datasets across a wider range of marine animals at the population level and (2) intergenerational mutational changes between parents and offspring in crosses of aquaculture species to quantify mutation rates.
Keywords: dn/ds ratios, high fecundity, larval mortality, life history, mutation, oyster, segregation distortion.
Introduction
Marine fish and invertebrates have long been noted for their remarkable genetic diversity. Population genetic studies using allozyme or DNA markers have repeatedly demonstrated markedly higher diversity in comparisons of marine versus terrestrial species or marine versus freshwater fish (e.g., Smith and Fujio 1982; Ward et al. 1994; Bazin et al. 2006), which suggests something potentially unique about the biology or ecology of marine animals (Williams 1975; Bazin et al. 2006; Harrang et al. 2013; Plough et al. 2016). Explanations for such extreme diversity in marine animals have often emphasized the highly dispersive nature of marine environments (few physical barriers, strong ocean currents) and the long-lived dispersive larval stage of most fish and invertebrate species, which facilitate potentially high gene flow and the establishment of large populations capable of sustaining high levels of allelic or nucleotide variation (e.g., Smith and Fujio 1982; Mitton 1993; Ward et al. 1994; Small et al. 2007; Hellberg 2009).
Though the assumption of large population size may hold for some marine animal species, as a general explanation for high levels of observed diversity overall, it is insufficient for a few reasons (e.g., Harrang et al. 2013). First, features of marine animal life history and reproductive biology, particularly high variance in reproductive success among parents, can dramatically decrease the effective population size of marine species relative to census numbers (Nunney 1995; Hedrick 2005; Hedgecock and Pudovkin 2011). Indeed, a number of recent population genetic studies of fish and shellfish find that effective population sizes of marine species can be orders of magnitude below their census size and thus are not universally "large" (e.g., Hedgecock 1994; Turner et al. 1999; Turner et al. 2002; Hedgecock and Pudovkin 2011). Second, population size is only one-half of the equation when considering the expected level of polymorphism in a population at equilibrium. Theta, the expected level of diversity at equilibrium under the neutral theory (Nei 1987), is a function not only of effective population size but also of mutation rate. Thus, a higher rate of mutation in marine animals could also contribute to the high polymorphism observed in marine populations in nature (e.g., Tsagkogeorga et al. 2012); however, this has not been sufficiently examined in the literature.
Previous work on the population genetics of marine animals has revealed important insights about connectivity and adaptive change within high gene flow species (e.g., Hedgecock et al. 2007; Hauser and Carvalho 2008; Hellberg 2009; Gagnaire et al. 2015), but a number of questions remain regarding the biological and demographic factors underlying high genetic diversity in these species. First, is higher diversity in marine animal species associated with a higher mutation rate and higher genetic load? Second, how do the life-history characteristics of marine animals impact diversity and shape how genetic load is expressed in populations? Recent advances in sequencing technology and the availability of genome-scale datasets for non-model marine species are providing novel insights into these questions, such as, the number and effect of mutations segregating in the Pacific oyster genome (e.g., Plough and Hedgecock 2011) or the distribution of effects for recent deleterious mutations in the flat oyster (e.g., Harrang et al. 2013). In this review, I examine the evidence for a higher genetic load in marine animals from the 40+ years of literature on segregation distortion in experimental crosses, which includes a comprehensive survey of recent linkage mapping data from marine (aquaculture) animals. I also examine evidence from studies of natural populations, recapping the heterozygote fitness correlation (HFC) literature in bivalves as well as addressing the small but significant molecular evolution literature for marine animals. In addition to reviewing the literature, I provide a background on the genetic load concept, discuss how newer sequencing technologies are advancing the field, and suggest what the next steps should be to continue advancing research in this field.
Genetic Load: Definitions and A Brief History
The origin of the genetic load concept can be traced to Haldane (1937), in which he examined the loss of population fitness associated with mutation-selection balance. Haldane argued that populations routinely harbor individuals with suboptimal fitnesses relative to an expected population maximum and that the number of these less-fit individuals would remain more or less constant despite the action of selection because the allelic variants underlying fitness loss are being continuously replenished via mutation (i.e., mutational load) or because specific combinations of those alleles may be more or less advantageous (i.e., segregation load). Haldane illustrated this mathematically, showing that the expected mean population fitness (at equilibrium) for a single locus with a normal and deleterious allele (where the frequency of the deleterious allele, q, is expected to be quite rare, i.e., q ≪ 1) is: , which simplifies to at mutation-selection balance (see Wallace 1991; Agrawal and Whitlock 2012 for a review of calculations). The important and perhaps surprising result here is that the reduction in mean fitness at a locus depends only upon the rate of mutation and is thus independent of its effect size (i.e., Load or L ≈ µ). Under the assumption of no linkage disequilibrium or epistasis, this result can be extrapolated across the genome, and the total mean average fitness, , is estimated as the product of mean fitnesses at each locus,
where is the mean fitness with respect to locus , and is the mutation rate for that locus. A similar result was arrived at by Crow (1958) as well as by Muller (1950), in his classic paper on the role of mutation in disease and defect among human populations, which is one of the publications to use the term "genetic load". Crow (1958) would formalize the definition of genetic load as "the proportional decrease in average fitness (or other measurable quantity) of a population relative to the genotype possessing the maximum or optimum value," resulting in the equation or = , following the convention that maximum fitness = 1.00. Overall, this relatively simple but powerful theoretical result (Load ≈ mutation rate) would spur novel work by theorists and empiricists to quantify and compare loads across populations and species, to estimate and compare mutation rates across species (e.g., humans and Drosophila), and to consider the theoretical limits of load burden that could be absorbed by populations.
Inbreeding load—a practical measure of the mutational load across species
In the decades that followed Haldane’s original paper, the theory of genetic load would be widely tested and debated among empirical and theoretical geneticists (e.g., Wallace 1956, 1970, 1991; Mukai et al. 1972; Agrawal and Whitlock 2012) but it would ultimately recede from the front lines of population genetic research in the 1980s as workers, now armed with more advanced molecular techniques, could more directly estimate genetic variation at the protein and then DNA level. Perhaps the most challenging aspect of the load concept was that it was difficult, in practice, to measure load in natural populations. For example, how does one find and determine the fitness of a mutation-free, "optimal" reference genotype in a natural population, especially when it is very unlikely to exist in the first place? Though largely limited in its practical utility, load theory did provide a mathematical and theoretical framework to estimate the sources or components of fitness reduction compared to some base or "wild" (e.g., heterozygous) population. One of the most powerful experimental avenues for this work was the balanced-lethal system in Drosophila (e.g., Greenberg and Crow 1960, Simmons and Crow 1977), in which mutations or entire chromosomes from a wild population are made homozygous identical-by-descent through a series of controlled crosses with specialized stocks, and their viability is compared to heterozygotes or an index of relative heterozygote viability among crosses. The balancer technique (available only for Drosophila and a few other model systems; e.g., Steiner 1956; Simmons and Crow 1977; Herman 1978; Edgley et al. 1995) uses strains or stocks with special chromosomes that have recombination-suppressing inversions and morphological markers that allow researchers to assay the viability of the "captured" wild chromosome by the relative frequency of marked (heterozygous) versus non-marked (homozygous) flies in the offspring. Based on the relative viability of different chromosomes from nature made homozygous, the total impact of these mutants could then be partitioned into various components of load (e.g., lethal load vs. detrimental load) and the dominance of mutations (i.e., their effects on heterozygote viability) quantified. The balanced lethal system in Drosophila facilitated substantial progress in understanding the magnitude of mutation rates for various classes of mutants, their dominance, and the relative effects of mild versus lethal mutations on overall fitness (Simmons and Crow 1977).
For researchers working on species that lack a balanced lethal system, mutational (inbreeding) load cannot be examined for entire chromosomes made homozygous identical-by-descent, but crosses among relatives can be carried out to examine the relative reduction in offspring fitness upon inbreeding, that is, . Because many deleterious mutations are largely "hidden" from selection in the heterozygous state, mating of close relatives exposes the fitness effects of a portion of these recessive mutations in the homozygous state, and the comparison of inbred versus outbred fitness, particularly the viability of early life stages, provides a measure of the recessive mutational load in a given population or species. This approach was formalized into a regression framework by Morton et al. (1956) in calculating the number of "lethal equivalents," which were defined as: “…a group of mutant genes of such number that, if dispersed in different individuals, would cause on average one death, e.g., one lethal mutant or two mutants each with a 50% probability of death.” By performing linear regression of log survival across levels of inbreeding, the investigator can estimate the "concealed" load (the slope, B, or reduction in fitness with increasing inbreeding) and the "expressed" load (A, the intercept; Malogolowkin-Cohen et al. 1964; Anderson 1992; Lynch and Walsh 1998).
The experimental framework for measuring inbreeding load, primed by the original load theory, has guided much of the empirical work on genetic load over the last half-century and thus, much of the literature reviewed herein. In the following sections, a synthesis of trait- and molecular marker-based studies of inbreeding load provide a comprehensive view of differences among taxa, and then a survey of the molecular evolution literature provides a complementary view of genetic load from investigations of polymorphism in natural populations.
Trait-based analysis of inbreeding load—lethal equivalents
Lethal equivalents (LEs) have been measured for a wide variety of plants and animals since Morton et al. (1956) and thus provide an important and robust source of data to compare levels of genetic load across diverse taxa. Lynch and Walsh (1998; Tables 10.4 and 10.6) compiled LE estimates per gamete across a number of species and I have replotted these data, arranging them by broad taxonomic group (e.g., mammals, birds, angiosperms, Drosophila; Figure 1). Included in this figure are novel estimates of LE for a variety of marine fish, invertebrates, and salmon, calculated from published data on inbreeding depression (ID) for early survival or growth rate using a maximum likelihood method (Kalinowski and Hedrick 1998) or molecular marker-based estimates of the inbreeding load (see below; Supplementary Table S1). The literature search for additional ID studies was extensive, selective, and representative of a variety of marine animals, but was not exhaustive. Studies were only considered if they (1) provided raw or averaged data and (2) measured fitness of early life-history traits in lab-based experiments (see Supplementary Table S1, File S1 for more details). Estimates of LEs derived from molecular marker data (oysters) were made by counting the number of independent lethal loci per haploid parental genome within families, using partial (Launey and Hedgecock 2001) or whole-genome linkage data (quantitative trait locus [QTL] mapping of distortion; Plough and Hedgecock 2011, see next section). Specifically, LEs were tallied as the sum of the selection coefficients (often 0.9–1.0) calculated for each independent lethal or nearly lethal mutation detected. For example, two loci, each with a selection coefficient of 0.80 together would count as 1.6 LE. Note that molecular-based approaches may substantially underestimate the number of LEs because only the most lethal mutations are detected and the cumulative effect of many weakly deleterious alleles is likely to be missed. Furthermore, only ∼25% of the recessive lethal alleles carried by a grandparent become homozygous identical by descent in the F2 generation (the typical cross examined) and thus many lethal alleles potentially remain hidden in the heterozygous state.
Figure 1.
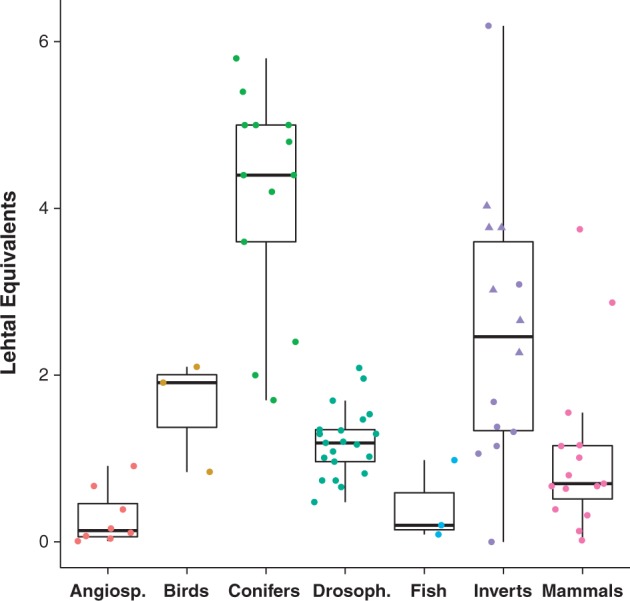
Lethal equivalent (LE) data for a variety of marine and non-marine taxa, plotted by broad taxonomic group. Estimates are per gamete and come primarily from Lynch and Walsh (1998) as well as other sources (see Supplementary Table S1, File S1). Individual data points are plotted over box-and-whisker plots that display the interquartile range and the median (thick black line) of LE estimates in each group. Triangles in the marine invertebrate group represent LE estimates that are calculated from molecular marker segregation data; LE are tallied as the sum of the selection coefficients at each independent recessive deleterious locus detected across the genome (Plough and Hedgecock 2011, Table 2; Launey and Hedgecock 2001, Table 4). LE estimates from Launey and Hedgecock (2001) derive from low-density microsatellite marker data with linkage information, adjusted for overall expected map length, which was estimated to be 1000 cm. Here, these LE estimates are recalculated (Supplementary Table S1) using the more accurate total map distance that has recently been estimated (∼600 cm; Hedgecock et al. 2015).
Figure 1 shows that marine invertebrates have the widest range of LEs, but appear to have the highest estimates among animal taxa. Within the invertebrate grouping, oysters have the highest estimates (top four data points; Supplementary Table S1), some of which are molecular-based (triangles); however, there is also an estimate of ∼0 (no ID for larval survival in the Eastern oyster; Mallet and Haley 1983). The two trait-based data points for oysters come from the Eastern oyster Crassostrea virginica and it is not clear why such different levels of inbreeding load would be observed within the same species (0 vs. 6.02; Longwell and Stiles 1973; Mallet and Haley 1983). The molecular-based estimates, on the other hand, appear to group more closely (range 2.67–4.02; Figure 1, Supplementary Table S1). Overall, LE estimates for oysters appear to be most similar in magnitude to conifers, which have the highest estimates across the taxa examined. Long-lived conifers share a number of life-history and population biology characteristics with bivalves, such as high early mortality, early expression of inbreeding load, and high fecundity, which have been argued to result in a higher load (Williams 1975; Carr and Dudash 2003; Plough and Hedgecock 2011). Though the differences in load across taxa are intriguing, it is important to acknowledge how varied the data are in terms of quality, quantity, and approach, particularly among the studies of marine animals (Supplementary Table S1). For example, some LE estimates are based on fitness data measured across many families and at 4 or more inbreeding levels, whereas other studies use just 2 levels of inbreeding (outbred and full sib mating, f = 0.25) from a single crossing experiment. Moreover, fitness is measured for a variety of related but very different traits across studies, including larval viability, egg hatching rate, and relative genotype viability in the molecular-based studies. Thus, it is quite difficult in practice to directly compare load or LE estimates among taxa. Nevertheless, these data provide a rough view of the differences among taxonomic groups, and suggest that oysters and bivalves may have a higher inbreeding load than other animals.
Molecular marker-based measures of genetic load in marine animals
Recent analyses of inbreeding load in marine animals have turned to molecular marker-based methods to uncover the number and effect of deleterious mutations carried by a set of parents. Among marine species, most available data come from studies of bivalves, perhaps due to the ease with which bivalves can be crossed and reared in the laboratory and the early adoption of molecular marker technologies (allozymes) among workers in the field. Similar to the LE approach (Morton et al. 1956), molecular-based estimates of inbreeding load require crosses to be made between related individuals (often full siblings from an F1 cross), to expose the effects of homozygosis for identical-by-descent deleterious recessive alleles on offspring viability. However, the magnitude of load is measured not as phenotypic decline over successive levels of inbreeding, but as the deviation from expected Mendelian ratios of inheritance (segregation distortion) in the counts of genotypes at one or more molecular markers segregating in the offspring (e.g., Mallet et al. 1985; Bierne et al. 1998; Launey and Hedgecock 2001; Carr and Dudash 2003; Plough and Hedgecock 2011). The deficiencies of genotypes (relative to Mendelian expectation) at a given point in offspring development are either the result of differential survival of gametes (gametic selection) or zygotes (zygotic selection), though temporal studies of the Pacific oyster indicate that most if not all of the early selection is zygotic (see below; Plough and Hedgecock 2011). The observation of differential mortality among genotypes (genotype-dependent mortality) implies viability selection either at the focal marker or, more likely, at a closely linked, deleterious locus, the magnitude of which depends on the recombination distance (c) between marker and mutation and the strength of selection (s) against that mutation (two-locus linked selection model; Hedrick and Muona 1990; Launey and Hedgecock 2001).
Marker segregation data in inbred crosses
Distorted segregation ratios were first reported for marine bivalves in the 1970s from inbred crosses that produced substantial deficiencies of homozygous genotypes in offspring, indicating selection against recessive deleterious alleles (e.g., Wada 1975; Wilkins 1976; Beaumont et al. 1983). Various explanations for the observed distortions were proposed, including gametic selection (e.g., meiotic drive) and zygotic selection (e.g., Mallet et al. 1985; Foltz 1986), but null alleles could generally be ruled out because they could easily be detected as unexpected genotypes segregating in the offspring (e.g., Plough and Hedgecock 2011). Over the next 4 decades, similar observations of significant and sometimes extreme levels of segregation distortion were made primarily in inbred, but also outbred, crosses for a variety of marine bivalves including the blue mussel, eastern oyster, Pacific oyster, and European flat oyster (e.g., Beaumont et al. 1983, 1988, 1990; Gaffney and Scott 1984; Mallet et al. 1985; Foltz 1986; Hu et al. 1993; Hu and Foltz 1996; McGoldrick and Hedgecock 1997; Bierne et al. 1998; Launey and Hedgecock 2001; Bucklin 2003, Lallias et al. 2007a, 2007b; Lallias et al. 2009; Sauvage et al. 2010; Plough and Hedgecock 2011; Harrang et al. 2015). The proportion of markers exhibiting segregation distortion varies across studies, with some experiments exhibiting distortion in greater than 75% of markers assayed (Bierne et al. 1998), but commonly, ∼15–50% of markers assayed within a family are distorted. Moreover, the frequency of distortion is exasperated in parents from related aquaculture stocks or with higher levels of inbreeding, which connects the observed load and distortion from molecular data to inbreeding level (e.g., Wilkins 1976; McGoldrick and Hedgecock 1997).
Interestingly, segregation distortion is also observed in crosses of unrelated parents, though the patterns of selection are quite different and the results of outbred crossing experiments have been published less frequently. Mallet et al. (1985) was one of the first studies to report significant heterozygote deficiencies in the offspring of pair-crossing experiments of wild-caught blue mussels Mytilus edulis. After considering a number of explanations for heterozygote deficiencies, the authors concluded that they most likely were explained by zygotic selection. A more recent study of segregation in the offspring of four outbred pair-crosses of the Pacific oyster also revealed substantial deficiencies of heterozygous genotypes in ∼40% of typed markers (Plough et al. 2016). That wild pair-crosses show characteristic deficiencies of heterozygous genotypes implies a load affecting early viability that is expressed in the absence of inbreeding, which would not be expected if most harmful mutations are primarily recessive (only expressed when made homozygous identical-by-descent). Partial dominance was demonstrated for a number of deleterious alleles identified in inbred Pacific oyster families (see below; e.g., Launey and Hedgecock 2001, Plough and Hedgecock 2011), which suggests that at least some portion of the load may reduce fitness in outbred crosses.
Researchers using molecular markers have also revealed details about inbreeding and genetic load in natural populations of marine bivalves through studies of heterozygosity fitness correlation (HFC; e.g., David 1998, Szulkin et al. 2010). Correlations between multi-locus heterozygosity (MLH; the proportion of heterozygous markers for a given individual) and fitness traits (e.g., growth, size) have been found in a variety of marine bivalves, which signals some level of inbreeding variance or drift (non-random mating) within a population (e.g., Singh and Zouros 1978; Koehn and Shumway 1982; Koehn and Gaffney 1984; David 1998). Though not as powerful an approach to quantify genetic load as genotyping markers in controlled crosses, HFCs in natural populations have the advantage of yielding estimates of both the potential genetic load and its actual impact in a population and can be performed in species for which crossing experiments may be impractical. The fairly consistent presence of HFCs across marine bivalves, though often of quite small magnitude, was initially surprising because marine bivalves are generally thought to have large randomly mating populations with little opportunity for inbreeding. Although confusion with HFC terminology and underlying theoretical mechanisms, as well as issues related to power and significance have limited the scope of this literature in recent years (e.g., Szulkin et al. 2010), the population-level HFC data in bivalves does support the idea that these species may have an elevated genetic load.
Advances in mapping of load in marine bivalves
Subsequent studies of marker segregation in marine bivalves have employed a greater number of markers and linkage or QTL approaches to more precisely estimate the number, location, and effect of deleterious mutations in the genome. Bierne et al. (1998) and Launey and Hedgecock (2001) made the first systematic attempts to estimate the total number of harmful recessive mutations in the genome, accounting for the proportion of the genome assayed by independent marker segregations and linkage information among markers. With data at 7 microsatellite markers, Bierne et al. (1998) estimated between 15 and 38 deleterious mutations, whereas Launey and Hedgecock (2001), using 19 markers and a linked-selection model (Hedrick and Muona 1990), estimated between 8 and 14 mutations in the wild (grandparent) founders. With 1st-generation microsatellite linkage maps, Plough and Hedgecock (2011) and Plough (2012) employed a QTL mapping approach to scan the genome for deleterious mutations (termed viability loci or vQTL in the QTL mapping context), using the viability model of Luo and Xu (2003). These studies revealed a similar number of mutations (11–15 vQTL) scattered throughout the genome, some of which had significant dominance effects. By genotyping oysters throughout the life cycle (e.g., larval, juvenile, adult), Plough and Hedgecock (2011) also demonstrated that this load was first "expressed" (i.e., caused significant viability selection) during the larval stages and at metamorphosis to the juvenile stage, but was not gametic. In other words, inbreeding load was expressed early in the life cycle. Further, Plough (2012) showed that diet influenced the fitness effect of individual mutations. Rearing offspring in a more stressful, low-quality diet produced a significant increase both in selection against and dominance of deleterious alleles at vQTL. Finally, these studies attempted to estimate the total "cost" of deleterious mutations in the partially inbred offspring by calculating the total mortality associated with viability selection at all identified deleterious loci. By first testing for and ruling out interactions (epistasis) among vQTL, Plough and Hedgecock (2011) and Plough (2012) were then able to calculate the cumulative, egg-to-adult "genetic mortality" from the product of average relative survival across all independent viability loci. In these experiments, mortality attributed to viability selection ranged from 85% to 99% across the families examined. Thus, the real-world effect of high inbreeding load in marine bivalves is to significantly reduce the early survival of offspring from inbred pair-crosses.
Load in Outbred Crosses of Marine Animals: Aquaculture Data
The previous paragraphs have summarized numerous reports of segregation distortion in the progeny of inbred crosses of marine bivalves, and some data on HFCs in natural populations, with much work focused specifically on mussels or oysters. These studies establish that marine bivalves may possess a very high load of partially recessive deleterious mutations; however, they provide less information on the broader question of whether genetic load is higher across marine animals in general, and whether or not this load has serious fitness or adaptive consequences in natural populations. Fortunately, a great wealth of marker segregation data for marine animals, including fish and crustaceans, can be found in the recent aquaculture literature in the form of genetic linkage maps for outbred (F1) families using hundreds to thousands of markers. These maps are usually the first step in generating genomic resources for a species and are used for QTL mapping of economically important production traits. The number of genome-wide marker datasets for linkage analyses has increased substantially over the last decade due to the availability of novel marker technologies and high-throughput genotyping approaches that make large-scale genotyping of non-model species possible (e.g., Luikart et al. 2003; Davey et al. 2011). Examining the expression of genetic load (i.e., viability selection) in the progeny of outbred crosses of marine animals begins to address the question of how a high load in marine animals might directly impact natural population fitness (early offspring mortality), at least in an experimental context. If some proportion of the load in these species is only partially recessive, as has been shown in Pacific oysters (Launey and Hedgecock 2001; Plough and Hedgecock 2011; Plough 2012; Plough et al. 2016) it would be detected as a significant viability selection in the outbred offspring of experimental pair-crosses.
To compile and summarize available genome-wide segregation data from marine animals, I performed a Web of Science (WOS) search for articles with titles containing words about segregation/linkage mapping and names of marine animal groups (e.g., clam, crustacean or sea bass; see Supplementary Table S2 for the complete list of studies and more details of the search criteria). The initial search resulted in the identification of 89 published research articles, ∼20 of which were removed because they examined segregation data from inbred (F2) families, or were off-topic (non-marine animal, or no report of linkage or segregation data). An additional 12 linkage studies of marine fish and shellfish not found in the WOS search were added; these studies were missed initially because they had specific common or scientific names in the title. Finally, a maximum of 3 studies per species were retained to reduce redundancy. Within bivalves, a few species had many published linkage studies (e.g., Pacific oyster, Zhikong scallop, and Bay scallop), so culling to 3 per species reduced the apparent number of studies in this group. Overall, 67 studies from 45 species were examined, comprising a variety of marine animal groups including crustaceans, fish, echinoderms, gastropods, and bivalve mollusks. The primary reporting metric was the proportion of distorted marker loci within a cross or family, significant at the nominal, α = 0.05, level. The tally of distorted markers was done at α = 0.05 because not all studies explicitly stated what significance threshold they used to report or remove loci and in the absence of any information, it was assumed that significance threshold was at α = 0.05 level. Some studies only reported the number of distorted markers after Bonferroni or FDR correction (noted in Supplementary Table S2); so, estimates may be conservative in some cases.
Two major trends emerged from the review of segregation distortion data in outbred crosses of marine animals. First, marine bivalves appear to have the highest proportion of distorted markers among the marine animal groups examined (Figure 2, Supplementary Table S2). The average proportion of distorted markers reported for bivalves was 24.74%, whereas averages for crustaceans and fish were lower (18.2% and 14.3%, respectively). The difference in the proportion of distorted markers between fish and bivalves was statistically significant (Wilcoxon rank sum test; P = 0.034). Interestingly, there was a wide range in the level of distortion reported across bivalve species and, in some cases, substantial variation within species (Figure 2, Supplementary Table S2). The highest proportion of distorted markers among bivalves was reported for the Bay scallop (60%; Wang et al. 2012), the Pacific oyster (27–49%; Li and Guo 2004; Hedgecock et al. 2015; Plough et al. 2016) the flat oyster (32%; Lallias et al. 2007a) and the Zhikong scallop (35%, 6%, and 30%—Wang et al. 2003, 2005; Zhan et al. 2009, respectively). The high variability in distortion among studies of Zhikong scallop is not easy to explain. In the two studies with the most contrasting results (35% and 6% distortion), AFLP (amplified fragment length polymorphisms) markers were used and similar numbers of progeny were examined (51 and 60 in Wang et al. 2003 and 2005, respectively), so differences in power or marker class do not explain this. No effect of marker type on distortion was detected across the studies examined. Generally, the proportion of distorted markers was somewhat similar across studies of the same species (e.g., 10–15% in Asian sea bass, Wang et al. 2007, 2015 and 7–21% in Tiger shrimp, Staelens et al. 2008; Baranski et al. 2014), which would be expected if the level of distortion was associated with the biology of the species and not with marker type or other sources of error. However, some variation among estimates within species could be attributed to different husbandry techniques or rearing conditions, the latter of which has been shown to affect the magnitude of segregation distortion in Pacific oysters (Plough 2012). Among crustaceans, studies of Pacific white shrimp showed the greatest proportion of distorted loci (39%; Zhang et al. 2007) and for fish, the half smooth-tongued sole demonstrated the largest proportion of distorted markers (up to 33%, Liao et al 2009), though one estimate was only 4% (Diopere et al. 2014). A few fish had very low or negligible numbers of markers with segregation distortion (white croaker, Dor et al. 2014; red drum, Hollenbeck et al. 2015; Japanese eel, Kai et al. 2014). Unfortunately, segregation results were reported for only a single echinoderm (of 2 studies retrieved; Yan et al. 2013), so it is difficult to infer any patterns for this group. Many data points were also missing for marine fish despite a large number of linkage studies examined (18 out of 38 studies; Figure 2, Supplementary Table S1). Clustering of distorted loci on specific linkage groups was reported for 4 fish species, (Asian sea bass, Wang et al. 2007; brill, Hermida et al. 2014; smooth sole, Liao et al. 2009; red drum, Portnoy et al. 2010) but was not addressed explicitly in most studies. Overall, segregation distortion in wild crosses appears to be lower in fish and crustaceans compared with bivalves (Figure 2).
Figure 2.
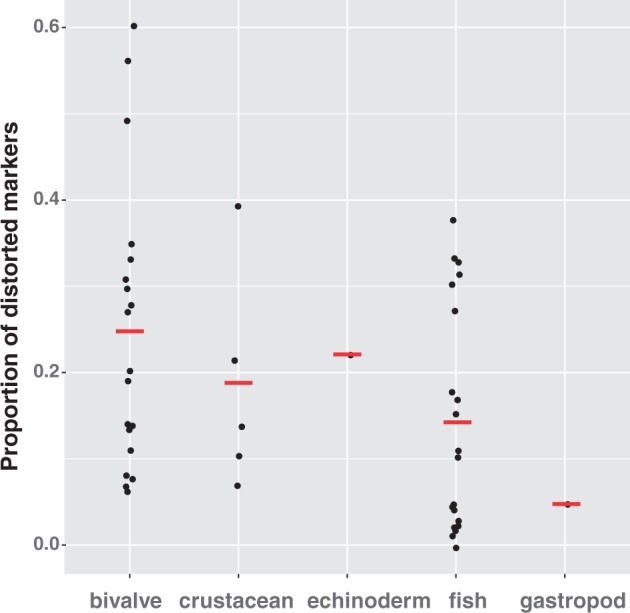
Proportion of markers exhibiting segregation distortion (α = 0.05) in linkage mapping studies of marine animals in outbred crosses. Red bars represent the mean of the individual observations for each group.
The second major finding from the review of linkage data is that segregation distortion routinely goes unreported and/or distorted markers are removed before analysis in studies of fish and crustaceans, but not bivalves. For example, approximately half of the studies reviewed for fish and crustaceans failed to report segregation distortion or removed distorted markers before analysis (Figure 3). This was particularly pronounced in studies with single nucleotide polymorphism (SNP) data generated via next-generation sequencing methods (i.e., RADseq), in which distorted markers were treated as genotype error or marker artifact, and culled at the same stage of data filtering that low-coverage markers or markers with excessive missing data were removed (e.g., Palaiokostas et al. 2015, Wang et al. 2015). The number of distorted markers that were removed was reported in some cases (e.g., Hubert et al. 2010) but often, a quantitative estimate of segregation distortion was not possible (Figure 3). This is in sharp contrast to marker segregation studies of bivalves, for which the research community is highly attuned to issues surrounding non-Mendelian inheritance and segregation distortion was always addressed or reported explicitly. In some cases, removal of markers with segregation distortion may be necessary as distortion can produce erroneous marker order and map lengths (e.g., Lorieux et al. 1995, Lorieux 2012; Hedgecock et al. 2015). Moreover, cases of segregation distortion could be caused by systematic genotype errors, and the culling of markers with questionable inheritance patterns may be appropriate (e.g., Pompanon et al. 2005). Still, it appears that a number of studies do not treat instances of segregation distortion as relevant biological phenomena, and a reporting bias may depress observations in the literature for marine fish.
Figure 3.

Proportion of studies that reported segregation distortion results, plotted by broad marine animal group.
Overall, based on the 67 studies examined, segregation distortion appears to be a common feature in crosses of marine animals, particularly bivalves, which implies a potentially large load of mutations affecting early viability. However, while bivalves appear to have the highest levels of segregation distortion among marine animals, high variation within this group and the lack of linkage or segregation studies in most other marine taxa makes it difficult to draw any definitive conclusions at this point.
Estimating Genetic Load from Molecular Data in Natural Populations
An alternative approach to estimate the influx of deleterious mutations (mutational load) at the population level is to quantify the rates of diversity in different classes of polymorphisms (e.g., synonymous vs. non-synonymous mutations) in natural population samples. Polymorphism of non-synonymous mutations is expected to be lower relative to synonymous or silent mutations, assuming amino acid changes generally have negative effects on fitness and are removed by purifying selection (depending on population size among other parameters; Kimura 1983). Thus, by examining the ratio of diversity (e.g., nucleotide diversity, : Nei 1987) at amino acid changing versus silent SNPs (dn/ds ratios), one can infer how selection is acting on mutations in a population (e.g., Fay et al. 2001; Eyre-Walker et al. 2006; Eyre-Walker and Keightley 2007). Further, by categorizing SNPs according to their minor allele frequency (e.g., low, moderate, or common), the ratio of non-synonymous to silent diversity within frequency classes can be used to infer the proportion of silent, weakly deleterious, and strongly deleterious mutations (Fay et al. 2001; Harrang et al. 2013). Averaged across many or all coding regions in the genome, the dn/ds ratio can tell the investigator something about the mutation pressure in a given species: organisms with higher load (whatever the cause) would be expected to have a greater number of weak to moderately deleterious mutations segregating within populations (excess rare variation), which would manifest as a higher dn/ds ratio (Fay et al. 2001). It is important to note that dn/ds ratios were originally developed in a phylogenetic context to estimate the ratio of fixed non-synonymous to synonymous changes between highly diverged lineages (species), the magnitude of which (e.g., above or below unity) is used to infer the type of selection acting on that particular locus (e.g., Kimura 1977; Goldman and Yang 1994). In a population genetics context, where polymorphisms may not be fixed among populations, the interpretation of the dn/ds ratio is not straightforward for inferring selection at a particular gene (e.g., Kryazhimskiy and Plotkin 2008; Mugal et al. 2014). However, in the context of inferring the fitness effects of new mutations and genetic loads, these ratios can be used in a comparative framework to examine the distributions of mutation fitness effects.
Studies of polymorphism with multi-marker dn/ds datasets (at least 10s of loci) are relatively scarce for marine animals, but the few datasets that are available suggest that a high load of weakly selected mutations may be present in these species. In an analysis of polymorphism across 35 gene regions in the flat oyster Ostrea edulis, Harrang et al. (2013) found a relatively modest silent diversity (π ds = 0.0067), but a high non-synonymous diversity (dn = 0.0025), which produces a remarkable dn/ds ratio of 0.38, the highest such estimate among animals currently reported in the literature. The dn/ds ratios appear to be somewhat similar across other oyster species, though perhaps are not as extreme as O. edulis. Using data from 41 coding genes spanning ∼10.5 kb of the Pacific oyster Crassostrea gigas genome, Sauvage et al. (2007) estimated an average dn/ds ratio of 0.16; silent diversity (π) was relatively high (ds = 0.035), but non-synonymous diversity was also rather substantial (dn = 0.0056). In another study of SNP variation in C. gigas, Kim et al. (2011) estimated a 10-fold higher silent versus non-synonymous (π silent = 0.0254, π non-synonymous = 0.0023), resulting in a dn/ds ratio of 0.09. It is important to note that these 2 studies of C. gigas used hatchery stocks (Sauvage et al. 2007) or a mixture of wild and hatchery oysters (Kim et al. 2011), thus these estimates may not reflect the true level of polymorphism in a "natural" oyster population. The relatively high dn/ds values observed in bivalves contrast with data from another highly polymorphic marine invertebrate, the sea squirt Ciona, which has similar levels of non-synonymous diversity, but very high silent diversity (e.g., Small et al. 2007; Tsagkogeorga et al. 2012) resulting in much lower dn/ds ratios (Ciona intestinalis sp. B = 0.05; Tsagkogeorga et al. 2012). Overall, bivalves (or at least the oyster species examined so far) appear to have a high segregating load of deleterious mutations, reflected by relatively higher non-synonymous diversity.
To put the high non-synonymous diversity of marine bivalves into biological context, it is instructive to compare these data to similar indices from a variety of other marine and non-marine taxa. In their analysis of non-synonymous diversity in the flat oyster O. edulis, Harrang et al. (2013) compiled and compared diversity data (dn/ds values) from the plant, terrestrial animal, and model animal literature. These data are replotted here in Figure 4 by broad taxonomic group, with some additional data points for marine animals included. Estimates of dn and ds (π) for the blue crab Callinectes sapidus are taken from Yednock and Neigel (2014) and data from the 2 aforementioned C. gigas studies are also plotted (Sauvage et al. 2007; Kim et al. 2011). Overall, it appears that marine animals (red squares) group more closely with terrestrial plants (green circles) than they do with other terrestrial animals (blue triangles) in terms of dn/ds diversity data (Figure 4). Plotting simple least square regression lines of dn versus ds for each group (dashed lines colored according to group), "slopes" appear to be steeper for the plants and marine animals compared with terrestrial animals, which reflects greater non-synonymous diversity relative to silent diversity. Bivalves appear to have particularly elevated average non-synonymous diversity. One of the polymorphism datasets for C. gigas shows the highest value of dn across all taxa (0.0056; Sauvage et al. 2007) and the dn/ds ratio for the flat oyster O. edulis is the largest among all other animal species examined (0.38). However, data from the blue crab show a much more modest level of non-synonymous diversity, and data for C. intestinalis show a very large silent diversity and much lower relative non-synonymous diversity. Based on these data, bivalves appear to stand out somewhat among marine animals, but marine animals do group fairly well together overall, with a slightly higher slope (dn/ds) compared to terrestrial animals. As noted above, however, data from the 2 studies of Pacific oyster SNP diversity may be biased by the inclusion of hatchery stock, and the data for blue crab are based on just 5 loci (Yednock and Neigel 2014). Thus, more studies of marine animals with large or genome-scale datasets will be needed before any definitive conclusions about diversity can be drawn from these data.
Figure 4.
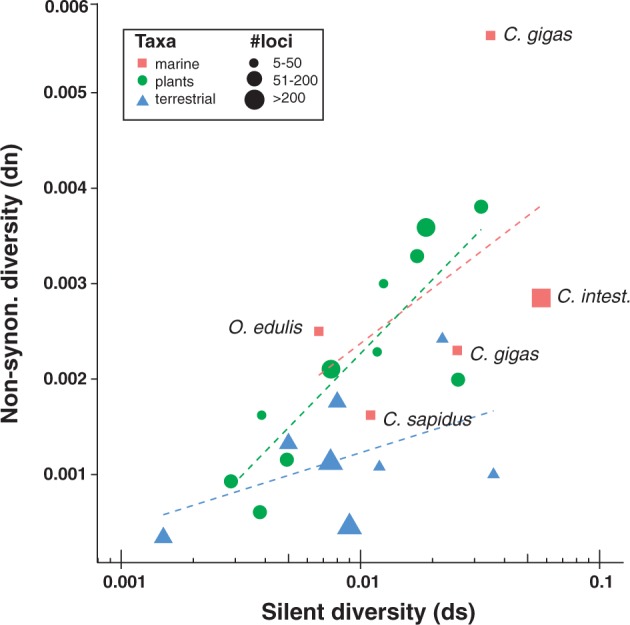
Plot of dn/ds ratios for terrestrial animals, plants, and marine animals (adapted from Harrang et al. 2013). Points are colored based on species group membership; size of the point (all shapes) represents the number of genes or gene regions examined, which are classified into 3 ranges (small, medium, and large datasets; 5–50, 51–200, > 200 loci, respectively). Least square regression lines are plotted and color-coded for each species group. C. intest. is the sea quirt Ciona intestinalis.
One possible explanation for the extreme dn/ds ratios observed in oysters, which imply a larger mutational load for this species, is a chronically low effective population size. Given that purifying selection is expected to be more efficient in larger populations (Nei 1987), populations with lower effective size may retain weakly deleterious variants at higher frequency for a longer duration, thus increasing the dn/ds ratio. Indeed, a number of population genetic studies of oysters have demonstrated sweepstakes reproductive success and a much lower effective population size than expected given census estimates (e.g., Hedgecock 1994; Hedgecock et al. 2007; Taris et al. 2007; Lallias et al. 2010). A lower effective size and substantial mutation pressure could drive a larger number of weakly deleterious mutations to segregate in populations, increasing the dn/ds ratio. For O. edulis, this idea is supported by examining diversity across frequency classes of SNPs, which reveals that approximately 33% of segregating mutations are weakly deleterious (cf. estimates for humans and Drosophila, ∼20–23%; Fay et al. 2001; Harrang et al. 2013).
An alternative explanation for the high dn/ds ratios in oysters is that the populations examined are actually very far from demographic equilibrium and that the excess of low frequency deleterious variants is due to demographic forces such as a bottleneck or recent population expansion. While these are valid hypotheses, Harrang et al. (2013) did not detect any systematic deviation from mutation–drift equilibrium (Wright–Fisher model) in diversity-based tests such as Tajima’s D, thus, a demographic explanation may not be sufficient. Moreover, oysters (and other marine animals) may not adhere to assumptions of the Wright–Fisher model due to their high fecundity life-history strategy, which can result in a high variance in reproductive success among parents (Eldon and Wakeley 2006, 2009; Sargsyan and Wakeley 2008; Hedgecock and Pudovkin 2011; Harrang et al. 2013). Alternatives to the Wright–Fisher or Kingman coalescent model framework that account for multi-furcating genealogies and multiple merger coalescent events may better reflect the reproductive biology of highly fecund marine animals (Eldon and Wakeley 2006; Steinrücken et al. 2013; Tellier and Lemaire 2014). Under the multiple merger coalescent, an excess of high-frequency and low-frequency variants (singletons) is actually expected at equilibrium and may not be a true signal of population bottleneck (e.g., Hoban et al. 2013) or recent expansion as would be indicated by a negative Tajima’s D under a standard coalescent model. Recent population genetic analyses of the putatively selected Ckma locus in Atlantic cod using a multiple merger coalescent framework demonstrated a much better-fit than the Kingman coalescent, modeling the excess of singletons and capturing the relatively fast coalescent timescales expected (Árnason and Halldórsdóttir 2015).
Overall, examining molecular diversity data in a limited number of marine animals suggests that bivalves (oysters) may have elevated levels of non-synonymous diversity compared with other marine and terrestrial taxa. However, diversity data are quite variable within groups and even within the same species (i.e., the Pacific oyster), which highlights the need for more datasets. Potential differences in effective population size among marine invertebrate species may in part explain differences in silent (neutral) diversity (e.g., Ciona spp. vs. O. edulis), and the lower effective size of the oyster species examined may contribute to the increased number of low frequency deleterious variants segregating in their populations. However, until more large-scale (e.g., tens to hundreds of genes) datasets of molecular diversity are available for marine invertebrates and fish species, the molecular evolution literature will be too limited to draw any major conclusions about differences in load among animal or marine taxa.
Summing It Up: A Higher Load in Marine Animals?
Reviewing 40+ years of literature on segregation distortion in experimental inbred crosses of marine bivalves, genome-wide linkage datasets from a range of marine aquaculture species, and empirical molecular evolution literature for marine animals, paints an interesting but largely incomplete picture of genetic load in marine animals. Overall, genetic load appears to be higher in marine versus terrestrial animals, but a general lack of data for many marine taxa (e.g., echinoderms), substantial variation within taxa, and a possible reporting bias of segregation distortion in studies of marine fish (Figure 3), complicates the emergence a clear trend from the available literature. What does emerge, however, and quite clearly, is that marine bivalves, and perhaps oysters in particular, stand apart from most marine species in their apparent level of mutational load. Marine bivalves consistently show higher inbreeding load, higher segregation distortion, and, from the few datasets available, elevated non-synonymous variation relative to neutral diversity (Sauvage et al. 2007; Kim et al. 2011; Harrang et al. 2013). Marine bivalves have received far greater and focused attention on topics like genetic load, segregation distortion, and molecular diversity compared with other marine animals, so we may yet see similar patterns emerging in other marine species groups as more studies are conducted. The finding of a high load in marine bivalves is consistent with predictions made by Williams (1975) in his "elm-oyster" model, in which he drew parallels between the population biology of oysters and long-lived, highly fecund plants. Williams hypothesized that the high and variable early mortality observed in these species resulted in part from a high segregating load. Indeed, across-species comparisons of LEs (Figure 1) and dn/ds ratios (Figure 4) illustrate that bivalves group more closely with plants and are rather distinct from terrestrial animals and even other marine animals.
Potential origins of a high load in marine bivalves
Based on our understanding of marine bivalve biology and life history, two major factors could explain the higher load observed in these organisms: (1) chronically low-effective population size driven by a sweepstakes life history and (2) substantially elevated mutation pressure. These potential causes are not mutually exclusive and, in nature, may interact to increase the load observed in bivalves. Considering effective size and life history first, the idea is that a chronically lower effective size (Ne) could allow deleterious mutations to be retained for longer time periods or at higher frequencies than would be expected if the Ne was larger (closer to census size, Nc), where selection would be more efficient at removing deleterious mutations (Nei 1987). Reduced effective size in marine bivalves could be driven by characteristic features of their life history, such as prolific fecundity and substantial mortality of the larval stages, which can result in high variance in reproductive success among parents and a reduction in the effective population size relative to census size (e.g., Hedrick 2005; Hedgecock and Pudovkin 2011). Indeed, a lower relative effective size and evidence of sweepstakes reproductive success have been documented in multiple oyster species (e.g., Hedgecock 1994; Hedgecock et al. 2007; Taris et al. 2007; Lallias et al. 2010; Hedgecock and Pudovkin 2011). Thus, perhaps high fecundity, which is associated with low and variable recruitment in marine fish as well (e.g., Rickman et al. 2000), could be a driver of low effective size that may contribute to higher genetic load in these species. Fecundity may also affect the mutation rate more directly. Plough et al. (2016) hypothesized that the very large number of meiosis required to produce millions of eggs in oysters could increase the likelihood of clustered germline mutations that would elevate the initial frequency and perhaps residence time of new deleterious mutations (similar to the male-driven evolution concept; e.g., Ellegren and Fridolfsson 1997; Makova and Li 2002). Support for this idea will require laboratory experiments to assess more directly the nature and rate of mutational change in high fecundity species.
If a chronically reduced effective size driven by a high fecundity life history is linked to higher load in marine bivalves, then we might expect a similar phenomenon in other highly fecund marine animals such as marine fish. This idea does not appear to be strongly supported based on the data in this review, however. Whereas many of the marine fish with linkage or segregation data are highly fecund (e.g., Atlantic cod, turbot, red drum), they generally demonstrate much lower levels of segregation distortion than bivalves (Figure 2, Supplementary Table S2). For example, Atlantic cod has fecundity in the millions (1–7 million eggs/female; Pinhorn 1984; Wroblewski et al. 1999) and thus high potential for sweepstakes reproductive success, but exhibits minimal segregation distortion (Hubert et al. 2010). Similarly, red drum Sciaenops ocellatus has very high fecundity (tens of millions of eggs/female; Overstreet 1983), shows some evidence of sweepstakes reproductive success (Turner et al. 1999), and has a reduced effective population size (Turner et al. 1999, 2002), but exhibited almost no segregation distortion in crosses (1–5%; Portnoy et al. 2010; Hollenbeck et al. 2015). To the extent that segregation distortion in wild pair-crosses is indicative of the magnitude of genetic load for a given species (see next section and Plough et al. 2016), the lack of distortion in a number of highly fecund fish suggests that fecundity may not be a primary driver of genetic load, or at least not the only factor. However, the marker segregation and genetic mapping literature for marine fish (from which this inference is primarily drawn) appear to be somewhat biased toward non-reporting or culling of distorted markers, so more data is needed before we can conclusively rule out fecundity as a major factor underlying elevated genetic load in marine animals.
The other potential explanation for high genetic load observed in marine bivalves is a much higher mutation rate relative to other terrestrial and marine animals. This explanation takes us back to the beginning of the review, where it is suggested that the relatively higher genetic diversity observed in marine animals might be driven not just by large population size but also by an increased rate of mutation. Though there is currently very little empirical data on mutation rates for marine bivalves, recent work in the Pacific oyster suggests that the mutation rate in this species may be elevated. After finding high levels of segregation distortion in a study of outbred crosses of the Pacific oyster, Plough et al. (2016) attempted to estimate the mutation rate that would account for the frequency of numerous, partly dominant deleterious mutations in the families examined (∼ 7.25 lethal alleles per parent). Using classical, mutation-selection equilibrium theory for partially dominant lethal alleles, they estimated a per gene mutation rate of ∼ 3.6 × 10−4 or a genomic mutation rate, U, of 1.81, which is approximately 90 times that of Drosophila, but quite similar to estimates of the mutation rate in conifers (10−4 to 10−5; Lande 1994). This estimate establishes, at the very least, the biological plausibility of a mutation rate that would account for the presence of the high load observed in wild oysters. Nevertheless, an empirical estimate of mutation rate will be needed for oysters and other marine bivalves to verify this.
As to the potential source(s) of elevated mutation pressure in oysters (and possibly other marine bivalves), one can only speculate at this point, but transposable element (TE) activity appears to be an intriguing possibility. Novel classes of active TEs unique to bivalves have been reported previously (e.g., Gaffney et al. 2003) and the draft genome of the Pacific oyster revealed abundant repetitive sequences and active transposable elements, which could shape genome variation and contribute to high rates of structural variation and polymorphism observed in oysters (Zhang et al. 2012). The potential effect of TEs on indel variation in the Pacific oyster genome may be profound. Re-sequencing of a wild oyster aligned to the reference genome identified thousands of deletions 100 bp or greater, more than of 80% of which overlapped with known transposable elements (Zhang et al. 2012). Thus, TE activity could contribute to an increased rate of large-effect deleterious mutations in critical genes or regulatory regions. Future work examining the relationship between TE activity and proximal viability mutations and segregation distortion could be a very fruitful area of future research on this topic.
Alternative explanations
Finally, it is important to consider some alternative interpretations of the widespread segregation distortion observed in crosses of marine bivalves, the analysis of which forms the basis of much of the inference of high load in this review. As summarized in previous sections, a high mutational load in bivalves can be inferred from observations of substantial deviations from Mendelian inheritance (segregation distortion) at markers across the genome, which implies fitness or viability differences among genotypes due to selection against linked deleterious mutations (e.g., Plough and Hedgecock 2011). Yet, while this explanation appears to be supported by a number of studies (e.g., Bierne et al. 1998; Launey and Hedgecock 2001; Lallias et al. 2007a, 2007b; Plough and Hedgecock 2011; Plough et al. 2016), other types of selection that are related to extrinsic or environmental factors may be important as well. One intriguing possibility is that the selection observed in the offspring actually reflects a dynamic of local adaptation or the lack of local adaptation for a given set of genotypes. Bivalves have a widely dispersing larval stage that promotes high gene flow among populations, but once settled as juveniles, individuals are fixed in a certain location for the remainder of their life and must contend with local or highly variable environmental conditions. Thus, perhaps the selection that we are observing in the early life stages is related to fitness differences or fitness mismatch between the parental environment and the offspring environment. A modeling study by Yeaman (2015) found that local adaptation could develop in populations with high gene flow, driven primarily by alleles of small effect that could withstand "swamping" (high gene flow) and cause divergence in different environments, given sufficient genetic variance and genetic redundancy in the underlying trait. Though the selection observed in crossing experiments of bivalves is quite strong and involves alleles of rather large effect, the idea that incompatibilities between genotypes and the environment could drive the observed patterns of selection is interesting. If distinct genotypes are more or less fit in different environments, one could hypothesize that the production of such a diverse array of genotypes in the offspring of high fecundity bivalves is a response to the variable and unpredictable nature of recruitment and the eventual "fixed" environment of the settled juvenile, that is, a "bet-hedging" strategy. Though this is difficult to demonstrate empirically, some evidence of genotype–environment interaction was observed among viability loci (i.e., among different genotypes) in Pacific oysters reared in different diet environments (Plough 2012). A potentially complex dynamic of selection such as this could be playing out during recruitment of bivalve species and could contribute to observed patterns of chaotic genetic patchiness that are often described in genetic studies of marine invertebrate populations (e.g., Eldon et al. 2016). Future studies of the dynamics of selection during the larval period with field or mesocosm-based sampling could begin to address this.
Future Directions
It is an exciting time to be studying the evolutionary genetics of marine animals as genomic data becomes less expensive and easier to generate (e.g., Andrews and Luikart 2014) and fundamental population genetic theory is being revised to accommodate the extreme life history and demographic features of high fecundity marine species (e.g., Sargsyan and Wakeley 2008; Eldon and Wakeley 2009; Tellier and Lemaire 2014). Driven by the availability of genomic datasets for non-model marine species and advances in our understanding of the underlying causes of genetic load in marine species, I suggest two major research foci going forward. First, there is a definite need for more studies of molecular diversity focusing on coding regions at the genome scale, to increase the available data on dn/ds ratios, site frequency spectra (SFS), and the distribution of mutation fitness effects (Eyre-Walker and Keightley 2007) in marine species. Though RAD sequencing is a powerful technique to survey diversity across the genome in virtually any species (e.g., Davey et al. 2011; Andrews et al. 2016), it largely profiles non-coding regions, and therefore provides little information on the frequency and polymorphism of amino acid changing mutations. Other approaches, including re-sequencing of whole genomes, (Andrews and Luikart 2014), non-model exon-capture techniques (e.g., Cosart et al. 2011; Bi et al. 2012), or the analysis of RNAseq data across multiple (>15) individuals, will provide much needed data to address this gap. There is also the potential to mine the vast, online repositories of RNAseq data for SNP variation, and tools exist to estimate synonymous and non-synonymous diversity from pooled samples (e.g., Nelson et al. 2015). With more genome-scale data on the molecular diversity of marine species, especially from taxa that are poorly represented currently (e.g., fish, echinoderms, annelids, crustaceans, gastropods), more robust taxonomic comparisons of genetic load can be made across marine animals groups and to other non-marine taxa.
The second major research focus should be to generate high resolution, genome-wide segregation data for a select number of taxonomically diverse "target" marine animal species, employing QTL mapping methodologies to characterize viability loci and sequence comparison between parents and offspring to estimate mutation rates more directly. This work could be focused on a select group of marine animal species for which husbandry protocols are well established and the generation of many offspring (hundreds to thousands) would be routine. However, this effort should not simply be limited to oysters, which may turn out to be a rather extreme case, but must also examine other bivalves (e.g., blue mussels, quahogs, dwarf surf clam), marine fish (e.g., cod, turbot, pollock) and crustaceans (e.g., Pacific white shrimp, blue crab, American lobster). High-resolution linkage maps (tens of thousands of markers) are now relatively routine to generate using RADseq data of parents and offspring; however, care should be taken with linkage map construction because it can be complicated significantly by many loci and segregation distortion (e.g., Lorieux et al. 1995; Lorieux 2012; Hedgecock et al. 2015). Statistical software to detect and characterize the number and effect viability loci through QTL mapping is available and open source (e.g., Luo and Xu 2003; Hu and Xu 2009; Zhan and Xu 2011, see Plough et al. 2016).
To examine de novo mutations and their frequency distribution in the offspring (e.g., clustered; Gao et al. 2011), sequences at RADseq loci could be compared between parents and offspring to detect any single base changes, deletions, or insertions, following methods and techniques developed for analysis of parent–offspring trios or quartets in human disease research (e.g., Roach et al. 2010; Sanders et al. 2012). Human parent–offspring sequencing studies often employ exon or whole-genome sequencing, approaches that are possible for some species with a sequenced genome like the Pacific oyster (though impractical for large numbers of offspring) but not for most non-"model" marine species. Instead, generation of genotype data with a RADseq approach would strike a balance between sequencing effort and cost, producing relatively unbiased genome-wide coverage at a cost that would allow the genotyping of hundreds of offspring, which increases the likelihood of detecting rare, novel variants. Paired-end RADseq would be particularly appropriate here, as the assembly of paired-end reads recovers larger overlapping stretches of the genome that may traverse coding regions. RADseq data sets may already be available from some of the linkage map data reviewed here (e.g., Kai et al. 2014; Wang et al. 2015; Palaiokostas et al. 2015) and these data could be reanalyzed for this purpose. Overall, novel work focused on either an experimental or molecular evolution approach in multiple, tractable, marine animals, will significantly advance our understanding of genetic load in these species and help continue the very exciting and theory-challenging work up to now.
Supplementary Material
Acknowledgments
Thanks to Dennis Hedgecock and two anonymous reviewers for constructive comments that improved the quality of this manuscript. Thanks also to Nicolas Bierne for the invitation to contribute to this issue and for sharing compiled dn/ds data in marine and non-marine species. Finally, thanks to the Deerbrook Cheritable Trust and University of Maryland for funding. This is contribution 5216 from the University of Maryland Center for Environmental Science.
Funding
Partial funding for this study was provided by the Deerbrook Cheritable Trust (Grant number DCT 15-30).
Supplementary Material
Supplementary material can be found at http://www.cz.oxfordjournals.org/
References
- Agrawal AF, Whitlock MC, 2012. Mutation load: The fitness of individuals in populations where deleterious alleles are abundant. Ann Rev Ecol Evol System 43:115–135. [Google Scholar]
- Anderson N, 1992. Lethal equivalents and genetic load. Plant Breed Rev 10:93–127. [Google Scholar]
- Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA, 2016. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet 17:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews KR, Luikart G, 2014. Recent novel approaches for population genomics data analysis. Mol Ecol 23:1661–1667. [DOI] [PubMed] [Google Scholar]
- Árnason E, Halldórsdóttir K, 2015. Nucleotide variation and balancing selection at the CKMAgene in Atlantic cod: analysis with multiple merger coalescent models. PeerJ 3:e786.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranski M, Gopikrishna G, Robinson NA, Katneni VK, Shekhar MS. et al. , 2014. The development of a high density linkage map for black tiger shrimp (Penaeus monodon) based on cSNPs. PLoS ONE 9:e85413.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazin E, Glémin S, Galtier N, 2006. Population size does not influence mitochondrial genetic diversity in animals. Science 312:570–572. [DOI] [PubMed] [Google Scholar]
- Beaumont AR, Beveridge CM, Barnett EA, Budd MD, 1990. Genetic studies of laboratory–reared Mytilus edulis. III. Scored loci act as markers for genotype-specific mortalities which are unrelated to temperature. Mar Biol 106:227–233. [Google Scholar]
- Beaumont AR, Beveridge CM, Barnett EA, Budd MD, Smyth-Chamosa M, 1988. Genetic studies of laboratory reared Mytilus edulis. I. Genotype specific selection in relation to salinity. Heredity 61:389–400. [Google Scholar]
- Beaumont AR, Beveridge CM, Budd MD, 1983. Selection and heterozygosity within single families of the mussel Mytilus edulis (L). Mar Biol Lett 4:151–161. [Google Scholar]
- Bi K, Vanderpool D, Singhal S, Linderoth T, Moritz C. et al. , 2012. Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genom 13:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne N, Launey S, Naciri-Graven Y, Bonhomme F, 1998. Early effect of inbreeding as revealed by microsatellite analyses on Ostrea edulis larvae. Genetics 148:1893–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucklin KA, 2003. Analysis of the Genetic Basis of Inbreeding Depression in the Pacific OysterCrassostrea gigas [PhD Dissertation]: University of California, Davis, CA.
- Carr DE, Dudash MR, 2003. Recent approaches into the genetic basis of inbreeding depression in plants. Philos Trans R Soc Lond B 358:1071–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosart T, Beja-Pereira A, Chen S, Ng SB, Shendure J. et al. , 2011. Exome-wide DNA capture and next generation sequencing in domestic and wild species. BMC Genom 12:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF, 1958. Some possibilities for measuring selection intensities in man. Hum Biol 30:1–13. [PubMed] [Google Scholar]
- Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM. et al. , 2011. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510. [DOI] [PubMed] [Google Scholar]
- David P, 1998. Heterozygosity-fitness correlations: new perspective on old problems. Heredity 80:531–537. [DOI] [PubMed] [Google Scholar]
- Diopere E, Maes GE, Komen H, Volckaert FAM, Groenen MAM, 2014. A genetic linkage map of sole Solea solea: a tool for evolutionary and comparative analyses of exploited (flat) fishes. PLoS ONE 9:e115040.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor L, Shirak A, Gorshkov S, Band MR, Korol A. et al. , 2014. Construction of a microsatellites-based linkage map for the white grouper Epinephelus aeneus. G3 4:1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgley ML, Baillie DL, Riddle DL, Rose AM, 1995. Genetic balancers. Method Cell Biol 48:147–184. [PubMed] [Google Scholar]
- Eldon B, Wakeley J, 2006. Coalescent processes when the distribution of offspring number among individuals is highly skewed. Genetics 172:2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldon B, Wakeley J, 2009. Coalescence times and Fst under a skewed offspring distribution among individuals in a population. Genetics 181:615–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldon B, Riquet F, Yearsley J, Jollivet D, Broquet T, 2016. Current hypotheses to explain genetic chaos under the sea. Current Zoology 62:551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Fridolfsson AK, 1997. Male-driven evolution of DNA sequences in birds. Nat Genet 17:182–184. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD, 2007. The distribution of fitness effects of new mutations. Nat Rev Genet 8:610–618. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Woolfit M, Phelps T, 2006. The distribution of fitness effects of new deleterious amino acid mutations in humans. Genetics 173:891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu CI, 2001. Positive and negative selection on the human genome. Genetics 158:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DW, 1986. Segregation and linkage studies of allozyme loci in pair crosses of the oyster Crassostrea virginica. Biochem Genet 24:941–956. [DOI] [PubMed] [Google Scholar]
- Gaffney PM, Pierce JC, Mackinley AG, Titchen DA, Glenn WK, 2003. Pearl, a novel family of putative transposable elements in bivalve mollusks. J Mol Evol 56:308–316. [DOI] [PubMed] [Google Scholar]
- Gagnaire PA, Broquet T, Aurelle D, Viard F, Souissi A. et al. , 2015. Using neutral, selected, and hitchhiker loci to assess connectivity ofmarine populations in the genomic era. Evol Appl 8:769–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney PM, Scott TM, 1984. Genetic heterozygosity and production traits in natural and hatchery populations of bivalves. Aquaculture 42:289–302. [Google Scholar]
- Gao JJ, Pan X-R, Hu J, Ma L, Wu J-M. et al. , 2011. Highly variable recessive lethal or nearly lethal mutation rates during germ–line development of male Drosophila melanogaster. Proc Natl Acad Sci USA 108:15914–15919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman N, Yang Z, 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11:725–736. [DOI] [PubMed] [Google Scholar]
- Greenberg R, Crow JF, 1960. A comparison of the effect of lethal and detrimental chromosomes from Drosophila populations. Genetics 45:1153–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS, 1937. The effect of variation on fitness. Am Nat 71:337–349. [Google Scholar]
- Hauser L, Carvalho GR, 2008. Paradigm shifts in fisheries genetics: ugly hypotheses slain by beautiful facts. Fish Fisheries 9:333–362. [Google Scholar]
- Harrang E, Heurtebise S, Faury N, Robert M,, Arzul I. et al. , 2015. Can survival of European flat oysters following experimental infection with Bonamia ostreae be predicted using QTLs? Aquaculture 448:521–530. [Google Scholar]
- Harrang E, Lapègue S, Morga B, Bierne N, 2013. A high load of non–neutral amino–acid polymorphisms explains high protein diversity despite moderate effective population size in a marine bivalve with sweepstakes reproduction. G3 (Bethesda) 3:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock D, 1994. Does variance in reproductive success limit effective population sizes of marine organisms? In: Beaumont AR, editor. Genetics and evolution of aquatic organisms. London: Chapman & Hall, 122–134. [Google Scholar]
- Hedgecock D, Barber PH, Edmands S, 2007. Genetic approaches to measuring connectivity. Oceanography 20:70–79. [Google Scholar]
- Hedgecock D, Gracey A, Shin G, Van Den Berg JD, Samanta MP, 2015. Second-generation linkage maps for the Pacific oyster Crassostrea gigas reveal errors in assembly of genome scaffolds. G3 (Bethesda) 5:2007–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock D, Launey S, Pudovkin AI, Naciri Y, Lapègue S. et al. , 2007. Small effective number of parents (N-b) inferred for a naturally spawned cohort of juvenile European flat oysters Ostrea edulis. Mar Biol 150:1173–1182. [Google Scholar]
- Hedgecock D, Pudovkin AI, 2011. Sweepstakes reproductive success in highly fecund marine fish and shellfish: a review and commentary. Bull Mar Sci 87:971–1002. [Google Scholar]
- Hedrick P, 2005. Large variance in reproductive success and the Ne/N ratio. Evolution 59:1596–1599. [PubMed] [Google Scholar]
- Hedrick PW, Muona O, 1990. Linkage of viability genes to marker loci in selfing organisms. Heredity 64:67–72. [Google Scholar]
- Hellberg ME, 2009. Gene flow and isolation among populations of marine animals. Ann Rev Ecol Evol System 40:291–310. [Google Scholar]
- Herman RK, 1978. Crossover suppressors and balanced recessive lethals in Caenorhabdilis elegans. Genetics 88:49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermida M, Rodriguez-Ramilo ST, Hachero-Cruzado I. et al. , 2014. First genetic linkage map for comparative mapping and QTL screening of brill Scophthalmus rhombus. Aquaculture 420:S111–S120. [Google Scholar]
- Hoban SM, Mezzavilla M, Gaggiotti OE, Benazzo A, van Oosterhout C. et al. , 2013. High variance in reproductive success generates a false signature of a genetic bottleneck in populations of constant size: a simulation study. BMC Bioinform 14:309.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck CM, Portnoy DS, Gold JR, 2015. A genetic linkage map of red drum Sciaenops ocellatus and comparison of chromosomal syntenies with four other fish species. Aquaculture 435:265–274. [Google Scholar]
- Hu YP, Foltz DW, 1996. Genetics of scnDNA polymorphisms in juvenile oysters Crassostrea virginica: characterizing the inheritance of polymorphisms in controlled crosses. Mol Mar Biol Biotechnol 5:123–129. [PubMed] [Google Scholar]
- Hu YP, Lutz RA, Vrijenhoek RC, 1993. Overdominance in early-life stages of an American oyster strain. J Hered 84:254–258. [Google Scholar]
- Hu Z, Xu S, 2009. PROC QTL—A SAS procedure for mapping quantitative trait loci. Int J Plant Genom 200:141234.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert S, Higgins B, Borza T, Bowman S, 2010. Development of a SNP resource and a genetic linkage map for Atlantic cod Gadus morhua. BMC Genom 11:191.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai W, Nomura K, Fujiwara A, Nakamura Y, Yasuike M. et al. , 2014. A ddRAD–based genetic map and its integration with the genome assembly of Japanese eel (Anguilla japonica) provides insights into genome evolution after the teleost–specific genome duplication. BMC Genom 15:233.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski ST, Hedrick PW, 1998. An improved method for estimating inbreeding depression in pedigrees. Zool Biol 17:481–497. [Google Scholar]
- Kim WJ, Jung H, Gaffney PM, 2011. Development of type I genetic markers from expressed sequence tags in highly polymorphic species. Mar Biotechnol 13:127–132. [DOI] [PubMed] [Google Scholar]
- Kimura M, 1977. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 267:275–276. [DOI] [PubMed] [Google Scholar]
- Kimura M, 1983. The Neutral Theory of Molecular Evolution. New York: Cambridge University Press. [Google Scholar]
- Koehn RK, Shumway SE, 1982. A genetic/physiological explanation for differential growth rate among individuals of the American Oyster Crassostrea virginica (Gmelin). Mar Biol Lett 3:35–42. [Google Scholar]
- Koehn RK, Gaffney PM, 1984. Genetic heterozygosity and growth rate in Mytilus edulis. Mar Biol 82:1–7. [Google Scholar]
- Kryazhimskiy S, Plotkin JB, 2008. The poplation genetics of dN/dS. PLoS Genet 4:e1000304.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallias D, Beaumont AR, Haley CS, Boudry P, Heurtebise S. et al. , 2007a. A first-generation genetic linkage map of the European flat oyster Ostrea edulis (L.) based on AFLP and microsatellite markers. Anim Genet 38:560–568. [DOI] [PubMed] [Google Scholar]
- Lallias D, Gomez-Raya L, Haley CS, Arzul I, Heurtebise S. et al. , 2009. Combining two-stage testing and interval mapping strategies to detect QTL for resistance to bonamiosis in the European flat oyster Ostrea edulis. Mar Biotechnol 11:570–584. [DOI] [PubMed] [Google Scholar]
- Lallias D, Lapegue S, Hecquet C, Boudry P, Beaumont AR, 2007b. AFLP-based genetic linkage maps of the blue mussel Mytilus edulis. Anim Genet 38:340–349. [DOI] [PubMed] [Google Scholar]
- Lallias D, Taris N, Boudry P, Bonhomme F, Lapegue S, 2010. Variance in the reproductive success of flat oyster Ostrea edulis L. assessed by parentage analyses in natural and experimental conditions. Genet Res 92:175–187. [DOI] [PubMed] [Google Scholar]
- Launey S, Hedgecock D, 2001. High genetic load in the Pacific oyster Crassostrea gigas. Genetics 159:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Guo XM, 2004. AFLP-based genetic linkage maps of the Pacific oyster Crassostrea gigas Thunberg. Mar Biotechnol 6:26–36. [DOI] [PubMed] [Google Scholar]
- Liao XL, Ma HY, Xu GB, Shao CW, Tian YS. et al. , 2009. Construction of a genetic linkage map and mapping of a female-specific DNA marker in halfsmooth tongue sole Cynoglossus semilaevis. Mar Biotechnol 11:699–709. [DOI] [PubMed] [Google Scholar]
- Longwell AC, Stiles SS, 1973. Gamete cross incompatibility and inbreeding in the commercial American oyster Crassostrea virginica Gmelin. Cytologia (Tokyo) 38:521. [DOI] [PubMed] [Google Scholar]
- Lorieux M, 2012. MapDisto: fast and efficient computation of genetic linkage maps. Mol Breed 30:1231–1235. [Google Scholar]
- Lorieux M, Perrier X, Goffinet B, Lanaud C, González-de-León D, 1995. Maximum-likelihood models for mapping genetic markers showing segregation distortion. 2. F2 populations. Theor Appl Genet 90:81–89. [DOI] [PubMed] [Google Scholar]
- Luikart G, England PR, Tallmon D, Jordan S, Taberlet P, 2003. The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet 4:981–994. [DOI] [PubMed] [Google Scholar]
- Lynch M, Walsh JB, 1998. Genetics and analysis of quantitative traits. Sunderland: Sinauer Associates, Inc. [Google Scholar]
- Luo L, Xu S, 2003. Mapping viability loci using molecular markers. Heredity 90:459–467. [DOI] [PubMed] [Google Scholar]
- Makova KD, Li W-H, 2002. Strong male-driven evolution of DNA sequences in humans and apes. Nature 416:624–626. [DOI] [PubMed] [Google Scholar]
- Mallet AL, Haley LE, 1983. Effects of inbreeding on larval and spat performance in the American oyster. Aquaculture 38:521–533. [Google Scholar]
- Mallet AL, Zouros E, Gartner-Kepkay KE, Freeman KR, Dickie LM, 1985. Larval viability and heterozygote deficiency in populations of marine bivalves: evidence from pair matings of mussels. Mar Biol 87:165–172. [Google Scholar]
- Malogolowkin-Cohen C, Levene H, Dobzhansky NP, Solima Simmons A, 1964. Inbreeding and the mutational and balanced loads in natural populations of Drosophila willistoni. Genetics 50:1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGoldrick DJ, Hedgecock D, 1997. Fixation, segregation and linkage of allozyme loci in inbred families of the Pacific oyster Crassostrea gigas (Thunberg): implications for the causes of inbreeding depression. Genetics 146:321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitton JB, 1993. Theory and data pertinent to the relationship between heterozygosity and fitness In: Thornhill NW, editor. The natural history of inbreeding and outbreeding: theoritical and empirical perspectives. Chicago: University of Chicago Press, 17–41. [Google Scholar]
- Morton NE, Crow JF, Muller HJ, 1956. An estimate of the mutational damage in man from consanguineous marriages. Proc Natl Acad Sci USA 42:855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugal CF, Wolf JBW, Kaj I, 2014. Why time matters: codon evolution and the temporal dynamics of dN/dS. Mol Biol Evol 31:212–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T, Chigusa SI, Mettler LE, Crow JF, 1972. Mutation rate and dominance of genes affecting viability in Drosophila. Genetics 72:333–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ, 1950. Our load of mutations. Am J Hum Genet 2:111–176. [PMC free article] [PubMed] [Google Scholar]
- Nei M, 1987. Molecular Evolutionary Genetics. New York: Columbia University Press. [Google Scholar]
- Nelson CW, Moncla LH, Hughes AL, 2015. SNPGenie: estimating evolutionary parameters to detect natural selection using pooled next–generation sequencing data. Bioinformatics 31:3709–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunney L, 1995. Measuring the ratio of effective population size to adult numbers using genetic and ecological data. Evolution 49:389–392. [DOI] [PubMed] [Google Scholar]
- Overstreet RM, 1983. Aspects of the biology of the red drum Sciaenops Ocellatus in the Mississippi. Gulf Research Reports (1 Suppl):45–68. [Google Scholar]
- Palaiokostas C, Bekaert M, Taggart JB, Gharbi K, McAndrew BJ. et al. , 2015. A new SNP-based vision of the genetics of sex determination in European sea bass Dicentrarchus labrax. Gen Sel Evol 47:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinhorn AT, 1984. Temporal and spatial variation in fecundity of atlantic cod Gadus morhua in Newfoundland Waters. J Northw Atl Fish Sci 5:161–170. [Google Scholar]
- Plough LV, 2012. Environmental stress increases selection against and dominance of deleterious mutations in inbred families of the Pacific oyster Crassostrea gigas. Mol Ecol 21:3974–3987. [DOI] [PubMed] [Google Scholar]
- Plough LV, Hedgecock D, 2011. Quantitative trait locus analysis of stage-specific inbreeding depression in the Pacific oyster Crassostrea gigas. Genetics 18:1473–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plough LV, Shin G, Hedgecock D, 2016. Genetic inviability is a major driver of type-III survivorship in experimental families of a highly fecund marine bivalve. Mol Ecol 25:895–910. [DOI] [PubMed] [Google Scholar]
- Pompanon F, Bonin A, Bellemain E, Taberlet P, 2005. Genotyping errors: causes consequences, and solutions. Nat Rev Genet 6:847–859. [DOI] [PubMed] [Google Scholar]
- Portnoy DS, Renshaw MA, Hollenbeck CM, Gold JR, 2010. A genetic linkage map of red drum Sciaenops ocellatus. Anim Genet 41:630–641. [DOI] [PubMed] [Google Scholar]
- Rickman S, Dulvy N, Jennings S, Reynolds J, 2000. Recruitment variation related to fecundity in marine fishes. Can J Fish Aquat Sci 57:116–124. [Google Scholar]
- Roach JC, Glusman G, Smit AF, Huff CD, Hubley R. et al. , 2010. Analysis of genetic inheritance in a family quartet by whole–genome sequencing. Science 328:636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ. et al. , 2012. De novo mutations revealed by whole–exome sequencing are strongly associated with autism. Nature 485:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargsyan O, Wakeley J, 2008. A coalescent process with simultaneous multiple mergers for approximating the gene genealogies of many marine organisms. Theor Pop Biol 74:104–114. [DOI] [PubMed] [Google Scholar]
- Sauvage C, Bierne N, Lapègue S, Boudry P, 2007. Single nucleotide polymorphisms and their relationship to codon usage bias in the Pacific oyster Crassostrea gigas. Gene 406:13–22. [DOI] [PubMed] [Google Scholar]
- Sauvage C, Boudry P, de Koning DJ, Haley CS, Heurtebise S. et al. , 2010. QTL for resistance to summer mortality and OsHV-1 load in the Pacific oyster Crassostrea gigas. Anim Genet 41:390–399. [DOI] [PubMed] [Google Scholar]
- Simmons MJ, Crow JF, 1977. Mutations affecting fitness in Drosophila populations. Ann Rev Genet 11:49–78. [DOI] [PubMed] [Google Scholar]
- Singh SM, Zouros E, 1978. Genetic variation associated with growth rate in the American oyster Crassostrea virginica. Evolution 32:342–353. [DOI] [PubMed] [Google Scholar]
- Small KS, Brudno M, Hill MM, Sidow A, 2007. Extreme genomic variation in a natural population. Proc Natl Acad Sci USA 104:5698–5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PJ, Fujio Y, 1982. Genetic variation in marine teleosts: high variability in habitat specialists and low variability in habitat generalists. Mar. Biol 69:7–20. [Google Scholar]
- Staelens J, Rombaut D, Vercauteren I, Argue B, Benzie J, Vuylsteke M, 2008. High-density linkage maps and sex-linked markers for the black tiger shrimp Penaeus monodon. Genetics 179:917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinrücken M, Birkner M, Blath J, 2013. Analysis of DNA sequence variation within marine speciesusing Beta–coalescents. Theor Popul Biol 87:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner E, 1956. New aspects of the balanced lethal mechanism in Oenothera. Genetics 41:486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulkin M, Bierne N, David P, 2010. Heterozygosity-fitness correlations: a time for reappraisal. Evolution 64:1202–1217. [DOI] [PubMed] [Google Scholar]
- Taris N, Batista FM, Boudry P, 2007. Evidence of response to unintentional selection for faster development and inbreeding depression in Crassostrea gigas larvae. Aquaculture 272:S69–S79. [Google Scholar]
- Tellier A, Lemaire C, 2014. Coalescence 2.0: a multiple branching of recent theoretical developments and their applications. Mol Ecol 23:2637–2652. [DOI] [PubMed] [Google Scholar]
- Tsagkogeorga G, Cahais V, Galtier N, 2012. The population genomics of a fast evolver: high levels of diversity, functional constraint and molecular adaptation in the tunicate Ciona intestinalis. Genome Biol Evol 4:740–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TF, Richardson LR, Gold JR, 1999. Temporal genetic variation of mitochondrial DNA and the female effective population size of red drum Sciaenops ocellatus in the northern Gulf of Mexico. Mol Ecol 8:1223–1229. [DOI] [PubMed] [Google Scholar]
- Turner TF, Wares JP, Gold JR, 2002. Genetic effective size is three orders of magnitude smaller than adult census size in an abundant, estuarine-dependent marine fish Sciaenops ocellatus. Genetics 162:1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada KT, 1975. Electrophoretic variants of leucine aminopeptidase of the Japanese pearl oyster Pinctada fucata (Gould). Bull Natl Pearl Res Lab Jpn 19:2152–2156. [Google Scholar]
- Wallace B, 1956. Studies on irradiated populations of Drosophila melanogaster. J Genet 54:280–293. [Google Scholar]
- Wallace B, 1970. Genetic Load: Its Biological and Conceptual Aspects. Englewood Cliffs (NJ: ): Prentice-Hall. [Google Scholar]
- Wallace B, 1991. Fifty Years of Genetic Load: An Odyssey. Ithaca: Cornell University Press. [Google Scholar]
- Wang S, Bao Z, Pan P, Zhang Q, 2003. AFLP linkage map of an intraspecific cross in Chlamys farreri. J Shellfish Res 23:491–499. [Google Scholar]
- Wang LL, Song LS, Chang YQ, Xu W, Ni D. et al. , 2005. A preliminary genetic map of Zhikong scallop Chlamys farreri Jones et Preston 1904. Aqua Res 36:643–653. [Google Scholar]
- Wang CM, Zhu ZY, Lo LC, Feng F, Lin G. et al. , 2007. A microsatellite linkage map of Barramundi Lates calcarifer. Genetics 175:907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li L, Zhang S-D, Zheng H-P, Zhang G, 2012. Microsatellite segregation distortion analysis of the out bred, inbred and selfed families of the Bay scallop Argopecten irradians. Mar Sci 36:109–115. [Google Scholar]
- Wang L, Wan ZY, Bai B, Huang SQ, Chua E. et al. , 2015. Construction of a high–density linkage map and fine mapping of QTL for growth in Asian seabass. Sci Rep 5:16358.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RD, Woodwark M, Skibinski DOF, 1994. A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J Fish Biol 44:213–223. [Google Scholar]
- Williams GC, 1975. Sex and Evolution. Princeton: Princeton University Press. [Google Scholar]
- Wilkins NP, 1976. Enzyme polymorphisms in the European oyster Ostrea edulis L. In: Persoone G, Jaspers E, editors. Proceedings of the 10th European symposium on Marine Biology Vol. 1 – Mariculture. Wetteren, Belgium: Universa Press, 549–563.
- Wroblewski JS, Hiscock HW, Bradbury IR, 1999. Fecundity of Atlantic cod Gadus morhua farmed for stock enhancement in Newfoundland bays. Aquaculture 171:163–180. [Google Scholar]
- Yan J, Jing J, Mu X, Du H, Tian M. et al. , 2013. A genetic linkage map of the sea cucumber Apostichopus japonicus based on microsatellites and SNPs. Aquaculture 404:1–7. [Google Scholar]
- Yeaman S, 2015. Local adaptation by alleles of small effect. Am Nat 186:1–16. [DOI] [PubMed] [Google Scholar]
- Yednock BK, Neigel JE, 2014. Detecting selection in the blue crab Callinectes sapidus using DNA sequence data from multiple nuclear protein-coding genes. PLoS ONE 9:e99081.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan A, Hu J, Hu X, Hui M, Wang M, Peng W, Huang X, Wang S, Lu W, Sun C, Boa Z. et al. , 2009. Construction of microsatellite-based linkage maps and identification of size-related quantitative trait loci for Zhikong scallop Chlamys farreri. Anim Genet 40:21–831. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yang C, Zhang Y, Li L, Zhang X. et al. , 2007. A genetic linkage map of Pacific white shrimp Litopenaeus vannamei: sex-linked microsatellite markers and high recombination rates. Genetica 131:37–49. [DOI] [PubMed] [Google Scholar]
- Zhang G, Fang X, Guo X, Li L, Luo R. et al. , 2012. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490:49–52. [DOI] [PubMed] [Google Scholar]
- Zhan H, Xu S, 2011. Generalized linear mixed model for segregation distortion analysis. BMC Genom 12:97.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.