


Abstract
Biological functions frequently require protein–protein interactions that involve secondary and tertiary structural perturbation. Here we study protein–protein dissociation and reassociation dynamics in insulin, a model system for protein oligomerization. Insulin dimer dissociation into monomers was induced by a nanosecond temperature-jump (T-jump) of ~8 °C in aqueous solution, and the resulting protein and solvent dynamics were tracked by time-resolved X-ray solution scattering (TRXSS) on time scales of 10 ns to 100 ms. The protein scattering signals revealed the formation of five distinguishable transient species during the association process that deviate from simple two-state kinetics. Our results show that the combination of T-jump pump coupled to TRXSS probe allows for direct tracking of structural dynamics in nonphotoactive proteins.
Graphical abstract
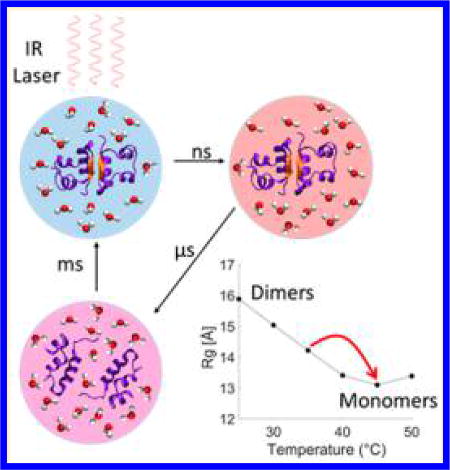
The function and behavior of a protein under physiological conditions are heavily tied to its structure, which can be strongly dependent on the local environment.1 For example, control over protein association and dissociation can be achieved by altering factors such as local pH, temperature and concentration.2–4 These external factors can cause proteins to transiently assume different conformations or alter the protein binding affinities to adapt to the new conditions.5–7 Understanding the protein responses to the external factors and mapping out the protein structural transformation pathways induced by these factors are important for unraveling mechanisms of complex biological processes.8
Insulin is a 51-residue protein that has been extensively studied as a model system for protein–protein interactions and protein oligomerization because of its ability to form a wide range of oligomers, such as tetramers and hexamers.9,10 The smallest insulin oligomer is a dimer formed by association of the C-terminal end of the B chains into an intermolecular β-sheet. The stability of insulin–insulin binding interactions can be tuned by varying the environmental conditions, such as concentration, temperature, and pH.11 In aqueous mixtures with ethanol at low pH values, the dimer–monomer equilibrium is strongly temperature-dependent and shifts from predominantly dimers at 25 °C to predominantly monomers at 50 °C.9,12 Thus a sudden perturbation to the thermal equilibrium by a laser-induced temperature jump (T-jump) can drive a transient increase in monomer population, allowing for the study of insulin association dynamics via its reverse process of dissociation, which is a first-order process. Such an approach avoids having long-distance protein diffusion in solution as the rate-limiting processes and therefore simplifies the extraction of the oligomerization structural dynamics. Recently, Zhang et al. investigated the dynamics of insulin association by two-dimensional IR (2D-IR) spectroscopy utilizing laser-induced T-jump experiments.9,12 The 2D-IR results suggest the formation of an intermediate dimer conformation on a 5–150 µs time scale, followed by melting of the intermolecular β-sheet and dissociation within 250–1000 µs.12 However, questions remain about the nature of the transient dimer structure, whether any other structural intermediate states exist, and whether T-jumps can provide information about a possible long-lived encounter complex as the system cools back to equilibrium.
In this work, we utilize T-jump-induced time-resolved X-ray solution scattering (TRXSS) to directly probe both protein and solvent structural changes. TRXSS enables direct tracking of protein dynamics as well as solvent hydrodynamics and may provide additional and complementary secondary and tertiary structural information to that derived by IR spectroscopy.13 Furthermore, in contrast with IR-based techniques, TRXSS does not require the use of deuterated solvent and protein, and thus it allows for interrogation of protein dynamics in protonated water environment. Previous TRXSS studies have focused on direct laser excitation of photoactive proteins, such as hemoglobin and photoactive yellow protein.8,14–23 Here we extended the TRXSS technique to study nonphotoactive proteins by employing indirect solvent excitation. We report the existence of several new intermediate states and show that the system dynamics can be disentangled through a direct comparison of the experimental data with signals calculated using protein structures obtained from crystallographic data. Furthermore, we observe the existence of a restructured monomer state that appears prior to the reassociation of dimers as the system cools back down to equilibrium.
Temperature-dependent static X-ray scattering measurements and UV circular dichroism (UV-CD) were carried out between 15 and 50 °C. Both techniques confirm that a temperature increase is able to dissociate insulin dimers to monomers, as previously reported (see the Supporting Information (SI) for details).9,12 The scattering results show that under the experimental conditions the radius of gyration (Rg) of insulin at room temperature is 15.8 Å, a value that is in the range attributed to dimeric insulin.11 As the temperature is increased from room temperature to 45 °C the Rg decreases to 13.1 Å, in the range reported for monomers.11 Kratky curve analysis was carried out to verify the folding state of the dimers and monomers.24 The analysis indicates that both dimers and monomers predominantly assume folded conformations in the entire range of investigated temperatures and that only two primary species (dimer and monomer) are adopted by the protein in the temperature range of 25–50 °C. Furthermore, it was found that insulin monomers in EtOH–water mixture adopt a more flexible conformation than the compact one that is adopted in acetic acid at room temperature (see the SI for details). The UV-CD measurements provide further evidence of the temperature dependence of the insulin dimer stability through the disappearance of signals associated with the intermolecular β-sheet (see the SI).11,25
T-jump induced TRXSS experiments were performed to study the insulin dimer dissociation and association dynamics on time scales of 10 ns to 100 ms following T-jumps from three initial temperature conditions, 25, 30, and 35 °C. The T-jump was induced by exciting the sample with 7 ns laser pulses at 1.443 µm wavelength, which corresponds to an overtone of O–H stretch. TRXSS signals were obtained by subtracting the scattering signals at positive time delays from the scattering signal 5 µs before laser excitation. The protein-associated signal was separated from the buffer-associated signal (i.e., the hydrodynamic response) by fitting the higher angle region of the data with pure buffer heating signal (see Figure 1A, details are available in the SI). At early time scales following the T-jump, changes in the buffer density are known to cause artifacts when performing optical T-jump experiments;26 however, TRXSS allows for direct tracking of the temperature and density changes in the buffer (see Figure 1B). The buffer-associated signal was compared with signals obtained from static temperature measurements on pure buffer, and it was found that the T-jump magnitude was ~8 °C (see the SI). From the hydrodynamics analysis, it is apparent that both temperature and density stabilize in ~300 ns after the laser excitation and retain their values to within ~10% variation up to 1 ms time delay. TRXSS patterns recorded at 1 s showed no detectable difference scattering signal compared with the starting sample solution before the laser pulse, indicating that the cool-down process is complete on these time scales (see the SI).
Figure 1.

(A) Protein sample TRXSS signal at 5 µs time delay fit with the signal from T-jump in pure buffer. (B) Temperature (red) and density (black) hydrodynamic response of the buffer derived from fitting of pure buffer TRXSS signal to protein sample TRXSS data as a function of time from an initial temperature of 35 °C.
Characteristic insulin-associated TRXSS signals for T-jumps from initial temperature of 35 °C are presented in Figure 2A (for full time series, see the SI). The evolution of the TRXSS signal depicts the changes in the protein conformation during dissociation and reassociation. The analysis of the TRXSS signal was divided into the small-angle region (SAXS) at q < 0.2 Å−1, providing information on tertiary structure, and the wide-angle region (WAXS) at q > 0.2 Å−1, providing information on secondary structure. For the SAXS region, we observed different kinetic behavior between the q-range of q < 0.04 Å−1 and 0.04 Å−1 < q < 0.2 Å−1 (Figure 2B, top), particularly in the rise time of the signals at ~1 µs. The plateau regions observed in the integrated signal imply the formation of stable transient structures, suggesting the existence of an intermediate that arises with the T-jump and, by comparing the rise times of both SAXS regions, the formation of at least two more intermediates by 1 µs. Furthermore, the increase in the integrated signal intensity on the time scale of hundreds of microseconds reveals the existence of an additional intermediate, which is also accompanied by the emergence of features in the WAXS region. The WAXS signals indicate that this intermediate presents substantial changes in the protein secondary structure. Finally, the only other process observed from SAXS signal integration is that the magnitude of the integral decreases as the temperature of the system decreases back to thermal equilibrium.
Figure 2.

(A) Characteristic TRXSS difference curves at various time delays after T-jump, presented as time series. (B) (Top) Normalized integrated signal in the region of q < 0.04 Å−1 (magenta) and 0.04 Å−1 < q < 0.2 Å−1 (black). (Bottom) Population dynamics derived from kinetic analysis for associated species along with the scaled temperature derived from fitting of pure buffer TRXSS signal to the protein TRXSS signal. (C) Species-associated TRXSS difference patterns from kinetic analysis; the signal at q > 0.2 Å−1 is multiplied by a factor of 10 to magnify the observed WAXS features. The legend is shared for both B and C panels.
Global analysis was performed on the protein associated TRXSS data based on established data analysis procedures.17,18,27–31 The singular value decomposition (SVD)-derived components were analyzed using a kinetic model to extract physically meaningful TRXSS patterns and dynamics that are associated with particular species (see the SI for details). Our results show the presence of five transient states. Their species-associated kinetic evolutions and TRXSS signals are shown in Figure 2B,C, respectively.
The early time delay data show the formation of an intermediate state (D*) on the time scale of the instrument response (~7 ns). Because this early state presents a loss of SAXS intensity and emerges concurrently with the T-jump process (Figure 2C), it is ascribed to a “hot” dimer state in which solvation shell rearrangement and protein thermal expansion takes place due to thermal weakening of hydrogen bonding. Following the initial solvent thermalized state, two consecutive states, D1 and D2, emerge on the time scales of 310 and 900 ns, respectively. The extracted scattering signals for these states represent a loss of scattering intensity in the SAXS region without any features in the WAXS region (see Figure 2C). The D1-associated TRXSS signal contains a positive feature below 0.05 Å−1, which is estimated to comprise a ~10% change in the forward scattering relative to the ground state. This feature is attributed to an increase in the protein mass due to solvent uptake by the dimer (i.e., increase in solvent accessible surface), whereas overall the decrease in SAXS scattering is attributed to slight volume expansion of the dimer.17,31 In addition, the scattering intensity loss in the region of q < 0.2 Å−1 in D2 implies that the dimer adopts a significantly more expanded state; however the lack of WAXS features in both states implies conservation of the protein secondary structure, specifically the intramolecular β-sheet, responsible for retaining the dimer form of insulin. Overall, the scattering signal from the D2 state indicates that the internal rearrangement of the monomer units leads the protein to adopt a stable tertiary conformation in which the overall size of the protein is expanded.
Following the early time dynamics, we observed the emergence of another state (2M) 240 µs after the T-jump, characterized by a further loss of SAXS intensity and the appearance of strong WAXS features (Figure 2B) indicative of changes in the protein secondary structure. By comparing the 2M-associated TRXSS signal to the signal calculated from steady-state scattering curves taken at 45 and 35 °C, which represents the expected signal for dissociation of dimers to monomers, this intermediate is assigned to the dissociated state (Figure 3). The good agreement between the steady-state and time-resolved curves in the WAXS region allows definitive assignment of the observed WAXS features to the loss of the intermolecular β-sheet. We note that the curves have slight disagreement in the SAXS region, which is likely due to differences in the change in solvation in the experiment done with 8 °C T-jump and the steady-state measurement at 10 °C higher than the starting temperature. Finally, we have also compared the 2M associated signal corresponding to dissociated monomers with the theoretical signal calculated using insulin dimer and monomer structures from the PDB database (entries 2A3G for dimer32 and 2JV1 for monomer33) (see the SI for details). The theoretical and TRXSS curves appear to be similar: The negative dip at q < 0.2 Å−1, as well as oscillatory features in the WAXS region, are qualitatively reproduced by theory, providing additional evidence of the assignment to a dissociated state. Discrepancies between theoretical curves and the TRXSS data are likely due to the differences in protein environment in crystal and solution forms, such as pH, temperature, buffer composition, and crystal packing.
Figure 3.

Comparison of 2M state TRXSS signal with signal calculated from steady-state scattering between 45 and 35 °C and theoretical calculation from crystal structure. The signals are magnified ×70 in the q > 0.2 Å region for clarity.
The final intermediate (2M′) emerges with a lifetime of 8 ms and decays on longer time scales from 10 to 100 ms as the system temperature cools back to the pre-T-jump equilibrium. The decay of 2M′ was successfully reproduced by a stretched exponential function exp[−(t/τ)β]; however, because the formation and decay of this intermediate appears during the system cool down, the observed lifetimes do not represent physically meaningful values. The 2M′-associated signal has a large SAXS signal that is similar to the dissociated 2M state, indicating that the system is still largely in the monomer (dissociated) state. However, the decay of the WAXS features indicates that the monomers go through an internal rearrangement, which brings their secondary structure to a conformation similar to that in the dimer state (see the SI). Comparison of calculated difference radial distribution functions for 2M and 2M′ states shows that the intensity of the 2M′ signal at distances below 25 Å is smaller in comparison with 2M, while the two signals have the same shape and intensity at larger distances (see the SI for details). The latter confirms that structural rearrangement indeed appears within the insulin monomer state prior to the formation of the dimers, although the signal may represent multiple conformations of the monomer rather than one specific structural species. Previous 2D-IR experiments have suggested that the reassociation process of insulin monomers is much slower than the diffusion limit, suggesting that the rate-determining step represents a so-called induced fit step, where the coupled protein folding and binding processes have to search for a correct conformation that will result in the dimer formation.12 This suggestion is confirmed by our measurements, as the population of the relatively long-lived 2M′ state with a reorganized structure within the monomer conformation appears to be a necessary step before the final relaxation to the ground (dimer) state during the cool-down process.
Our findings in regards to dissociation can be summarized with a schematic representation of the free-energy surface of the dimer in its hot state shown in Figure 4. Immediately after the T-jump the dimer population, in its hot D* state, is in a nonequilibrium state. As the system evolves with time, two kinetic intermediates are populated in rapid succession, leading to the formation of a long-lived expanded dimer intermediate (D2). Finally, the dimer dissociates (2M) with changes in the secondary structure, including the loss of the intermolecular β-sheet. The following slow process of system cooling and reassociation to form the dimer requires the monomers to diffuse and associate while also shifting the thermal equilibrium. As the previous 2D-IR study indicated, the process of dimer association is not diffusion-limited but rather shows evidence that another intermediate state represents the rate-limiting step in the association process. The 2M′ state revealed by TRXSS experiments indicates that dimer association is preceded by internal reorganization of monomers changing their conformation to one close to the monomer structure within the dimer. The subsequent association dynamics are likely to involve the states observed during the dissociation process in the reverse order (D2 followed by D1).
Figure 4.

Free-energy surfaces for insulin dissociation reaction as a result of T-jump. The red lines represent the nonequilibrium population immediately after T-jump. The yellow line shows the ensemble pathway toward dissociation as the system is driven back toward equilibrium.
T-jump TRXSS measurements were also carried out starting from lower initial temperatures of 25 and 30 °C, where the initial state includes a higher dimer to monomer ratio. The results showed conservation of all intermediate states, including dissociation to 2M and the formation of restructured monomers 2M′ (see the SI). The kinetic analysis of these data sets shows that the process of dissociation of the D2 intermediate into insulin monomers approximately doubles its lifetime when the ground-state temperature is set to 25 °C (see the SI). In contrast, the formation processes of both D1 and D2 intermediates have similar lifetimes at all temperatures, implying that these processes are already at their speed limit. The appearance of all of the same intermediate states at different temperatures further supports the conclusion that insulin association deviates from two-state kinetics regardless of the initial temperature conditions.
Comparison of TRXSS results to the previous 2D-IR study reveals a substantially different picture of the system structural kinetics. Specifically, at early times 2D-IR results demonstrated a single disordered dimer species appearing on time scales of 5–150 µs, in contrast with TRXSS, which suggests intermediates form much faster than 5 µs.12 Although both TRXSS and 2D-IR methods probe structural dynamics, 2D-IR probes more localized dynamics by following the amide-I vibrations of the protein. In contrast, TRXSS provides information about the global structure of the protein without details of local atomic structure. Because structural dynamics of the protein can vary on different length scales, the experimentally observed time required for formation of intermediates can also be different. Additionally, the 2D-IR experiments were performed on deuterated samples where both the mechanisms and the time scales of reaction processes can be significantly altered.9 Finally, the existence of a 2M′ state was postulated by the 2D-IR study; however, no specific evidence was found for it. Here TRXSS results clearly indicate the presence of a restructured monomer state, leading to the formation of the dimer.
To conclude, this study shows the utility of combining ultrafast T-jumps with time-resolved X-ray scattering to track conformational changes in protein complexes. The ability to indirectly induce protein interactions and structural changes by exciting the solvent medium allows for time-resolved X-ray studies on proteins that do not have light-activated functions, such as insulin. The use of T-jump provides insight into temperature sensitive or thermally activated processes, such as protein association, which would otherwise be obscured by complex bimolecular interactions, which are subject to rate-limiting processes. In addition, our results show that TRXSS can provide additional information on structural dynamics to vibrational spectroscopic techniques, like 2D-IR, without the need for deuterated solvents. In the case of insulin dimers, TRXSS has revealed the existence of new structural intermediates involved in the process of association, demonstrating the complexity of protein–protein interactions and deviation from simple kinetic models.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health, under contract no. R01-GM115761. This research used resources of the APS, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of BioCARS was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number R24GM111072. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Time-resolved setup at Sector 14 was funded in part through a collaboration with Philip Anfinrud (NIH/NIDDK). Optical equipment used for IR beam delivery at BioCARS was purchased with support from the Fraser lab at University of California San Francisco. We acknowledge Robert W. Henning (BioCARS) for his assistance in performing the TRXSS experiments. We also acknowledge Guy Macha (BioCARS) for his assistance in designing the sample holder. Portions of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the APS. DND-CAT is supported by Northwestern University, E.I. DuPont de Nemours & Co., and The Dow Chemical Company. Data were collected using an instrument funded by the National Science Foundation under Award Number 0960140.
Footnotes
ASSOCIATED CONTENT
- Experimental methods, UV-CD and static X-ray scattering results, details of TRXSS fitting and analysis procedures, and results for T-jump experiments from various initial temperature conditions. (PDF)
The authors declare no competing financial interest.
References
- 1.Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. 2015;54(31):8896–8927. doi: 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nooren IMA, Thornton JM. Structural Characterisation and Functional Significance of Transient Protein-Protein Interactions. J. Mol. Biol. 2003;325(5):991–1018. doi: 10.1016/s0022-2836(02)01281-0. [DOI] [PubMed] [Google Scholar]
- 3.Schwarze S, Zwettler FU, Johnson CM, Neuweiler H, Boles JO. The N-Terminal Domains of Spider Silk Proteins Assemble Ultrafast and Protected from Charge Screening. Nat. Commun. 2013;4:179–182. doi: 10.1038/ncomms3815. [DOI] [PubMed] [Google Scholar]
- 4.Sato D, Ohtomo H, Yamada Y, Hikima T, Kurobe A, Fujiwara K, Ikeguchi M. Ferritin Assembly Revisited: A Time- Resolved Small-Angle X-Ray Scattering Study. Biochemistry. 2016;55(2):287–293. doi: 10.1021/acs.biochem.5b01152. [DOI] [PubMed] [Google Scholar]
- 5.Schuck P. On the Analysis of Protein Self-Association by Sedimentation Velocity Analytical Ultracentrifugation. Anal. Biochem. 2003;320(1):104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 6.Akiyama S, Takahashi S, Kimura T, Ishimori K, Morishima I, Nishikawa Y, Fujisawa T. Conformational Landscape of Cytochrome c Folding Studied by Microsecond-Resolved Small- Angle X-Ray Scattering. Proc. Natl. Acad. Sci. U. S. A. 2002;99(3):1329–1334. doi: 10.1073/pnas.012458999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ries J, Schwarze S, Johnson CM, Neuweiler H. Microsecond Folding and Domain Motions of a Spider Silk Protein Structural Switch. J. Am. Chem. Soc. 2014;136(49):17136–17144. doi: 10.1021/ja508760a. [DOI] [PubMed] [Google Scholar]
- 8.Oang KY, Kim JG, Yang C, Kim TW, Kim Y, Kim KH, Kim J, Ihee H. Conformational Substates of Myoglobin Intermediate Resolved by Picosecond X-Ray Solution Scattering. J. Phys. Chem. Lett. 2014;5(5):804–808. doi: 10.1021/jz4027425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganim Z, Jones KC, Tokmakoff A. Insulin Dimer Dissociation and Unfolding Revealed by Amide I Two-Dimensional Infrared Spectroscopy. Phys. Chem. Chem. Phys. 2010;12(14):3579–3588. doi: 10.1039/b923515a. [DOI] [PubMed] [Google Scholar]
- 10.Attri AK, Fernández C, Minton AP. pH-Dependent Self-Association of Zinc-Free Insulin Characterized by Concentration- Gradient Static Light Scattering. Biophys. Chem. 2010;148(1):28–33. doi: 10.1016/j.bpc.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uversky VN, Garriques LN, Millett IS, Frokjaer S, Brange J, Doniach S, Fink AL. Prediction of the Association State of Insulin Using Spectral Parameters. J. Pharm. Sci. 2003;92(4):847–858. doi: 10.1002/jps.10355. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X-X, Jones KC, Fitzpatrick A, Peng CS, Feng C-J, Baiz CR, Tokmakoff A. Studying Protein–Protein Binding through T-Jump Induced Dissociation: Transient 2D IR Spectroscopy of Insulin Dimer. J. Phys. Chem. B. 2016;120(23):5134–5145. doi: 10.1021/acs.jpcb.6b03246. [DOI] [PubMed] [Google Scholar]
- 13.Cammarata M, Levantino M, Schotte F, Anfinrud Pa, Ewald F, Choi J, Cupane A, Wulff M, Ihee H. Tracking the Structural Dynamics of Proteins in Solution Using Time-Resolved Wide-Angle X-Ray Scattering. Nat. Methods. 2008;5(10):881–886. doi: 10.1038/nmeth.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levantino M, Schirò G, Lemke HT, Cottone G, Glownia JM, Zhu D, Chollet M, Ihee H, Cupane A, Cammarata M. Ultrafast Myoglobin Structural Dynamics Observed with an X-Ray Free-Electron Laser. Nat. Commun. 2015;6:6772. doi: 10.1038/ncomms7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Kim KH, Kim JG, Kim TW, Kim Y, Ihee H. Anisotropic Picosecond X-Ray Solution Scattering from Photo-selectively Aligned Protein Molecules. J. Phys. Chem. Lett. 2011;2(5):350–356. doi: 10.1021/jz101503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graber T, Anderson S, Brewer H, Chen YS, Cho HS, Dashdorj N, Henning RW, Kosheleva I, Macha G, Meron M, et al. BioCARS: A Synchrotron Resource for Time-Resolved X-Ray Science. J. Synchrotron Radiat. 2011;18(4):658–670. doi: 10.1107/S0909049511009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho HS, Dashdorj N, Schotte F, Graber T, Henning R, Anfinrud P. Protein Structural Dynamics in Solution Unveiled via 100-Ps Time-Resolved X-Ray Scattering. Proc. Natl. Acad. Sci. U. S. A. 2010;107(16):7281–7286. doi: 10.1073/pnas.1002951107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KH, Muniyappan S, Oang KY, Kim JG, Nozawa S, Sato T, Koshihara SY, Henning R, Kosheleva I, Ki H, et al. Direct Observation of Cooperative Protein Structural Dynamics of Homodimeric Hemoglobin from 100 ps to 10 ms with Pump-Probe X-Ray Solution Scattering. J. Am. Chem. Soc. 2012;134(16):7001–7008. doi: 10.1021/ja210856v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim TK, Lorenc M, Lee JH, Lo Russo M, Kim J, Cammarata M, Kong Q, Noel S, Plech A, Wulff M, et al. Spatiotemporal Reaction Kinetics of an Ultrafast Photoreaction Pathway Visualized by Time-Resolved Liquid X-Ray Diffraction. Proc. Natl. Acad. Sci. U. S. A. 2006;103(25):9410–9415. doi: 10.1073/pnas.0601958103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ihee H, Wulff M, Kim J, Adachi S. Ultrafast X-Ray Scattering: Structural Dynamics from Diatomic to Protein Molecules. Int. Rev. Phys. Chem. 2010;29(3):453–520. [Google Scholar]
- 21.Ihee H. Visualizing Solution-Phase Reaction Dynamics with Time-Resolved X-Ray Liquidography. Acc. Chem. Res. 2009;42(2):356–366. doi: 10.1021/ar800168v. [DOI] [PubMed] [Google Scholar]
- 22.Kim TW, Lee JH, Choi J, Kim KH, van Wilderen LJGW, Guerin L, Kim Y, Jung YO, Yang C, Kim J, et al. Protein Structural Dynamics of Photoactive Yellow Protein in Solution Revealed by Pump-Probe X-Ray Solution Scattering. J. Am. Chem. Soc. 2012;134(6):3145–3153. doi: 10.1021/ja210435n. [DOI] [PubMed] [Google Scholar]
- 23.Berntsson O, Diensthuber RP, Panman MR, Björling A, Hughes AJ, Henry L, Niebling S, Newby G, Liebi M, Menzel A, et al. Time-Resolved X-Ray Solution Scattering Reveals the Structural Photoactivation of a Light-Oxygen-Voltage Photoreceptor. Structure. 2017;25(6):933–938. e3. doi: 10.1016/j.str.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad A, Millett IS, Doniach S, Uversky VN, Fink AL. Partially Folded Intermediates in Insulin Fibrillation. Biochemistry. 2003;42(39):11404–11416. doi: 10.1021/bi034868o. [DOI] [PubMed] [Google Scholar]
- 25.Strickland EH, Mercola D. Near-Ultraviolet Tyrosyl Circular Dichroism of Pig Insulin Monomers, Dimers, and Hexamers. Dipole-Dipole Coupling Calculations in the Monopole Approximation. Biochemistry. 1976;15(17):3875–3884. doi: 10.1021/bi00662a035. [DOI] [PubMed] [Google Scholar]
- 26.Hamm P, Helbing J, Bredenbeck J. Two-Dimensional Infrared Spectroscopy of Photoswitchable Peptides. Annu. Rev. Phys. Chem. 2008;59(1):291–317. doi: 10.1146/annurev.physchem.59.032607.093757. [DOI] [PubMed] [Google Scholar]
- 27.Oang KY, Yang C, Muniyappan S, Kim J, Ihee H. SVD-Aided Pseudo Principal-Component Analysis: A New Method to Speed up and Improve Determination of the Optimum Kinetic Model from Time-Resolved Data. Struct. Dyn. 2017;4(4):044013. doi: 10.1063/1.4979854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt M, Pahl R, Srajer V, Anderson S, Ren Z, Ihee H, Rajagopal S, Moffat K. Protein Kinetics: Structures of Intermediates and Reaction Mechanism from Time-Resolved X-Ray Data. Proc. Natl. Acad. Sci. U. S. A. 2004;101(14):4799. doi: 10.1073/pnas.0305983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KH, Oang KY, Kim J, Lee JH, Kim Y, Ihee H. Direct Observation of Myoglobin Structural Dynamics from 100 ps to 1 Microsecond with Picosecond X-Ray Solution Scattering. Chem. Commun. 2011;47(1):289–291. doi: 10.1039/c0cc01817a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moffat K. The Frontiers of Time-Resolved Macromolecular Crystallography: Movies and Chirped X-Ray Pulses. Faraday Discuss. 2003;122:65–77. doi: 10.1039/b201620f. [DOI] [PubMed] [Google Scholar]
- 31.Cho HS, Schotte F, Dashdorj N, Kyndt J, Henning R, Anfinrud PA. Picosecond Photobiology: Watching a Signaling Protein Function in Real Time via Time-Resolved Small- and Wide-Angle X-Ray Scattering. J. Am. Chem. Soc. 2016;138(28):8815–8823. doi: 10.1021/jacs.6b03565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith GD, Pangborn WA, Blessing RH. The Structure of T 6 Bovine Insulin. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2005;61(11):1476–1482. doi: 10.1107/S0907444905025771. [DOI] [PubMed] [Google Scholar]
- 33.Bocian W, Sitkowski J, Bednarek E, Tarnowska A, Kawęcki R, Kozerski L. Structure of Human Insulin Monomer in Water/ acetonitrile Solution. J. Biomol. NMR. 2008;40(1):55–64. doi: 10.1007/s10858-007-9206-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.