

Abstract
Mice are the most commonly used species in preclinical research on the pathophysiology of metabolic diseases. Although they are extremely useful for identifying pathways, mechanisms and genes regulating glucose and energy homeostasis, the specificities of the various mouse models and methodologies used to investigate a metabolic phenotype can have a profound impact on experimental results and their interpretation. This review aims to: (1) describe the most commonly used experimental tests to assess glucose and energy homeostasis in mice; (2) provide some guidelines regarding the design, analysis and interpretation of these tests, as well as for studies using genetic models; and (3) identify important caveats and confounding factors that must be taken into account in the interpretation of findings.
Keywords: Clamp, Energy balance, Genetic background, Glucose tolerance, Metabolic phenotyping, Mouse, Review
Introduction
Owing to their ease of breeding, relatively low housing costs, amenability to genetic manipulations, and generally better acceptance for use as laboratory animals than larger species, rodents play an essential role in preclinical research in general, and in diabetes research in particular. Spontaneous, chemically-, surgically- or environmentally-induced mouse models of obesity, type 1 and type 2 diabetes have greatly contributed to our understanding of the underlying mechanisms of these diseases. The advent of gene-targeting methodologies, together with the sequencing of the mouse genome, have enabled scientists to study the biological function and pathological role of genes implicated in metabolic regulation. Yet, we often question whether findings obtained in mice are relevant to human pathophysiology. This question is legitimate and the answer is far from simple. In our opinion, mice are extremely valuable tools in diabetes research, as long as we acknowledge their limitations. The question is not so much whether mouse models are useful in diabetes research but rather under which conditions they can provide relevant information. With this question in mind, we have attempted: (1) to review the most commonly used experimental tests to assess glucose and energy homeostasis in mice; (2) to provide some guidelines regarding the design, analysis and interpretation of these tests; and (3) to identify important caveats and confounding factors that must be taken into account when drawing conclusions from the data generated. We certainly do not pretend to set the rules for the field, but simply propose guidelines stemming from our experience with our own research programmes and with the Rodent Metabolic Phenotyping core facility (Metabolic Disease Innovate Solutions) that we have established at the Montréal Diabetes Research Center (Centre de Recherche du Centre Hospitalier de l’Université de Montréal [CRCHUM], Montréal, QC, Canada). Such points of view are inherently debatable and open for discussion, which we hope this review will stimulate. We have not discussed the different types of animal models available for metabolic physiology and diabetes studies, topics that have been extensively reviewed by others [1–3]. Not covered, either, are the factors that drive reproducibility in preclinical research, an extremely important and topical issue recently and eloquently discussed by Drucker [4] and Flier [5].
Assessment of glucose homeostasis in mice
GTTs and insulin tolerance tests
GTTs and insulin tolerance tests (ITTs) are first-line experiments to assess glucose and insulin tolerance, respectively, since they are relatively easy to perform and minimally invasive. They consist of measuring blood glucose levels in response to a bolus administration of glucose (GTT) or insulin (ITT). Although they do not require special skills or previous surgery, and they enable rapid investigation of the impact of an intervention on glucose homeostasis, interpretation of these tests can be strongly influenced by variable experimental conditions and data analysis. Further, tolerance tests provide limited mechanistic insights to explain a metabolic phenotype.
Considerations when conducting tolerance tests
Several variables have to be considered when performing tolerance tests, including nutritional state (fasted vs fed), time of the day, route of administration, glucose/insulin dose and the level of restraint required for blood sampling. These experimental conditions have been tested and discussed in detail [6–11].
A general recommendation that has emerged is to subject animals to a short fast (~6 h); avoiding prolonged (overnight) fasting is especially important in mice because of their high metabolic rate and the fact that they consume most of their daily calories during the dark cycle. The body weight loss and metabolic stress induced by an overnight fast depletes liver glycogen stores and increases insulin-stimulated glucose transport, which can have confounding effects [8, 12].
The recommended doses of glucose and insulin to detect robust differences between experimental groups are 1 to 2 g/kg (i.p. or oral) and 0.5 to 1 U/kg, respectively (Table 1), depending on the strain. It is important to keep in mind that, during an oral GTT, the rate of glucose clearance depends on gastric emptying, glucose intestinal absorption and the potentiating effect of incretins on insulin secretion. The i.p. or i.v. routes of administration bypass these processes.
Table 1.
Recommendations for tests and data presentation when assessing glucose homeostasis in mice
| Test | Measures | Dose | Recommendations | Suggested index |
|---|---|---|---|---|
| GTT | Glucose tolerance | Glucose: 1–2 g/kg (i.p. or oral) Normalise dose to lean mass when possible |
5–6 h fast Report glucose levels as a function of time and insulin and/or C-peptide levels at 0, 15 and 30 min |
AUC glucose |
| ITT | Insulin resistance | Insulin: 0.5–1 U/kg (i.p.) Normalise dose to lean mass when possible |
5–6 h fast Report glucose levels as a function of time (normalised and non-normalised to basal time 0 values) |
KITT |
| HIEC | Insulin sensitivity | Insulin priming bolus: none or low dose (<20 mU/kg) Insulin infusion: 1 –20 mU kg−1 min−1 |
5–6 h fast Arterial blood sampling when possible Report glucose levels as a function of time as well as GIR, basal and steady-state insulin levels |
M/I, ISI |
| HGC | Insulin secretion | No priming glucose bolus Target BG: 10–18 mmol/l |
5–6 h fast Arterial blood sampling when possible Report GIR, glucose and insulin levels as a function of time and C-peptide levels during the steady state Arginine bolus at the end of the clamp to measure maximal insulin secretion |
AUC insulin, AIRmax, M/I, DI, insulin clearance |
| FS IVGTT | Insulin secretion, sensitivity and GE | Glucose: 1 g/kg (i.v.) | 5–6 h fast Report glucose and insulin levels as a function of time |
AUC glucose, ISI, GE, AIRg |
AIRg, acute insulin response to glucose; AIRmax, maximal arginine-induced insulin response; BG, blood glucose; GE, glucose effectiveness; ISI, insulin sensitivity index
Because glucose disposal is mostly dependent on lean mass, it is also important to consider whether glucose or insulin dose should be adjusted based on body weight or lean body mass (this has been discussed in detail [7, 10, 11]). When tolerance tests are performed in animals of different body weight (mostly owing to differences in fat mass), adjusting glucose or insulin dose based on body weight introduces a bias, worsening glucose intolerance in obese models [6, 13]. For this reason, it is more accurate to normalise glucose or insulin dose based on lean mass. However, this requires lean mass to be measured before the test using MRI, a stressful manipulation that requires restraint or anaesthesia and may introduce confounding effects. Moreover, this approach is limited in the context of longitudinal studies that require repeated GTTs or ITTs, during a diet intervention for instance. A potential alternative is to use a fixed dose of glucose or insulin based on the body weight of the control group [6, 13], as performed in humans.
GTT/ITT data analysis
Analysis of data from GTTs and ITTs is relatively straightforward. It should always be performed on raw data rather than percentages of the basal value, to avoid misinterpretation in cases where the experimental groups have different basal glucose or insulin levels. Changes in glucose levels over time during a GTT or ITT should be analysed using two-way ANOVA (and not at a given time point), and AUC should be presented for GTT to draw reliable conclusions (Table 1). It is important to substract the basal value when calculating the AUC. The rate of glucose disappearance can also be calculated as the slope of the decreasing line of blood glucose levels (KITT) during an ITT [14].
Basal glucose and insulin levels measured at time 0 of GTTs have been extensively used to calculate indexes of insulin sensitivity in mice, including HOMA. This index was initially established to empirically estimate insulin sensitivity in epidemiological and clinical studies. The HOMA equation is derived from human studies and is defined by the product of blood glucose and blood insulin levels in the fasted state, divided by a normalising factor derived from ‘normal’ insulin and glucose levels in an ‘ideal’ individual. In rodents, this index is poorly predictive of insulin sensitivity [15] and its use is generally not recommended. It is, however, conceivable that the HOMA index might be useable with derivation of an equation specific to mice and/or mouse strains in fasted conditions.
Application of murine-derived GTT/ITT data
The only solid conclusions that can be drawn from GTT and ITT are relating to glucose tolerance and insulin resistance, respectively. Measuring insulin and/or C-peptide levels during a GTT might provide valuable information on insulin secretion as long as blood can be sampled without stressing the animals. A higher AUC for glucose and/or a lower rate of glucose disappearance vs a control group studied side-by-side indicates glucose intolerance. Likewise, a lower rate of glucose disappearance during an ITT is indicative of insulin resistance. However, the converse is not true: a lack of differences between the groups during an ITT does not mean that the animals have similar levels of insulin sensitivity; ITTs are not sensitive enough to draw this conclusion. Insulin sensitivity can only be reliably assessed at steady-state levels of glucose and insulin (see below).
Tolerance tests are poor predictors of the glucoregulatory mechanisms underlying a particular phenotype. For instance, many studies show normal or minor changes in glucose tolerance despite a significant decrease or even complete absence of insulin secretion during a GTT [16–23], while the opposite is also true [24]. The main variable contributing to this discrepancy between insulin secretion and glucose excursion is glucose effectiveness, a measure that represents the capacity of glucose to regulate its own disposal independently of insulin. This can be estimated using a frequently-sampled IVGTT (FS IVGTT; described in detail below). During a GTT, ~50% of glucose disposal is independent of insulin secretion or sensitivity in rodents and humans [16, 25]. Importantly, glucose effectiveness is regulated by various metabolic signals, including gut hormones [26], and is altered in rodent models of obesity and diabetes [27]. Thus, glucose tolerance during a GTT results from insulin secretion, sensitivity and glucose effectiveness. Since gut signals (e.g. glucagon-like peptide- 1 [GLP-1], fibroblast growth factor 19 [FGF19]) regulate glucose effectiveness [26], the relative contribution of glucose effectiveness to glucose tolerance is dependent on the route of glucose administration (i.p. vs oral).
ITTs were initially designed to measure the activity of the hypothalamic–pituitary–adrenal (HPA) axis in response to insulin-induced hypoglycaemia (during the 90–120 min period post-injection) in humans and were adapted to measure insulin sensitivity using frequent sampling during the 15 min period following insulin injection [28]. Most ITTs performed in mice use the early and late responses to insulin to estimate insulin sensitivity. Considering that the half-life of insulin is ~10 min, glucose excursions at later time points mostly depend on the counteregulatory response to hypoglycaemia (glucagon, catecholamnies and cortisol) rather than changes in insulin sensitivity. In addition, the hypoglycaemia threshold triggering counterregulation may vary from one mouse model to another. Thus, it is more accurate to only consider the response to insulin within the first 20 min for analysis and KITT estimation [28].
For all reasons described above, it is essential to accurately report the experimental conditions of tolerance tests (fasting duration, glucose and insulin doses, administration route, blood sampling site, use of restraint), conduct rigorous data analysis, and interpret the results with caution before drawing robust conclusions.
In summary, tolerance tests can generate valuable results but suffer from limited sensitivity. More powerful tests to interrogate specific mechanisms, such as insulin secretion, sensitivity and glucose effectiveness, are described in the following sections.
Insulin and glucose clamps
The hyperinsulinaemic–euglycaemic clamp
Also known as the ’insulin clamp’, the hyperinsulinaemic–euglycaemic clamp (HIEC) was developed by DeFronzo et al [29] to measure insulin sensitivity in humans and was subsequently adapted for use in conscious mice. The procedure consists of inducing a state of stable hyperinsulinaemia by perfusing a fixed and constant dose of insulin concomitantly with a variable glucose infusion to maintain euglycaemia. In these conditions, the glucose infusion rate (GIR) directly reflects whole-body glucose disposal. The M/I index of insulin sensitivity is calculated as the ratio of GIR (M) to circulating insulin levels (I) during the steady-state period. Additional information can be obtained using isotopic glucose tracers during clamp to estimate tissue- and organ-specific insulin sensitivity (glucose uptake and endogenous glucose production).
Key considerations when conducting HIEC tests
Several key methodological variables must be considered when performing HIEC tests in mice (Table 1). These include the nutritional state, blood sampling sites and initial insulin priming and infusion rates, as discussed by Ayala et al [12]. For instance, blood sampling from the tail significantly increases circulating catecholamines, leading to decreased insulin sensitivity compared with arterial sampling, which requires less restraint. In addition, insulin sensitivity is increased after 18 h fasting compared with a 5 h fast. Finally, the insulin priming dose and infusion rates affect insulin clearance, as well as hepatic vs peripheral insulin action.
Other important issues often arise in the design of a clamp. For example, our core facility recently performed clamps in transgenic mice with reduced fasting blood glucose levels compared with controls. In the transgenic group, glucose levels were clamped to the level in the control group. However, alternatives, such as using the average blood glucose of both groups, would have been acceptable, as long as they are precisely reported.
More complicated is the issue of insulin levels, since these cannot be measured extemporaneously during the clamp and, therefore, are not adjusted in real time. By design, insulin levels during the steady-state period of the HIEC should be similar between groups. In practice, however, this is not always the case, even if endogenous insulin secretion is suppressed by co-infusion of somatostatin. This is due to differences in insulin clearance. For instance, we [30] and others [31] have shown that lipopolysaccharide-induced endotoxaemia dramatically increases insulin levels during HIEC, potentially because of decreased hepatic insulin clearance. Similarly, glucose and lipid co-infusion in rats decreases the clearance of exogenous human insulin during a subsequent HIEC [32]. Such differences in insulin levels during the clamp make comparisons between groups very difficult unless the insulin dose is adapted in the experimental group to match blood levels of insulin in the control group [31] or comparisons are only performed between groups with similar levels of circulating insulin. Alternatively, one can circumvent differences in insulin levels by calculating the sensitivity index, which takes into account changes in insulin levels (clamped – basal) rather than absolute values [33]. In all cases, it is essential that basal and clamped glucose and insulin values are reported [34].
The hyperglycaemic clamp
The hyperglycaemic clamp (HGC), also known as the ‘glucose clamp’, is considered the gold standard for measuring glucose-stimulated insulin secretion (GSIS) in vivo. It consists of infusing a variable rate of glucose (i.v.) to maintain hyperglycaemia at a target value (usually between 12–and 18 mmol/l) and measuring insulin and C-peptide levels during this steady-state hyperglycaemia. In addition, a bolus of arginine can be performed at the end of the clamp to assess the maximal arginine-induced insulin response (AIRmax).
Importantly, the clamp can be designed as a one-step or two-step protocol; the latter allows for measurement of beta cell glucose responsiveness at different levels of hyperglycaemia (e.g. 8 and 16 mmol/l) during the same test, as we have previously described [35]. The different measures obtained during steady-state hyperglycaemia can be used to estimate insulin clearance (C-peptide/insulin), M/I and the disposition index (DI; an index of beta cell function based on insulin secretion corrected for insulin sensitivity, calculated by multiplying the M/I index by C-peptide levels). However, M/I and DI estimations are limited because they are based on the assumption that, in a given mouse model, the relationship between insulin sensitivity and secretion is hyperbolic [36]. Since it is technically difficult to perform both a HIEC and HGC in the same animal (both are typically terminal experiments), M/I and DI are typically obtained from the same HGC. Therefore, they are not independent from each other and should be interpreted with caution [37].
Key considerations when conducting HGC tests
As with HIEC, several experimental variables should be considered when designing HGC studies, including nutritional state or blood sampling sites (Table 1). For example, compared with fed animals, GIR and insulin secretion in 16 h fasted mice is decreased [38].
HGC vs GTT
The sensitivity of HGC vs GTT is highlighted by studies on the role of the fatty acid receptor G-protein-coupled receptor 40 (GPR40; also known as free fatty acid receptor 1 [FFA1]) in GSIS. For example, using oral GTT, IP- or IVGTT, we [19, 39] and others [40, 41] found that glucose tolerance and insulin secretion were similar in chow-fed GPR40-deficient mice compared with wild-type (WT) littermates, while, in comparison, using HGC, we found that insulin secretion was impaired in GPR40 knockout (KO) mice [38].
Although clamp methodology is sensitive and powerful, it is technically challenging. Dual venous and arterial catheterisation in mice is difficult to perform and often results in loss of animals (because of occluded or leaky catheters). For this reason, some groups only use a venous catheter for infusions and collect blood samples from the tail. As aforementioned, blood sampling from the tail, which involves animal restraint, directly impacts stress levels and glucoregulatory responses and, thus, introduces confounding factors. In addition, obese models (diet-induced and genetic) do not tolerate surgery well and may not recover pre-surgery body weights, potentially affecting insulin sensitivity and/or secretion. Thus, it is essential to precisely report the experimental conditions used for clamp studies (blood sampling site, restraint used, insulin dose and levels, blood glucose profile and GIR) [34]. If clamps cannot be performed under optimised conditions, GTT and ITT remain valuable alternative methodologies to generate interpretable results, as long as the experimental conditions are well reported and the data analysed accurately.
FSIVGTT
The FSIVGTT, initially developed by Bergman et al [42] in humans, provides an indirect measure of insulin sensitivity based on glucose and insulin levels obtained during the test. The FSIVGTT has been used in dogs and rats but was only recently validated in mice [20, 27]. In addition to measuring insulin sensitivity and secretion, glucose effectiveness can be estimated from FSIVGTTs. As such, it represents a powerful method for concomitantly measuring the three processes involved in glucose tolerance in the same animal. Alonso et al [27] observed strong heterogeneity in the first phase of GSIS during FSIVGTT within the same BL/6J mouse substrain. They also showed strong differences in glucose effectiveness in pathological models (diet-induced obesity [DIO] and ob/ob mice). Although this test has many advantages, the challenging dual catheterisation (venous and arterial) and frequent blood sampling make it difficult to use routinely in mice.
In Fig. 1, we provide a step-by-step flowchart for selecting the appropriate in vivo tests, which can be followed by isolation of tissues or organs (e.g. pancreatic islets or muscle) for ex vivo studies.
Fig. 1.
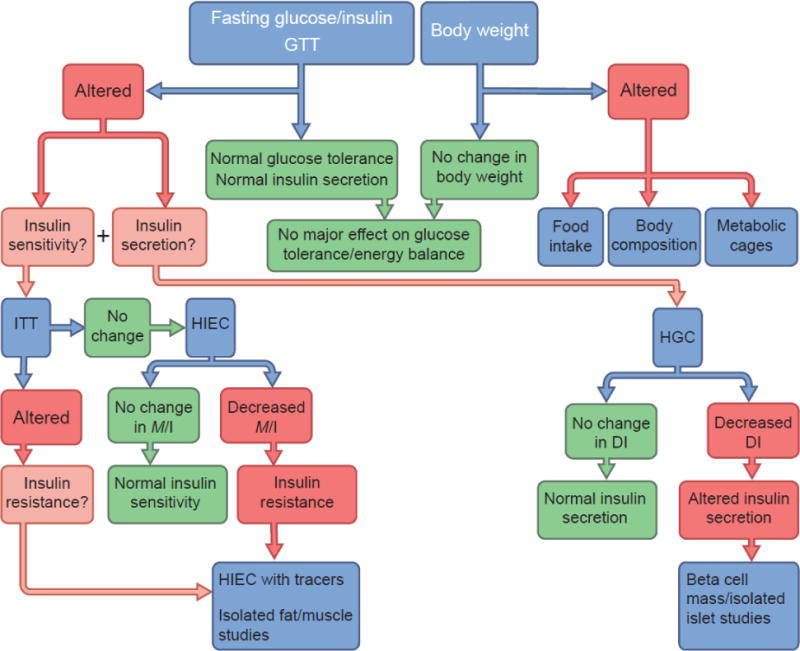
Proposed flowchart for the investigation of glucose and energy homeostasis in mice. The first simple step for metabolic phenotyping is to measure body weight, fasting blood glucose and plasma insulin levels and to perform a GTT that includes insulin measurements. If body weight is affected, we refer the reader to the guidelines proposed by Tschöp et al [43] to investigate energy balance in rodents. If glucose tolerance is found to be altered, changes in insulin sensitivity and/or secretion can be tested using ITTs and/or the gold standard, HGC, respectively. However, ITT is not very sensitive and, hence, a lack of change in insulin tolerance does not rule out insulin resistance. HIECs can be used to interrogate insulin sensitivity at the whole-body level or in a tissue-specific manner using glucose tracers. ITT, HIEC and HGC tests can be combined with ex vivo studies (e.g. analysis of insulin secretion in isolated islets, glucose transport in tissues, beta cell mass measurements) to better define the underlying mechanisms
Assessment of energy homeostasis in mice
Energy metabolism
Alterations in glucose homeostasis often result from changes in energy balance and body composition. Therefore, investigating whole-body energy homeostasis is critical to understanding the mechanisms underlying changes in glucoregulatory responses. Several types of metabolic cages are now available for in-depth analysis of energy balance through measurements of energy expenditure, respiratory quotient, locomotor activity and food intake.
Although indirect calorimetry technology (and its sensitivity) has significantly improved, there is no single approach to mouse metabolic phenotyping and several considerations must be taken into account to generate reliable results and meaningful interpretations. These considerations have been recently discussed and guidelines have been proposed for metabolic phenotyping [43–45]. Here we summarise some key aspects that require consideration.
First, housing conditions (temperature, single- vs group housing, position of the cage on the rack and type of diet) can dramatically affect animal energy homeostasis. For instance, recent studies highlighted the impact of conventional housing temperature at ~22°C vs ~30°C (which is thermoneutral for mice) [46] on feeding [47], glucoregulatory responses [48] and DIO [49]. Second, body composition has a major impact on energy expenditure analysis. Most studies normalise energy expenditure to total body or lean mass, which may introduce confounding factors. Although still debated [44], one recommendation is to analyse group and body composition effects on metabolic rates using ANCOVA [43, 50, 51]. Third, accurate measurement and analysis of food intake should take into account several variables, including the type of diet, pattern of feeding behaviour, and spillage and foraging. Commercial low-fat diets (purified) are more reliable controls for high-fat diet (HFD) studies because of their standardised composition compared with ‘chow’ diets, the dietary composition of which can differ from batch to batch. When using HFDs, the nature and type of fat should also be considered. Although food intake can be accurately measured manually, automated systems enable us to track circadian feeding patterns, frequency and quantity of food ingested. Reporting daily food intake values that have been averaged over a period of time is not informative, especially when animals are group housed. Cumulative food intake, calorie efficiency (amount of body weight gained based on calories consumed over time) and loss of calories in the faeces are also important variables to measure. Finally, paired and yoked feeding, both of which allow us to circumvent differences in diurnal feeding pattern between groups, are powerful strategies to determine whether or not changes in energy balance and body weight are dependent on the amount of calories consumed. Also important to consider are the acclimation period to new cages or to changes in husbandry conditions (single vs group housing) and the nutrient content of bedding [52].
Considering the number of variables that must be taken into account and controlled for, metabolic phenotyping is a complex and challenging endeavour. For this reason, it is essential to report detailed descriptions of procedures and protocols, perform accurate data analysis and interpret the results in a given experimental context. Finally, when studying energy metabolism in a genetically-modified model, it is important to keep in mind that the gene of interest may regulate sensorial (taste, smell, sight), locomotor and/or gastrointestinal functions, thereby affecting feeding behaviour; these must be considered to avoid misinterpretation of the phenotype [53].
Some caveats and confounding factors in metabolic studies using mice
As described in the previous sections, most tests used to examine glucose and energy homeostasis in rodents were initially developed in humans and have rarely been validated in rodents. Yet, major differences exist between these species, such as metabolic rates, contribution of glucose effectiveness to glucose disposal, and feeding behaviour, influencing the design and interpretation of these tests. One of the major advantages of using mice lies in their highly standardised breeding and housing conditions and genetic background. This homogeneity increases the power of an experiment by lowering the inter-individual variability as compared with human studies, in which lifestyle, ethnicity and genetic variations may have a strong impact on a given response. On the other hand, the relatively standardised experimental conditions used for laboratory animals may also introduce some bias or confounding factors.
Age and sex
For cost-related reasons, most studies in the field of metabolism (except, of course, on aging) are performed in young animals (8–12 weeks). Yet, obesity and type 2 diabetes are strongly associated with aging, and several differences have been reported between young and old rodents. For example, the beta cell replicative capacity strongly decreases with age in mice [54, 55]. Further, we [56, 57] have shown that young rats do not develop insulin resistance in response to nutrient infusion (in contrast to older animals), suggesting that adaptive responses to nutrient surfeit are age-dependent. Even HFD studies often underestimate the impact of age since the typical length of HFD is 8–16 weeks, starting in 5–6-week-old animals. For these reasons, studies pertaining to chronic metabolic diseases should ideally be performed in older animals and/or using short- (4–6 weeks) and long-term (12–20 weeks) dietary interventions so as not to overlook potential age-dependent adaptive mechanisms.
Likewise, for reasons related to housing cost and difficulty to synchronise and/or test the cycle phase in female mice, many metabolic studies only include male mice. This introduces a bias since oestrogen is well known to have beneficial effects on metabolism. Oestrogen acts on the brain and peripheral tissues to reduce food intake while increasing energy expenditure, and enhance insulin sensitivity and beta cell function (reviewed in [58]). Together, these effects likely contribute to the well-known protection against the development of obesity, insulin resistance and hyperglycaemia in HFD-fed female vs male mice, and the sexual dimorphism of metabolic diseases in humans [59]. In addition, there are significant differences in drug/xenobiotic pharmacokinetics in male vs female animals [60]. The importance of including male and female animals has been recently recognised by the National Institutes of Health to improve reproducibility in preclinical research [61], and guidelines have been proposed to study the mechanisms of sex differences in metabolic research [62].
Environment
Although mice are typically bred and housed under highly controlled environmental conditions, many environmental factors can affect their physiology and behaviour including diets, husbandry, handling, noise/vibrations, temperature and circadian rhythm (reviewed in [47]). A prime example of this is the protection from diabetes observed in NOD mice when housed on the top of the rack compared with those housed on the lower rack shelf [63].
Amongst environmental factors, the impact of the gut microbiota on energy homeostasis and the aetiology of metabolic diseases is receiving increasing attention [3]. Sequencing of the gut microbiota and interventions altering gut microflora have shown that microbiota composition modulates insulin sensitivity and the response to DIO. In addition, elegant studies recently showed that the interaction between the microbiome and the host is dependent on the genetic background, leading to a complex crosstalk between microbiota and host in disease development (reviewed in [64]). This is a highly complex issue that, under most circumstances, is impossible to control for experimentally. However, the potential impact of the microbiota should be taken into account when otherwise unexplainable differences in the response to a particular manipulation are observed between, for example, mice from the same strain and background but from different vendors, or experiments performed in the same mouse colony but in different animal facilities.
Genetic background and breeding strategies
Type 2 diabetes and obesity have a highly complex genetic architecture (reviewed in [65]), with individual SNPs only contributing a minor fraction to the overall risk. Thus, one may question the relevance of using inbred animals as models for these polygenic diseases. On the one hand, one would want to examine the role of individual genes or variants in a model that is as close as possible to the genetic variety of humans (keeping in mind that outbred animals are still not as genetically diverse as humans). On the other hand, keeping everything else as equal as possible facilitates the identification of a phenotype associated with the gene of interest. For this reason, genetic studies in mice are often performed in inbred strains. However, many studies have shown that the impact of a genetic manipulation is highly influenced by the animal’s genetic background. For instance, db mutations only lead to obesity in the C57BL/6J background but induce obesity and chronic hyperglycaemia in C57BLKS/J animals [66]. A recent study by Sittig et al [67] showed that the same genetic mutation in 30 genetic backgrounds can lead to dramatically different, sometimes completely divergent, phenotypes. Of particular interest, only three out of 30 mouse strains harbouring a null mutation of Tcf7l2 (a genetic risk factor for type 2 diabetes [65]) showed significant reduction in blood glucose levels measured throughout the day. In addition, Tcf7l2 mutant mice showed behavioural phenotypes that varied depending on the strain. While raising new questions on the importance of gene–gene interactions, these findings highlight important limitations of studies in genetically-modified inbred animals. Were financial and timing aspects not limiting, it would be ideal to study the impact of genetic interventions in both inbred and outbred animals, or at least in several different inbred strains. This type of approach would strengthen the outcomes and conclusions of studies. The clustered regularly interspaced short palindromic repeats (CRISPR) technology has opened new avenues for generating gene mutations more quickly and could thus be helpful in this regard.
Despite the limitations of inbred strains, phenotypic differences between strains have been useful for the identification of new genes or gene polymorphisms involved in beta cell function or glucose homeostasis using transcriptomic analyses [68], linkage studies allowing genetic mapping of qualitative and quantitative trait loci, or genome-wide association studies (GWAS) (reviewed in [69]).
Inbred strains display major differences in glucoregulatory responses and susceptibility to DIO. For example, dilute brown non-agouti (DBA) mice show heightened insulin secretory response to glucose and body weight gain when fed a HFD compared with C57BL/6, Friend virus B-type (FVB) or A/J mice [17, 68, 70]. The C57BL/6 strain has been widely used because of its high susceptibility to develop obesity and hyperglycaemia under HFD compared with other strains. However, there are several BL/6 sub-strains, including the commonly used BL/6J (often from Jackson Laboratories) and BL/6N (often from Charles River and Taconic). Several studies have now demonstrated important metabolic differences between these two sub-strains that strongly influence the development of obesity and diabetes. The BL/6J mice harbour a mutation in the Nnt gene that is responsible for impaired GSIS under chow-fed conditions [21, 71]. Importantly, BL/6 mice heterozygous for the Nnt mutation (BL/6NJ) are characterised by impaired GSIS compared with BL/6NN animals on a HFD [22], although they show reduced body weight gain and glucose intolerance compared with BL/6J [72]. These findings have major implications when using the cre-loxP system. Most engineered cre mice are available at the Jackson depository and maintained on the BL/6J background. In contrast, floxed mice are usually generated using BL/6N- or 129-derived embryonic stem cells. The resulting cre-floxed animals have mixed BL/6J and BL/6N or 129 genetic backgrounds. Uncontrolled genetic background and unknown status of the Nnt mutation (JJ, NJ or NN) will undoubtedly affect beta cell function and thus introduce confounding factors, strong phenotypic heterogeneity and difficulties in replicating findings. This is a major issue in the field that has been recently discussed by Fontaine and Davis [73]. In the literature, there are several examples of inconsistent or opposite mouse phenotypes that are likely related to genetic background. For instance, Steneberg et al [74] initially reported that mice deficient in GPR40 were protected from obesity-induced hyperinsulinaemia and glucose intolerance under HFD conditions whereas mice overexpressing GPR40 in the pancreas were prone to diabetes, suggesting that antagonism of GPR40 could be beneficial for the treatment of diabetes. In contrast, subsequent studies by us [39] and others [40, 41] using three independent GPR40 KO strains (backcrossed with BL/6N or BL/6J) and a transgenic BL/6J mouse overexpressing GPR40 in beta cells [75] led to opposite conclusions. More recently, discrepant results were reported regarding the anti-inflammatory role of the n-3 fatty acid receptor GPR120 (also known as free fatty acid receptor 4 [FFA4]) during high-fat-feeding; Oh et al [76] found that the anti-inflammatory and beneficial effects of dietary n-3 on glucose homeostasis were absent in GPR120 KO mice of an unspecified genetic background fed with a HFD. In contrast, two subsequent studies showed that dietary n-3 were equally effective in GPR120 KO mice and WT littermates on a mixed (129 and BL/6J) or pure (BL/6N) background [77, 78]. Finally, studies on the role of uncoupling protein 2 (UCP2) in insulin secretion also led to diametrically opposite conclusions (reviewed in [79]). The initial UCP2 KO mice generated on a mixed background (BL/6J and 129) showed improved GSIS. However, when these animals were backcrossed with inbred strains (BL/6J, AJ or 129), a decrease in GSIS was observed [79]. Interestingly, Seshadri et al recently demonstrated that UCP2 expression is temporally regulated in beta cells throughout the day and that this pattern is required for the temporal control of glucose-induced ATP production, normal rhythms of GSIS and, ultimately, glucose tolerance [80]. We believe that it is important to encourage authors and editors to adequately report breeding strategies and detailed genetic backgrounds. An ideal approach would be to phenotype experimental animals on pure and mixed backgrounds and report potential differences.
Different breeding schemes can be used to generate experimental whole-body or conditional KO animals using the cre-loxP system. In all circumstances, control animals must be littermates, implying that homozygous KO should never be crossed together to generate experimental mice. Accordingly, it is not acceptable to use WT controls purchased from a supplier; WT controls must be littermates. The absence of a phenotype in female KO animals does not preclude a phenotype from arising during gestation. Therefore, it is safer to use heterozygous female KO mice for breeding. Cre homozygosity may introduce confounding factors and/or cre-associated toxicity [81] and, so, it is recommended to cross cre-negative female mice with cre-hemizygous male mice [82]. Finally, it is important to consider multiparity in the breeding scheme and the size of the litter that may influence the metabolic phenotype of the offspring [83].
Additional issues with the cre-loxP system and inducible models
The cre-loxP strategy involves flanking the endogenous gene or a portion of it with loxP sites, which can affect gene expression. A well-known example is the Glut4 (also known as Scl2a4)-floxed mouse in which the recombined Glut4-floxed gene was expressed at lower levels, specifically in adipose tissues, compared with WT controls [84]. This could be owing to the loxP sequences and/or the neoR selection cassette that is part of the construct. It is therefore critical to verify the expression of the recombined floxed gene and to excise the neoR cassette used for embryonic stem cell selection using flippase. Also, the genome contains degenerate loxP sites that can be targeted by cre, leading to off-target recombination of genes in a tissue-specific manner, as described in the heart using the αMyHC-cre mouse (B6.FVB-Tg(Myh6-cre)2182Mds/J) [81].
There are also issues regarding the efficiency, specificity and phenotype of cre transgenics, which have been recently discussed in the context of beta cell- and brain-specific cre lines [85, 86]. For example, the Nestin-cre (B6.Cg-Tg(Nes-cre)1Kln/J) that targets the brain is known to leak into multiple peripheral tissues. In addition, Nestin-cre [85] and RIP-cre (B6.Cg-Tg(Ins2-cre)25Mgn/J) [87] mice are phenotypically different to their WT littermates. Other examples of ectopic expression include the Pdx1-cre (Tg(Pdx1-cre)89.1Dam) and RIP-cre strains, which are designed for pancreas and beta cell KO, respectively, but leak in the brain [85, 86, 88]. The leakiness of cre expression in RIP-cre strains might be caused by the truncated insulin promoter in the transgene construct. Indeed, the full-length insulin promoter used in the MIP-creERT (B6.Cg-Tg(Ins1-cre/ERT)1Lphi/J) strain restricts cre expression to the beta cell [88, 89]. A strategy to circumvent the leakiness issue is to generate a ‘knockin’ cre strain such that cre expression is under the control of an endogenous promoter, with the caveat that this deletes one of the two endogenous alleles, which may result in lower gene expression. This strategy has recently been used to express cre from the endogenous Ins1 gene, resulting in faithful cre expression in beta cells without leakiness into the brain [90], and from the proglucagon promoter, resulting in cre expression in GLP-1-producing alpha cells, intestinal L cells and neurons [91]. Cre efficiency can be influenced by the breeding or genetic background of a mouse. For instance, cre expression in Albumin-cre mice (B6.Cg-Tg(Alb-cre)21Mgn/J) is silenced with an increasing number of generations [92]. Along the same lines, cre-mediated excision can be affected if the cre-driving promoter is sensitive to changes in metabolic status or diet. Douglas et al [93] recently reported that GFAP-creER (B6.Cg-Tg(GFAP-cre/ERT2)505Fmv/J)/Ikkb (also known as Ikbkb)-floxed mice had minimal gene deletion in hypothalamic astrocytes when chow fed, while the efficiency of cre-mediated excision of inhibitor of kappaB kinase β (IKKβ) was dramatically enhanced after HFD.
Recent studies have shown that several cre lines used in beta cell research ectopically express the human growth hormone (hGH) minigene, commonly used as a polyadenylation sequence in transgene constructs [89, 94]. Expression and secretion of hGH by mouse beta cells has a significant impact on beta cell function and mass and protects animals from streptozotocin (STZ)-induced hyperglycaemia [89, 94]. This also has implications in other cre models as it was shown that hGH expression in the hypothalamus is responsible for the metabolic phenotype in Nestin-cre mice [95].
Finally, it is important to consider that drugs used to induce cre expression, such as tamoxifen and doxycycline, have off-target effects that may be toxic [82, 96–98]. As such, it is recommended to test potential adverse effects and administer all experimental groups with the lowest tamoxifen/doxycycline doses suitable, using the most appropriate route of administration (e.g. oral tamoxifen is considered less toxic than i.p. injections).
For all aforementioned reasons, it is strongly recommended to include all three cre, floxed and WT control groups in tissue-specific KO studies, or alternatively to verify, at the outset, that these control groups have a similar phenotype. Also, including heterozygous KO animals can help strengthen conclusions by establishing a relationship between gene dosage and phenotype severity. It is essentially impossible to avoid all the confounding variables associated with mouse genetic studies but most of them can, and should be, controlled for.
Conclusions
In this review, we attempted to address the question, under which conditions can mouse models provide relevant information for metabolic research? We focused on some aspects that we consider of particular importance, while deliberately not covering others that are just as important but were recently discussed elsewhere, such as sample size, randomisation and, more generally, applying similarly rigorous standards to the design, execution and reporting of preclinical studies to those used in clinical research [4, 5].
The pitfalls and limitations highlighted herein should certainly not discourage the use of mice for metabolic studies. They are an extremely useful model, especially at early stages of the discovery process, be it for functional genomics, target identification and validation, in vivo validation of test compounds or early toxicology studies. Yet, these pitfalls and limitations should be known, acknowledged and, whenever possible, circumvented or controlled for. In addition to the recommendations summarised in the text box, the reporting of animal experiments should be transparent, accurate and complete. In that sense, we encourage editors and reviewers to request that detailed information be provided by authors regarding the design and interpretation of studies using mice. Diabetologia has recently implemented a mandatory preclinical checklist to that effect.
Finally, and perhaps most importantly, before broad conclusions can be made regarding the role of a particular gene or impact of given experimental manipulations, it is essential that the findings are reproduced using complementary approaches, i.e. genetic loss- and/or gain-of-function, genetic rescue and pharmacological tools (inhibitors, antagonists and agonists), when available, and in different animal models rather than a single mouse strain.
The research environment and publication process have evolved in such a way that favours depth over breadth and encourages (even incentivizes) overinterpretation of the findings and inflation of the conclusions so as to convince the reader or reviewer of the translational potential of the work [99]. As a first step towards reversing this dangerous trend, may we all (including the authors) agree to: (1) provide detailed and accurate reporting of the methodologies; (2) acknowledge the caveats and limitations of a particular rodent model or metabolic test; and (3) avoid such sentences as ‘these findings identify a novel pathway for the treatment of metabolic diseases’ from our future publications?
Supplementary Material
Considerations for genetic model generation and metabolic phenotyping.
Design
Include older animals for studies pertaining to the pathophysiology of metabolic diseases
Include male and female mice
Test the impact of genetic mutations on different backgrounds
Control for Nnt mutations when using C57BL/6 mice
Breeding
Set up a breeding scheme that produces all genotypes as littermates (no KO breeding)
Use the same background for crosses between different lines (e.g. cre × floxed)
Use heterozygous KO female mice for breeding (whole-body KO models)
Use cre hemizygous males and females not carrying cre for breeding (cre-loxP models)
Eliminate the neoR cassette using flippase if flippase recognition target (FRT) sequences are part of the recombined gene
Controls
Use littermates as controls
Verify lack of effect of loxP sites on endogenous gene expression
Control for possible phenotype of the cre line
Verify tissue specificity of cre-mediated recombination
Verify stability of recombination overtime (generations)
Verify that the experimental manipulation (e.g. DIO) does not affect cre-mediated recombination
Abbreviations
- CRCHUM
Centre de Recherche du Centre Hospitalier de l’Université de Montréal
- DI
Disposition index
- DIO
Diet-induced obesity
- FSIVGTT
Frequently-sampled IVGTT
- GIR
Glucose infusion rate
- GLP-1
Glucagon-like peptide- 1
- GPR40
G-protein-coupled receptor 40
- GPR120
G-protein-coupled receptor 120
- GSIS
Glucose-stimulated insulin secretion
- HFD
High-fat diet
- HGC
Hyperglycaemic clamp
- HIEC
Hyperinsulinaemic–euglycaemic clamp
- I
Circulating insulin levels
- ITT
Insulin tolerance test
- KITT
The slope of the decreasing line of blood glucose levels
- KO
Knockout
- UCP2
Uncoupling protein 2
- WT
Wild-type
References
- 1.Lee AW, Cox RD. Use of mouse models in studying type 2 diabetes mellitus. Expert reviews in molecular medicine. 2011;13:e1. doi: 10.1017/S1462399410001729. [DOI] [PubMed] [Google Scholar]
- 2.King A, Bowe J. Animal models for diabetes: understanding the pathogenesis and finding new treatments. Biochemical pharmacology. 2016;99:1–10. doi: 10.1016/j.bcp.2015.08.108. [DOI] [PubMed] [Google Scholar]
- 3.Leulier F, MacNeil LT, Lee WJ, et al. Integrative physiology: at the crossroads of nutrition, microbiota, animal physiology, and human health. Cell Metab. 2017;25:522–534. doi: 10.1016/j.cmet.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drucker DJ. Never waste a good crisis: confronting reproducibility in translational research. Cell Metab. 2016;24:348–360. doi: 10.1016/j.cmet.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Flier JS. Irreproducibility of published bioscience research: diagnosis, pathogenesis and therapy. Molecular metabolism. 2017;6:2–9. doi: 10.1016/j.molmet.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab. 2008;295:E1323–E1332. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- 7.McGuinness OP, Ayala JE, Laughlin MR, Wasserman DH. NIH experiment in centralized mouse phenotyping: the Vanderbilt experience and recommendations for evaluating glucose homeostasis in the mouse. Am J Physiol Endocrinol Metab. 2009;297:E849–E855. doi: 10.1152/ajpendo.90996.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ayala JE, Samuel VT, Morton GJ, et al. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Disease models & mechanisms. 2010;3:525–534. doi: 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pacini G, Omar B, Ahren B. Methods and models for metabolic assessment in mice. Journal of diabetes research. 2013;2013:986906. doi: 10.1155/2013/986906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowe JE, Franklin ZJ, Hauge-Evans AC, King AJ, Persaud SJ, Jones PM. Metabolic phenotyping guidelines: assessing glucose homeostasis in rodent models. The Journal of endocrinology. 2014;222:G13–G25. doi: 10.1530/JOE-14-0182. [DOI] [PubMed] [Google Scholar]
- 11.Lee HY, Jeong KH, Choi CS, International Mouse Phenotyping Consortium In-depth metabolic phenotyping of genetically engineered mouse models in obesity and diabetes. Mammalian genome. 2014;25:508–521. doi: 10.1007/s00335-014-9520-4. [DOI] [PubMed] [Google Scholar]
- 12.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes. 2006;55:390–397. doi: 10.2337/diabetes.55.02.06.db05-0686. [DOI] [PubMed] [Google Scholar]
- 13.Omar BA, Pacini G, Ahren B. Impact of glucose dosing regimens on modeling of glucose tolerance and β-cell function by intravenous glucose tolerance test in diet-induced obese mice. Physiological reports. 2014;2:e12011. doi: 10.14814/phy2.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lundbaek K. Intravenous glucose tolerance as a tool in definition and diagnosis of diabetes mellitus. British medical journal. 1962;1:1507–1513. doi: 10.1136/bmj.1.5291.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S, Muniyappa R, Yan X, et al. Comparison between surrogate indexes of insulin sensitivity and resistance and hyperinsulinemic euglycemic clamp estimates in mice. Am J Physiol Endocrinol Metab. 2008;294:E261–E270. doi: 10.1152/ajpendo.00676.2007. [DOI] [PubMed] [Google Scholar]
- 16.Best JD, Kahn SE, Ader M, Watanabe RM, Ni TC, Bergman RN. Role of glucose effectiveness in the determination of glucose tolerance. Diabetes care. 1996;19:1018–1030. doi: 10.2337/diacare.19.9.1018. [DOI] [PubMed] [Google Scholar]
- 17.Andrikopoulos S, Massa CM, Aston-Mourney K, et al. Differential effect of inbred mouse strain (C57BL/6, DBA/2, 129T2) on insulin secretory function in response to a high fat diet. The Journal of endocrinology. 2005;187:45–53. doi: 10.1677/joe.1.06333. [DOI] [PubMed] [Google Scholar]
- 18.Aston-Mourney K, Wong N, Kebede M, et al. Increased nicotinamide nucleotide transhydrogenase levels predispose to insulin hypersecretion in a mouse strain susceptible to diabetes. Diabetologia. 2007;50:2476–2485. doi: 10.1007/s00125-007-0814-x. [DOI] [PubMed] [Google Scholar]
- 19.Latour MG, Alquier T, Oseid E, et al. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes. 2007;56:1087–1094. doi: 10.2337/db06-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morton GJ, Matsen ME, Bracy DP, et al. FGF19 action in the brain induces insulin-independent glucose lowering. The Journal of clinical investigation. 2013;123:4799–4808. doi: 10.1172/JCI70710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fergusson G, Ethier M, Guevremont M, et al. Defective insulin secretory response to intravenous glucose in C57Bl/6J compared to C57Bl/6N mice. Molecular metabolism. 2014;3:848–854. doi: 10.1016/j.molmet.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Attane C, Peyot ML, Lussier R, et al. Differential insulin secretion of high-fat diet-fed C57BL/6NN and C57BL/6NJ mice: implications of mixed genetic background in metabolic studies. PloS one. 2016;11:e0159165. doi: 10.1371/journal.pone.0159165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korsgren E, Korsgren O. Glucose effectiveness: the mouse trap in the development of novel ss-cell replacement therapies. Transplantation. 2016;100:111–115. doi: 10.1097/TP.0000000000000900. [DOI] [PubMed] [Google Scholar]
- 24.Wong N, Blair AR, Morahan G, Andrikopoulos S. The deletion variant of nicotinamide nucleotide transhydrogenase (Nnt) does not affect insulin secretion or glucose tolerance. Endocrinology. 2010;151:96–102. doi: 10.1210/en.2009-0887. [DOI] [PubMed] [Google Scholar]
- 25.Kahn SE, Prigeon RL, McCulloch DK, et al. The contribution of insulin-dependent and insulin-independent glucose uptake to intravenous glucose tolerance in healthy human subjects. Diabetes. 1994;43:587–592. doi: 10.2337/diab.43.4.587. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz MW, Seeley RJ, Tschop MH, et al. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature. 2013;503:59–66. doi: 10.1038/nature12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alonso LC, Watanabe Y, Stefanovski D, et al. Simultaneous measurement of insulin sensitivity, insulin secretion, and the disposition index in conscious unhandled mice. Obesity. 2012;20:1403–1412. doi: 10.1038/oby.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughey CC, Wasserman DH, Lee-Young RS, Lantier L. Approach to assessing determinants of glucose homeostasis in the conscious mouse. Mammalian genome. 2014;25:522–538. doi: 10.1007/s00335-014-9533-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. The American journal of physiology. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 30.Pepin E, Higa A, Schuster-Klein C, et al. Deletion of apoptosis signal-regulating kinase 1 (ASK1) protects pancreatic beta-cells from stress-induced death but not from glucose homeostasis alterations under pro-inflammatory conditions. PloS one. 2014;9:e112714. doi: 10.1371/journal.pone.0112714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mulligan KX, Morris RT, Otero YF, Wasserman DH, McGuinness OP. Disassociation of muscle insulin signaling and insulin-stimulated glucose uptake during endotoxemia. PloS one. 2012;7:e30160. doi: 10.1371/journal.pone.0030160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moulle VS, Vivot K, Tremblay C, Zarrouki B, Ghislain J, Poitout V. Glucose and fatty acids synergistically and reversibly promote beta cell proliferation in rats. Diabetologia. 2017;60:879–888. doi: 10.1007/s00125-016-4197-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steil GM, Volund A, Kahn SE, Bergman RN. Reduced sample number for calculation of insulin sensitivity and glucose effectiveness from the minimal model. Suitability for use in population studies. Diabetes. 1993;42:250–256. doi: 10.2337/diab.42.2.250. [DOI] [PubMed] [Google Scholar]
- 34.Wasserman DH, Ayala JE, McGuinness OP. Lost in translation. Diabetes. 2009;58:1947–1950. doi: 10.2337/db09-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamontagne J, Jalbert-Arsenault E, Pepin E, et al. Pioglitazone acutely reduces energy metabolism and insulin secretion in rats. Diabetes. 2013;62:2122–2129. doi: 10.2337/db12-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 37.Fontes G, Ghislain J, Benterki I, et al. The ΔF508 mutation in the cystic fibrosis transmembrane conductance regulator is associated with progressive insulin resistance and decreased functional β-cell mass in mice. Diabetes. 2015;64:4112–4122. doi: 10.2337/db14-0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alquier T, Peyot ML, Latour MG, et al. Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes. 2009;58:2607–2615. doi: 10.2337/db09-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kebede M, Alquier T, Latour MG, Semache M, Tremblay C, Poitout V. The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding. Diabetes. 2008;57:2432–2437. doi: 10.2337/db08-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lan H, Hoos LM, Liu L, et al. Lack of FFAR1/GPR40 does not protect mice from high-fat diet-induced metabolic disease. Diabetes. 2008;57:2999–3006. doi: 10.2337/db08-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuda-Nagasumi K, Takami-Esaki R, Iwachidow K, et al. Lack of GPR40/FFAR1 does not induce diabetes even under insulin resistance condition. Diabetes Obes Metab. 2013;15:538–545. doi: 10.1111/dom.12065. [DOI] [PubMed] [Google Scholar]
- 42.Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. The Journal of clinical investigation. 1981;68:1456–1467. doi: 10.1172/JCI110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tschöp MH, Speakman JR, Arch JR, et al. A guide to analysis of mouse energy metabolism. Nature methods. 2011;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Speakman JR. Measuring energy metabolism in the mouse - theoretical, practical, and analytical considerations. Frontiers in physiology. 2013;4:34. doi: 10.3389/fphys.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moir L, Bentley L, Cox RD. Comprehensive energy balance measurements in mice. Current protocols in mouse biology. 2016;6:211–222. doi: 10.1002/cpmo.13. [DOI] [PubMed] [Google Scholar]
- 46.Ganeshan K, Chawla A. Warming the mouse to model human diseases. Nature reviews Endocrinology. 2017;13:458–465. doi: 10.1038/nrendo.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toth LA. The influence of the cage environment on rodent physiology and behavior: Implications for reproducibility of pre-clinical rodent research. Experimental neurology. 2015;270:72–77. doi: 10.1016/j.expneurol.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Dudele A, Rasmussen GM, Mayntz D, Malte H, Lund S, Wang T. Effects of ambient temperature on glucose tolerance and insulin sensitivity test outcomes in normal and obese C57 male mice. Physiological reports. 2015;3:e12396. doi: 10.14814/phy2.12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cui X, Nguyen NL, Zarebidaki E, et al. Thermoneutrality decreases thermogenic program and promotes adiposity in high-fat diet-fed mice. Physiological reports. 2016;4:e12799. doi: 10.14814/phy2.12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Butler AA, Kozak LP. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes. 2010;59:323–329. doi: 10.2337/db09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaiyala KJ, Morton GJ, Leroux BG, Ogimoto K, Wisse B, Schwartz MW. Identification of body fat mass as a major determinant of metabolic rate in mice. Diabetes. 2010;59:1657–1666. doi: 10.2337/db09-1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ambery AG, Tackett L, Penque BA, Hickman DL, Elmendorf JS. Effect of corncob bedding on feed conversion efficiency in a high-fat diet-induced prediabetic model in C57Bl/6J mice. Journal of the American Association for Laboratory Animal Science. 2014;53:449–451. [PMC free article] [PubMed] [Google Scholar]
- 53.Davies J. Regulation, necessity, and the misinterpretation of knockouts. BioEssays: news and reviews in molecular, cellular and developmental biology. 2009;31:826–830. doi: 10.1002/bies.200900044. [DOI] [PubMed] [Google Scholar]
- 54.Rankin MM, Kushner JA. Adaptive β-cell proliferation is severely restricted with advanced age. Diabetes. 2009;58:1365–1372. doi: 10.2337/db08-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in β-cell proliferation restricts the capacity of β-cell regeneration in mice. Diabetes. 2009;58:1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fontes G, Zarrouki B, Hagman DK, et al. Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass. Diabetologia. 2010;53:2369–2379. doi: 10.1007/s00125-010-1850-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zarrouki B, Benterki I, Fontes G, et al. Epidermal growth factor receptor signaling promotes pancreatic β-cell proliferation in response to nutrient excess in rats through mTOR and FOXM1. Diabetes. 2014;63:982–993. doi: 10.2337/db13-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34:309–338. doi: 10.1210/er.2012-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palmer BF, Clegg DJ. The sexual dimorphism of obesity. Mol Cell Endocrinol. 2015;402:113–119. doi: 10.1016/j.mce.2014.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Della Torre S, Maggi A. Sex differences: a resultant of an evolutionary pressure? Cell Metab. 2017;25:499–505. doi: 10.1016/j.cmet.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 61.McCullough LD, de Vries GJ, Miller VM, Becker JB, Sandberg K, McCarthy MM. NIH initiative to balance sex of animals in preclinical studies: generative questions to guide policy, implementation, and metrics. Biology of sex differences. 2014;5:15. doi: 10.1186/s13293-014-0015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mauvais-Jarvis F, Arnold AP, Reue K. A guide for the design of pre-clinical studies on sex differences in metabolism. Cell Metab. 2017;25:1216–1230. doi: 10.1016/j.cmet.2017.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ader DN, Johnson SB, Huang SW, Riley WJ. Group size, cage shelf level, and emotionality in non-obese diabetic mice: impact on onset and incidence of IDDM. Psychosomatic medicine. 1991;53:313–321. doi: 10.1097/00006842-199105000-00005. [DOI] [PubMed] [Google Scholar]
- 64.Ussar S, Fujisaka S, Kahn CR. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Molecular metabolism. 2016;5:795–803. doi: 10.1016/j.molmet.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grarup N, Sandholt CH, Hansen T, Pedersen O. Genetic susceptibility to type 2 diabetes and obesity: from genome-wide association studies to rare variants and beyond. Diabetologia. 2014;57:1528–1541. doi: 10.1007/s00125-014-3270-4. [DOI] [PubMed] [Google Scholar]
- 66.Alarcon C, Boland BB, Uchizono Y, et al. Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes. 2016;65:438–450. doi: 10.2337/db15-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sittig LJ, Carbonetto P, Engel KA, Krauss KS, Barrios-Camacho CM, Palmer AA. Genetic background limits generalizability of genotype-phenotype relationships. Neuron. 2016;91:1253–1259. doi: 10.1016/j.neuron.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cruciani-Guglielmacci C, Bellini L, Denom J, et al. Molecular phenotyping of multiple mouse strains under metabolic challenge uncovers a role for Elovl2 in glucose-induced insulin secretion. Molecular metabolism. 2017;6:340–351. doi: 10.1016/j.molmet.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kebede MA, Attie AD. Insights into obesity and diabetes at the intersection of mouse and human genetics. Trends Endocrinol Metab. 2014;25:493–501. doi: 10.1016/j.tem.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berglund ED, Li CY, Poffenberger G, et al. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes. 2008;57:1790–1799. doi: 10.2337/db07-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freeman H, Shimomura K, Horner E, Cox RD, Ashcroft FM. Nicotinamide nucleotide transhydrogenase: a key role in insulin secretion. Cell Metab. 2006;3:35–45. doi: 10.1016/j.cmet.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 72.Nicholson A, Reifsnyder PC, Malcolm RD, et al. Diet-induced obesity in two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity. 2010;18:1902–1905. doi: 10.1038/oby.2009.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fontaine DA, Davis DB. Attention to background strain is essential for metabolic research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes. 2016;65:25–33. doi: 10.2337/db15-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005;1:245–258. doi: 10.1016/j.cmet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 75.Nagasumi K, Esaki R, Iwachidow K, et al. Overexpression of GPR40 in pancreatic β-cells augments glucose-stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes. 2009;58:1067–1076. doi: 10.2337/db08-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bjursell M, Xu X, Admyre T, et al. The beneficial effects of n-3 polyunsaturated fatty acids on diet induced obesity and impaired glucose control do not require Gpr120. PloS one. 2014;9:e114942. doi: 10.1371/journal.pone.0114942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Paerregaard SI, Agerholm M, Serup AK, et al. FFAR4 (GPR120) signaling is not required for anti-inflammatory and insulin-sensitizing effects of omega-3 fatty acids. Mediators of inflammation. 2016;2016:1536047. doi: 10.1155/2016/1536047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pi J, Collins S. Reactive oxygen species and uncoupling protein 2 in pancreatic β-cell function. Diabetes Obes Metab 12 Suppl. 2010;2:141–148. doi: 10.1111/j.1463-1326.2010.01269.x. [DOI] [PubMed] [Google Scholar]
- 80.Seshadri N, Jonasson ME, Hunt KL, et al. Uncoupling protein 2 regulates daily rhythms of insulin secretion capacity in MIN6 cells and isolated islets from male mice. Molecular metabolism. 2017;6:760–769. doi: 10.1016/j.molmet.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pugach EK, Richmond PA, Azofeifa JG, Dowell RD, Leinwand LA. Prolonged cre expression driven by the alpha-myosin heavy chain promoter can be cardiotoxic. Journal of molecular and cellular cardiology. 2015;86:54–61. doi: 10.1016/j.yjmcc.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matthaei KI. Genetically manipulated mice: a powerful tool with unsuspected caveats. J Physiol. 2007;582:481–488. doi: 10.1113/jphysiol.2007.134908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rebholz SL, Jones T, Burke KT, et al. Multiparity leads to obesity and inflammation in mothers and obesity in male offspring. Am J Physiol Endocrinol Metab. 2012;302:E449–E457. doi: 10.1152/ajpendo.00487.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Abel ED, Peroni O, Kim JK, et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 85.Harno E, Cottrell EC, White A. Metabolic pitfalls of CNS cre-based technology. Cell Metab. 2013;18:21–28. doi: 10.1016/j.cmet.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 86.Magnuson MA, Osipovich AB. Pancreas-specific cre driver lines and considerations for their prudent use. Cell Metab. 2013;18:9–20. doi: 10.1016/j.cmet.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee JY, Ristow M, Lin X, White MF, Magnuson MA, Hennighausen L. RIP-cre revisited, evidence for impairments of pancreatic β-cell function. J Biol Chem. 2006;281:2649–2653. doi: 10.1074/jbc.M512373200. [DOI] [PubMed] [Google Scholar]
- 88.Wicksteed B, Brissova M, Yan W, et al. Conditional gene targeting in mouse pancreatic β-cells: Analysis of ectopic cre transgene expression in the brain. Diabetes. 2010;59:3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oropeza D, Jouvet N, Budry L, et al. Phenotypic characterization of MIP-creERT1Lphi mice with transgene-driven islet expression of human growth hormone. Diabetes. 2015;64:3798–3807. doi: 10.2337/db15-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkila E, Ferrer J. Ins1(cre) knock-in mice for beta cell-specific gene recombination. Diabetologia. 2015;58:558–565. doi: 10.1007/s00125-014-3468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shiota C, Prasadan K, Guo P, Fusco J, Xiao X, Gittes GK. Gcg creERT2 knockin mice as a tool for genetic manipulation in pancreatic alpha cells. Diabetologia. 2017 doi: 10.1007/s00125-017-4425-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schulz TJ, Glaubitz M, Kuhlow D, et al. Variable expression of cre recombinase transgenes precludes reliable prediction of tissue-specific gene disruption by tail-biopsy genotyping. PloS one. 2007;2:e1013. doi: 10.1371/journal.pone.0001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Douglass JD, Dorfman MD, Fasnacht R, Shaffer LD, Thaler JP. Astrocyte IKKbeta/NF-kappaB signaling is required for diet-induced obesity and hypothalamic inflammation. Molecular metabolism. 2017;6:366–373. doi: 10.1016/j.molmet.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brouwers B, de Faudeur G, Osipovich AB, et al. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab. 2014;20:979–990. doi: 10.1016/j.cmet.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Declercq J, Brouwers B, Pruniau VP, et al. Metabolic and behavioural phenotypes in Nestin-cre mice are caused by hypothalamic expression of human growth hormone. PloS one. 2015;10:e0135502. doi: 10.1371/journal.pone.0135502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Koitabashi N, Bedja D, Zaiman AL, et al. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ Res. 2009;105:12–15. doi: 10.1161/CIRCRESAHA.109.198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morimoto M, Kopan R. rtTA toxicity limits the usefulness of the SP-C-rtTA transgenic mouse. Dev Biol. 2009;325:171–178. doi: 10.1016/j.ydbio.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bersell K, Choudhury S, Mollova M, et al. Moderate and high amounts of tamoxifen in αMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Disease models & mechanisms. 2013;6:1459–1469. doi: 10.1242/dmm.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kaelin WG., Jr Publish houses of brick, not mansions of straw. Nature. 2017;545:387. doi: 10.1038/545387a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.