

Abstract
INTRODUCTION
The boronic acid is a functional group of enormous utility in materials science, chemosensor development, and drug discovery. In medicinal chemistry, boronic acids have been harnessed as a replacement for various structural motifs (a bioisostere) to improve the potency or pharmacokinetic profiles of lead compounds. However, the widespread incorporation of alkyl boronic acids has been largely hampered by the challenges associated with their preparation. Consequently, only two alkyl boronic acids are currently in clinical use, namely Velcade and Ninlaro. Few methods are capable of delivering alkyl boronates from readily available starting materials; most exhibit modest functional group compatibility. Indeed, boronate motifs are often installed at the early stage of a synthesis and thus consume disproportionate effort from the standpoint of planning and manipulation in multistep processes.
RATIONALE
Alkyl carboxylic acids, as the most variegated chemical building blocks on Earth, are present in a myriad of natural products and medicines. They represent an ideal precursor to boronic acids. Previous efforts from our laboratory revealed that, through the intermediacy of simple redox-active esters (RAEs, e.g., N-hydroxyphthalimide esters), alkyl carboxylic acids could be harnessed as convenient alkyl halide surrogates in metal-catalyzed decarboxylative cross-coupling reactions with carbon nucleophiles, using the same activating principles as amide bond formation. It was therefore surmised that such reactivity could be exploited in a decarboxylative borylation process wherein structurally diverse and ever-present carboxylic acids could be converted directly into high-value boronic acids.
RESULTS
Through the exclusive use of N-hydroxyphthalimide RAEs, a simple means to convert carboxylic acids into boronate esters was enabled with an inexpensive nickel catalyst. This reaction was broad in scope (>40 examples) and demonstrated excellent functional group compatibility (tolerating alkyl/aryl halides, amides/carbamates, alcohols, ketones, and olefins), and high levels of diastereoselectivity, allowing transformations of densely functionalized drug molecules (e.g., vancomycin and Lipitor) and natural products (e.g., enoxolone) into the analogous boronic acids. This method’s unique capacity to access α-amino boronic acids from native peptides not only allowed the concise syntheses of both Velcade and Ninlaro, it also enabled the expedient discovery of three highly potent human neutrophil elastase (HNE) inhibitors, the most potent of which has shown improved in vitro inhibitory activities (IC50 = 15 pM, Ki = 3.7 pM) relative to leading candidates previously tested in clinical trials. Enzymatic and pharmacokinetic studies indicated high functional stability in physiologically relevant media.
CONCLUSION
The nickel-catalyzed decarboxylative cross-coupling of RAEs enables substitution of ubiquitous alkyl carboxylic acids with boronate esters using an inexpensive boron source: B2pin2 (Bpin = pinacol boronate). This process provides simple and practical access to complex boronic acids that were heretofore difficult to prepare. The wide diversity of useful reactivity that is exclusive to boronic acids, such as cross-coupling, oxidation, amination, and homologation, will open distinct possibilities in retrosynthetic analysis. This work may also accelerate the discovery and development of new boron-containing therapeutics.
Graphical abstract
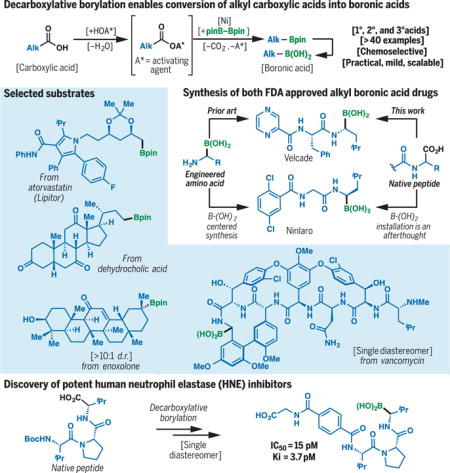
Boronic acids and their esters are of paramount importance to all facets of chemical science. Although their popularization has largely been spurred by the incredible utility of the Suzuki coupling (1), boronic acids have, to date, found countless applications in fields far outside of cross-coupling, such as materials science (2), chemosensor development (3), and drug discovery (4, 5). Boronic acids display unique chemical and biological properties as a result of their Lewis acidity, their propensity to reversibly engage various nucleophiles (e.g., alcohols and amines), and their ability to form hydrogen bonds. For example, in materials science, the reversible covalent bonding of boronic acids has enabled the development of self-assembled nanomaterials, hydrogels, and macromolecular saccharide sensors (2, 3). In medicinal chemistry, boronic acids have been harnessed as bioisosteres, where they replace structural motifs of similar physical and chemical properties, such as carboxylic acids (6), to alter the physiochemical properties of lead candidates (4, 5). The reversible covalent binding properties allow tuning of the amount of time the drug remains on the target at an active dose as evidenced by its pharmacodynamic activity rather than by affinity, while avoiding permanent covalent adducts with off-target proteins that could lead to associated toxicity.
Alkyl boronic acids, and α-amino boronic acids in particular, have attracted considerable attention as potent protease inhibitors (7). Currently, two alkyl boronic acids are approved by the FDA for various oncology indications: Ninlaro (1) (Fig. 1A) and Velcade (49). However, efforts to fully exploit the vast potential of these promising medicinal scaffolds are often hampered by the challenges associated with their preparation; relatively few complex alkyl boronic acids have been synthesized for biological evaluations (4). Unlike carboxylic acids, which are ubiquitous in nature and inexpensive, boronic acids are almost entirely derived through synthesis. The retrosynthetic analysis of alkyl boronic acids can itself be a deterrent to their incorporation into drug candidates. General methods to forge alkyl C–B bonds include hydroboration of alkenes (8, 9), Miyaura borylation of alkyl halides (10–14), transmetallation (e.g., with alkyl organolithium species) (15), and conjugate addition (16, 17). α-Amino boronic acids are typically accessed through metal-catalyzed addition of diboron species onto imines (18), where elegant asymmetric variants have been reported (19). Although these approaches have been highly enabling, few of them use naturally occurring or readily available starting materials; many of these methods also possess modest functional group compatibility. Path-pointing advances in metal-catalyzed C–H activation highlight the possibilities of transforming bonds directly into alkyl boronates at a late stage of a synthesis (20). Currently, however, alkyl boronic acids are usually installed at an early stage when few reactive functionalities are present. Thus, as illustrated with 1 (Fig. 1A), the conventional approach focuses all strategic attention on the means by which the boron atom will be incorporated, even though this represents <5% of the total molecular weight of 1 (21). The synthesis of an engineered amino acid is therefore required. Any systematic examination of the structure-activity relationship of lead compounds would require each analog to be made individually.
Fig. 1. Development of the decarboxylative borylation reaction.
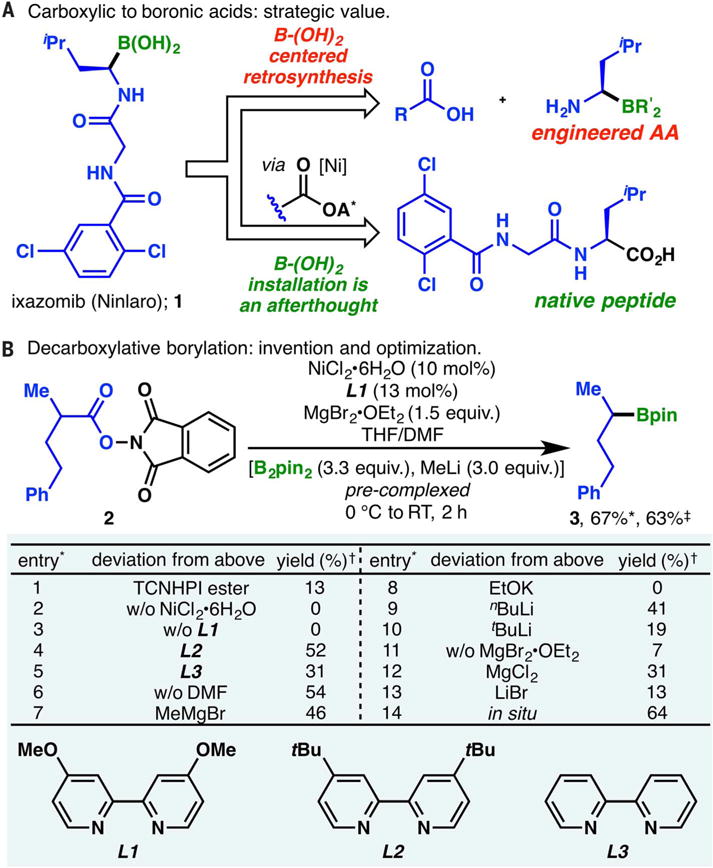
(A) Strategic value of the decarboxylative borylation illustrated through the retrosynthetic analysis of Ninlaro (1). (B) Development and optimization of the decarboxylative borylation reaction. Footnotes: *Reaction conducted on 0.10 mmol scale. †Yield by gas chromatography. ‡Isolated yield.
In contrast, the direct transformation of a carboxylic acid–containing native peptide into the corresponding boronic acid at a late stage constitutes a far easier and more logical approach. Given the sheer number of alkyl carboxylic acids in feedstock chemicals, natural products, and drug molecules, this transformation could provide the unique opportunity to expediently procure a myriad of previously difficult-to-access boronic acids as versatile building blocks, functional materials, and potent medicines.
Here, we present a simple method for nickel-catalyzed decarboxylative borylation that is mild, scalable, and general across a range of primary, secondary, tertiary, peptidic, and even natural product–derived substrates. A diverse array of boronates that would otherwise require lengthy de novo synthesis was furnished directly from the corresponding carboxylic acids. This method’s capacity to transform native peptides into α-amino boronic acids has led to the discovery of three potent small-molecule elastase inhibitors.
Development of the decarboxylative borylation reaction
Recent efforts in our laboratory revealed redox-active esters (RAEs; e.g., N-hydroxyphthalimide ester 2) derived from alkyl carboxylic acids as convenient surrogates for alkyl halides in nickel-or iron-catalyzed cross-coupling reactions. These versatile intermediates, most commonly used in amide bond–forming reactions, have enabled practical means of C–C bond formation in various modalities, including decarboxylative Negishi (22, 23), Suzuki (24), and Kumada (25) couplings, as well as Giese reactions (26). Although RAEs have yet to be used in carbon-heteroatom cross-coupling reactions, our earlier discoveries, coupled with Dudnik and Fu’s (10) pioneering work on nickel-catalyzed Miyaura borylation of alkyl halides (11–14), prompted us to investigate the possibility of harnessing them for C–B bond formation, thereby achieving the direct conversion of alkyl carboxylic acids into boronic acid derivatives.
Realization of this transformation required considerable experimentation. Figure 1B provides the optimal reaction parameters alongside an abbreviated picture of the optimization process on 2-methyl-4-phenylbutanoic acid. N-hydroxyphthalimide (NHPI) ester (2) proved to be the optimal substrate for borylation with B2pin2 (Bpin = pinacol boronate); other RAEs such as tetrachloro-NHPI (TCNHPI) ester were less effective (entry 1). The inexpensive combination of NiCl2•6H2O and bipyridine ligand L1 emerged as the best catalyst system after an exhaustive screening; the use of alternative catalysts (table S4) or ligands (entries 3 to 5; see table S5) had deleterious effects. The choice of solvent was critical: A binary mixture of tetrahydrofuran (THF) and dimethylformamide (DMF) gave the optimal result; lower yields were observed in the absence of DMF (entry 6). Premixing methyl lithium with B2pin2 was necessary to activate the diboron species toward transmetallation; numerous other activating agents surveyed (e.g., entries 7 to 10) were less effective, affording borylation products in lower yields, if at all. Magnesium salts were also indispensable to the reaction: In the absence of the MgBr2•OEt2, virtually no product was obtained (entry 11), whereas other Lewis acidic additives surveyed (entries 12 and 13) gave lower yields. Although its precise role is unclear, MgBr2•OEt2 is hypothesized to promote the transmetallation of boronate species onto the metal catalyst (27, 28). Bpin ester product 3 could be accessed directly from the carboxylic acid in comparable yields using a one-pot procedure wherein a RAE was formed in situ, in a similar vein to amide coupling (entry 14; see also Fig. 2). Overall, the reaction was found to proceed smoothly over the course of 2 hours (1 hour at 0°C and 1 hour at room temperature).
Fig. 2. Scope of the Ni-catalyzed decarboxylative borylation reaction of redox-active esters.
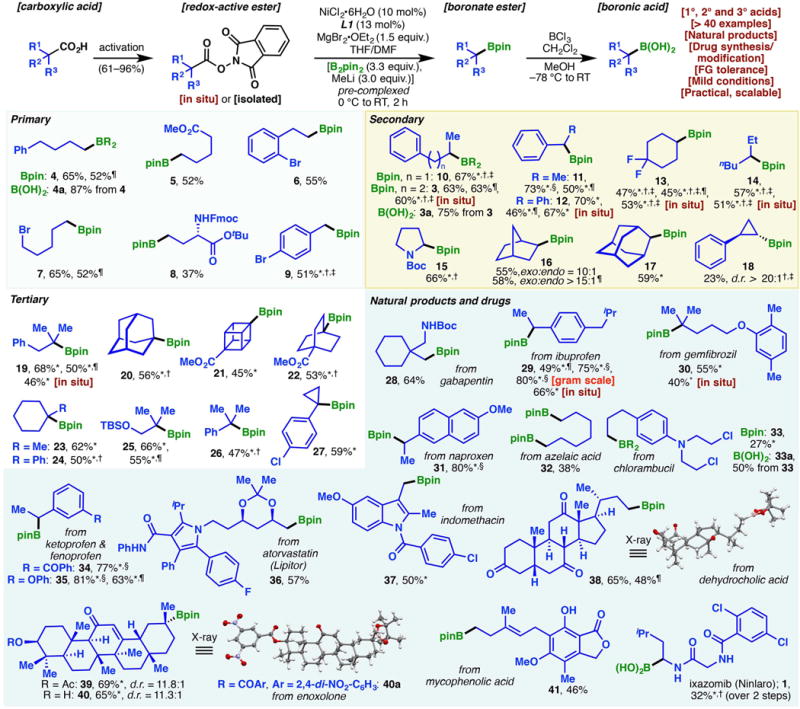
Standard reaction conditions: Redox-active NHPI ester (1.0 equiv), NiCl2•6H2O (10 mol %), L1 (13 mol %), MgBr2•OEt2 (1.5 equiv), [B2pin2 (3.3 equiv), MeLi (3.0 equiv)] precomplexed,THF/DMF (2.5:1), 0°C to room temperature, 2 hours. Footnotes: *Using THF as the solvent. †Using L2 as the ligand. ‡Using tetrachloro-NHPI (TCNHPI) ester. §Using 1.0 equiv of MgBr2•OEt2. ¶Using 2.5 mol % of NiCl2•6H2O and 3.3 mol % of ligand (L1 or L2). (See supplementary materials for experimental details.)
Scope of the decarboxylative borylation reaction
With the optimized conditions in hand, we next explored the scope of this methodology. RAEs derived from a broad selection of primary, secondary, and tertiary carboxylic acids were all found to be viable substrates (Fig. 2). These encompass acyclic, cyclic, caged, bridgehead, fluoroalkyl, and benzylic acids, which were transformed to the corresponding Bpin esters smoothly. Scalability of the reaction was evident through the preparation of 29 in gram scale. Additionally, 12 of the products (3, 4, 7, 11, 12, 13, 16, 19, 25, 29, 35, 38) were obtained in comparable yields when only 2.5 mol % of nickel catalyst (3.3 mol % of ligand) was used, further attesting to the adaptability of this method in a process setting.
Because the methyl lithium was premixed with B2pin2 to form the [B2pin2Me]Li complex, strongly nucleophilic/basic organometallic species were sequestered from the substrate: A gamut of functionalities such as ethers (30, 31, 35, 37, 41), esters (5, 8, 21, 22, 39, 41), carbamates/amides (1, 8, 15, 28, 36, 37), ketones (34, 38, 39, 40), olefins (39, 40, 41), and alcohol/phenol (40, 41) were left unscathed under the mild reaction conditions. Indeed, even the highly base-sensitive fluorenylmethyloxycarbonyl group was tolerated (see 8). The compatibility with alkyl bromides (7) and chlorides (33) points to the orthogonality of this reaction to halide-based Miyaura borylations. Enoxolone-derived boronates 39 and 40 were obtained in similar yields, which suggests that the free hydroxyl group had minimal influence on the reaction. The discrete isolation of RAEs, as alluded to earlier, is not necessary. Tertiary and secondary boronate esters can be prepared directly from carboxylic acids when RAEs are generated in situ. This one-pot procedure also pertains to some primary substrates, albeit in lower yields.
Although some of the products presented herein (e.g., 9, 16, 17, 19, 20, 23) can be synthesized from the analogous halides via Miyaura borylation reactions (10–14), organohalides are often not commercially available and require extraneous steps to prepare (usually from the corresponding alcohols). Conversely, the use of readily available carboxylic acids largely circumvents this problem. A great majority of products in Fig. 2 are derived from commercially available acids. For instance, 21 was conveniently prepared from a cubane-based carboxylic acid, whereas the reported synthesis of the analogous bromide enlisted a harsh Hunsdiecker reaction (Br2 and HgO) on the same acid (29). Furthermore, the scope of this borylation protocol can be extended to amino acid derivatives to furnish α-amino boronate esters such as 15. The synthesis of 15 through halide-based Miyaura borylation is simply not feasible; the corresponding α-amino halide starting material would be unstable. In this regard, the decarboxylative borylation strategy allows explorations of previously elusive chemical space.
The prevalence of alkyl carboxylic acids is demonstrated by their presence in more than 450 marketed drug molecules (30). To this end, the impressive chemoselectivity of this reaction offers the unique opportunity to pursue late-stage modifications of bioactive molecules that are densely adorned with reactive functionalities. More than 10 carboxylate-containing drug molecules or natural products have been successfully converted into Bpin esters (28 to 41), which would otherwise only be accessible through multistep functional group interconversions or de novo syntheses.
Synthetic applications of the decarboxylative borylation reaction
The Bpin esters can be conveniently hydrolyzed into the corresponding boronic acids (e.g., 1, 3a, 4a, 33a) (Fig. 2). This allows the transformation of carboxylic acids into their borono-bioisosteres to identify compounds with superior potency or pharmacokinetic properties. Alternatively, boronate esters could be diversified into a variety of structural motifs (31–33). As an illustrative example, the Lipitor-derived Bpin ester (36) can be expediently elaborated into the corresponding carbamate (36a) (34) or alcohol (36b) upon treatment with appropriate oxidants (Fig. 3A). Under conditions reported in (35), 36c and 36d were directly accessed through reaction with aryllithium species. Decarboxylative borylation could also convert RAEs, which are electrophiles in cross-couplings, into Bpin esters that serve as nucleophiles in Suzuki reactions (e.g., 36 to 36e and 36 to 36f) (36). This “umpolung” approach is particularly strategic in the case of 36e, as 2-pyridylboronic acid or organozinc species are often not viable Suzuki/Negishi coupling substrates, owing to a lack of stability.
Fig. 3. Applications of the decarboxylative borylation reaction.
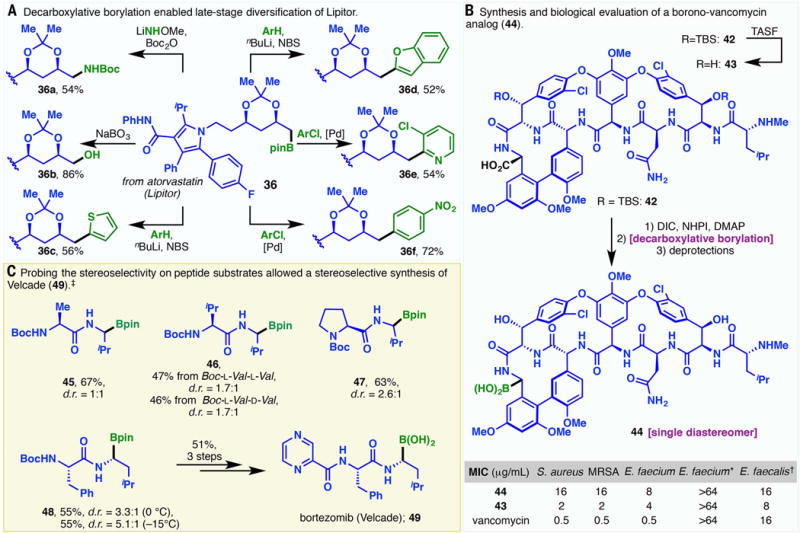
(A) Late-stage diversification of Lipitor. (B) Synthesis and biological evaluation of 44, a boronic acid analog of vancomycin. (C) Probing the stereoselectivity of peptide substrates and the ensuing stereoselective synthesis of Velcade (49). Footnotes: *VRE(VanA). †VRE(VanB). ‡Yield and diastereoselectivity refer to the decarboxylative borylation of the corresponding RAE. See supplementary materials for experimental details.
Moreover, selective decarboxylative borylation at the C terminus of native peptides allowed rapid access to coveted α-amino boronic acids, which are privileged medicinal chemistry motifs (18, 37). Ninlaro (1), for example, was obtained in three steps from a simple peptide (Fig. 2). This opens up a distinct dimension to the study of peptide-based therapeutics: In perhaps the most striking example, vancomycin was converted into a boronic acid analog (44) through the decarboxylative borylation of 42 (Fig. 3B) (38). This process proceeded smoothly in the presence of four methylated phenoxy groups, two tert-butyldimethylsilyl ethers, two aryl chlorides, six secondary amides, one primary amide, one secondary amine, and seven epimerizable stereocenters. Although 44 showed less activity than the parent acid 43 (Fig. 3B and table S12), such remarkable chemo-selectivity still attests to the potential utility of this reaction.
The unpredictable stereoselectivity of radical processes often presents a hurdle to their broad adoption in late-stage modifications of drug leads or natural products. Complex α-amino boronic acid 44 was obtained as a single diastereomer in this radical-based decarboxylative borylation reaction. This result prompted us to investigate the stereoselectivity of the decarboxylative borylation on several dipeptides (Fig. 3C). We found that increased steric bulk on the N-terminal residue resulted in better diastereoselectivity: Both diastereomers were furnished in almost equal quantities for 45, whereas higher selectivities were observed for 46 and 47. Meanwhile, 46 was obtained in the same diastereomeric ratio from (tert-butoxycarbonyl)-L-valine-L-valine and (tert-butoxycarbonyl)-L-valine-D-valine. Lower reaction temperatures could also be used to enhance the stereoselectivity. At −15°C, 48 was furnished in greater than 5:1 diastereomeric ratio, enabling a stereoselective synthesis of Velcade (49) in a short sequence.
Discovery of potent human neutrophil elastase inhibitors
By wedding the rich medicinal potential of boronates to the ubiquity of alkyl carboxylic acids, the decarboxylative borylation reaction has the potential to open up new vistas in drug development. For example, application of the decarboxylative borylation reaction to readily available tripeptides allowed the expedient preparations of 50, 51, and 52, which were formed as single diastereomers and were found to be potent inhibitors of human neutrophil elastase (HNE) (Fig. 4, A and B). Notably, the carboxylic acid precursor to 50 (50b) was found to be devoid of any inhibitory activities, whereas 50 and 51 displayed substantially enhanced potency relative to their trifluoromethyl ketone congeners (50a and 51a), which have been examined in phase II clinical trials for lung diseases such as cystic fibrosis (CF) (39–45). HNE, a highly active serine protease, plays a pivotal role in the immune response, tissue remodeling, and onset and resolution of inflammation by breaking down mechanically important structures of the body’s cellular matrix as well as proteins of foreign origin (46). Although five generations of HNE inhibitors have been evaluated clinically in multiple inflammatory lung diseases (e.g., CF, emphysema, and bronchiectasis), none have been sufficiently efficacious in humans to make a significant impact in these conditions (46).
Fig. 4. Discovery of novel human neutrophil elastase (HNE) inhibitors.
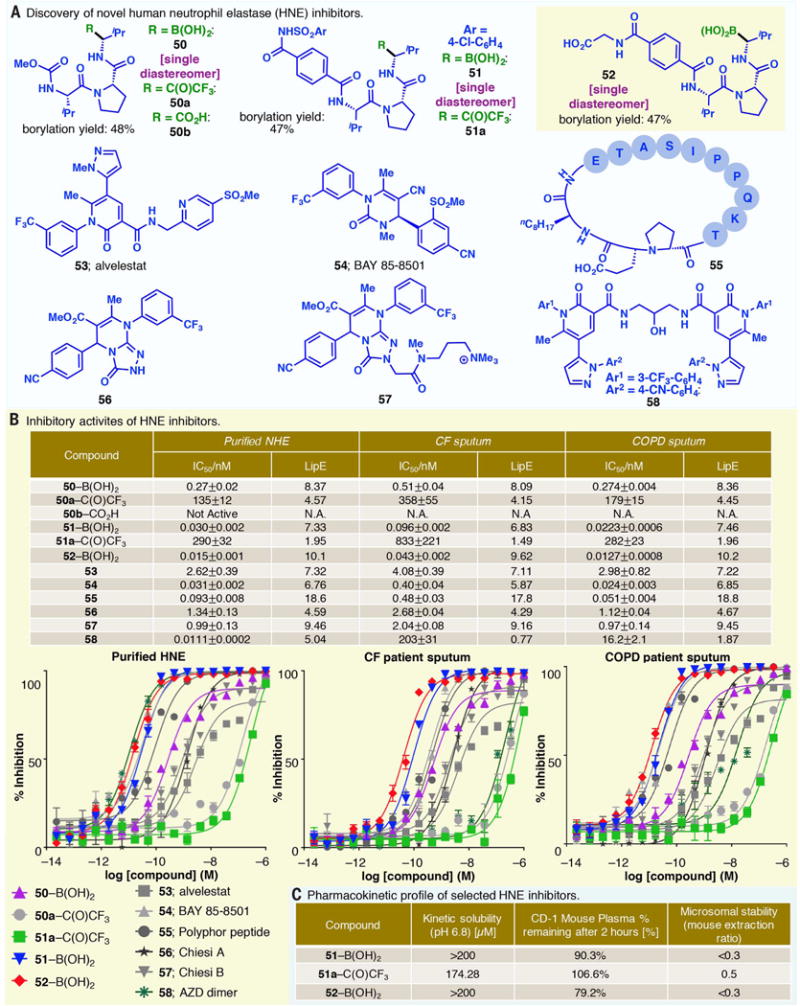
(A) Structures of selected elastase inhibitors. (B) IC50 values (nM) of selected elastase inhibitors (average ± SD, n = 3 plotted, representative of three independent, triplicate experiments). A nonlinear three-parameter log inhibitor curve was used to calculate the IC50 values. Curve fit statistics: Purified HNE, R2 ≥ 0.95; CF patient sputum, R2 ≥ 0.93; COPD patient sputum, R2 ≥ 0.93. (C) ADME profile of 51, 51a, and 52.
Toward this end, 52 exhibited an IC50 value of 15 pM, where IC50 is the concentration of an inhibitor when the response (or inhibition of cleavage of the HNE substrate) is reduced by half (Ki = 3.7 pM where Ki refers to the concentration of inhibitor at which the reaction rate is half of the maximum reaction rate under saturating substrate conditions) against purified HNE; 51 exhibited an IC50 of 30 pM (Ki = 34 pM) against purified HNE (Fig. 4B). The IC50 values were determined head-to-head with other preclinically and clinically validated HNE inhibitors (53–57), including BAY 85-8501 (54, a leading clinical candidate with reported Ki = 80 pM) (47), 55 (an analog in a series of peptide-based elastase inhibitors currently in clinical trials) (48), as well as 56 and 57 (reported by Chiesi Pharmaceuticals) (49). Additionally, 51 and 52 retained much of their inhibitory activities in sputum samples of CF and chronic obstructive pulmonary disease (COPD) patients, underscoring the potency of these compounds in the context of a more pathophysiologically relevant environment than the traditional biochemical assay. Conversely, although dimeric compound 58 from AstraZeneca (IC50 = 11 pM, Ki = 2.7 pM) (50) and BAY 85-8501 (54) displayed low IC50 values, their potencies diminished in CF patient sputum. Comparison of the lipophilic efficiency (LipE) values in COPD sputum revealed that the superior potency of 52 is not driven by increased lipophilicity (10.2 for 52 versus 9.46 for 57) (51).
Additionally, the IC50 value of 52 was found to remain unchanged with increasing incubation times (5 to 60 min), whereas that of 58, a noncovalent inhibitor, exhibited a factor of 55 increase in potency under the same conditions. These data retain the profile that is expected: Compound 52 is behaving like a partial mechanism-based inhibitor (or a covalent reversible inhibitor), likely as a result of the potentially slow off-rate of the α-amino boronic acid. This correlates with tighter binding and potentially long residence time, as seen in other amino boronic acid compounds and unlike the many reversible elastase inhibitors (e.g., 58) (52). Clinically, this mechanism has been proven successfully through Velcade (49), which inhibits the catalytic site of the 26S proteasome. In this example, covalent reversible bonding between the boronate and the nucleophilic oxygen results in a slow disassociation rate (53, 54). Because many of the early clinical elastase inhibitors are mechanism-based inhibitors that act through a covalent manner with the enzyme’s active site or nonreactive, reversible inhibitors (such as 54, BAY 85-8501, one of the most potent HNE inhibitors reported to date), the high potency of 52 and the inherent mechanism of the amino boronic acids could help to improve the limited clinical efficacy demonstrated to date. Through this “hybrid” enzymatic inhibitory approach (based on Fischer’s lock-and-key model and Ehrlich’s pharmacophore model), boronic acids such as 52, which combine a rapid, potent binding with a slow off-rate, may effectively restore the protease-antiprotease balance in a clinical setting. They could therefore be tuned rapidly toward lung-specific clinical applications.
To further evaluate the therapeutic potential of 51 and 52, we probed the in vitro adsorption, distribution metabolism, and excretion properties (ADME properties) to determine whether any deleterious effects of the boronate replacements of the trifluoromethyl ketone would be revealed (Fig. 4C). These amino boronic acids displayed kinetic solubility comparable to that of the trifluoromethyl ketone analog (51a). Substantial proportions of 51 and 52 (90.3% and 79.2%, respectively) were found to be intact in CD-1 mouse plasma after 2 hours. The metabolic stability exhibited by 51 and 52 was similar to that of the trifluoromethyl ketone 51a. 51 and 51a also demonstrated similar levels of permeability in human Caco-2 cells (see table S19). These data suggest that the novel boronates simply improve potency without changing the drug-like properties of their ketone congeners.
Method summary
Procedurally, the conversion of redox-active esters into boronate esters was achieved in three stages: the preparation of the catalyst mixture, the preparation of the [B2pin2Me]Li complex, and the nickel-catalyzed decarboxylative borylation reaction. The following is an abbreviated experimental protocol with a graphical guide (Fig. 5, from the gram-scale decarboxylative borylation of ibuprofen-derived RAE). See the supplementary materials for comprehensive information on the commercial source and purity of chemicals or variations in experimental details for different substrate classes.
Fig. 5. A graphical guide to the decarboxylative borylation reaction.

(A) General transformation and materials for the reaction. (B) Addition of reagents. (C) Observations during the reaction. (D) Work-up and purification of the reaction mixture.
Preparation of NiCl2•6H2O/ligand stock solution or suspension
A flask charged with NiCl2•6H2O (1.0 equiv) and ligand (L1 or L2, 1.3 equiv) was evacuated and backfilled with argon three times. After addition of THF (NiCl2•6H2O concentration, 0.025 M) or DMF (NiCl2•6H2O concentration, 0.050 M), the resulting mixture was stirred at room temperature overnight (or until no granular NiCl2•6H2O was observed) to afford a green solution or suspension (for an example, see Fig. 5A, center left).
Preparation of [B2pin2Me]Li complex
MeLi (1.6 M in Et2O, 1.0 equiv) was added to a solution of B2pin2 (1.1 equiv) in THF (B2pin2 concentration, 1.1 M) at 0°C under argon. The reaction mixture was warmed to room temperature and stirred for 1 hour to afford a milky white suspension (Fig. 5A, center right).
Ni-catalyzed decarboxylative borylation
A flask charged with the redox-active ester (1.0 equiv) and MgBr2•OEt2 (1.5 equiv) was evacuated and backfilled with argon three times (Fig. 5A, left). Catalyst solution or suspension (containing 10 mol % NiCl2•6H2O and 13 mol % ligand) was added via a syringe (Fig. 5B, left). When a catalyst suspension/solution in DMF was used, an additional portion of THF (twice the volume of the DMF suspension/solution needed) was added to the reaction vessel before addition of the catalyst mixture. (This process can be exothermic on large scales, and cooling by ice/water bath may be necessary.) The resulting mixture was stirred vigorously until no visible solid was observed at the bottom of the reaction vessel (Fig. 5B, center); this process was found to be accelerated by sonication. This mixture was cooled to 0°C before a suspension of [B2pin2Me]Li in THF (3 equiv) was added in one portion (Fig. 5B, right). After stirring for 1 hour at 0°C (Fig. 5C, left), the reaction was warmed to room temperature and stirred for another 1 hour (Fig. 5C, center). When thin-layer chromatography (TLC) analysis indicated the completion of the reaction (Fig. 5C, right), the reaction was quenched with aqueous HCl (0.1 M) or saturated aqueous NH4Cl and extracted with diethyl ether (Et2O) or ethyl acetate (EtOAc). Alternatively, on larger scales, as is the case shown in Fig. 5, the reaction mixture was directly poured onto Et2O (Fig. 5D, left) and the resulting suspension was filtered through a pad of silica gel and celite (Fig. 5D, center). Purification by flash column chromatography (Fig. 5D, right) afforded the desired Bpin ester.
Supplementary Material
Acknowledgments
Supported by a China Scholarship Council postdoctoral fellowship (C.L.); a Shanghai Institute of Organic Chemistry, Zhejiang Medicine Co., and Pharmaron postdoctoral fellowship (J.W.); a Crohn’s and Colitis Foundation of America fellowship (S.Y.); a Deutsche Forschungsgemeinschaft postdoctoral fellowship (M.T.); NIH grant GM-118176; and Bristol-Myers Squibb. We thank D. L. Boger and A. Okano for insightful discussions and generous donation of vancomycin; F. E. Romesberg for help with antibacterial assays; D.-H. Huang and L. Pasternack for assistance with nuclear magnetic resonance (NMR) spectroscopy; and A. L. Rheingold, C. E. Moore, and M. Gembicky for x-ray analysis. Crystallographic data for compounds 28, 38, and 40a are available free of charge from the Cambridge Crystallographic Data Centre under reference numbers CCDC 1525341, 1525343, and 1533051, respectively. Experimental procedures, frequently asked questions, extensive optimization data, biological testing protocols, 1H NMR spectra, 13C NMR spectra, and MS data are available in the supplementary materials. A provisional U.S. patent application on this work has been filed (application number 62474181), where P.S.B., C.L., J.W., A.K.C., M.K., S.Y., and K.A.J. are listed as inventors.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/356/6342/eaam7355/suppl/DC1
Materials and Methods
NMR spectra
References (55–69)
REFERENCES AND NOTES
- 1.Suzuki A. Cross-coupling reactions of organoboranes: An easy way to construct C-C bonds (Nobel Lecture) Angew Chem Int Ed. 2011;50:6722–6737. doi: 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]
- 2.Brooks WLA, Sumerlin BS. Synthesis and applications of boronic acid-containing polymers: From materials to medicine. Chem Rev. 2016;116:1375–1397. doi: 10.1021/acs.chemrev.5b00300. [DOI] [PubMed] [Google Scholar]
- 3.Bull SD, et al. Exploiting the reversible covalent bonding of boronic acids: Recognition, sensing, and assembly. Acc Chem Res. 2013;46:312–326. doi: 10.1021/ar300130w. [DOI] [PubMed] [Google Scholar]
- 4.Trippier PC, McGuigan C. Boronic acids in medicinal chemistry: Anticancer, antibacterial and antiviral applications. MedChemComm. 2010;1:183. doi: 10.1039/c0md00119h. [DOI] [Google Scholar]
- 5.Draganov A, Wang D, Wang B. The future of boron in medicinal chemistry: Therapeutic and diagnostic applications. In. In: Schwarz J, editor. Atypical Elements in Drug Design. Springer; 2014. pp. 1–27. [DOI] [Google Scholar]
- 6.Ballatore C, Huryn DM, Smith AB., 3rd Carboxylic acid (bio) isosteres in drug design. ChemMedChem. 2013;8:385–395. doi: 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smoum R, Rubinstein A, Dembitsky VM, Srebnik M. Boron containing compounds as protease inhibitors. Chem Rev. 2012;112:4156–4220. doi: 10.1021/cr608202m. [DOI] [PubMed] [Google Scholar]
- 8.Brown HC. Hydroboration. Benjamin/Cummings; 1980. [Google Scholar]
- 9.Vogels CM, Westcott SA. Recent advances in organic synthesis using transition metal-catalyzed hydroborations. Curr Org Chem. 2005;9:687–699. doi: 10.2174/1385272053765060. [DOI] [Google Scholar]
- 10.Dudnik AS, Fu GC. Nickel-catalyzed coupling reactions of alkyl electrophiles, including unactivated tertiary halides, to generate carbon-boron bonds. J Am Chem Soc. 2012;134:10693–10697. doi: 10.1021/ja304068t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atack TC, Lecker RM, Cook SP. Iron-catalyzed borylation of alkyl electrophiles. J Am Chem Soc. 2014;136:9521–9523. doi: 10.1021/ja505199u. [DOI] [PubMed] [Google Scholar]
- 12.Bedford RB, et al. Iron-catalyzed borylation of alkyl, allyl, and aryl halides: Isolation of an iron(I) boryl complex. Organometallics. 2014;33:5940–5943. doi: 10.1021/om500847j. [DOI] [Google Scholar]
- 13.Yang CT, et al. Alkylboronic esters from copper-catalyzed borylation of primary and secondary alkyl halides and pseudohalides. Angew Chem Int Ed. 2012;51:528–532. doi: 10.1002/anie.201106299. [DOI] [PubMed] [Google Scholar]
- 14.Ito H, Kubota K. Copper(I)-catalyzed boryl substitution of unactivated alkyl halides. Org Lett. 2012;14:890–893. doi: 10.1021/ol203413w. [DOI] [PubMed] [Google Scholar]
- 15.Brown HC, Cole TE. Organoboranes. 31. A simple preparation of boronic esters from organolithium reagents and selected trialkoxyboranes. Organometallics. 1983;2:1316–1319. doi: 10.1021/om50004a009. [DOI] [Google Scholar]
- 16.Lee KS, Zhugralin AR, Hoveyda AH. Efficient C-B bond formation promoted by N-heterocyclic carbenes: Synthesis of tertiary and quaternary B-substituted carbons through metal-free catalytic boron conjugate additions to cyclic and acyclic α,β-unsaturated carbonyls. J Am Chem Soc. 2009;131:7253–7255. doi: 10.1021/ja902889s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schiffner JA, Müther K, Oestreich M. Enantioselective conjugate borylation. Angew Chem Int Ed. 2010;49:1194–1196. doi: 10.1002/anie.200906521. [DOI] [PubMed] [Google Scholar]
- 18.Andrés P, Ballano G, Calaza MI, Cativiela C. Synthesis of α-aminoboronic acids. Chem Soc Rev. 2016;45:2291–2307. doi: 10.1039/C5CS00886G. [DOI] [PubMed] [Google Scholar]
- 19.Beenen MA, An C, Ellman JA. Asymmetric copper-catalyzed synthesis of α-amino boronate esters from N-tert-butanesulfinyl aldimines. J Am Chem Soc. 2008;130:6910–6911. doi: 10.1021/ja800829y. [DOI] [PubMed] [Google Scholar]
- 20.Mkhalid IA, Barnard JH, Marder TB, Murphy JM, Hartwig JF. C-H activation for the construction of C-B bonds. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 21.Olhava EJ, Danca MD. 7442830. US patent. 2008
- 22.Qin T, et al. A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science. 2016;352:801–805. doi: 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cornella J, et al. Practical Ni-catalyzed aryl-alkyl cross-coupling of secondary redox-active esters. J Am Chem Soc. 2016;138:2174–2177. doi: 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, et al. Nickel-catalyzed cross-coupling of redox-active esters with boronic acids. Angew Chem Int Ed. 2016;55:9676–9679. doi: 10.1002/anie.201605463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toriyama F, et al. Redox-active esters in Fe-catalyzed C-C coupling. J Am Chem Soc. 2016;138:11132–11135. doi: 10.1021/jacs.6b07172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin T, et al. Nickel-catalyzed barton decarboxylation and Giese reactions: A practical take on classic transforms. Angew Chem Int Ed. 2017;56:260–265. doi: 10.1002/anie.201609662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatakeyama T, et al. Iron-catalyzed Suzuki-Miyaura coupling of alkyl halides. J Am Chem Soc. 2010;132:10674–10676. doi: 10.1021/ja103973a. [DOI] [PubMed] [Google Scholar]
- 28.Bedford RB, et al. Expedient iron-catalyzed coupling of alkyl, benzyl and allyl halides with arylboronic esters. Chemistry. 2014;20:7935–7938. doi: 10.1002/chem.201402174. [DOI] [PubMed] [Google Scholar]
- 29.Al Hussainy R, et al. Design, synthesis, radiolabeling, and in vitro and in vivo evaluation of bridgehead iodinated analogues of N-2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl-N-(pyridin-2-yl)cyclohexanecarboxamide (WAY-100635) as potential SPECT ligands for the 5-HT1A receptor. J Med Chem. 2011;54:3480–3491. doi: 10.1021/jm1009956. [DOI] [PubMed] [Google Scholar]
- 30.Lassalas P, et al. Structure property relationships of carboxylic acid isosteres. J Med Chem. 2016;59:3183–3203. doi: 10.1021/acs.jmedchem.5b01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt J, Choi J, Liu AT, Slusarczyk M, Fu GC. A general, modular method for the catalytic asymmetric synthesis of alkylboronate esters. Science. 2016;354:1265–1269. doi: 10.1126/science.aai8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xi Y, Hartwig JF. Diverse asymmetric hydrofunctionalization of aliphatic internal alkenes through catalytic regioselective hydroboration. J Am Chem Soc. 2016;138:6703–6706. doi: 10.1021/jacs.6b02478. [DOI] [PubMed] [Google Scholar]
- 33.Molander GA, Ellis N. Organotrifluoroborates: Protected boronic acids that expand the versatility of the Suzuki coupling reaction. Acc Chem Res. 2007;40:275–286. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 34.Mlynarski SN, Karns AS, Morken JP. Direct stereospecific amination of alkyl and aryl pinacol boronates. J Am Chem Soc. 2012;134:16449–16451. doi: 10.1021/ja305448w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonet A, Odachowski M, Leonori D, Essafi S, Aggarwal VK. Enantiospecific sp2-sp3 coupling of secondary and tertiary boronic esters. Nat Chem. 2014;6:584–589. doi: 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]
- 36.Laulhé S, Blackburn JM, Roizen JL. Selective and serial Suzuki-Miyaura reactions of polychlorinated aromatics with alkyl pinacol boronic esters. Org Lett. 2016;18:4440–4443. doi: 10.1021/acs.orglett.6b02323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dembitsky VM, Srebnik M. Synthesis and biological activity of α-aminoboronic acids, amine-carboxyboranes and their derivatives. Tetrahedron. 2003;59:579–593. doi: 10.1016/S0040-40200201618-6. [DOI] [PubMed] [Google Scholar]
- 38.McAtee JJ, Castle SL, Jin Q, Boger DL. Synthesis and evaluation of vancomycin and vancomycin aglycon analogues that bear modifications in the residue 3 asparagine. Bioorg Med Chem Lett. 2002;12:1319–1322. doi: 10.1016/S0960-894X0200130-0. [DOI] [PubMed] [Google Scholar]
- 39.Brown FJ, et al. Design of orally active, non-peptidic inhibitors of human leukocyte elastase. J Med Chem. 1994;37:1259–1261. doi: 10.1021/jm00035a004. [DOI] [PubMed] [Google Scholar]
- 40.Veale CA, et al. Orally active trifluoromethyl ketone inhibitors of human leukocyte elastase. J Med Chem. 1997;40:3173–3181. doi: 10.1021/jm970250z. [DOI] [PubMed] [Google Scholar]
- 41.Bernstein PR, Gomes BC, Kosmider BJ, Vacek EP, Williams JC. Nonpeptidic inhibitors of human leukocyte elastase. 6. Design of a potent, intratracheally active, pyridone-based trifluoromethyl ketone. J Med Chem. 1995;38:212–215. doi: 10.1021/jm00001a028. [DOI] [PubMed] [Google Scholar]
- 42.Burkhart JP, et al. Inhibition of human neutrophil elastase. 3. An orally active enol acetate prodrug. J Med Chem. 1995;38:223–233. doi: 10.1021/jm00002a003. [DOI] [PubMed] [Google Scholar]
- 43.Edwards PD, et al. Discovery and biological activity of orally active peptidyl trifluoromethyl ketone inhibitors of human neutrophil elastase. J Med Chem. 1997;40:1876–1885. doi: 10.1021/jm960819g. [DOI] [PubMed] [Google Scholar]
- 44.Hemmi K, Shima I, Imai K, Tanaka H. EP0494071. European patent. 1992
- 45.Kinoshita T, Nakanishi I, Sato A, Tada T. True interaction mode of porcine pancreatic elastase with FR136706, a potent peptidyl inhibitor. Bioorg Med Chem Lett. 2003;13:21–24. doi: 10.1016/S0960-894X0200852-1. [DOI] [PubMed] [Google Scholar]
- 46.von Nussbaum F, Li VMJ. Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: Into clinical testing with pre-adaptive pharmacophores. Bioorg Med Chem Lett. 2015;25:4370–4381. doi: 10.1016/j.bmcl.2015.08.049. [DOI] [PubMed] [Google Scholar]
- 47.von Nussbaum F, et al. Freezing the bioactive conformation to boost potency: The identification of BAY 85-8501, a selective and potent inhibitor of human neutrophil elastase for pulmonary diseases. ChemMedChem. 2015;10:1163–1173. doi: 10.1002/cmdc.201500131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gombert FO, et al. WO2015096873. patent application. 2015
- 49.Blench TJ, et al. WO2013037809. patent application. 2013
- 50.Bergström L, Lundkvist M, Lönn H, Sjö P. WO2008030158. patent application. 2008
- 51.Shultz MD. The thermodynamic basis for the use of lipophilic efficiency (LipE) in enthalpic optimizations. Bioorg Med Chem Lett. 2013;23:5992–6000. doi: 10.1016/j.bmcl.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 52.Zervosen A, et al. Unexpected tricovalent binding mode of boronic acids within the active site of a penicillin-binding protein. J Am Chem Soc. 2011;133:10839–10848. doi: 10.1021/ja200696y. [DOI] [PubMed] [Google Scholar]
- 53.Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 54.Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: An expanding army attacking a unique target. Chem Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.