

Abstract
BACKGROUND
Increased protein phosphatase-1 (PP1) in heart failure (HF) induces molecular changes deleterious to the cardiac cell. Inhibiting PP1 through the overexpression of a constitutively active inhibitor-1 (I-1c) has been shown to reverse cardiac dysfunction in a model of ischemic HF.
OBJECTIVES
We sought to determine the therapeutic efficacy of a re-engineered adeno-associated viral vector carrying I-1c (BNP116.I-1c) in a preclinical model of nonischemic HF, and to assess thoroughly the safety of BNP116.I-1c gene therapy.
METHODS
Volume-overload HF was created in Yorkshire swine by inducing severe mitral regurgitation (MR). One month after MR induction, pigs were randomized to intracoronary delivery of either BNP116.I-1c (n = 6) or saline (n = 7). Therapeutic efficacy and safety were evaluated 2 months after gene delivery. Additionally, 24 naïve pigs received different doses of BNP116.I-1c for safety evaluation.
RESULTS
At 1 month after MR induction, pigs developed HF as evidenced by increased left ventricular (LV) end-diastolic pressure and LV volume indexes. Treatment with BNP116.I-1c resulted in improved LV ejection fraction (−5.9 ± 4.2% vs. 5.5 ± 4.0%; p < 0.001) and adjusted dP/dt maximum (−3.39 ± 2.44 s-1 vs. 1.30 ± 2.39s-1; p = 0.007). Moreover, BNP116.I-1c-treated pigs also exhibited a significant increase in left atrial (LA) ejection fraction at 2 months after gene delivery (−4.3 ± 3.1% vs. 7.5 ± 3.1%; p = 0.02). In vitro I-1c gene transfer in isolated LA myocytes from both pigs and rats exhibited increased calcium transient amplitude, consistent with its positive impact on LA contraction. We found no evidence of adverse electrical remodeling, arrhythmogenicity, activation of a cellular immune response, or off-target organ damage by BNP116.I-1c gene therapy in pigs.
CONCLUSIONS
Intracoronary delivery of BNP116.I-1c was safe and improved contractility of the left ventricle and atrium in a large animal model of nonischemic HF.
Keywords: chimeric adeno-associated virus, contractility, intracoronary gene therapy, inhibitor-1, mitral regurgitation
Despite advances in medical care of chronic cardiovascular disorders, the 5-year mortality rate of patients with heart failure (HF) remains unacceptably high (1). Novel therapeutic approaches, such as gene therapy and cell therapy, hold the promise of complementing if not replacing existing therapies for HF (2).
Abnormal calcium handling plays a central role in the pathophysiology of the failing heart (3,4). The key drivers of cardiomyocyte calcium handling are the sarcoplasmic reticulum Ca2+ ATPase (SERCA2a) together with phospholamban (PLN). Together, they play a pivotal role in mediating intracellular calcium homeostasis. Decreased SERCA2a expression and activity are strongly associated with HF (5). Inhibitor-1 (I-1), which decreases the activity of protein phosphatase 1 (PP1), is one of the upstream regulators of the SERCA2a-PLN complex and linked to the β-adrenoceptor system (6). Upon protein kinase A phosphorylation, I-1 activation enhances SERCA2a activity by increasing phosphorylation of its endogenous inhibitor PLN (7). It has been shown that, in failing hearts, I-1 messenger ribonucleic acid (mRNA), protein, and phosphorylation levels are reduced by 50%, 60%, and 80%, respectively (8). Accordingly, gene transfer of constitutively active I-1 (I-1c) was proposed as an HF therapy to augment PLN phosphorylation and SERCA2a activity and to increase cardiac contractility. Indeed, promising beneficial effects in alleviating HF have been shown in experimental small animal studies (9–11). To translate this approach to the clinic, we previously examined I-1c gene transfer in pigs using 2 different viral vectors: 1) adeno-associated virus (AAV) type 9 (12); and 2) BNP116, which is a chimeric vector of AAV-2 and 8 that has strong muscle tropism (13). In our studies, we demonstrated that I-1c gene transfer improved left ventricular (LV) function in pigs with ischemic HF secondary to myocardial infarction. The efficacy and safety of this approach in nonischemic HF, however, remain unknown. Around 30% to 40% of HF with reduced ejection fraction (EF) is reported to be of a nonischemic origin (14) and prior reports have shown that ischemic and nonischemic HF exhibit different profiles of calcium handling abnormalities (15,16).
To eventually apply BNP116.I-1c gene therapy to a clinical HF population composed of diverse etiologies, in this study we aimed to: 1) determine if BNP116.I-1c gene therapy is also effective in nonischemic HF; and 2) thoroughly examine the safety of BNP116.I-1c gene therapy.
METHODS
ANIMAL PROCEDURES AND STUDY PROTOCOL
This study was composed of 2 independent experiments that addressed the efficacy, safety, and bio-distribution of our gene therapy approach. Efficacy-safety experiments were conducted in HF pigs at the Icahn School of Medicine at Mount Sinai (Figure 1) and the safety-biodistribution study was conducted in naïve pigs at MPI Research Inc (Figure 2). The animal protocols complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and standards of United States regulatory agencies. They were approved by the Institutional Animal Care and Use Committee of the Icahn School of Medicine at Mount Sinai and MPI Research Inc.
FIGURE 1. Design of Efficacy-safety Study and MR Model.
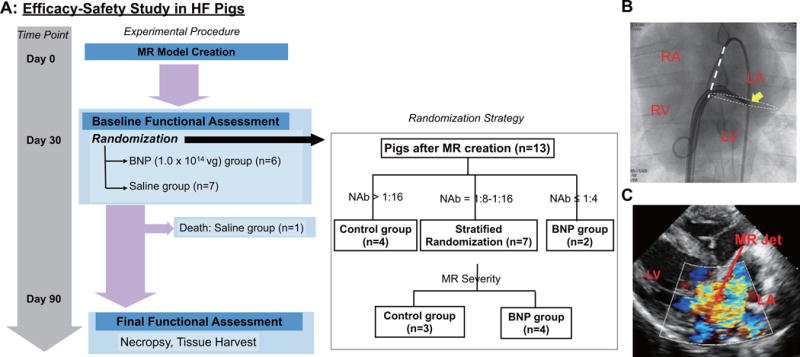
(A) Per the experimental protocol for the efficacy-safety study, cardiac performance was evaluated 1 month after mitral regurgitation (MR) induction of heart failure (HF) and the pigs were assigned to receive either BNP116.I-1c (BNP) or saline injection. Evaluation of function, hemodynamics, and arrhythmogenicity occurred 2 months after injection. Tissue samples were collected for histology and biodistribution analysis. (B) MR model was created by cutting the chordae-tendinae of the mitral valve (MV) in Yorkshire pigs via transseptal approach. Arrow indicates the tip of the biopsy catheter positioned at the postero-lateral part of the basal left ventricular (LV) for severing the cordae tendiane. (C) Representative echocardiographic color image shows the heart after MR creation. LA = left atrial; RA = right atrial; RV = right ventricular; vg = vector genome.
FIGURE 2. Design of Safety-biodistribution Study.
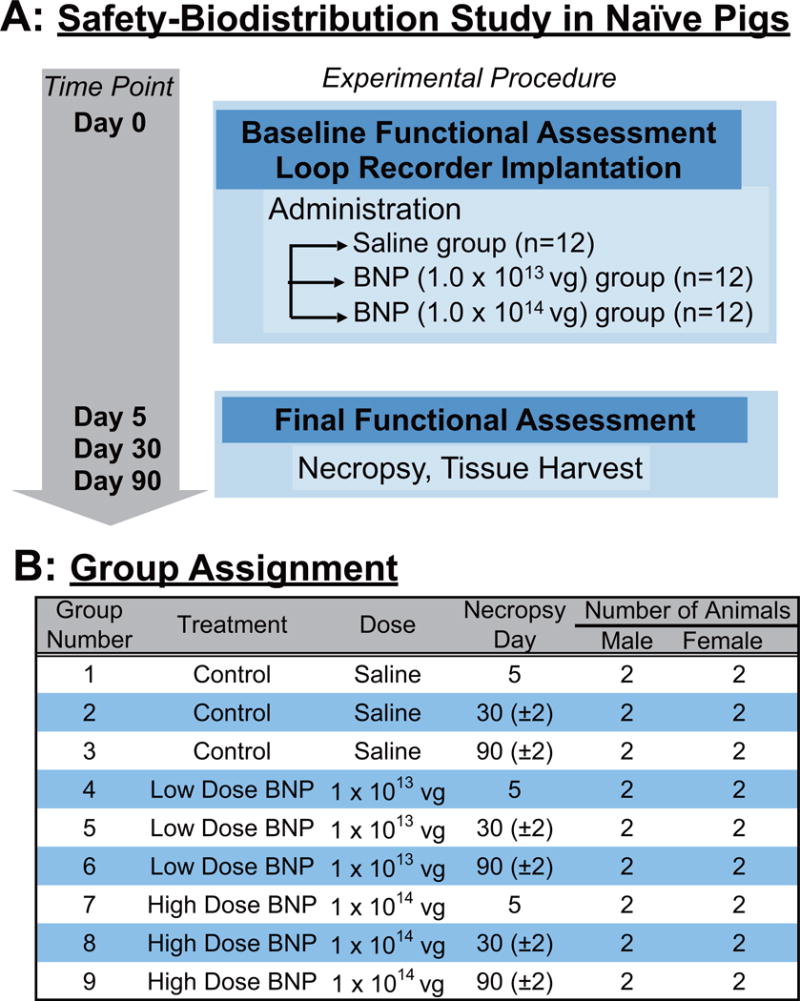
(A) In the experimental protocol for the safety-biodistribution study in healthy pigs, animals were (B) grouped by vector dose and euthanization time points. Abbreviations as in Figure
For the efficacy-safety study, nonischemic HF was induced by creating mitral regurgitation (MR) in female Yorkshire pigs (18 to 22 kg) as previously described (17), except for using a transseptal approach in this study. Cardiac performance was evaluated at 1 month after MR induction and the pigs received either BNP116.I-1c (1 × 1014 vector genomes [vg]) or the equivalent volume of saline through the coronary arteries (18). The titer of neutralizing antibodies in the sera before MR induction was used as a primary selection criterion for assigning treatment groups, since high neutralizing antibodies have been shown to reduce the gene transduction efficacy (19). The cutoff value of neutralizing antibodies for inclusion in the BNP116.I-1c treatment arm was set to 1:16, based on the initial screening and previous data (13). Thus, animals with high neutralizing antibody titers were assigned to the control group, whereas animals with titer ≤1:4 were assigned to the treatment group (Figure 1, Online Figure 1). When animals had a neutralizing antibody titer between 1:4 and 1:16, we applied a stratified randomization approach guided by the severity of MR as assessed by echocardiography. Two months after injection (3 months after MR creation), cardiac performance was re-evaluated to determine gene transfer efficacy. Pigs were subsequently euthanized and tissue samples were collected for histology and molecular analyses. Based on the expected I-1c effect on cardiac contractility and our previous findings (12,13), we defined the change in LV-adjusted rate of rise of pressure (dP/dt) maximum as the primary efficacy endpoint. Secondary efficacy endpoints included changes in echocardiographic volumetric and functional parameters of the LV and the left atrium (LA), pressure catheter-derived LV systolic and diastolic parameters, and PLN phosphorylation levels in the postmortem tissue analysis. Study sample size (n = 6 per group) was determined by using an alpha value of 0.05, a power of 80% to detect a 4s−1 difference in adjusted dP/dt maximum change with the expected 2.2% SD with 1 control per treatment animal. Scheme of this experimental protocol is shown in Figure 1.
In the safety-biodistribution study, healthy Yorkshire crossbred swine (27 to 37 kg) were subjected to the vector injection and its impact on cardiac function, arrhythmogenicity, blood chemistry, immune response, and biodistribution were examined at different time points after gene delivery (Figure 2). Three treatment groups of 2 male and 2 female pigs underwent the same intracoronary infusion procedure as the HF experiment. The experimental groups comprised a control plus 2 doses: saline; 1 × 1013 vg; or 1 × 1014 vg. Animals were sacrificed at 5, 30, or 90 days after vector delivery, making a total of 9 groups. Complete methods and protocols for both efficacy-safety study in HF pigs and safety-biodistribution study in naïve pigs are provided in the Online Appendix.
STATISTICAL ANALYSIS
Data are expressed as mean ± SD or median and interquartile range (IQR). The Shapiro-Wilk normality test was conducted to check the normality of the distribution. The unpaired Student t test was used to compare differences between 2 groups, and the Mann-Whitney U test was used where appropriate. Comparisons between 2 time points were performed by paired Student t test, whereas group differences between the time points were performed using repeated-measures analysis of variance (ANOVA). Fisher exact test was used to compare the 2 categorical variables between the groups. A p value < 0.05 was considered statistically significant.
RESULTS
Fourteen of 27 pigs that underwent the MR induction procedure survived to 1 month (52% survival), reflecting the severity of the HF phenotype in this model, as reported previously (17). One pig with only mild MR at 1 month was excluded from the study. At this point in time, pigs had chronic congestive HF, as evidenced by increased LV end-diastolic pressure and LV dilation (Online Table 1). These 13 pigs were assigned to receive either BNP116.I-1c (1.0 × 1014 vg; n = 6) or saline (control group; n = 7) (Figure 1). We employed a stratified randomization procedure that was guided by the severity of MR to reduce baseline differences between the groups; however, the titers of neutralizing antibodies (Online Figure 1) precluded a completely balanced stratified randomization and the pigs in the BNP116.I-1c group tended to be sicker despite any significant differences between the groups (as described previously in Methods and Figure 1). One pig in the saline group died 1 week after gene delivery. This pig’s post-mortem examination revealed a large amount of plural and pericardial effusion, suggesting that the cause of death was cardiac pump failure rather than the delivery procedure per se.
IMPACT OF INTRACORONARY BNP116.I-1C THERAPY
To determine the impact of BNP116.I-1c gene therapy on LV function in nonischemic HF, we evaluated cardiac function, volumes, and MR severity using a high-fidelity pressure-volume catheter, right heart catheterization, and echocardiography. Prior to gene transfer, functional and volumetric parameters were not significantly different between the BNP116.I-1c and control (saline injected) groups (Table 1). Exclusion of the control pig that died prematurely did not result in significant differences in baseline characteristics between the 2 groups, except for greater LV end-diastolic pressure in the BNP116.I-1c group (Online Table 2). The effective regurgitant orifice area (EROA), an established surrogate measure to assess MR severity, was measured echocardiographically by the proximal isovelocity surface area method (20). There was no difference in EROA between baseline and follow-up in either group (Figure 3). LVEF improved significantly in pigs treated with BNP116.I-1c by an average of +5.5% (p = 0.02), whereas it decreased by −5.8% (p = 0.02) in the control group (p = 0.001 for time × group interaction by repeated measures ANOVA) (Figure 3, Online Figure 2, Online Table 3). The control group exhibited a strong trend towards increased LV end-systolic volume index over the course of the study (+5.3 ml/m2; p = 0.06, before vs. 2 months after saline delivery). In contrast, I-1c-treated pigs exhibited a stable LV end-systolic volume index over the same time-course (−1.8 ml/m2; p = 0.55, before vs. 2 months after gene therapy) (p = 0.08 for time × group interaction by repeated measures ANOVA) (Figure 3, Online Figure 2, Online Table 3). Significant differences were documented between the saline and BNP116.I-1c groups for changes in LV dP/dt maximum (p = 0.006), and adjusted LV dP/dt maximum (p = 0.007) (Figure 3) consistent with findings in the previous studies in pigs (13,17). Pressure-volume loop-derived LV stroke work at 2 months after gene therapy was higher in the BNP116.I-1c group than the saline-treated controls (6.9 ± 1.0 mm Hg × 1 vs. 5.2 ± 1.6 mm Hg × 1; p = 0.04) (Figure 3). In contrast to significant improvement in LV contractility after BNP116.I-1c gene therapy, changes in cardiac lucitropy, as evaluated by dP/dt minimum and tau, were not different between the groups (Figure 3, Online Table 3). Similarly, change in LV end-diastolic pressure was similar between the groups. There was no difference in the heart weight between the groups after gene therapy (saline 204 ± 27 g vs. BNP116.I-1c 218 ± 44 g; p = 0.51).
Table 1.
LV and LA characteristics before gene transfer
| Control | BNP116.I-1c | P-value | |
|---|---|---|---|
| (Saline, n=7) | (1.0 × 1014 vg, n=6) | ||
| BW (kg) | 24.0 ± 4.0 | 22.8 ± 3.3 | 0.57 |
| 2D-Echocardiography | |||
| MR Severity | 4 (3, 4) | 4 (3, 4) | 0.63* |
| EROA (cm2) | 0.19 ± 0.10 | 0.23 ± 0.11 | 0.58 |
| RF (%) | 43.9 ± 17.1 | 50.0 ± 13.4 | 0.49 |
| TMF ratio (E/A) | 1.63 ± 0.87 | 2.08 ± 1.15 | 0.44 |
| LV mass (g) | 75.5 ± 10.0 | 82.2 ± 14.0 | 0.34 |
| LA dimension (mm) | 37.8 (35.7, 40.8) | 38.8 (37.4, 41.5) | 0.63* |
| 3D-Echocardiography | |||
| LVEF(%) | 72.8 ± 5.5 | 72.6 ± 4.2 | 0.95 |
| LVEDV(ml) | 71.0 ± 8.4 | 69.7 ± 17.9 | 0.87 |
| LVESV(ml) | 19.5 ± 5.4 | 19.3 ± 7.0 | 0.96 |
| LA-maximum volume (ml) | 40.3 ± 17.2 | 36.6 ± 8.1 | 0.64 |
| LA-minimum volume (ml) | 18.2 (11.2, 21.9) | 14.5 (12.8, 28.5) | 0.95* |
| LA-total EF(%) | 54.1 ± 7.3 | 50.7 ± 10.3 | 0.50 |
| LA-active EF(%) | 26.3 ± 7.7 | 22.7 ± 7.1 | 0.40 |
| LA-passive EF(%) | 37.1 ± 12.7 | 36.2 ± 12.8 | 0.90 |
| Invasive measurements | |||
| mPAP (mmHg) | 20.4 ± 5.5 | 20.7 ± 7.8 | 0.95 |
| mLAP (mmHg) | 11.7 ± 7.3 | 13.5 ± 8.2 | 0.69 |
| CI (L/min/m2) | 3.7 ± 1.0 | 3.3 ± 0.9 | 0.48 |
| LVP peak (mmHg) | 79.4 ± 14.5 | 81.9 ± 16.3 | 0.59 |
| LVEDP (mmHg) | 12.2 ± 2.3 | 14.1 ± 2.1 | 0.15 |
| HR (bpm) | 88 ± 20 | 75 ± 17 | 0.37 |
| LV dP/dt max (mmHg/sec) | 1351 ± 390 | 1299 ± 405 | 0.82 |
| adjusted LV dP/dt max (1/sec) | 20.0 ± 2.5 | 18.9 ± 2.7 | 0.47 |
| LV dP/dt min (mmHg/sec) | −1442 ± 565 | −1260 ± 580 | 0.58 |
| Tau (msec) | 50.0 (46.1, 53.3) | 54.2 (44.3, 79.3) | 0.37* |
p-value using the Mann-Whitney U test, bpm = beat per minute, BW= body weight, MR = mitral regurgitation, EROA = Effective regurgitant orifice area, RF = regurgitant fraction, TMF= trans-mitral flow, LV = left ventricle, LA = left atrium, EF = ejection fraction, EDV = end-diastolic volume, ESV = end-systolic volume, mLAP = mean left atrial pressure, mPAP = meanpulmonary arterial pressure, HR = heart rate, CI = cardiac index, LVP peak = LV peak pressure,LVEDP = LV end-diastolic pressure, LV dP/dt max = LV dP/dt maximum, LV dP/dt min = LVdP/dt minimum,
FIGURE 3. Impact of BNP116.I-1c on the LV.
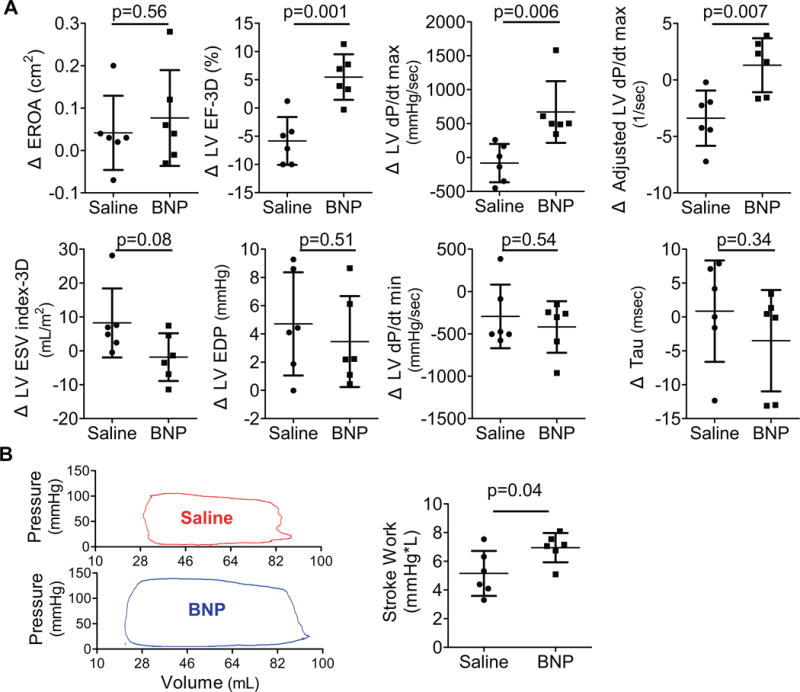
(A) In evaluating the change from baseline at 2 months, EROA remained similar in both groups while contractility parameters (LV dP/dt max, adjusted LV dP/dt max, and LVEF-3D) showed significant improvement only in the BNP-treated group. Meanwhile, diastolic functional parameters (LVEDP, dP/dt min, and Tau) did not show significant differences between the groups. (B) Pressure-volume loop-derived stroke work was also higher in the BNP group 2 months after gene therapy. The round shape of the representative loops indicates MR during the isovolumic phases. Absolute changes of each animal are shown in dots. Bars = mean ± SD. dP/dt = rate of rise of LV pressure; EROA= effective regurgitant orifice area; LVEDP = LV enddiastolic pressure; LVEF-3D = LV ejection fraction measured by 3-dimensional echocardiography; LVESV-3D = LV end-systolic volume measured by 3-dimensional echocardiography; other abbreviations as in Figure 1.
We then examined the cardiac tissues to evaluate if improved LV systolic function was associated with expected molecular changes after I-1c gene transfer. LV tissue samples from BNP116.I-1c-treated animals presented an average of 2.1 ± 1.9 × 104 copies of I-1c mRNA per μg RNA (Figure 4). As previously reported, I-1c protein detection by Western blot in cardiac tissue remained challenging (21) (Online Figure 3); thus, we examined PLN phosphorylation in these tissue samples by Western blot as a surrogate marker of I-1c expression. The analyses revealed a significant decrease in the ratio of phosphorylated PLN to total PLN in the saline group relative to the BNP116.I-1c-treated heart with some interanimal variability, possibly due to the severity of HF induced by chronic MR. Meanwhile, the relative protein expression of SERCA2a was not different between the groups (Figure 4). Interestingly, there was a significant correlation between the phosphorylated-to-total PLN ratio and the adjusted dP/dt maximum at the final time point (R = 0.65; p = 0.02) (Figure 4).
FIGURE 4. Impact of I-1c Gene Transfer on I-1c.
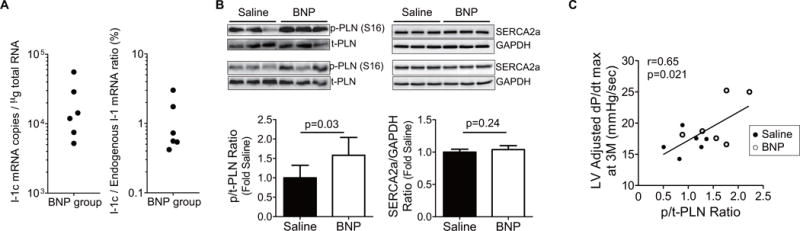
The impact of constitutively active inhibitor-1 (I-1c) is seen in (A) messenger ribonucleic acid (mRNA) copy numbers and its relative amount against endogenous I-1 in LV free wall tissue from BNP-treated animals, and (B) immunoblots of total phospholamban (t-PLN), phosphorylated-PLN at Ser16 (p-PLN), SERCA2a, and GAPDH protein in the LV free wall tissues. The relative p-PLN level was determined through normalization to t-PLN level. The significantly higher p-PLN-to-t-PLN ratio in the LV from BNP-treated pigs suggested reduced SERCA2a inhibitory effect. (C) The ratio of p-PLN-to-t-PLN exhibited significant correlations with adjusted dP/dt maximum at the time of tissue collection, suggesting that increased PLN phosphorylation is the responsible mechanism for increased cardiac contractility. GAPDH = glyceraldehyde-3-phosphate dehydrogenase; SERCA2a = sarcoplasmic reticulum Ca2+ ATPase; other abbreviations as in Figures 1 and 3.
IMPACT OF BNP116.I-1C THERAPY ON THE LA
The LA is under continuous exposure to increased volume and pressure load from regurgitant flow in the heart with MR, an effect that promotes atrial remodeling leading to atrial fibrillation. To examine the impact of MR and I-1c gene therapy on the LA, we examined the changes in LA function and remodeling using 3-dimensional (3D) echocardiography (Figure 5). Maximum and minimum LA volumes were not different between the groups at 2 months after gene therapy (maximum LA volume index: 50.5 ± 14.6 ml/m2 vs. 70.8 ± 37.4 ml/m2; p = 0.24; minimum LA volume index: 24.4 ± 6.0 ml/m2 vs. 31.9 ± 24.4 ml/m2; p = 0.48; saline vs BNP116.I-1c, respectively), and there was a linear relationship between EROA and maximum LA volume (R = 0.62; p = 0.03). Because mitral valve opening and LV filling precedes onset of LA contraction, the LA has 2 phases of systole. From the mitral valve opening to the LA contraction is the passive emptying phase; the start of the LA contraction to the mitral valve closing is the active emptying phase. These emptying functions can be calculated as passive LA-EF and active LA-EF respectively, while the sum of the 2 is the total LA-EF (see Online Appendix for detailed methods). Changes in total and active LA-EF from before to 2 months after gene therapy were significantly improved in the BNP116.I-1c group compared to controls, suggesting an improvement of LA contraction with I-1c gene transfer. (Figure 5, Online Figure 4). To determine if improved LA-EF was due to a direct gene therapy effect on LA myocytes or an indirect effect from improved LV function, we isolated LA myocytes from rats and pigs and examined the impact of I-1c gene transfer on calcium transients in vitro. I-1c gene transfer resulted in increased calcium transient amplitudes in LA myocytes from both rats and pigs, supporting the direct effect of I-1c gene transfer on the LA (Figure 5). In the LA of pigs treated with BNP116.I-1c, I-1c mRNA was also expressed, albeit to a lower level compared to the LV (Online Figure 5). In examining PLN phosphorylation status, where there was higher variability among the samples, we found no significant difference between the groups, although there was a tendency for an increase in the phosphorylated-to-total PLN ratio in the BNP116.I-1c group (Online Figure 5). The large variability may be due to sampling, because the MR jet was directly hitting the LA posterior wall in some pigs, which was the location from which we collected tissue. Interestingly, the ratios of total PLN against SERCA2a or glyceraldehyde-3-phosphate dehydrogenase were generally lower in the LA compared to the LV, indicating that a smaller fraction of SERCA2a is inhibited by PLN in the LA of pig hearts (Online Figure 6), possibly reducing the potential impact of I-1c expression in LA myocytes.
FIGURE 5. Impact of I-1c Gene transfer On LA Contractility.
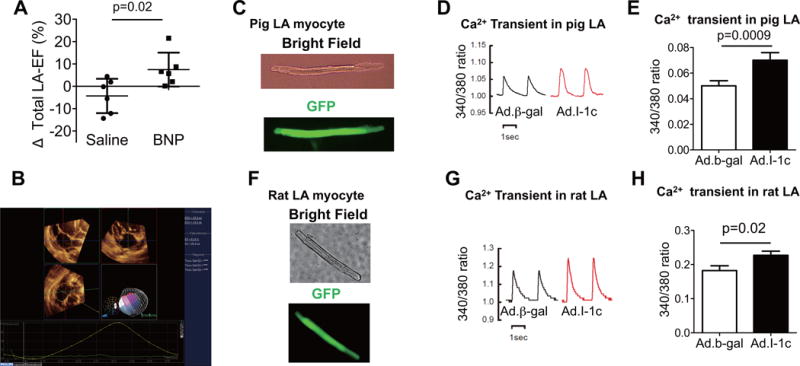
(A) Change in LA-EF (Δ) between baseline and 2 months after gene therapy was significantly higher in BNP groups. (B) Representative image of LA 3-dimensional echocardiographic analysis. Impact of I-1c gene transfer (using recombinant adenoviral vectors) on is seen in isolated LA myocytes from pigs (C through E) and rats (F through H). The recombinant adenoviral vectors for I-1c and the control vector (β-gal) also containing green fluorescent protein (GFP) allowed identification of infected cells. Adenoviral gene transfer of I-1c in isolated LA myocytes resulted in increased calcium transient in both pig and rat LA myocytes as assessed by the ratio of Fura-2 emissions at 340 nm and 380 nm (340:380 ratio). Absolute changes of each animal are shown in plots. The bars represent mean ± SD. Other abbreviations as in Figures 1, 3, and 4.
I-1c GENE TRANSFER AND CARDIAC ARRHYTHMIAS
To translate I-1c gene therapy to the clinic, its side effects must be minimal. Augmenting cardiac contractility has the potential of increasing the incidence of malignant arrhythmias, a major drawback for β-agonists. To evaluate whether I-1c gene transfer is associated with increased arrhythmogenicity, we conducted a programmed electrical stimulation study in the pigs at 2 months after gene therapy. None of the pigs developed ventricular arrhythmias (Figure 6) in contrast to the high inducibility in pigs with ischemic HF (~80%) (data not shown). We also performed rapid right atrial pacing to assess susceptibility to atrial arrhythmias and found a trend towards lower atrial arrhythmia propensity in HF pigs treated with BNP116.I-1c compared to controls. To examine further the impact of BNP116.I-1c gene transfer on LV arrhythmogenicity, we used an established ex vivo optical mapping technique (22) to examine detailed electrophysiological properties of the LV tissues. None of the tissues exhibited onset of ventricular tachycardia or fibrillation upon highfrequency stimulation. Moreover, BNP116.I-1c gene therapy did not alter key electrophysiological properties, including transmural conduction velocity, action potential duration (APD), and APD heterogeneity at basal or rapid rates (Figure 6).
FIGURE 6. Cardiac Arrhythmogenicity.
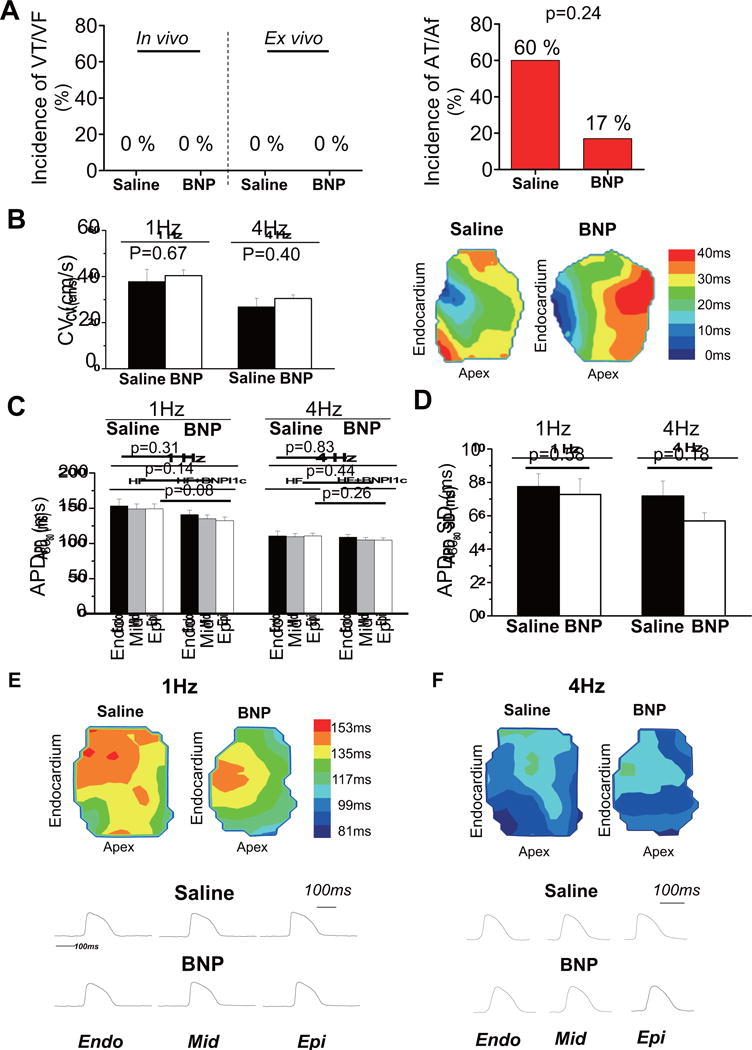
(A) No ventricular tachycardia (VT) or ventricular fibrillation (VF) was induced in either in vivo and ex vivo electrophysiological testing in either groups, while atrial tachycardia (AT) or atrial fibrillation (AF) was inducible in vivo in 1 pig after BNP116.I-1c treatment (n = 6) and 3 in the saline-treated group (n = 5). (B) No significant differences in average conduction velocity were observed between the 2 groups at 1 Hz or 4 Hz or in representative depolarization isochrone maps, indicating that BNP116.I-1c therapy did not affect cardiac excitation. In analyzing action potential duration (APD), (C) there were no significant differences in average APD measured at 80% of repolarization (APD80) measured from the subendocardial (Endo), mid-myocardial (Mid), and subepicardial (Epi) layers, nor was a difference seen in (D) transmural APD heterogeneity indexed by SD and range of APD80 values across the transmural wall at 1 Hz and 4 Hz. (E and F) In examples of representative action potentials and transmural APD contour maps at 1 Hz and 4 Hz, BNP and saline groups presented transmural action potential signals of similar morphology. Other abbreviations as in Figure 1.
SAFETY-BIODISTRIBUTION STUDY IN NORMAL PIGS
To further establish cardiac BNP116.I-1c gene therapy safety, we delivered BNP116.I-1c into naïve pigs at 1.0 × 1013 to 1014 vg per animal and examined acute and chronic impacts of vector delivery (Figure 2). No apparent changes in cardiac function were noted, and no events attributable to fatal arrhythmias were documented after vector delivery (Online Figure 7, Online Tables 4 and 5). The number of vector genomes in the major organs was determined in these pigs. The results indicated that the vector was cleared from the blood within 5 days after the injection (Online Figure 8, Online Table 6). Cardiac samples, including the coronary arteries, retained the vg copies up to 90 days after vector delivery with 30 to 150 times higher amounts in 1.0 × 1014 vg treated animals compared to those receiving 1.0 × 10 vg. Detailed blood chemistry revealed no changes that would suggest off-target effects (Online Table 7). As expected, the titer of neutralizing antibody against BNP116 was increased post-vector delivery (Online Figure 9).
SAFETY OF INTRACORONARY DELIVERY OF HIGH-DOSE BNP116.I-1c
One concern using a high dose of viral vectors is invoking a cellular immune response (23). After the delivery of 1.0 × 1014 vg per animal, we evaluated the tissue for the presence of an immune response at a relatively high dose compared to the previous studies in animals and humans (12–13,17,24). LV samples were stained by anti-CD3 antibody to evaluate the presence of T-cell immune response against BNP116.I-1c, which was negative in both the efficacy-safety and safety-biodistribution studies (Online Figure 10). Additionally, we found no pathological histology changes in off-target organs including liver, kidney, and lung in the BNP116.I-1c-treated pigs (Online Figure 10).
DISCUSSION
The main finding of the present study was that intracoronary delivery of BNP116.I-1c at a dose of 1.0 × 1014 vg was safe and improved LV systolic function in a large animal model of nonischemic HF (Central Illustration). Furthermore, LA function – which is impaired by mitral regurgitation – was improved in the BNP116.I-1c-treated pigs, likely due to a direct effect on atrial myocytes. Importantly, these effects were not associated with atrial or ventricular arrhythmogenicity, which are major concerns for inotropic therapies in HF. Together with previously reported efficacy of BNP116.I-1c in an ischemic HF pig model (13), our results supported testing of this novel therapeutic approach in clinical trials for patients with ischemic and nonischemic HF.
CENTRAL ILLUSTRATION. Inhibitor-1 Gene Therapy.
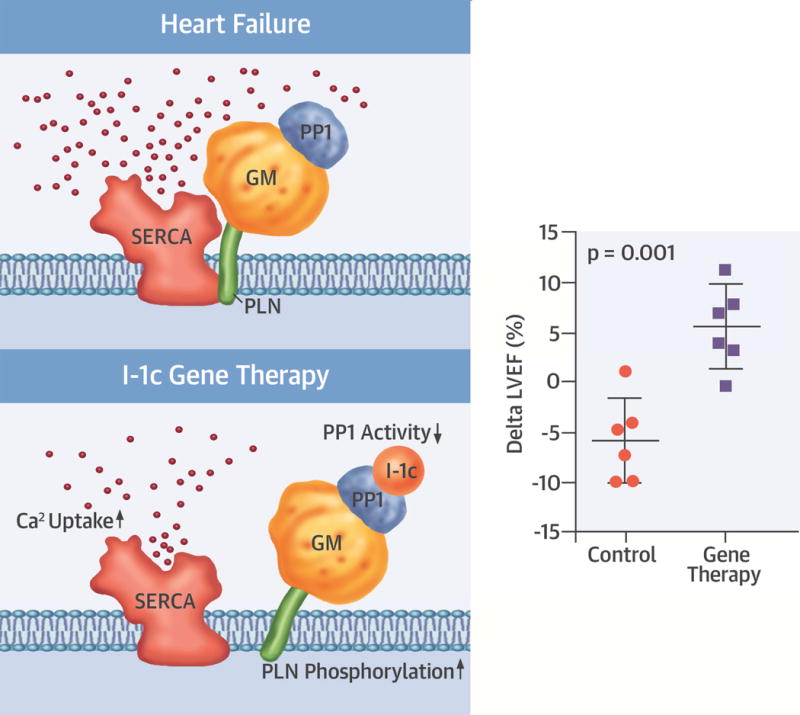
Inhibitor-1 (I-1) binding to protein phosphatase-1 (PP1) results in its inhibition, which leads to increased phospholamban (PLN) phosphorylation and sarcoplasmic reticulum Ca2+ ATPase (SERCA 2a) activity. Gene transfer of constitutively active I-1 (I-1c) increased both left ventricular and atrial function in a pig model of nonischemic heart failure. GM = PP1 regulatory subunit; LVEF = left ventricular ejection fraction.
Mechanistically, the increased PLN phosphorylation in the LV myocardium after I-1c gene transfer, together with its correlation to adjusted dP/dt maximum, suggested that I-1c gene transfer improves cardiac contractility through increased SERCA2a activity. Beyond its impact on the LV, we also demonstrated improved LA contractility in vitro using adenoviral gene transfer and in vivo using 3D echocardiography. Previous studies have focused on LV function with minimal attention to potential changes in LA function. In HF, the LA plays an important role in maintaining cardiac output and its contribution to cardiac output becomes larger than in nonfailing hearts (25). There is also growing evidence that LA dysfunction can actively contribute to HF symptoms and disease progression (25,26). More importantly, MR is a major risk factor for atrial fibrillation, the most common sustained arrhythmia encountered in clinical practice, and a major cause of stroke and HF. Therefore, improving both LA and LV function might more positively impact HF and prevent its progression.
Increased probability of arrhythmias is a common concern for any therapy that augments cardiomyocyte contractility. To determine if I-1c gene transfer is proarrhythmic, we conducted in vivo and ex vivo programmed electrical stimulation protocols. We also examined the electrophysiological substrate in ex vivo perfused porcine preparations using high-resolution optical action potential mapping to determine the effects of I-1c gene therapy on key electrophysiological parameters and whether they are mechanistically linked to arrhythmias in nonischemic HF. In pigs with MR-induced HF, I-1c gene transfer did not promote programmed stimulation induced ventricular arrhythmias. Moreover, I-1c gene therapy did not alter key conduction or repolarization properties. Finally, I-1c gene therapy caused a clear trend towards reduced susceptibility to pacing-induced AF in vivo.
Reduced expression of SERCA2a has been consistently reported in human and experimental models of HF (3). Accordingly, cardiac gene therapy was proposed to correct this condition in recent clinical trials using AAV1, such as in the CUPID (Calcium Upregulation by Percutaneous Administration of Gene Therapy in Patients With Cardiac Disease) trial. Despite promising efficacy demonstrated in a phase I/IIa study (24), the most recent phase IIb trial failed to reduce clinical events associated with HF (27). Post-trial analysis revealed that at an AAV1.SERCA2a dose of 1.0 × 10 vg, very low levels of viral genome were detected in the heart at the time of cardiac transplantation or ventricular assist device placement (27). Thus, there seemed to be a challenge in effectively transducing the human heart with the tools used in the CUPID trial.
Several strategies are proposed to increase transgene efficacy (28); the practical ones are: 1) using a different vector with a better cardiac tropism; 2) increasing the dose; and 3) modifying the vector delivery method. In the current study, we addressed the first 2 strategies to improve the transduction efficacy from that of CUPID. First, BNP116 is a synthetic AAV vector generated by site-directed mutagenesis on the heparan sulfate receptor footprint (29). In a mouse study, this chimeric vector of AAV2 and 8 demonstrated significant muscle tropism including the heart and reduced transduction of the liver (29). Second, we employed a vector dose of 1.0 × 1014 vg, which is 10 times higher than that used in CUPID IIb and previous preclinical studies (13,27). While achieving 30 to 150 times more vector genome retention at 90 days compared to the 1.0 × 1013 vg, we did not find any evidence of T-cell infiltration in the cardiac tissue and no apparent immune reaction has been detected in the histology, addressing potential concerns of cellular immune reaction at higher doses. Meanwhile, the third approach, the gene delivery method, was not altered from previous method (27) and we used a slow antegrade intracoronary delivery method. Although this approach has been reported to result in lower transduction efficacy compared to more invasive approaches (30), the procedure is simple, requires little time, and is feasible for most interventional cardiologists, all important aspects to ascertain safety of gene delivery to the heart. Importantly, the LA can be targeted with this approach as the vectors are also perfused into the LA branch of the coronary artery, in contrast to other methods, such as direct intramyocardial injection and retrograde coronary sinus injection.
The amount of mRNA detected in the pig heart was, on average, approximately 1% of endogenously expressed I-1. This accounted for a rough estimate of 0.24 to 1.7 I-1c mRNA per cardiomyocytes (see Online Appendix for calculation). Although very small, this seemed to be sufficient to increase the phosphorylation levels of PLN and improve cardiac function, likely due to the altered properties of I-1c compared to I-1. I-1c is a truncated form of I-1 with an amino acid substitution, resulting in its ability to maintain constitutive activity. It may also increase the stability of the protein from degradation. Thus far, the difficulty in detecting I-1c expression by western blot in cardiac cells (Online Figure 3) prevented us from testing this hypothesis.
To examine the safety of this new therapeutic approach, we thoroughly evaluated the potential negative impact of BNP116.I-1c therapy in HF pigs and determined the potential toxicity of high-dose vector delivery in normal pigs. None of the pigs presented with clear evidence of fatal arrhythmias as assessed by implantable loop recorders. Electrocardiograms did not show apparent changes after gene transfer in normal pigs; also, there was no detrimental effect on cardiac function in naïve pigs after BNP116.I-1c treatment. Additionally, detailed examination of off-target major organs, including brain, liver, lung, kidney, and more, exhibited no clear abnormalities of histology or blood chemistry. These results ensured the safety of the vectors for clinical examination.
CLINICAL IMPLICATIONS
Despite the high prevalence of nonischemic HF (14), therapies targeting this population are very limited. Cardiac resynchronization therapy is one of the few effective approaches for treating nonischemic HF and improving LVEF, which is correlated with the improved prognosis (31). Unfortunately, however, not all patients are candidates for this therapy. Meanwhile, recent clinical study using biopsy samples from nonischemic HF patients demonstrated that patients who exhibited improved LVEF after β-blocker therapy had significant increase in calcium cycling-related genes (32), suggesting targeting calcium cycling in nonischemic HF is a rational approach. Considering that remaining cardiomyocytes in nonischemic HF patients are likely more than patients with ischemic etiology, targeting dysfunctional myocytes and improving their function through gene therapy may provide new opportunities for this patient population.
STUDY LIMITATIONS
Due to the small sample size and limited study length, this study might have been underpowered to demonstrate the differences in some parameters, such as LV remodeling. However, we have demonstrated improved contractility consistent with previous studies, supporting the beneficial impact of BNP116.I-1c for treating HF. It is expected that it would require twice the size of the study (13 pigs per arm) to detect a difference in LV endsystolic volume index, a marker of cardiac remodeling, using an alpha value of 0.05, a power of 80% with differences found in the current study. Nonischemic HF was induced by MR and LVEF was preserved in treated pigs. Whether our result is applicable to other types or more advanced forms of nonischemic HF remains to be studied. In the meantime, MR is a common presentation in both ischemic and nonischemic HF without an effective therapy other than surgery. Of note, MR is considered one of the major contributors of HF progression (33), and borderline LVEF in nonischemic MR already implies overt HF (34). Finally, the endpoints of our study were set as a change in contractility parameters and not in clinical outcomes. Therefore, the impact of BNP116.I-1c on HF prognosis and quality of life in patients needs to be examined in future studies.
CONCLUSIONS
Our findings indicated that gene therapy using BNP116.I-1c with a dose of 1.0 × 1014 vg through the coronary arteries was safe and improved LV and LA contractility in a clinically relevant animal model of nonischemic HF. In addition to the HF study, dose-escalation experiments in naïve pigs established the safety of this vector without any evidence of increased arrhythmias and immune responses in the heart. Together with previously reported efficacy of this gene therapy approach in an ischemic HF swine model, our results support initiation of clinical trials to test this therapeutic approach to improve HF human patients.
Supplementary Material
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE
Gene therapy with BNP116.1-1c improved cardiac function in an animal model of nonischemic heart failure.
TRANSLATIONAL OUTLOOK IMPLICATIONS
Clinical studies of BNP116.I-1c gene therapy are needed to assess its impact on cardiac function and outcomes in patients with heart failure.
CONDENSED ABSTRACT.
Intracoronary gene therapy of constitutively active inhibitor-1 (I-1c) using a novel cardiotropic gene delivery vector, BNP116, improved left ventricular and left atrial contractility in a pig model of nonischemic heart failure induced by mitral regurgitation. Safety of this new therapeutic approach was thoroughly evaluated in naïve pigs and we found no evidence of adverse side effects including arrhythmogenicity and off-target pathological changes. These data prompt examination of BNP116.I-1c therapy in heart failure patients.
Acknowledgments
This study was supported by NIH P50 HL112324, R01 HL119046, R01 HL117505, R01HL128099, R01 HL129814, R01HL131404, & R43HL108581 (R. J. H.), HL26057, & HL64018 (E.G. K) and a Transatlantic Leducq Foundation grant. We would like to acknowledge the Gene Therapy Resource Program (GTRP) of the National Heart, Lung, and Blood Institute, National Institutes of Health for providing some of the gene vectors used in this study. K.I is supported by a grant from American Heart Association 17SDG33410873.
ABBREVIATIONS AND ACRONYMS
- AAV
adeno-associated virus
- EF
ejection fraction
- HF
heart failure
- I-1
inhibitor-1
- I-1c
constitutively active inhibitor-1
- LA
left atrial
- LV
left ventricular
- MR
mitral regurgitation
- PLN
phospholamban
- SERCA2a
sarcoplasmic reticulum Ca2+ ATPase
- vg
vector genome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: RJH, EK, RJS are co-founders of Nanocor. Other authors do not have anything to disclose.
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2015;385:812–24. doi: 10.1016/S0140-6736(14)61889-4. [DOI] [PubMed] [Google Scholar]
- 3.Kawase Y, Hajjar RJ. The cardiac sarcoplasmic/endoplasmic reticulum calcium ATPase: a potent target for cardiovascular diseases. Nat Clin Pract Cardiovasc Med. 2008;5:554–65. doi: 10.1038/ncpcardio1301. [DOI] [PubMed] [Google Scholar]
- 4.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–60. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hajjar RJ. Potential of gene therapy as a treatment for heart failure. J Clin Invest. 2013;123:53–61. doi: 10.1172/JCI62837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicolaou P, Hajjar RJ, Kranias EG. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J Mol Cell Cardiol. 2009;47:365–71. doi: 10.1016/j.yjmcc.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neumann J, Eschenhagen T, Jones LR, et al. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol. 1997;29:265–72. doi: 10.1006/jmcc.1996.0271. [DOI] [PubMed] [Google Scholar]
- 8.El-Armouche A, Rau T, Zolk O, et al. Evidence for protein phosphatase inhibitor-1 playing an amplifier role in beta-adrenergic signaling in cardiac myocytes. FASEB J. 2003;17:437–9. doi: 10.1096/fj.02-0057fje. [DOI] [PubMed] [Google Scholar]
- 9.Chen G, Zhou X, Florea S, et al. Expression of active protein phosphatase 1 inhibitor-1 attenuates chronic beta-agonist-induced cardiac apoptosis. Basic Res Cardiol. 2010;105:573–81. doi: 10.1007/s00395-010-0106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pathak A, del Monte F, Zhao W, et al. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005;96:756–66. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 11.Nicolaou P, Rodriguez P, Ren X, et al. Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ Res. 2009;104:1012–20. doi: 10.1161/CIRCRESAHA.108.189811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fish KM, Ladage D, Kawase Y, et al. AAV9.I-1c delivered via direct coronary infusion in a porcine model of heart failure improves contractility and mitigates adverse remodeling. Circ Heart Fail. 2013;6:310–317. doi: 10.1161/CIRCHEARTFAILURE.112.971325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa K, Fish KM, Tilemann L, et al. Cardiac I-1c Overexpression With Reengineered AAV Improves Cardiac Function in Swine Ischemic Heart Failure. Mol Ther. 2014;22:1–8. doi: 10.1038/mt.2014.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart. 2007;93:1137–46. doi: 10.1136/hrt.2003.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sen L, Cui G, Fonarow GC, Laks H. Differences in mechanisms of SR dysfunction in ischemic vs. idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2000;279:H709–18. doi: 10.1152/ajpheart.2000.279.2.H709. [DOI] [PubMed] [Google Scholar]
- 16.Brillantes AM, Allen P, Takahashi T, Izumo S, Marks AR. Differences in cardiac calcium release channel (ryanodine receptor) expression in myocardium from patients with end-stage heart failure caused by ischemic versus dilated cardiomyopathy. Circ Res. 1992;71:18–26. doi: 10.1161/01.res.71.1.18. [DOI] [PubMed] [Google Scholar]
- 17.Kawase Y, Ly HQ, Prunier F, et al. Reversal of Cardiac Dysfunction After Long-Term Expression of SERCA2a by Gene Transfer in a Pre-Clinical Model of Heart Failure. J Am Coll Cardiol. 2008;51:1112–9. doi: 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe S, Leonardson L, Hajjar RJ, Ishikawa K. Cardiac Gene Delivery in Large Animal Models: Antegrade Techniques. Methods Mol Biol (Clifton, NJ) 2017;1521:227–35. doi: 10.1007/978-1-4939-6588-5_16. [DOI] [PubMed] [Google Scholar]
- 19.Calcedo R, Wilson JM. Humoral Immune Response to AAV. Front Immunol. 2013;4:341. doi: 10.3389/fimmu.2013.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zoghbi WA, Enriquez-Sarano M, Foster E, et al. Recommendations for evaluation of the severity of native valvular regurgitation with two-dimensional and Doppler echocardiography. J Am Soc Echocardiogr. 2003;16:777–802. doi: 10.1016/S0894-7317(03)00335-3. [DOI] [PubMed] [Google Scholar]
- 21.Qian J, Vafiadaki E, Florea SM, et al. Small heat shock protein 20 interacts with protein phosphatase-1 and enhances sarcoplasmic reticulum calcium cycling. Circ Res. 2011;108:1429–38. doi: 10.1161/CIRCRESAHA.110.237644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie C, Hu J, Motloch LJ, Karam BS, Akar FG. The Classically Cardioprotective Agent Diazoxide Elicits Arrhythmias in Type 2 Diabetes Mellitus. J Am Coll Cardiol. 2015;66:1144–56. doi: 10.1016/j.jacc.2015.06.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nathwani AC, Tuddenham EGD, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–65. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jessup M, Greenberg B, Mancini D, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–13. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoit BD. Left atrial size and function: role in prognosis. J Am Coll Cardiol. 2014;63:493–505. doi: 10.1016/j.jacc.2013.10.055. [DOI] [PubMed] [Google Scholar]
- 26.Welles CC, Ku IA, Kwan DM, Whooley MA, Schiller NB, Turakhia MP. Left atrial function predicts heart failure hospitalization in subjects with preserved ejection fraction and coronary heart disease: longitudinal data from the Heart and Soul Study. J Am Coll Cardiol. 2012;59:673–80. doi: 10.1016/j.jacc.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenberg B, Butler J, Felker GM, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387:1178–86. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- 28.Hajjar RJ, Ishikawa K. Introducing Genes to the Heart: All About Delivery. Circ Res. 2017;120:33–35. doi: 10.1161/CIRCRESAHA.116.310039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asokan A, Conway JC, Phillips JL, et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol. 2010;28:79–82. doi: 10.1038/nbt.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishikawa K, Aguero J, Naim C, Fish K, Hajjar RJ. Percutaneous approaches for efficient cardiac gene delivery. J Cardiovasc Transl Res. 2013;6:649–59. doi: 10.1007/s12265-013-9479-7. [DOI] [PubMed] [Google Scholar]
- 31.Ghani A, Delnoy P, Adiyaman A, et al. Predictors and long-term outcome of superresponders to cardiac resynchronization therapy. Clin Cardiol. 2017;40:292–9. doi: 10.1002/clc.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kao DP, Lowes BD, Gilbert EM, et al. Therapeutic Molecular Phenotype of beta-Blocker-Associated Reverse-Remodeling in Nonischemic Dilated Cardiomyopathy. Circ Cardiovasc Genet. 2015;8:270–83. doi: 10.1161/CIRCGENETICS.114.000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otto CM. Clinical practice. Evaluation and management of chronic mitral regurgitation. N Engl J Med. 2001;345:740–6. doi: 10.1056/NEJMcp003331. [DOI] [PubMed] [Google Scholar]
- 34.Enriquez-Sarano M, Akins CW, Vahanian A. Mitral regurgitation. Lancet. 2009;373:1382–94. doi: 10.1016/S0140-6736(09)60692-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.