

Abstract
Breast cancer bone metastasis develops as the result of a series of complex interactions between tumor cells, bone marrow cells, and resident bone cells. The net effect of these interactions are the disruption of normal bone homeostasis, often with significantly increased osteoclast and osteoblast activity, which has provided a rational target for controlling tumor progression, with little or no emphasis on tumor eradication. Indeed, the clinical course of metastatic breast cancer is relatively long, with patients likely to experience sequential skeletal-related events (SREs), often over lengthy periods of time, even up to decades. These SREs include bone pain, fractures, and spinal cord compression, all of which may profoundly impair a patient’s quality-of-life. Our understanding of the contributions of the host bone and bone marrow cells to the control of tumor progression has grown over the years, yet the focus of virtually all available treatments remains on the control of resident bone cells, primarily osteoclasts. In this perspective, our focus is to move away from the current emphasis on the control of bone cells and focus our attention on the hallmarks of bone metastatic tumor cells and how these differ from primary tumor cells and normal host cells. In our opinion, there remains a largely unmet medical need to develop and utilize therapies that impede metastatic tumor cells while sparing normal host bone and bone marrow cells. This perspective examines the impact of metastatic tumor cells on the bone microenvironment and proposes potential new directions for uncovering the important mechanisms driving metastatic progression in bone based on the hallmarks of bone metastasis.
Keywords: Osteolysis, Metastasis, Pathology, Breast cancer, Treatment
Introduction
The skeleton is a multi-functional tissue responsible for a variety of processes that are fundamental to life. These include support, strength, and mobility of the overall organism, protection of the internal organs, as well as the maintenance of calcium and phosphate homeostasis [1]. The dynamic nature of bone is the consequence of its constant remodeling of old or damaged bone by osteoclasts of the hematopoietic macrophage/monocyte lineage, followed by the formation of new mineralized bone matrix by osteoblasts of the mesenchymal lineage. Moreover, bone is an endocrine organ, whose cells are capable of regulating (and being regulated by) the central nervous system, energy and glucose metabolism, and gonadal function [2].
Bone-forming osteoblasts and bone-resorbing osteoclasts (and their precursors) reside in the bone marrow compartment throughout their maturation. In addition, these host bone cells along with other immune cells of the hematopoietic lineage are in close proximity within the bone marrow, where stem cell niches and microenvironments are established [3–5]. Indeed, the histology of the tumor–bone interface in both humans and mouse models of tumor bone colonization reveals much about the cellular content and context of the bone marrow in the presence of metastatic tumor cells (Fig. 1), including our ability to replicate different features of human bone metastatic disease in vivo. The mechanics of these niches in the bone marrow drives a significant interdependence between hematopoietic stem cell differentiation and mesenchymal progenitor cells [6, 7] and the coupling of bone resorption with bone formation [8].
Fig. 1.
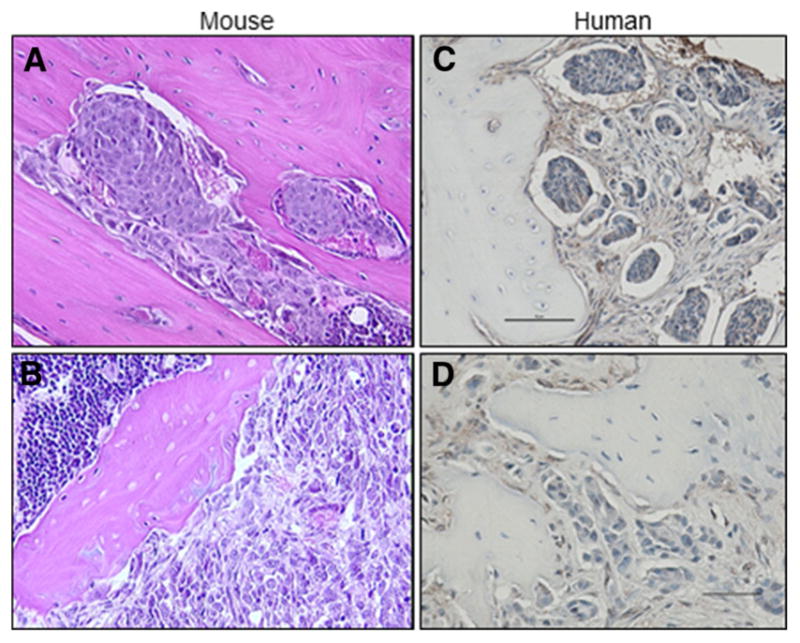
Histology of the tumor–bone interface. a, b Metastasis of human MDA-MET cells in the tibia of a nude mouse (40x). c, d Bone biopsy from patient with metastatic breast cancer. Bar = 50 μm
In the context of breast cancer bone metastasis, the bone marrow compartment provides not only a receptive growth factor-enriched environment but also favorable niches in which circulating/disseminated tumor cells can survive [8]. Following arrival in the bone marrow, tumor cells proliferate and interact with mesenchymal and hematopoietic progenitors at multiple stages of differentiation [9]. These interactions provide important signals for both tumor cells and host bone and bone marrow cells during tumor colonization of bone [4, 8], frequently (and often incorrectly) referred to as the “vicious-cycle” [10]. This term was originally associated with the underlying action of parathyroid hormone-related protein (PTHrP), a well-known regulator of tumor-associated bone destruction [10, 11] as well as the hypercalcemia of many cancer types [12]. It has been expanded to now include almost every tumor-derived factor implicated in breast cancer osteolysis. Interestingly, in many ways, the idea of a single responsible osteolytic agent has impeded progression towards a better understanding of the development of bone metastasis and has sustained the focus of potential treatments on managing the osteoclast, and not necessarily the tumor. The future of effective therapy (and perhaps cure) likely resides in the increased understanding of the common traits of bone metastatic breast cancer cells. Here we revisit the hallmarks of cancer [13, 14] and define the hallmarks of breast cancer bone metastasis; extravasation, disruption of bone homeostasis, secretion of osteolytic growth factors and others, engagement in bone niches, interactions with the host bone marrow microenvironment, and responses to the physical environment (Fig. 2), all culminating to promote breast cancer cell survival in the bone marrow, while highlighting some considerations for the future directions of the field.
Fig. 2.

Hallmarks of bone metastasis. Common features adopted by bone metastatic breast cancer cells that enable their survival in the bone marrow
Hallmarks of Breast Cancer Bone Metastasis
Survival in the Bone Marrow
The growth of disseminated tumor cells in subsequent metastatic locations is the primary cause of mortality for all cancer patients [15]. Despite being the focus of intense investigation, the molecular and cellular mechanisms that regulate the metastasis of disseminated tumor cells still remain largely unknown. Once resident in the bone marrow microenvironment, a select minority of metastatic tumor cells coordinate an extensive and complex series of molecular events that culminate in the colonization of the skeleton. The features that facilitate the survival of the arriving metastatic cells in the bone marrow allow for eventual manifestation as bone metastasis.
One of these features is the adoption of a quiescent phenotype, in which the tumor cells may enter a prolonged dormant or latent phase, eventually recurring as a clinically detectable metastasis [16]. What then defines a tumor cell as being dormant? A major impetus to the dormancy field has been the lack of a standardized definition of a dormant tumor cell. In general, a non-proliferative (e.g., Ki67 or BrdU negative) disseminated tumor cell that has not grown into a micrometastasis is often considered dormant [17–21], but there are several potential models for dormant tumor cell behavior: (1) very slow growing, (2) proliferative, but dying off at an equal rate, or (3) non-replicative until reactivated by the tumor microenvironment (e.g., angiogenic switch, immune surveillance) [22]. Importantly, these models may not be mutually exclusive; a slow-growing tumor cell may eventually be reactivated by the tumor microenvironment. Further, all of these models may exist simultaneously in various niches. In the bone marrow, an environment rich in growth factors, cytokines, and sinusoids/arterioles, any of these models may represent the behavior of a disseminated breast cancer cell.
There are likely to be many factors that enable meta-static tumor cells to become quiescent in the bone or any other distant site, but few have been identified [23]. In fact we know practically nothing about how tumor cells enter a dormant state. It has been suggested that circulating tumor cells adopt a dormant phenotype as a survival mechanism [24], but this is inconsistent with their ability to extravasate at a distant metastatic site. If a tumor cell becomes dormant while in circulation, it would almost certainly require an exit from dormancy to engage the endothelial niche and extravasate into the bone marrow. It is therefore reasonable to assume that dormant tumor cells detected in the bone marrow of patients acquired this dormant phenotype post-extravasation. The ability of the vascular niche, the first niche that the tumor cell encounters in the bone marrow, to induce a dormant phenotype is explored at length in the following section.
Of the factors identified that promote tumor cell exit from dormancy in the bone marrow [25], few ideas have been proposed that fit the patient model of solid tumor recurrence and survival in the bone marrow. Breast cancer patients that recur with bone metastatic disease present with bone metastases months to decades following tumor resection and primary/adjuvant therapy, and the latency period is particularly prolonged in patients with estrogen receptor (ER)-positive (ER+) disease [26]. This variation in the time to recurrence in patients may be due to multiple mechanisms of recurrence, or it may be due to the heterogeneous nature of the bone microenvironment. In a seminal paper from Tomasetti and Vogelstein [27], it was demonstrated that the lifetime risk of cancer in a particular tissue was correlated with the number of stem cell divisions for the tissue, suggesting that in addition to genetics and environmental influences, stochastic DNA replication errors or “bad luck” also drives tumorigenesis. If we apply this theory to bone metastatic breast cancer cells, is it possible that tumor cells exit dormancy and colonize the bone because of “bad luck” and that this may account for the broad range in patient time-to-relapse? We explore this concept in a later section.
Extravasation into the Bone Marrow Through the Vasculature/Lymphatics
The dissemination of tumor cells into the bone marrow must be facilitated by the vasculature and/or lymphatics and it is therefore likely that upon extravasation tumor cells must enter one of two phases: immediate proliferation and colonization of the bone marrow, or quiescence. Circulating tumor cells engage with the endothelial niche through adhesion molecules, chemokine-receptor interactions, and integrins [28], facilitating their entry into distant tissues. Are there factors secreted or expressed by endothelial cells that induce quiescence? Thrombospondin-1 produced by endothelial cells has been shown to induce dormancy in breast cancer cells that extravasate through established vasculature [29]. In contrast, sprouting neovasculature, which secretes TGFβ1 and periostin, promotes breast cancer cell growth. Thus, it would appear that if tumor cells extravasate through established vessels they will enter a quiescent state, while if they extravasate through sprouting neovasculature, they will immediately become proliferative. This is an intriguing idea since it would suggest that the use of clinical agents such as bevacizumab could be useful as an adjuvant therapy in early-stage breast cancer patients not to prevent dissemination to the bone marrow, but rather to prevent the early adoption of a proliferative phenotype upon dissemination. This would, however, require repurposing of bevacizumab which was withdrawn by the FDA for the metastatic breast cancer indication in 2011.
Breast cancer cells are capable of disseminating to the bone marrow through either the vasculature or lymphatics system, although it remains unclear how frequently breast cancer cells disseminate to the bone marrow through the lymphatics. This has clinical significance since lymph node involvement is frequently used to inform and direct the course of treatment in breast cancer patients, despite studies that have shown similar disease-free and overall survival five and eight years following diagnosis, regardless of the status of the patient sentinel lymph node or whether they received complete lymph node dissection [30–32].
VEGF-C, which is produced by many solid tumors including breast, has been identified as a potent inducer of tumor-associated lymphatics vessel formation in the human MCF7 breast cancer model [33] which is ER+ and does not induce significant osteolysis in mouse models of bone colonization [34–36]. A subsequent study examined whether VEGF-C protein expression in breast carcinoma samples (n = 51 tumors) was associated with bone marrow micrometastases found in iliac crest bone marrow aspirates. Among other parameters, no significant correlation between VEGF-C and the presence of micrometastases was observed [37]. VEGF-C has also been detected in ~ 45% of patients with osteosarcoma [38], and although one laboratory has identified several factors that regulate bone homeostasis including basic fibroblast growth factor (bFGF) [39], adiponectin [40], and leptin [41] as promoters of VEGF-C production in chondrosarcoma cells, VEGF-C is not correlated with metastasis [38]. Future studies, particularly in vivo studies that ablate lymphatic vessels in the bone marrow and examine the frequency of tumor dissemination to the bone marrow, may be key in determining whether lymphatic vessels are important for tumor cell dissemination and colonization of the bone marrow microenvironment.
Response to the Physical Microenvironment
Bone is a densely mineralized tissue with a high rigidity and modulus, making the skeleton an especially harsh environment for any tumor cell to establish and grow [42]. There is now ample evidence that breast cancer cells sense the modulus and mechanics of the local environment both in the primary tumor and at the bone metastatic site through integrin signaling. Cancer cells alter downstream signaling in response to mechanical changes in the environment, where increased stiffness of the environment typically promotes tumor progression [43–45]. Tumor cells seeded onto rigid substrates up-regulate factors that directly promote osteolysis through downstream osteoclast activation (e.g., via PTHrP). The expression of genes that further enhance production of osteolytic factors by tumor cells (e.g., GLI2, which we have termed “osteolytic drivers”) suggest that the physical modulus of the bone may fuel tumor-induced bone destruction [46]. This raises several important questions. First, do the tumor cells sense the stiffness of the bone matrix, or are these mechanotransduction signals mediated through the bone lining cells? In vitro evidence strongly supports direct effects of tissue modulus on tumor cell gene expression in bone metastatic breast and lung cancer cells cultured alone on substrates of varying modulus [46–48]. It is difficult to test whether tumor cells come into direct contact with the bone matrix in vivo, but more importantly, must we know? Histological evidence suggests that tumor cells may come in direct contact with the bone surface at late stages of disease (Fig. 1b), but if blocking integrin signaling ablates this interaction, it may not be necessary to determine whether the mechanotransduction signals are mediated through tumor–bone or tumor–bone lining cell interactions [35, 36]. Second, how does the bone modulus influence the initial phases of bone colonization and the progression of tumor cells in the bone marrow? One study showed that MCF7 human breast cancer cells, which home to bone but do not induce extensive bone destruction [34–36], do not up-regulate expression of osteolytic genes when they are cultured on stiff substrates. This is in contrast to MDA-MB-231 bone metastatic cells, which rapidly up-regulate expression of both GLI2 and PTHrP [46]. This suggests that cells that are already aggressive in nature can be further stimulated to promote bone destruction, but that bone modulus likely does not provide the initial cue that enables a disseminated tumor cell to induce osteolysis.
In addition to bone modulus, breast cancer cells encounter a variety of other physical stimuli in the bone microenvironment including a number of cell types from various lineages, different pH levels [49], and fluctuating oxygen tensions [50]. In general the bone marrow is considered hypoxic, although oxygen tensions in the marrow fluctuate across the cortices and trabeculae, and according to some measurements have been reported to range from < 1 to 6% partial oxygen (pO2) [51–53]. The deepest sinusoidal regions of the bone marrow are predicted to be the most hypoxic, dipping to levels at or below ~ 1% pO2 [51]. It has been observed in mice that breast cancer cells home to the metaphyseal growth plate following intracardiac inoculation [10, 54], where there is an abundance of growth factors, cytokines, and most importantly, vasculature upon which the tumor cells presumably disseminate. Since most oxygen measurements have been derived from either optical measurements in mouse calvaria or by mathematical modeling, it is generally assumed, but as yet unproven, that this region of the growth plate is less hypoxic than the rest of the bone marrow, given the abundance of vasculature. Given the generally hypoxic environment of the bone marrow, what is the impact of hypoxia on bone disseminated tumor cells? One potential effect may be on their exit from dormancy. Leukemia inhibitor factor (LIF) receptor (LIFR) signaling in breast cancer cells has been shown to maintain a state of dormancy in the bone marrow [35]. Hypoxia down-regulates LIFR promoter activity, mRNA, and protein levels in human breast cancer cells; however, this only occurs when breast cancer cells are cultured in ~ 0.5% pO2. Importantly, we know that breast cancer cells encounter hypoxia in the bone marrow, even along the vascularized and theoretically less hypoxic growth plate, as indicated by staining for the hypoxia probe pimonidazole (Fig. 3), but current technologies do not enable determination of the precise oxygen levels encountered. Pimonidazole is estimated to detect oxygen levels ≤ 10 mm Hg (approximately 1.3% pO2), but this has not been specifically testedin bone [55, 56].
Fig. 3.
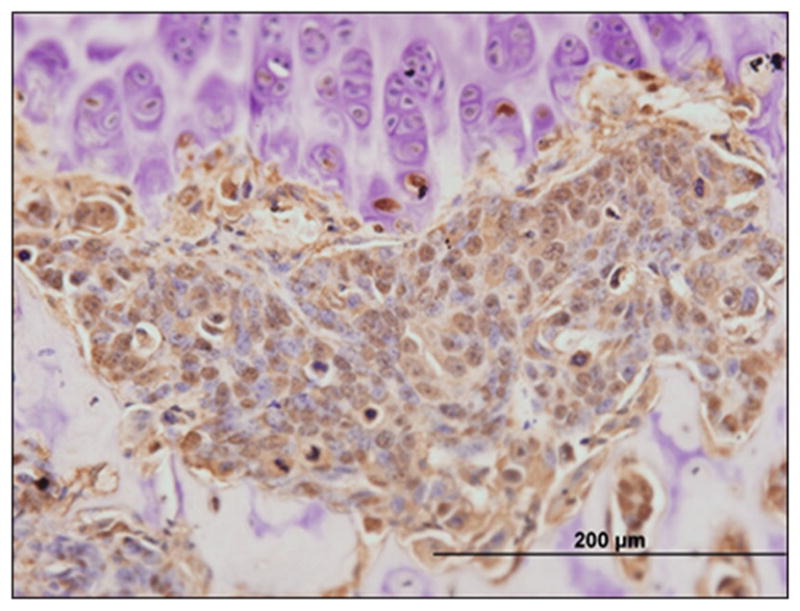
Breast cancer cells experience hypoxia in the bone marrow. A micrometastasis of human MCF7 cells in the tibia of a nude mouse stains for the hypoxia marker pimonidazole at the proximal metaphyseal growth plate. Brown staining = pimonidazole. Bar = 200 μm
Given that breast cancer cells likely disseminate into the bone marrow at the relatively oxygen-rich metaphyseal growth plate, it is unlikely that they will immediately experience such low levels of oxygen as to down-regulate LIFR and push these cells out of dormancy. Over time, however, as patients age and as the bone undergoes remodeling, it is reasonable to assume that the immediate environment surrounding a tumor cell will evolve. As it does, changes in the vasculature and cellular makeup may profoundly impact tumor cell availability to oxygen. We hypothesize that if a tumor cell happens to reside in an anoxic microenvironment (e.g., surrounding blood vessels die off due to injury or aging or proximity to a bone-remodeling unit with relatively high cellularity), there may be dips in oxygen below 0.5% pO2 that promote LIFR down-regulation on tumor cells and their spontaneous exit from dormancy. Thus, the “bad luck” hypothesis proposed by Tomasetti and Vogelstein [27] of tumor cell evolution in the primary site, independent of stem cell divisions, seems relevant here. Perhaps the spatial and cellular makeup of the adjacent, hyper-localized microenvironment surrounding a disseminated tumor cell may in part contribute to tumor cell exit from dormancy. This model partially accounts for the high degree of variability in the time to relapse in breast cancer patients who present with bone metastases after a period of remission, often measured in years. Further in vivo testing of this hypothesis is obviously required, and requires measurement of the precise levels of oxygen in real time. Technology that enables determination of the exact oxygen levels in multiple bone marrow compartments at high resolution will ultimately determine whether this hypothesis is correct. There are undoubtedly other impacts of the hypoxic microenvironment on tumor cells disseminated to the bone marrow, such as metabolic reprogramming [57], which is currently being explored by multiple investigators. Although the mechanisms remain ill-defined, it is clear that the physical components of the bone microenvironment (hypoxia, pH, and modulus) uniquely impact tumor cells disseminated within the skeleton.
Engagement with the Osteoblast/Osteoclast Niche(s)
It is important to recall that the bone destruction resulting from tumor osteolysis as well as any tumor-induced bone formation is entirely due to tumor-derived stimulation of resident bone cells; it is not related to any direct actions of the tumor cells themselves on the skeleton [8]. Upon dissemination to the bone marrow, it is well established that breast cancer cells interact with osteoblasts (and other resident cells) to induce osteolysis. These mechanisms were defined many years ago through the tumor-osteoblast PTHrP/PTHR1 signaling induction of (receptor activator of nuclear factor-κB ligand) RANKL in osteoblasts and subsequent osteoclastogenesis [58], but recent advances in imaging as well as detailed mechanistic analyses have begun to spatially and temporally characterize these interactions. Once resident in the bone marrow, the localization of tumor cells to the endosteal niche has been observed using live animal imaging for myeloma cells colonizing the bone marrow [59]. In this system, dormant myeloma cells are observed in close contact with the osteoblast niche, suggesting that osteoblasts promote a dormant phenotype in these cells. In contrast, others have shown that osteoblasts promote E-cadherin breast tumor cell exit from dormancy in the bone marrow through adherens junctions and N-cadherin expression on osteoblast lineage cells and that these effects are cell contact dependent [36]. This was functionally tested and it was shown that when E-cadherin is blocked in breast cancer cells, the proliferative advantage from osteoblasts is lost. While these two ideas appear to be in opposition, it is much more likely that this is a complex system and consideration must be given to the temporal and spatial context in which the tumor cells will encounter the osteoblast niche. It is possible that osteoblasts may promote dormancy in the earliest stages of extravasation into the bone marrow due to the expression of a particular factor and perhaps the absence of E-cadherin, if these cells have recently undergone EMT to progress through the metastatic cascade [60]. Likewise, it is possible that tumor cells may re-express E-cadherin by undergoing MET in the bone marrow and this would change the nature of the osteoblast–tumor cell interaction. The ability to observe tumor cell dissemination to the bone marrow in real time via intravital imaging has significantly advanced our understanding of the physical interactions between tumor cells and the bone microenvironment; we are now tasked with defining the molecular mechanisms that direct these interactions and enable tumor cells to engage the osteoblast/osteoclast niche [59, 61].
Interactions with Marrow-Resident Cells
The interactions between host bone cells and the immune system are only now being unraveled [62]. The majority of immune cells (B, T, NK, marrow-derived suppressor cells, macrophages, and neutrophils) as well as immune cytokines are involved to a greater and lesser extent in modulating or fine tuning bone homeostasis and turnover. Thus, it is not surprising that following development of a primary tumor, the subsequent establishment and growth in the bone/bone marrow is associated with major changes that divert immune cells from attacking to supporting tumor survival and progression [62, 63].
The cellular milieu of the tumor microenvironment is occupied by resident and recruited macrophages, dendritic cells, T cells, as well as natural killer cells and megakaryocytes [64]. Among these, tumor-associated macrophages [65] and T cells are frequently the prominent leukocytes present at both the primary and metastatic sites [66, 67]. These cells can exert profound effects during cancer progression and the net effect of these diverse cellular activities regulates bone remodeling [67–69]. Yet, the contribution of host immune cells to the microenvironment of osteolytic bone metastases has only recently begun to receive investigative attention. In fact, many previous mechanistic models of metastatic tumor progression proposed by many investigators have been derived from experiments utilizing immune compromised animal models. Thus, the more recent and welcome use of immune-replete and syngeneic animal models of tumor growth have driven greater elucidation of specific host immune cell contributions to the tumor microenvironment in bone, as well as in other metastatic sites [61].
Although the processes of bone remodeling (resorption and formation) are performed by osteoblasts and osteoclasts, the cells that orchestrate this complex process and seem to coordinate the activities of both osteoclasts and osteoblasts are osteocytes. These mesenchymal-derived terminally differentiated, bone matrix-entombed cells of the osteoblastic lineage comprise > 95% of total bone cells [70]. Osteocyte dendrites have been shown to have direct contact via gap junctions with other osteocytes, osteoblasts, and lining cells along the bone surface, as well as directly reaching into the bone marrow [71]. Importantly, like both osteoblasts and T cells, osteocytes can activate the differentiation and function of osteoclasts through their production of RANKL the essential factor for osteoclast formation [72, 73].
The demonstration that osteocytes produce significant levels of RANKL confirms the role of the osteocyte in the regulation of bone resorption as well as the overall maintenance of bone metabolism. It is now widely held dogma that osteocytes interact extensively with the cells of the bone marrow microenvironment, which directly implicates these cells in the regulation of the bone marrow, including during the arrival and progression of metastatic tumor cells. Indeed, in multiple myeloma patient bone biopsies, viable osteocyte number is significantly less than non-myeloma patients, which suggests that changes in osteocyte viability may precede the arrival of tumor cells [74]. How alterations in osteocyte viability and number, as well as other marrow-resident immune cells may affect breast cancer bone metastasis continues to be an obvious feature but remains a largely underexplored question.
Expression/Secretion of Factors that Promote Osteolysis: Development of New Therapeutics
The tumor microenvironment, which develops and changes with influences from both primary tumor as well as with metastatic tumor cell burden and the activation of host responses, is a critical component of bone metastasis [75]. Metastatic tumor cells regulate the local bone and bone marrow compartments via the secretion of growth factors and cytokines such as PTHrP, transforming growth factor beta (TGFβ), interleukin 8 (IL-8), IL-6, and tumor necrosis factor alpha (TNFα) (along with many others) that have been shown to directly or indirectly activate the recruitment, differentiation, and activity of macrophages, osteoclasts, fibroblasts, endothelial cells, and their progenitors [4, 5, 75]. Bone metastatic breast cancer cells may also express osteolytic “drivers” or transcription factors such as GLI2 and connective tissue growth factor (CTGF) that enhance secretion of osteolytic growth factors and cytokines [50]. Not content only with driving bone resorption, metastatic breast cancer cells also inhibit osteoblast differentiation through the RUNX2-dependent expression of sclerostin [76, 77]. This action disrupts the coupling of formation and resorption to facilitate unimpeded osteoclastic bone resorption resulting in the profound skeletal destruction characteristic of bone metastasis. Despite decades of research to uncover the molecular mechanisms that drive tumor progression and colonization in the bone marrow, few advances in knowledge have translated to clinical progress. This is in part due to the recurrence of signaling pathways with known roles in tumor etiology (e.g., TGFβ, WNT, Hedgehog) that appear unlikely to translate into FDA-approved therapies in the near future. We thus support several alternative approaches to therapeutically target osteolytic factors and drivers, which is to (1) alter the delivery of drugs into the bone metastatic site to enhance bioavailability and reduce off-target effects, (2) utilize broad agents that hit multiple targets, and (3) delve into personalized medicine.
In the last decade, we have significantly advanced our understanding of the signaling pathways, cellular compartments, and environmental factors that regulate tumor-induced osteolysis, and yet breast cancer-specific mortality for patients with bone metastasis has not significantly improved since the introduction of bisphosphonates and denosumab, which are now recommended as standard of care for patients with early or advanced post-menopausal breast cancer [78]. However, these therapeutic approaches which decrease skeletal-related events and in post-menopausal breast cancer patients increase overall survival [79] are still directly targeting tumor osteolysis and not tumor cells. The targeting of osteolytic factors (e.g., PTHrP) and bone resorption has provided much needed advances in patient care, but alternative approaches that translate in vivo tumor biology findings to breast cancer patients with bone metastatic disease are needed.
Targeting Wnt signaling [47, 80, 81], TGFβ signaling [82, 83], and Hedgehog signaling [84–86] in mouse models of bone colonization have all provided mechanistic insight, but will require creative delivery to be effective and prevent off-target effects and toxicity in patients. It is worth noting that the issue of bioavailability in the bone is more likely to be the limiting factor in targeting Wnt and Hedgehog signaling, since these pathways are physiologically switched off post-development and are frequently activated in tumor cells [87]. Advances in the field of nanoparticles hold promise for this avenue since several groups have identified effective inhibitors to the TGFβ [83, 88] and Hedgehog signaling pathways [89] that with nanoparticle encapsulation enable tumors to selectively uptake these inhibitors and prevent bone destruction.
An alternative approach to targeting bone disseminated tumor cells is to tackle all newly identified pathways through drug repurposing [90], and target multiple pathways at once. Interleukins have long been identified as osteolytic factors, including IL-8 and IL-6 [91–93], and neutralizing antibodies against IL-6 and its receptor in particular have been developed for clinical use in recent years. Siltuximab, tocilizumab, and sarilumab are all antagonists to the IL-6/IL-6 receptor signaling pathway and are FDA-approved for treatment of rheumatoid arthritis [94]. To date, only one clinical trial has been initiated to test the efficacy of tocilizumab in metastatic breast cancer, but given the mechanistic importance of IL-6 in tumor-induced bone disease, these inhibitors hold promise for use in bone metastatic breast cancer.
LIFR, which is a member of the IL-6 cytokine family and was identified as a pro-dormancy factor in breast cancer cells, was recently found to be regulated through an epigenetic mechanism that is targetable through the use of histone deacetylase (HDAC) inhibitors. The HDAC inhibitor valproic acid significantly stimulated not just LIFR mRNA levels, but 9 of 12 previously published pro-dormancy genes [35], suggesting HDAC inhibitors may be effective in promoting a chronic state of dormancy. There are currently four HDAC inhibitors that are FDA-approved, and many others that are currently in phase I–III clinical trials, for the treatment of other tumor types and diseases, including chronic treatment of epilepsy and migraines [95]. Further, several groups are investigating the use of HDAC inhibitors in bone metastatic breast [96], prostate cancer, and osteosarcoma with reports that these inhibitors block tumor-induced osteolysis. [96]. With these drugs already FDA-approved, the approval process for the treatment of metastatic breast, prostate, or osteosarcoma would be greatly shortened and may rapidly translate into a therapeutic benefit for these patients.
Perhaps the antithesis to the idea of repurposing drugs that hit multiple targets with less specificity is to approach the treatment of bone metastasis from the field of personalized medicine. Would it be of greater benefit to sequence a bone metastasis sample from every patient to obtain a specific genetic signature and personalized treatment program? The first hurdle is to address the complexity of intra-patient tumor heterogeneity. Is a breast cancer bone metastasis biopsy representative of other bone metastatic lesions? This may be particularly problematic in prostate cancer, where patients frequently present with a combination of osteolytic and osteoblastic disease [97], but mixed lesions also occur in breast cancer patients [98]. The second hurdle is that patients who have had the primary tumor resected and do not have detectable bone metastases remain at risk of developing bone metastases, but we have no effective way of detecting dormant disseminated tumor cells in these patients, nor do we know if the primary tumor is reflective of tumor cells that now reside dormant in the bone marrow compartment. In this case, it would be possible to sequence the primary tumor, ideally post-treatment, to extract the molecular makeup of metastatic tumor cells that have undergone the same treatment regimen, but experienced a microenvironment distinct from the disseminated tumor cells. In this regard, some of the molecular features of the tumor cell should remain intact, but it will be impossible to know which ones to target. In patients with clinically detectable bone metastatic disease, a biopsy or tumor sample obtained during palliative surgery would provide a more accurate genetic signature, but would also be far more difficult to treat given the extent of disease progression. Importantly, personalized medicine and treatment plans are becoming increasingly available [99] and as a field we should capitalize on these technologies. This capability will be ideal when it becomes possible to detect individual tumor cells and/or dormant micrometastases in patient bone marrow and to analyze these at the single cell level.
These targeting strategies rely on the secretion of osteolytic factors and expression of osteolytic drivers, which remain a key hallmark of bone metastatic breast cancer cells. Until other physical and cellular mechanisms are identified, this feature will likely continue to drive forward therapeutic advances that target bone metastatic cells.
Disruption of Normal Bone Homeostasis
Tumor cells arriving and residing in the bone marrow can remodel the bone and bone marrow into a permissive environment favoring tumor cell expansion [100], similar to tumor cells residing in the primary site [101]. Indeed, circulating tumor cells can even re-seed the original primary tumor site [102–104]. Since the adult human skeleton comprises some 206 bones, the tumor burden in the skeleton is likely the greatest of any individual tissue, and hence understanding the interactions and disruption of the skeleton by tumor cells is a major research imperative.
Numerous interactions exist between bone cells and resident bone marrow cells that are necessary to maintain normal endocrine homeostasis [8, 105]. As such, a variety of putative bone-targeted treatment opportunities targeting cancer cells resident in bone exist, but are largely under-explored. The requirement to maintain bone homeostasis and protect (as much as possible) bone mass and strength, in the face of oncologic treatments that kill tumor cells, is fundamental. Recent studies have described significant negative effects of standard chemotherapeutic agents on bone healing, suggesting that the specific treatments (or the tumor cells themselves) may have altered the behavior and/or differentiated activity of osteoclasts, osteoblasts, and/or osteocytes [106–108].
The identification of the fundamental contribution of the osteocyte to the control of bone homeostasis that is the basis of normal bone physiology [109] has spawned increased interest in the role of these cells in tumor progression and metastasis. As such significant efforts are now focusing on how the normal processes of bone resorption and bone formation controlled by osteocytes are disrupted by the formation of osteolytic and/or osteoblastic bone lesions. Indeed, some have even suggested that agents such as bisphosphonates which are highly effective in the adjuvant breast cancer bone metastasis setting exert their effects through osteocyte-mediated connexin 43 hemichannels [110]. If indeed as we suspect such mechanisms and the involvement of osteocytes are correct, then the discovery of new breast cancer bone metastasis treatment approaches will require significant re-thinking: shift away from osteoclasts and now incorporate osteocytes and the metastatic cells themselves.
The inability to maintain bone homeostasis in the presence of significant tumor burden leads to the collapse of the functional bone-remodeling compartment, ultimately resulting in patient fracture and increased morbidity. Interestingly, some bone metastatic patients never progress to this point, while others rapidly progress to a skeletal-related event. While estrogen receptor (ER) status may account for some of these differences (ER+ patients typically have longer latency and therefore may not develop clinically detectable bone metastases [111]), it remains unclear how early the functional bone-remodeling unit is disrupted by this process. Can a few disseminated tumor cells significantly disrupt “coupling”? How many tumor cells are required, and do these cells require certain features to disrupt this fundamental skeletal process? Studies are currently underway to investigate changes in the skeleton very early in the course of disease progression and to determine whether the uncoupling of osteoblasts and osteoclasts can lead to changes in the material properties that alter fracture risk early in the course of tumor dissemination to bone.
Thoughts and Opinions
The hallmarks of cancer as they were originally proposed [13, 14] focused on general features of all primary tumor types. In this perspective, we propose the hallmarks of bone metastasis (Fig. 2) as extravasation into the bone marrow through the vasculature/lymphatics, disruption of normal bone homeostasis, expression/secretion of osteolytic factors, interactions with marrow-resident cells, engagement with the osteoblast/osteoclast niche(s), and the response to the physical microenvironment. In the context of bone metastasis, these features provide bone metastatic cells with a significant growth and progression advantage.
It is important to recognize that the original hallmarks concept of Hanahan and Weinberg [13, 14] implies common features across tumor types, much the same as our proposal for bone metastasis suggests; however, it is critically important to remember that bone metastasis, like cancer, is not a single disease and that different tumor types metastasizing or inhabiting the bone and bone marrow may or may not share all the described pathways. For example, the ability to stimulate osteoclastic bone resorption is obviously a fundamental and shared hallmark of bone metastatic cancer cells, yet responses to the physical microenvironment may not be as universally shared or may present in different ways. Further, these traits are not mutually exclusive and are likely inextricably linked (e.g., the physical features of the bone may enable tumor cells to evolve, thus changing the tumor cell interactions with marrow-resident cells and the osteoblast/osteoclast niche).
Our refocused hallmarks model provides a roadmap and a flashlight for uncovering new potential treatment opportunities. However, these hallmarks do not fully address or answer the important questions of how these attributes are acquired or even why such alterations are undertaken in the first place, and will likely evolve with new discoveries in the field. Such fundamental questions must be answered and perhaps the answers reside in the still murky area of cancer stem cells and stem cell division [27] that may be resolved by transcriptomic or other “omic” profiling [27, 112] at the single cell level. We eagerly anticipate the collection of these and other data that will no doubt continue to revise and refocus our models and hallmarks of breast cancer bone metastasis.
Acknowledgments
This work was supported by NIH R01CA166060 (LJS) and NIH R00CA194198 (RWJ). Our efforts in this area are dedicated to the memory of the late Dr. Greg Mundy whose vision and leadership provided much of the stimulus for the field to study the bone marrow microenvironment of cancer.
Footnotes
Competing interests The authors Rachelle W. Johnson and Larry J. Suva declare no competing interests.
References
- 1.Suva LJ, Gaddy D, Perrien DS, Thomas RL, Findlay DM. Regulation of bone mass by mechanical loading: microarchitecture and genetics. Curr Osteoporos Rep. 2005;3:46–51. doi: 10.1007/s11914-005-0003-0. [DOI] [PubMed] [Google Scholar]
- 2.Reagan MR, Rosen CJ. Navigating the bone marrow niche: translational insights and cancer-driven dysfunction. Nat Rev Rheumatol. 2016;12:154–168. doi: 10.1038/nrrheum.2015.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–1201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 5.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou HS, Carter BZ, Andreeff M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol Med. 2016;13:248–259. doi: 10.20892/j.issn.2095-3941.2016.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makhoul I, Montgomery CO, Gaddy D, Suva LJ. The best of both worlds—managing the cancer, saving the bone. Nat Rev Endocrinol. 2016;12:29–42. doi: 10.1038/nrendo.2015.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 10.Guise TA, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98:1544–1549. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suva LJ, et al. A parathyroid hormone-related protein implicated in malignant hypercalcemia: cloning and expression. Science. 1987;237:893–896. doi: 10.1126/science.3616618. [DOI] [PubMed] [Google Scholar]
- 12.Mundy GR, Edwards JR. PTH-related peptide (PTHrP) in hypercalcemia. J Am Soc Nephrol. 2008;19:672–675. doi: 10.1681/ASN.2007090981. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Coleman RE, et al. Possible survival benefits from zoledronic acid treatment in patients with bone metastases from solid tumours and poor prognostic features-An exploratory analysis of placebo-controlled trials. J Bone Oncol. 2013;2:70–76. doi: 10.1016/j.jbo.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 17.Pantel K, Alix-Panabieres C, Riethdorf S. Cancer micrometastases. Nat Rev Clin Oncol. 2009;6:339–351. doi: 10.1038/nrclinonc.2009.44. [DOI] [PubMed] [Google Scholar]
- 18.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–456. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 19.Pantel K, Riethdorf S. Pathology: are circulating tumor cells predictive of overall survival? Nat Rev Clin Oncol. 2009;6:190–191. doi: 10.1038/nrclinonc.2009.23. [DOI] [PubMed] [Google Scholar]
- 20.Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009;9:302–312. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- 21.Price TT, et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med. 2016;8:340ra373. doi: 10.1126/scitranslmed.aad4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang XH, Giuliano M, Trivedi MV, Schiff R, Osborne CK. Metastasis dormancy in estrogen receptor-positive breast cancer. Clin Cancer Res. 2013;19:6389–6397. doi: 10.1158/1078-0432.CCR-13-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Croucher PI, McDonald MM, Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer. 2016;16:373–386. doi: 10.1038/nrc.2016.44. [DOI] [PubMed] [Google Scholar]
- 24.Alvarez-Cubero MJ, et al. Dormant circulating tumor cells in prostate cancer: therapeutic, clinical and biological implications. Curr Drug Targets. 2016;17:693–701. doi: 10.2174/1389450116666150309121346. [DOI] [PubMed] [Google Scholar]
- 25.Dasgupta A, Lim AR, Ghajar CM. Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Mol Oncol. 2017;11:40–61. doi: 10.1002/1878-0261.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomis RR, Alarcon C, Nadal C, Van Poznak C, Massague J. C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell. 2006;10:203–214. doi: 10.1016/j.ccr.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 27.Tomasetti C, Vogelstein B. Cancer risk: role of environment-response. Science. 2015;347:729–731. doi: 10.1126/science.aaa6592. [DOI] [PubMed] [Google Scholar]
- 28.Smith HA, Kang Y. The metastasis-promoting roles of tumor-associated immune cells. J Mol Med (Berl) 2013;91:411–429. doi: 10.1007/s00109-013-1021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghajar CM, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15:807–817. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giuliano AE, et al. Association of occult metastases in sentinel lymph nodes and bone marrow with survival among women with early-stage invasive breast cancer. JAMA. 2011;306:385–393. doi: 10.1001/jama.2011.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giuliano AE, et al. Locoregional recurrence after sentinel lymph node dissection with or without axillary dissection in patients with sentinel lymph node metastases: the American College of Surgeons Oncology Group Z0011 randomized trial. Ann Surg. 2010;252:426–432. doi: 10.1097/SLA.0b013e3181f08f32. discussion 432–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viehl CT, et al. Prognostic impact and therapeutic implications of sentinel lymph node micro-metastases in early-stage breast cancer patients. J Surg Oncol. 2011;103:531–533. doi: 10.1002/jso.21693. [DOI] [PubMed] [Google Scholar]
- 33.Karpanen T, et al. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res. 2001;61:1786–1790. [PubMed] [Google Scholar]
- 34.Thomas R, et al. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 1999;140:4451–4458. doi: 10.1210/endo.140.10.7037. [DOI] [PubMed] [Google Scholar]
- 35.Johnson RW, et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol. 2016;18:1078–1089. doi: 10.1038/ncb3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015;27:193–210. doi: 10.1016/j.ccell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoar FJ, Chaudhri S, Wadley MS, Stonelake PS. Co-expression of vascular endothelial growth factor C (VEGF-C) and c-erbB2 in human breast carcinoma. Eur J Cancer. 2003;39:1698–1703. doi: 10.1016/s0959-8049(03)00382-4. [DOI] [PubMed] [Google Scholar]
- 38.Park HR, Min K, Kim HS, Jung WW, Park YK. Expression of vascular endothelial growth factor-C and its receptor in osteosarcomas. Pathol Res Pract. 2008;204:575–582. doi: 10.1016/j.prp.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 39.Tzeng HE, Chang AC, Tsai CH, Wang SW, Tang CH. Basic fibroblast growth factor promotes VEGF-C-dependent lymphangiogenesis via inhibition of miR-381 in human chondrosarcoma cells. Oncotarget. 2016;7:38566–38578. doi: 10.18632/oncotarget.9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang CY, et al. Adiponectin promotes VEGF-C-dependent lymphangiogenesis by inhibiting miR-27b through a CaMKII/AMPK/p38 signaling pathway in human chondrosarcoma cells. Clin Sci (Lond) 2016;130:1523–1533. doi: 10.1042/CS20160117. [DOI] [PubMed] [Google Scholar]
- 41.Yang WH, et al. Leptin promotes VEGF-C production and induces lymphangiogenesis by suppressing miR-27b in human chondrosarcoma cells. Sci Rep. 2016;6:28647. doi: 10.1038/srep28647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin TJ. Manipulating the environment of cancer cells in bone: a novel therapeutic approach. J Clin Invest. 2002;110:1399–1401. doi: 10.1172/JCI17124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9:325–342. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- 44.Kwakwa KA, Sterling JA. Integrin alphavbeta3 signaling in tumor-induced bone disease. Cancers (Basel) 2017;9:84. doi: 10.3390/cancers9070084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwakwa KA, Vanderburgh JP, Guelcher SA, Sterling JA. Engineering 3D models of tumors and bone to understand tumor-induced bone disease and improve treatments. Curr Osteoporos Rep. 2017;15:247–254. doi: 10.1007/s11914-017-0385-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruppender NS, et al. Matrix rigidity induces osteolytic gene expression of metastatic breast cancer cells. PLoS ONE. 2010;5:e15451. doi: 10.1371/journal.pone.0015451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson RW, et al. Wnt signaling induces gene expression of factors associated with bone destruction in lung and breast cancer. Clin Exp Metastasis. 2014;31:945–959. doi: 10.1007/s10585-014-9682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Page JM, et al. Matrix rigidity regulates the transition of tumor cells to a bone-destructive phenotype through integrin beta3 and TGF-beta receptor type II. Biomaterials. 2015;64:33–44. doi: 10.1016/j.biomaterials.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amend SR, Pienta KJ. Ecology meets cancer biology: the cancer swamp promotes the lethal cancer phenotype. Oncotarget. 2015;6:9669–9678. doi: 10.18632/oncotarget.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson RW, Sowder ME, Giaccia AJ. Hypoxia and bone metastatic disease. Curr Osteoporos Rep. 2017;15:231–238. doi: 10.1007/s11914-017-0378-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spencer JA, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508:269–273. doi: 10.1038/nature13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys J. 2001;81:685–696. doi: 10.1016/S0006-3495(01)75733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harrison JS, Rameshwar P, Chang V, Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood. 2002;99:394. doi: 10.1182/blood.v99.1.394. [DOI] [PubMed] [Google Scholar]
- 54.Bendre MS, et al. Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer cells correlates with bone metastasis in vivo. Cancer Res. 2002;62:5571–5579. [PubMed] [Google Scholar]
- 55.Chou SC, Azuma Y, Varia MA, Raleigh JA. Evidence that involucrin, a marker for differentiation, is oxygen regulated in human squamous cell carcinomas. Br J Cancer. 2004;90:728–735. doi: 10.1038/sj.bjc.6601585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gross MW, Karbach U, Groebe K, Franko AJ, Mueller-Klieser W. Calibration of misonidazole labeling by simultaneous measurement of oxygen tension and labeling density in multicellular spheroids. Int J Cancer. 1995;61:567–573. doi: 10.1002/ijc.2910610422. [DOI] [PubMed] [Google Scholar]
- 57.Dupuy F, et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015;22:577–589. doi: 10.1016/j.cmet.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 58.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 59.Lawson MA, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun. 2015;6:8983. doi: 10.1038/ncomms9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Junankar S, et al. Real-time intravital imaging establishes tumor-associated macrophages as the extraskeletal target of bisphosphonate action in cancer. Cancer Discov. 2015;5:35–42. doi: 10.1158/2159-8290.CD-14-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mori G, D’Amelio P, Faccio R, Brunetti G. Bone-immune cell crosstalk: bone diseases. J Immunol Res. 2015;2015:108451. doi: 10.1155/2015/108451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roato I. Interaction among cells of bone, immune system, and solid tumors leads to bone metastases. Clin Dev Immunol. 2013;2013:315024. doi: 10.1155/2013/315024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lau YS, et al. RANKL-dependent and RANKL-independent mechanisms of macrophage-osteoclast differentiation in breast cancer. Breast Cancer Res Treat. 2007;105:7–16. doi: 10.1007/s10549-006-9438-y. [DOI] [PubMed] [Google Scholar]
- 66.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 67.Fournier PG, Chirgwin JM, Guise TA. New insights into the role of T cells in the vicious cycle of bone metastases. Curr Opin Rheumatol. 2006;18:396–404. doi: 10.1097/01.bor.0000231909.35043.da. [DOI] [PubMed] [Google Scholar]
- 68.Kamalakar A, et al. PTHrP(12-48) modulates the bone marrow microenvironment and suppresses human osteoclast differentiation and lifespan. J Bone Miner Res. 2017;32:1421–1431. doi: 10.1002/jbmr.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leblanc R, Peyruchaud O. The role of platelets and megakaryocytes in bone metastasis. J Bone Oncol. 2016;5:109–111. doi: 10.1016/j.jbo.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dallas SL, Prideaux M, Bonewald LF. The osteocyte: an endocrine cell… and more. Endocr Rev. 2013;34:658–690. doi: 10.1210/er.2012-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palumbo C, Ferretti M, Marotti G. Osteocyte dendrogenesis in static and dynamic bone formation: an ultrastructural study. Anat Rec A. 2004;278:474–480. doi: 10.1002/ar.a.20032. [DOI] [PubMed] [Google Scholar]
- 72.Nakashima T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–1234. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 73.Xiong J, et al. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giuliani N, et al. Increased osteocyte death in multiple myeloma patients: role in myeloma-induced osteoclast formation. Leukemia. 2012;26:1391–1401. doi: 10.1038/leu.2011.381. [DOI] [PubMed] [Google Scholar]
- 75.Suva LJ, Washam C, Nicholas RW, Griffin RJ. Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol. 2011;7:208–218. doi: 10.1038/nrendo.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gkotzamanidou M, et al. Sclerostin: a possible target for the management of cancer-induced bone disease. Expert Opin Ther Targets. 2012;16:761–769. doi: 10.1517/14728222.2012.697154. [DOI] [PubMed] [Google Scholar]
- 77.Mendoza-Villanueva D, Zeef L, Shore P. Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2/CBFbeta-dependent expression of the Wnt antagonist, sclerostin. Breast Cancer Res. 2011;13:R106. doi: 10.1186/bcr3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wilson C, Coleman R. Adjuvant bone-targeted therapies for postmenopausal breast cancer. JAMA Oncol. 2016;2:423–424. doi: 10.1001/jamaoncol.2015.5768. [DOI] [PubMed] [Google Scholar]
- 79.Early Breast Cancer Trialists’ Collaborative Group. Adjuvant bisphosphonate treatment in early breast cancer: meta-analyses of individual patient data from randomised trials. Lancet. 2015;386:1353–1361. doi: 10.1016/S0140-6736(15)60908-4. [DOI] [PubMed] [Google Scholar]
- 80.Taipaleenmaki H, et al. Antagonizing miR-218-5p attenuates Wnt signaling and reduces metastatic bone disease of triple negative breast cancer cells. Oncotarget. 2016;7:79032–79046. doi: 10.18632/oncotarget.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hassan MQ, et al. miR-218 directs a Wnt signaling circuit to promote differentiation of osteoblasts and osteomimicry of metastatic cancer cells. J Biol Chem. 2012;287:42084–42092. doi: 10.1074/jbc.M112.377515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yin JJ, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Biswas S, et al. Anti-transforming growth factor ss antibody treatment rescues bone loss and prevents breast cancer metastasis to bone. PLoS ONE. 2011;6:e27090. doi: 10.1371/journal.pone.0027090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnson RW, et al. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 2011;71:822–831. doi: 10.1158/0008-5472.CAN-10-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sterling JA, et al. The hedgehog signaling molecule Gli2 induces parathyroid hormone-related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res. 2006;66:7548–7553. doi: 10.1158/0008-5472.CAN-06-0452. [DOI] [PubMed] [Google Scholar]
- 86.Heller E, et al. Hedgehog signaling inhibition blocks growth of resistant tumors through effects on tumor microenvironment. Cancer Res. 2012;72:897–907. doi: 10.1158/0008-5472.CAN-11-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 88.Mohammad KS, et al. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011;71:175–184. doi: 10.1158/0008-5472.CAN-10-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cannonier SA, Gonzales CB, Ely K, Guelcher SA, Sterling JA. Hedgehog and TGFbeta signaling converge on Gli2 to control bony invasion and bone destruction in oral squamous cell carcinoma. Oncotarget. 2016;7:76062–76075. doi: 10.18632/oncotarget.12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Johnson RW, Schipani E, Giaccia AJ. HIF targets in bone remodeling and metastatic disease. Pharmacol Ther. 2015;150:169–177. doi: 10.1016/j.pharmthera.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bendre MS, et al. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone. 2003;33:28–37. doi: 10.1016/s8756-3282(03)00086-3. [DOI] [PubMed] [Google Scholar]
- 92.Zheng Y, et al. Targeting IL-6 and RANKL signaling inhibits prostate cancer growth in bone. Clin Exp Metastasis. 2014;31:921–933. doi: 10.1007/s10585-014-9680-3. [DOI] [PubMed] [Google Scholar]
- 93.Luo X, et al. Stromal-initiated changes in the bone promote metastatic niche development. Cell Rep. 2016;14:82–92. doi: 10.1016/j.celrep.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Williams SC. First IL-6-blocking drug nears approval for rare blood disorder. Nat Med. 2013;19:1193. doi: 10.1038/nm1013-1193. [DOI] [PubMed] [Google Scholar]
- 95.Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pratap J, et al. The histone deacetylase inhibitor, vorinostat, reduces tumor growth at the metastatic bone site and associated osteolysis, but promotes normal bone loss. Mol Cancer Ther. 2010;9:3210–3220. doi: 10.1158/1535-7163.MCT-10-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Larson SR, et al. Prostate cancer derived prostatic acid phosphatase promotes an osteoblastic response in the bone microenvironment. Clin Exp Metastasis. 2014;31:247–256. doi: 10.1007/s10585-013-9625-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Coleman RE. Skeletal complications of malignancy. Cancer. 1997;80:1588–1594. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1588::aid-cncr9>3.3.co;2-z. [DOI] [PubMed] [Google Scholar]
- 99.Suva LJ, Brander BE, Makhoul I. Update on bone-modifying agents in metastatic breast cancer. Nat Rev Endocrinol. 2011;7:380–381. doi: 10.1038/nrendo.2011.80. [DOI] [PubMed] [Google Scholar]
- 100.Kelly T, et al. Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Res. 2005;65:5778–5784. doi: 10.1158/0008-5472.CAN-05-0749. [DOI] [PubMed] [Google Scholar]
- 101.Ozdemir BC, et al. The molecular signature of the stroma response in prostate cancer-induced osteoblastic bone metastasis highlights expansion of hematopoietic and prostate epithelial stem cell niches. PLoS ONE. 2014;9:e114530. doi: 10.1371/journal.pone.0114530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim MY, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315–1326. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Padua D, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vilalta M, Rafat M, Giaccia AJ, Graves EE. Recruitment of circulating breast cancer cells is stimulated by radiotherapy. Cell Rep. 2014;8:402–409. doi: 10.1016/j.celrep.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Manolagas S. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 106.Martinez LM, et al. Changes in the peripheral blood and bone marrow from untreated advanced breast cancer patients that are associated with the establishment of bone metastases. Clin Exp Metastasis. 2014;31:213–232. doi: 10.1007/s10585-013-9622-5. [DOI] [PubMed] [Google Scholar]
- 107.Stine KC, et al. Cisplatin inhibits bone healing during distraction osteogenesis. J Orthop Res. 2014;32:464–470. doi: 10.1002/jor.22527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stine KC, et al. Nutlin-3 treatment spares cisplatin-induced inhibition of bone healing while maintaining osteosarcoma toxicity. J Orthop Res. 2016;34:1716–1724. doi: 10.1002/jor.23192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiong J, et al. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS ONE. 2015;10:e0138189. doi: 10.1371/journal.pone.0138189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bultynck G. The anti-metastatic micro-environment of the bone: importance of osteocyte Cx43 hemichannels. Biochim Biophys Acta. 2016;1866:121–127. doi: 10.1016/j.bbcan.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 111.Massague J, Batlle E, Gomis RR. Understanding the molecular mechanisms driving metastasis. Mol Oncol. 2017;11:3–4. doi: 10.1002/1878-0261.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Giustacchini A, et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat Med. 2017;23:692–702. doi: 10.1038/nm.4336. [DOI] [PubMed] [Google Scholar]