

Summary
The noncanonical IKK family member TANK-binding kinase 1 (TBK1) is activated by pro-inflammatory cytokines, but its role in controlling metabolism remains unclear. Here we report that the kinase uniquely controls energy metabolism. Tbk1 expression is increased in adipocytes of HFD-fed mice. Adipocyte-specific TBK1 knockout (ATKO) attenuates HFD-induced obesity by increasing energy expenditure; further studies show that TBK1 directly inhibits AMPK to repress respiration and increase energy storage. Conversely, activation of AMPK under catabolic conditions can increase TBK1 activity through phosphorylation, mediated by AMPK’s downstream target ULK1. Surprisingly, ATKO also exaggerates adipose tissue inflammation and insulin resistance. TBK1 suppresses inflammation by phosphorylating and inducing the degradation of the IKK kinase NIK, thus attenuating NFκB activity. Moreover, TBK1 mediates the negative impact of AMPK activity on NFκB activation. These data implicate a unique role for TBK1 in mediating bi-directional crosstalk between energy sensing and inflammatory signaling pathways in both over- and under-nutrition.
ETOC
Adipose tissue inflammation plays an important role in reducing energy expenditure in obesity
Introduction
Obesity results from a positive energy balance, when food intake exceeds energy expenditure. Although energy intake and expenditure are tightly coupled to ensure homeostasis (Hall et al., 2012), even a slight mismatch over time can result in massive weight gain and glucose intolerance. While short term nutritional overload is associated with increased energy expenditure in both rodents (Pazos et al., 2015) and humans (Rasmussen et al., 2007), sustained obesity produces a decrease in energy expenditure (Bachman et al., 2002; Choi et al., 2015; Coleman, 1978; Leibel et al., 1995; Ravussin et al., 1988; Roberts and Leibel, 1998). While caloric restriction also reduces energy expenditure, resulting in lower basal metabolic rate, in an attempt to defend body weight (Leibel et al., 1995). Thus, both over- and under-nutrition are associated with reduced energy expenditure.
A great deal of basal energy metabolism occurs in adipose tissue, due to both nonshivering thermogenesis and lipid oxidation (Guilherme et al., 2008). Among the mechanisms that control energy expenditure are activation of AMP-activated protein kinase (AMPK), a key regulator that senses energy status and responds by increasing lipid oxidation and mitochondrial biogenesis, while reducing lipogenesis and glycogenesis (Daval et al., 2006; Hardie, 2011). AMPK is a heterotrimeric protein activated by liver kinase B1 (LKB1) via phosphorylation at Thr172 in the AMPKα subunit (Shaw et al., 2004). The enzyme is also allosterically activated in response to energy stress, by sensing an increased AMP/ATP ratio (Hardie et al., 2012; Mihaylova and Shaw, 2011). Activation of AMPK increases lipid oxidation, represses lipogenesis, and significantly increases mitochondrial biogenesis through peroxisome proliferator-activated receptor gamma co-activator 1α (PGC1α) (Canto and Auwerx, 2009; Puigserver et al., 1998; Wan et al., 2014). AMPK can also increase autophagy via phosphorylation and activation of Unc-51 like autophagy activating kinase 1 (ULK1) (Kim et al., 2011). The AMPK/ULK1 axis is required for maintaining mitochondrial homeostasis, and also increases the browning of inguinal white adipose tissues (Mottillo et al., 2016). Moreover, studies have demonstrated a correlation between decreased AMPK activity and increased inflammation (Gauthier et al., 2011), while activation of AMPK decreases expression of proinflammatory mediators and attenuates inflammation (Huang et al., 2015; O’Neill and Hardie, 2013; Salminen et al., 2011). Interestingly, AMPK activity is reduced in both obese rodents (Martin et al., 2006) and patients (Viollet et al., 2010; Xu et al., 2012), explaining in part reduced energy expenditure in obesity.
Numerous studies in both rodent models and patients indicate that obesity generates a state of chronic inflammation in adipose tissue, which in turn leads to insulin resistance and type 2 diabetes (Reilly and Saltiel, 2017; Weisberg et al., 2003; Xu et al., 2003). This inflammation is generated by recruitment and activation of immune cells, especially proinflammatory macrophages (Lumeng and Saltiel, 2011; Weisberg et al., 2003). Monocyte chemotactic protein 1 (MCP1/Ccl2) is produced by adipocytes during obesity, and is required for macrophage infiltration (Kamei et al., 2006; Kanda et al., 2006). In turn, pro-inflammatory cytokines secreted by immune cells in adipose tissue disrupt insulin action in adipocytes (Hotamisligil et al., 1995; Hotamisligil et al., 1994; Hotamisligil et al., 1996).
The NFκB signaling pathway plays an important role in the development of inflammation and insulin resistance in adipose tissue (Arkan et al., 2005; Baker et al., 2011; Chiang et al., 2009). NFκB is activated by the canonical IκB kinases (IKKs), IKKα and IKKβ, which phosphorylate IκB, leading to its degradation and release of NFκB (Lawrence, 2009). Unlike IKKα and β, the noncanonical IKKs, IKKε and TBK1 are induced downstream of NFκB activation (Reilly et al., 2013). TBK1 can also be activated by phosphorylation in response to proinflammatory cytokines or other substances that bind to Toll-like receptors 3 and 4 (Perry et al., 2004), although the upstream kinase that phosphorylates TBK1 remains unknown. Previous studies from our group showed that the activities of IKKε and TBK1 are significantly increased in adipose tissue of HFD-fed mice (Reilly et al., 2013), and further that IKKε deficiency partially protects mice from HFD-induced systemic inflammation, insulin resistance and hepatic steatosis (Chiang et al., 2009). However, how TBK1 might influence glucose metabolism and energy homeostasis remains uncertain.
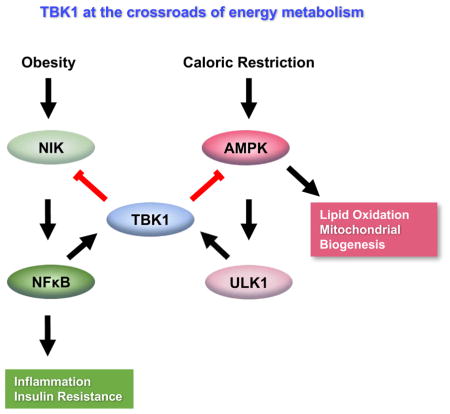
Here we report that Tbk1 expression and activity are significantly increased in mature adipocytes of HFD-fed mice. To investigate the roles of adipocyte TBK1, we generated adipocyte-specific TBK1 knockout (ATKO) mice, as whole-body knockout of TBK1 is lethal (Delhase et al., 2012). Adipose-specific TBK1 deficiency attenuates HFD-induced obesity, due to dramatically increased oxygen consumption and energy expenditure in white adipose tissue of obese but not normal weight mice. This was explained by increased activity of AMPK. In vitro studies revealed that AMPK is a direct substrate of TBK1, and further that TBK1 mediates the TNFα-induced repression of AMPK activity. At the same time, AMPK activation reciprocally increased the activity of TBK1; this effect is indirect and mediated by the AMPK downstream kinase ULK1, which directly phosphorylates TBK1 at Ser172. This infers a negative feedback loop in which AMPK activity is limited when activated, perhaps explaining in part the repression of energy expenditure during caloric restriction. Surprisingly, TBK1 KO in adipocytes led to increased activation of NFκB, with up-regulation of MCP1 production from adipocytes and macrophage infiltration into adipose tissue, accompanied by exaggerated insulin resistance in HFD-fed mice. The anti-inflammatory effect of TBK1 is mediated by phosphorylation of NIK, which results in its degradation and reduced activity of the atypical NFκB pathway. The loss of TBK1 thus substantially attenuates the AMPK-induced negative regulation of NFκB, showing that TBK1 is essential for the inhibitory crosstalk between AMPK and pro-inflammatory signaling. Taken together, these data reveal an unexpected role for TBK1 in both feedback and bi-directional crosstalk between energy sensing and inflammatory pathways in adipocytes to ensure energy homeostasis.
Results
Adipocyte-specific TBK1 deficiency increases energy expenditure and attenuates HFD-induced obesity
We previously demonstrated that TBK1 activity is induced by TNFα, and increased in adipose tissue of high fat diet (HFD)-fed mice (Reilly et al., 2013). We here showed that Tbk1 expression was significantly up-regulated in mature adipocytes derived from both inguinal (iWAT) and epididymal WAT (eWAT) in HFD-fed compared to normal diet (ND)-fed mice (Fig. 1A). We generated adipocyte-specific TBK1 knockout (ATKO) mice by crossing floxed TBK1 (Flox) mice with adiponectin-Cre mice. ATKO mice have greater than a 50% decrease of Tbk1 mRNA in both eWAT and iWAT, without any change of expression in the liver (Fig. 1B), reflecting complete reduction of TBK1 in adipocytes, with residual protein present in other adipose tissue cells (Jeffery et al., 2014).
Figure 1. Adipocyte-specific TBK1 deficiency increases energy expenditure and attenuates HFD-induced obesity.

(A) Tbk1 expression in mature adipocytes from iWAT and eWAT of 16 weeks ND or HFD-fed mice. N=3 from 9 mice. (B) Expression of Tbk1 in eWAT, iWAT and liver of Flox and ATKO mice. N=6–7. (C) Photo of dissected mice and eWAT from 16 weeks HFD-fed Flox and ATKO mice. (D) DEXA imaging of HFD-fed Flox and ATKO mice. (E) Body weight of 16 weeks HFD-fed Flox and ATKO mice. N=11–13. (F, G) Fat mass (F) or percentage of fat mass (G) in 16 weeks HFD-fed Flox and ATKO mice. N=10–11. (H) Tissues weight of HFD-fed Flox and ATKO mice. N=11–13. (I, K) OCR of 16 weeks ND (I) or HFD (K)-fed Flox and ATKO mice. N=5–6. (J, L) EE of ND (J) and HFD (L)-fed Flox and ATKO mice. N=5–6. Data are represented as mean±s.e.m. *, p<0.05. See also Figure S1.
Adipocyte-specific TBK1 knockout had no effect on body weight or composition in mice fed ND (Fig. S1A–S1E). However, TBK1 deficient mice tended to be leaner and have a lower body weight when fed HFD, although this difference did not reach statistical significance (Fig. 1C–1E). DEXA scanning revealed that ATKO mice have significantly less fat mass (Fig. 1D, F and G). In addition, weights of both eWAT and iWAT were significantly reduced in ATKO mice, with no difference in brown adipose tissue (BAT) or liver (Fig. 1C and 1H).
While TBK1 deficiency did not affect oxygen consumption rate (OCR) or energy expenditure (EE) in ND-fed mice (Fig. 1I and 1J), ATKO mice exhibited dramatically increased OCR and EE after HFD (Fig. 1K and 1L). Although HFD feeding inhibited OCR and EE in both Flox and ATKO mice, TBK1 deficiency significantly attenuated the reduction of OCR observed after HFD. TBK1 KO did not affect respiratory exchange rate (RER) or activity in either ND or HFD-fed mice (Fig. S1F–S1I). Moreover, adipocyte-specific TBK1 knockout had no effect on food intake (Fig. S1J) or body temperature (Fig. S1K) of ND or HFD-fed mice.
TBK1 deficiency up-regulates mitochondrial biogenesis by attenuating the inflammation-induced inhibition of AMPK activity in adipose tissues during HFD
Adipocyte size was significantly decreased in HFD-fed ATKO mice (Fig. 2A), suggesting that TBK1 deficiency attenuates HFD-induced adipocyte hypertrophy. Loss of TBK1 significantly increased ex vivo OCR in both WATs of HFD-fed mice (Fig. 2B). This increase was accompanied by a significant up-regulation of mitochondrial biogenesis genes such as Ppargc1a, as well as the mitochondrial genes Ndufs7, Cox4i1, Cox5b, Cox8a, Atp5d, all of which belong to different complexes in the respiratory chain (Fig. 2C and 2D). Moreover, expression of the thermogenic gene Ucp1 did not change (Fig. 2D), which is consistent with no major change in body temperature in ATKO mice. There was no effect on mitochondrial biogenic and thermogenic genes expression in BAT (Fig. S2A).
Figure 2. TBK1 deficiency up-regulates mitochondrial biogenesis via attenuating inflammation-induced inhibition of AMPK activity in adipose tissues of 16 weeks HFD-fed mice.

(A) Analysis of adipocyte size in eWAT of HFD-fed Flox and ATKO mice. N=4–5. (B) Ex vivo OCR of eWAT and iWAT HFD-fed Flox and ATKO mice. N=5–6. (C, D) Mitochondrial biogenic genes expression in eWAT (C) and iWAT (D) of HFD-fed Flox and ATKO mice. N=9. (E) Mitotracker staining of eWAT and iWAT of HFD-fed Flox and ATKO mice. Scale bar, 50 μm. N=4–6. (F) NAD/NADH ratio in eWAT and iWAT of HFD-fed Flox and ATKO mice. N=8– 10. (G, H) Immunoblot (IB) analysis of eWAT (G) and iWAT (H) in HFD-fed Flox and ATKO mice. N=5. (I) IB analysis of eWAT in ND or HFD-fed mice. N=6. Data are represented as mean±s.e.m. *, p<0.05. (J) IB analysis of 3T3-L1 adipocytes starved in low glucose DMEM, and treated with TNFα (10nM) for indicated time. (K) IB analysis of differentiated primary preadipocytes (Flox and ATKO) starved in low glucose DMEM, and treated with TNFα (10nM) for 45min. Data are represented as mean±s.d. *, p<0.05. See also Figure S2.
Recent studies have identified a creatine-driven pathway that increases EE and thermogenesis (Kazak et al., 2015), and mice lacking this cycle have reduced EE and greater HFD-induced obesity (Kazak et al., 2017). There was significant up-regulation of Ckmt1 expression in eWAT and Gamt expression in iWAT, suggesting increased activity of the creatine cycle in ATKO mice (Fig. S2B and S2C). Staining of WATs with MitoTracker also confirmed a substantial increase of mitochondria in adipocytes of HFD-fed ATKO mice (Fig. 2E). Likewise, NAD/NADH ratio was significantly increased in eWAT, and only slightly increased in iWAT from ATKO mice (Fig. 2F). The significant up-regulation of hormone sensitive lipase (HSL/Lipe) and adipose triglyceride lipase (ATGL/Pnpla2) in iWAT, but not eWAT (Fig. S2D and S2E) could lead to elevation of lipolysis and lipid oxidation in iWAT, potentially attenuating the elevation of NAD/NADH ratio produced by increased mitochondrial biogenesis. Taken together, our data indicate a mitochondria-driven increase of catabolism in WATs of ATKO mice.
Sympathetic activation of adipose tissue increases cAMP levels, and induces mitochondrial biogenesis (Wu et al., 1999). TBK1 KO did not affect cAMP levels in HFD-fed mice (Fig. S2F), nor was there a change in response to β-adrenergic stimulation (Fig. S2G and S2H), suggesting that TBK1 plays no role in controlling catecholamine sensitivity, unlike that proposed for IKKε (Mowers et al., 2013). While a slight increase of basal lipolysis in iWAT was observed because of up-regulated expression of Lipe and Pnpla2 (Fig. S2H), TBK1 deficiency did not affect serum triglycerides and non-esterified fatty acid (NEFA) concentrations (Fig. S2I and S2J). However, TBK1 deficiency substantially up-regulated AMPK Thr172 phosphorylation in both eWAT and iWAT, but not BAT, of HFD-fed (Fig. 2G and 2H, Fig. S2K), but not ND (Fig. S2M and S2N) mice. The lack of effect on BAT is likely due to the fact that TBK1 expression is not significantly induced in BAT that has lower inflammation (Fig. S2L). There was no difference in AMP/ATP or ADP/ATP ratios between HFD-fed ATKO and Flox mice (Fig. S2O and S2P).
HFD feeding significantly decreased AMPK Thr172 phosphorylation in adipose tissue (Fig. 2I). TNFα decreased AMPK Thr172 phosphorylation within 30min in adipocytes (Fig. 2J). TBK1 deficiency in both differentiated primary preadipocytes and murine embryonic fibroblasts (MEFs) abrogated inhibition of AMPK activation by TNFα (Fig. 2K, Fig. S2Q). Thus, since TBK1 levels and activity are increased during obesity, these in vivo and in vitro observations suggest that the decrease in AMPK activity observed in obesity is mediated by TBK1, which is elevated by HFD-induced adipose inflammation, and thus may represent a major mechanism of the obesitydependent reduction in AMPK activity in fat tissue.
TBK1 directly phosphorylates AMPK α-subunit to inhibit AMPK activity
We examined whether TBK1 interacts with AMPKα to inhibit its activity. Overexpression of TBK1 WT, but not TBK1 K38A kinase-dead mutant (KD), substantially decreased AMPKα1 Thr172 phosphorylation. AMPKα1 directly interacts with both TBK1 WT and TBK1 KD (Fig. 3A). Moreover, overexpression of TBK1 WT, but not TBK1 KD, resulted in a shift of AMPKα1 band (Fig. 3A). Lambda protein phosphatase treatment of immunoprecipitated AMPKα1 abolished the shift produced by TBK1 WT (Fig. 3A), indicating that TBK1 directly interacts with and phosphorylates the AMPKα1 subunit to inhibit its activation.
Figure 3. TBK1 directly phosphorylates AMPKα subunit to inhibit AMPK activity.

(A) IB analysis of whole cell lysate from HEK293T cells overexpressing HA-AMPKα1 with Flag- TBK1 WT or Flag-TBK1 kinase-dead mutant (KD) (left part). IB analysis of product from immunoprecipitation with HA-antibody treated with lambda protein phosphatase or not (right part). (B) Phosphopeptide derived from MS chromatogram showing AMPKα1 Ser459 and Ser476 phosphorylation (S+80). (C) Alignment of AMPKα1 (PRKAA1) and AMPKα2 (PRKAA2) sequence. (D) Alignment of AMPKα1 (PRKAA1) sequences in different species. Identified Ser phosphorylation sites are highlighted with red box. (E) IB analysis of HEK293T cells overexpressing HA-AMPKα1 WT or SS/AA or SS/DD mutant with Flag-AMPKβ1 and Myc-AMPKγ1. (F) Quantification of pAMPK/AMPK ratio in (E). Data are represented as mean±s.d. *, p<0.05. (G) Proposed model of TBK1-mediated repression of AMPK and EE. See also Figure S3 and Table S1.
The AMPKα1 protein from cells overexpressing AMPKα1/TBK1 WT and AMPKα1/TBK1 KD was subject to phospho-amino acid analysis by LC-MS/MS, revealing two phosphorylation sites (Ser459 and Ser476) on AMPKα1 from cells overexpressing AMPKα1 with TBK1 WT, but not the KD mutant (Fig. 3B, Fig. S3A–S3C, Table S1). Both Ser459 and Ser476 are conserved between AMPKα1 (PRKAA1) and AMPKα2 (PRKAA2) isoforms (Fig. 3C) and among species from Drosophila to human (Fig. 3D). We generated Ser/Ala nonphospho-mimetic (SS/AA) and Ser/Asp phospho-mimetic (SS/DD) mutants for both sites. Overexpression of HA-AMPKα1 WT or mutants with Flag-AMPKβ1 and Myc-AMPKγ1 demonstrated that the SS/AA mutation increased, while the SS/DD mutation decreased AMPKα1 Thr172 phosphorylation (Fig. 3E and 3F), suggesting that TBK1-induced phosphorylation of AMPKα1 Ser459 and Ser476 inhibits AMPK activation, in turn repressing energy expenditure in obesity (Fig. 3G). The TBK1 phosphorylation sites in the AMPKα subunit are close to, but distinct from inhibitory sites phosphorylated by Akt and GSK-3, both of which also inhibit AMPK activity (Suzuki et al., 2013). Both phosphorylation sites identified by our study are located close to β subunit-interacting domain and ST loop of the AMPKα subunit. Previous studies have shown that the interaction between AMPKα and β subunits affect AMPK Thr172 phosphorylation and activation (Garcia-Haro et al., 2010), and AMPKβ knockout causes a loss of AMPKα in fat tissue (Mottillo et al., 2016). Phosphorylation within the ST loop may induce a conformational change (Hawley et al., 2014), or recruit a phosphatase to decrease Thr172 phosphorylation (Suzuki et al., 2013).
TBK1 is stimulated by activation of AMPK
Since caloric restriction reduces energy expenditure to defend body weight (Martin et al., 2007), we sought to determine whether TBK1 might contribute to the reduction of energy expenditure during caloric restriction via repressing AMPK. We treated WT and AMPKα1/α2 knockout MEFs with the TLR4 agonist LPS or the AMPK activator AICAR. AICAR treatment resulted in activating phosphorylation of TBK1 at Ser172 in WT but not AMPKα1/α2 knockout MEFs, while LPS remained effective (Fig. 4A). Additionally, treatment of 3T3-L1 adipocytes with AICAR also increased TBK1 phosphorylation, and this increase was attenuated by pretreatment with the AMPK inhibitor compound C (Fig. 4B). Co-overexpression of AMPKα1 or α2 with AMPKβ1 and γ1 subunits increased TBK1 phosphorylation in HEK293T cells (Fig. S4A and S4B). These data indicate that activation of AMPK stimulates TBK1 activating phosphorylation in vitro.
Figure 4. Activation of AMPK induces TBK1 phosphorylation via ULK1 in vitro and in vivo.

(A) IB analysis of WT and AMPKα1/α2 KO MEFs treated with Vehicle, LPS (100ng/ml) or AICAR (500μM) for indicated time. (B) IB analysis of 3T3-L1 adipocytes pretreated with Vehicle, compound C (CC, 5μM) or Amlexanox (Amx, 50μM) for 1hr, and then treated with AICAR (500μM) for 45min. (C) IB analysis of eWAT from FL/FL and α1/α2 AKO mice (12–14 weeks old) intraperitoneally injected with Vehicle or AICAR (500mg/kg) for 45min. (D) IB analysis of 3T3-L1 adipocytes pretreated with Vehicle, Oxozeaenol (Oxo, 300nM) or SBI- 0206965 (SBI, 10μM) for 1hr, and then treated with AICAR (500μM) for 45min. (E) IB analysis of WT and ULK1 KO MEFs treated with Vehicle or AICAR (500μM) for 45min. (F) IB analysis of eWAT from FL/FL and ULK1 AKO mice (12–14 weeks old) intraperitoneally injected with Vehicle or AICAR (500mg/kg) for 45min. (G) IB of in vitro kinase assay with ULK1 active kinase and immunoprecipitated Flag-TBK1 WT or Flag-TBK1 KD. Immunoprecipitated Flag- TBK1 WT were pretreated with lambda-protein phosphatase before performing kinase assay. Each panel is a representative experiment that was repeated three times. (H) Proposed model of AMPK-induced activation of TBK1. See also Figure S4.
To examine AMPK-stimulated TBK1 phosphorylation in vivo, we generated adipocyte-specific AMPKα1/α2 double knockout mice (α1/α2 AKO). AICAR injection induced TBK1 Ser172 phosphorylation in adipose tissue of AMPK FL/FL mice, but not α1/α2 AKO mice (Fig. 4C). Moreover, we found that Tbk1 expression is significantly induced by fasting (Fig. S4C). Taken together, these data demonstrate that activation of AMPK under catabolic conditions produces TBK1 phosphorylation and activation both in vitro and in vivo, suggesting that TBK1 activity is increased by both inflammatory and catabolic signals.
We were unable to demonstrate a direct phosphorylation of TBK1 by recombinant AMPK, suggesting existence of an intermediate kinase. Since both TAK and ULK1 have been reported as downstream effectors of AMPK, we pretreated 3T3-L1 adipocytes with the TAK1 inhibitor Oxozeaenol (Oxo) or the ULK1 inhibitor SBI-0206965 (SBI), followed by treatment with AICAR. Inhibition of ULK1 with SBI-0206965 attenuated AICAR-induced TBK1 phosphorylation, while Oxo had no effect (Fig. 4D), suggesting that ULK1 mediates the phosphorylation of TBK1 stimulated by AMPK. Moreover, AICAR-stimulated TBK1 phosphorylation was completely abrogated in ULK1-deficient MEFs (Fig. 4E). We generated adipocyte-specific ULK1 knockout (ULK1 AKO) mice, and injected ULK1 FL/FL and ULK1 AKO mice with AICAR. While AICAR treatment stimulated TBK1 phosphorylation in adipose tissue of ULK1 FL/FL mice, loss of ULK1 abrogated the effect of AICAR (Fig. 4F). Additionally, an in vitro kinase assay was performed to test if ULK1 directly phosphorylates TBK1 Ser172. Immunoprecipitated TBK1 WT was pretreated with lambda protein phosphatase to reduce basal phosphorylation, and then incubated with ULK1. Active ULK1 directly phosphorylated both TBK1 WT and its KD mutant (Fig. 4G). These data conclusively demonstrate that ULK1 directly phosphorylates TBK1 Ser172 and mediates the activation of TBK1 phosphorylation produced by AMPK activation both in vitro and in vivo (Fig. 4H).
We sought to examine if the ULK1/TBK1 axis mediates negative feedback regulation of AMPK. 2hrs glucose starvation induced AMPK Thr172 phosphorylation in both WT and ULK1 KO MEFs, but was significantly higher in ULK1 KO cells (Fig. S4D). Additionally, glucose starvation increased TBK1 Ser172 phosphorylation in WT MEFs, but not in ULK1 KO MEFs (Fig. S4D). These results indicate that upon its phosphorylation and activation by AMPK, ULK1 initiates a negative feedback loop to regulate AMPK activity by phosphorylation and activation of TBK1. These data suggest that the ULK1/TBK1 axis normally limits AMPK activity to preserve energy storage.
Adipocyte-specific TBK1 knockout mice have exaggerated HFD-induced glucose intolerance and insulin resistance, along with increased adipose inflammation
TBK1 deficiency did not significantly affect fasting glucose or insulin levels in ND mice. Surprisingly, however, both fasting blood glucose and insulin tended to be slightly higher in HFD-fed ATKO mice (Fig. S5A and S5B). Glucose (GTT) and insulin tolerance tests (ITT) showed no difference between the genotypes in ND-fed mice (Fig. S5C and S5D). However, TBK1 deficiency exaggerated glucose intolerance and insulin resistance in HFD-fed mice (Fig. 5A and 5B), without significant effects on insulin levels during GTT (Fig. S5E). Because there was no difference in hepatic steatosis and liver gene expression in ATKO mice on HFD (Fig. S5F and S5G), we examined whether glucose intolerance was due to decreased insulin-stimulated glucose uptake in adipose tissues by in vivo glucose uptake assay. Adipose tissue glucose uptake was significantly decreased in both eWAT and iWAT of HFD-fed ATKO mice (Fig. 5C). This was accompanied by reduced insulin-stimulated Akt phosphorylation in adipose tissue of TBK1 deficient mice (Fig. 5D).
Figure 5. Adipocyte-specific TBK1 knockout mice have exaggerated HFD-induced glucose intolerance and insulin resistance, along with increased adipose inflammation.

(A) Glucose (1.2g/kg BW) tolerance test on 12 weeks HFD-fed Flox and ATKO mice. N=10–12. (B) Insulin (1.2U/kg BW) tolerance test on 12 weeks HFD-fed Flox and ATKO mice. N=10–12. (C) In vivo glucose uptake in eWAT and iWAT of 14 weeks HFD-fed Flox and ATKO mice. Each mouse was injected with 1.2g/kg glucose, 1U/kg insulin and 180uCi/kg 2-[C14]-deoxyglucose for 1hr. N=4–5. (D) IB analysis of eWAT in HFD-fed Flox and ATKO mice injected with PBS or 1.2 U/kg insulin. (E) Expression of macrophage marker genes in eWAT of 16 weeks ND or HFD-fed Flox and ATKO mice. N=6–8. (F) H&E (left) and F4/80 (right) staining of eWAT in HFD-fed Flox and ATKO mice. Scale bar, 100μm. (G, H) FACS analysis of F4/80+ cells in stromal vascular fraction (SVF) from eWAT of HFD-fed Flox and ATKO mice. N=4 from 10 mice. (H) is statistical analysis of (G). (I) Ccl2 (MCP1) expression in eWAT of 16 weeks HFD-fed Flox and ATKO mice. N=6–8. (J) Ccl2 expression in mature adipocyte fraction from eWAT of HFD-fed Flox and ATKO mice. N=6 from 12 mice. (K) Tnfa expression in eWAT of HFD-fed Flox and ATKO mice. N=6–8. Data are represented as mean±s.e.m. *, p<0.05. See also Figure S5.
Increased adipose inflammation and macrophage infiltration inhibit insulin-stimulated glucose uptake and result in insulin resistance (Hotamisligil et al., 1993; Weisberg et al., 2003). Adipose expression of macrophage markers F4/80 (Adgre1), CD11c (Itgax) and CD206 (Mrc1) were significantly induced by HFD feeding in Flox mice. However, TBK1 deficiency further increased expression of macrophage marker genes (Fig. 5E). H&E and F4/80 staining of adipose tissue showed a substantial increase of adipose macrophage infiltration in ATKO mice (Fig. 5F). Staining and FACS analysis of stromal vascular fractions (SVFs) from eWAT with F4/80 antibody revealed a significant increase of macrophages in HFD-fed ATKO mice (Fig. 5G and 5H). MCP1 (Ccl2) plays an essential role in the induction of adipose macrophage infiltration (Kanda et al., 2006). Ccl2 expression was significantly up-regulated in adipose tissue (Fig. 5I) and in mature adipocytes of HFD-fed ATKO mice (Fig. 5J). Further analysis revealed that TBK1 deficiency exaggerated HFD-induced expression of Tnfa in eWAT (Fig. 5K). Taken together, these data indicate that TBK1 deficiency in adipocytes increases the recruitment of proinflammatory macrophages into adipose tissue, releasing inflammatory cytokines such as TNFα to block insulin action.
Loss of TBK1 increases TNFα-induced NFκB activation in HFD-fed mice
To understand the underlying mechanism of increased inflammation in ATKO mice, we examined NFκB signaling in adipose tissues. Compared to Flox controls, there was an increase in p52, the active form of NFκB, as well as phosphorylation of p105 in eWAT and iWAT, but not BAT of HFD-fed ATKO mice or WATs of ND-fed ATKO mice (Fig. 6A and 6B, Fig. S6A, S6B and S6C). To determine whether the hyperactivation of NFκB caused by TBK1 deficiency was cell autonomous, we differentiated primary preadipocytes from Flox and ATKO mice and treated with TNFα for 2 or 8hrs. TBK1 knockout significantly increased TNFα-induced p52 generation and p105 phosphorylation (Fig. 6C and S6D). Furthermore, Ccl2 expression induced by 16hrs TNFα treatment was significantly increased in TBK1-deficient differentiated preadipocytes and MEFs (Fig. 6D and S6E), although TBK1 deficiency did not affect acute NFκB activation up to 1hr of TNFα treatment (Fig. S6F). Taken together, these data indicate that TBK1 knockout affects TNFα-induced NFκB activity only after 2hrs of treatment, strongly suggesting that the effects of TBK1 knockout are dependent on gene expression or protein degradation.
Figure 6. Loss of TBK1 increases TNFα-induced NFκB activation.

(A, B) IB analysis of eWAT (A) and iWAT (B) from HFD-fed Flox and ATKO mice. (C) IB analysis of differentiated primary preadipocytes treated with TNFα (10nM) for indicated time. (D) Ccl2 expression in differentiated primary preadipocytes treated with TNFα (5nM) for 16hrs. N=3. (E) IB analysis of WT, NKO (NIK KO), TKO (TBK1 KO) or DKO (NIK/TBK1 double KO) MEFs treated with TNFα (10nM) for 45min. (F) Ccl2 expression in WT, NKO, TKO or DKO MEFs treated with TNFα (5nM) for 16hrs. N=3. Data are represented as mean±s.d. *, p<0.05. (G) Proposed model of TBK1-mediated attenuation of NFκB activation. See also Figure S6.
NFκB-inducing kinase (NIK) phosphorylates and activates IKKα homodimers and to a lesser extent IKKα/β heterodimers. The activated IKKα/IKKα homodimer phosphorylates p100 to induce its cleavage and generate p52, while the activated IKKα/IKKβ heterodimer phosphorylates p105 (Sun, 2011; Zarnegar et al., 2008). A previous study suggested that TBK1 phosphorylates NIK, and induces its degradation to attenuate NFκB activation (Jin et al., 2012). Overexpression of TBK1 WT but not KD decreased NIK protein (Fig. S6G). Co-immunoprecipitation experiments confirmed that NIK directly interacts with both TBK1 WT and its KD mutant (Fig. S6H). Unfortunately, the currently available NIK antibody was not able to detect endogenous NIK without MG132 treatment. We used CRISPR/Cas9 to knock out NIK in immortalized WT and TBK1 KO MEFs (Fig. S6I). NIK knockout attenuated TNFα-induced activation of NFκB, and abrogated NFκB hyperactivation caused by TBK1 deficiency (Fig. 6E). NIK KO significantly attenuated TNFα-induced Ccl2 expression, and abrogated increased Ccl2 expression in TBK1 knockout cells (Fig. 6F). These results demonstrate that TBK1 attenuates TNFα-induced NFκB activation and Ccl2 expression via regulating NIK degradation, and strongly suggest that increased inflammation in TBK1 KO adipocytes is due to increased levels of NIK and hyperactivation of NFκB. Our study thus reveals a counter-inflammatory role of TBK1, which controls TNFα-induced NFκB activation in a negative feedback loop, and maintains inflammation at low levels, typical of obesity-induced inflammation (Fig. 6G).
TBK1 deficiency attenuates inhibition of NFκB signaling by AMPK
Activation of AMPK reduces production of inflammatory cytokines and attenuates inflammation (Huang et al., 2015; Salminen et al., 2011), although the underlying mechanism is unknown. Because TBK1 can be activated downstream of AMPK, and since loss of TBK1 abrogates the feedback inhibition of inflammation, we explored the hypothesis that TBK1 mediates the anti-inflammatory effect of AMPK activation. We pretreated adipocytes with AICAR, and then treated cells with TNFα to examine activation of NFκB signaling. AICAR attenuated TNFα-induced phosphorylation of p105 and IKKα/β in adipocytes (Fig. 7A), and decreased TNFα-induced Ccl2 expression (Fig. 7B). TBK1 deficiency abrogated the inhibitory effect of AICAR on TNFα-induced p105 and IKKα/β phosphorylation (Fig. 7C, Fig. S7A). AICAR-induced inhibition of Ccl2 expression was significantly attenuated in TBK1 knockout cells (Fig. 7D, Fig. S7B). To confirm that attenuation of the anti-inflammatory effects of AICAR is due to loss of TBK1, immortalized TBK1 KO MEFs were transfected to express GFP or TBK1. In TBK1 KO MEFs, expression of TBK1 restored inhibition of p105 and IKKα/β phosphorylation by AMPK activation (Fig. 7E). We conclude that TBK1 mediates the anti-inflammatory effects of AMPK activation via attenuation of NFκB activity (Fig. 7F). Taken together, our findings demonstrate that TBK1 operates at the intersection of two crucial pathways to mediate both the repression of energy expression by inflammation and the attenuation of inflammation by AMPK activation.
Figure 7. TBK1 deficiency attenuates inhibition of NFκB signaling by AMPK.

(A) IB analysis of 3T3-L1 adipocytes pretreated with Vehicle or AICAR (250μM) for 7hrs, and then treated with TNFα (10nM) for 45min. (B) Ccl2 expression in 3T3-L1 adipocytes pretreated with Vehicle or AICAR (250μM) for 7hrs, and then treated with TNFα (10nM) for 2hrs. *, p<0.05. (C) IB analysis of differentiated primary preadipocytes (Flox and ATKO) pretreated with Vehicle or AICAR (250μM) for 7hrs, and then treated with TNFα (10nM) for 45min. (D) Ccl2 expression in differentiated primary preadipocytes (Flox and ATKO) pretreated with Vehicle or AICAR (250μM) for 7hrs, and then treated with TNFα (10nM) for 2hrs. Data are represented as mean±s.d. *, p<0.05. (E) IB analysis of TKO MEFs overexpressing GFP or TBK1. MEFs were pretreated with Vehicle or AICAR (500μM) for 7hrs, and then treated with TNFα (10nM) for 45min. (F) Proposed model of TBK1-mediated anti-inflammatory effect of AMPK. See also Figure S7.
Discussion
We demonstrate here a previously unappreciated role for the protein kinase TBK1; upon its induction and activation during obesity and caloric restriction, TBK1 represses energy expenditure while also attenuating inflammation in adipocytes and surrounding cells in adipose tissue, thus operating at the intersection of inflammation and energy homeostasis. We conclude that TBK1 mediates both feedback and bi-directional inhibitory crosstalk between AMPK and inflammation (Fig. S7C). Numerous studies have demonstrated that obesity in both rodents and patients is associated with reduced energy expenditure (Hill et al., 2012). However, the mechanisms by which energy expenditure is reduced in obesity is unknown. Data shown here suggest that adipose tissue inflammation may play an important role, at least partially through the induction of TBK1.
Several studies have implicated a central role for the NFκB pathway in triggering and sustaining the deleterious effects of overnutrition on energy homeostasis (Baker et al., 2011). Although TBK1 displays significant sequence homology to the other IKKs (Clement et al., 2008), and although TBK1 is activated by pro-inflammatory stimuli, the kinase is completely dispensable for NFκB activation. Interestingly, we found that Tbk1 expression is significantly up-regulated in mature adipocytes of HFD-fed mice, suggesting that it might be an effector of inflammation. Moreover, the specific IKKε/TBK1 inhibitor amlexanox alleviates many of the effects of HFD feeding and genetic obesity, improving weight, insulin sensitivity, inflammation and hepatic steatosis (Reilly et al., 2013), along with a substantial, clinically relevant improvement in blood sugar and glucose hemostasis in a subset of obese type 2 diabetic patients with underlying inflammation (Oral et al., 2017). As we have already studied IKKε knockout mice (Chiang et al., 2009), these findings prompted us to create adipocyte-specific TBK1 KO mice to evaluate the role of TBK1 in metabolic homeostasis. Adipocyte-specific TBK1 KO ameliorates HFD-induced obesity via increasing oxygen consumption rates and energy expenditure, and like the IKKε/TBK1 inhibitor amlexanox (Reilly et al., 2013), has no effect on normal weight mice.
What is the mechanism by which attenuation of AMPK activity by TBK1 represses energy expenditure? Numerous studies have indicated that AMPK increases lipid oxidation, while inhibiting lipogenesis through reduced activity of mTORC1 (Gwinn et al., 2008), and increases mitochondrial biogenesis (Reznick and Shulman, 2006). Increased AMPK activity in HFD-fed ATKO mice increases expression of Pparc1a and other genes involved in respiration, including a substantial increase of mitochondrial biogenesis in adipocytes. Thus, these findings illuminate how obesity-dependent chronic inflammation reduces energy metabolism via TNFα-induced inhibition of AMPK activity in adipose tissues.
TBK1 Ser172 phosphorylation is increased by pro-inflammatory cytokines and ligands that bind to TLR3 and TLR4 (Perry et al., 2004), and structural studies show that TBK1 is activated by trans-autophosphorylation (Ma et al., 2012). We show here that TBK1 is surprisingly activated by AMPK via its downstream protein kinase ULK1. ULK1 is thus the first TBK1 kinase discovered. Although ULK1 is a key regulator of autophagy (Russell et al., 2013), the discovery of the AMPK/ULK1/TBK1 axis indicates that ULK1 may have other important functions. More importantly, under conditions in which TBK1 is expressed, AMPK activation may be self-limiting through this feedback loop. Moreover, TBK1 may limit energy expenditure during caloric restriction (Leibel et al., 1995), in which absolute restriction of food intake in patients resulted in reduced energy expenditure, in an attempt to defend body weight. This finding also has important implications for the discovery and development of AMPK activators for the treatment of metabolic disease, and suggests that a combination of AMPK activators with TBK1 inhibitors such as amlexanox might be synergistic.
Despite increasing energy expenditure, TBK1 knockout exaggerated HFD-induced glucose intolerance and insulin resistance, likely due to enhanced adipose inflammation and macrophage infiltration in HFD-fed ATKO mice (Hotamisligil et al., 1993; Weisberg et al., 2003). TBK1 deficiency increased TNFα-induced p105 phosphorylation and generation of p52 in a cell-autonomous manner, and up-regulated TNFα-induced Ccl2 expression. Increased MCP1 production is the primary signal for macrophage infiltration in adipose tissue in obesity (Kamei et al., 2006). Knockout of NIK by CRISPR/Cas9 confirmed that the TBK1 deficiency-induced hyper-response to TNFα is dependent on NIK. However, while NIK knockout did not completely abrogate TNFα-induced NFκB activation and Ccl2 expression, it completely abolished the difference between WT and TBK1 deficient cells, suggesting that TBK1 mainly controls the atypical pathway for NFκB activation. Moreover, these data also indicate that TBK1 does not affect the initiation of inflammation in obese mice, but limits its development, perhaps accounting for the low-grade nature of adipose tissue inflammation in obesity (Xu et al., 2003). In addition, studies have shown that extracellular ATP exerts a pro-inflammatory effect (Cauwels et al., 2014), synergistic with the effect of TNFα (Schnurr et al., 2000). Thus, in HFD-fed ATKO mice, increased AMPK-induced ATP synthesis and TNFα-stimulated ATP release (Lohman et al., 2015) may exacerbate adipose inflammation via increasing extracellular ATP. The increased ATP release might also explain why intracellular ATP in adipocytes of HFD-fed ATKO mice remains stable, despite a dramatic increase in mitochondrial number and activity.
It is important to note that the IKKε/TBK1 inhibitor amlexanox also increases energy expenditure in rodents (Reilly et al., 2013), and was effective in lowering blood sugar in a subset of obese patients with type 2 diabetes (Oral et al., 2017). Interestingly, analysis of gene expression changes in adipose tissue from responding patients revealed many of the same changes in mitochondrial respiration genes observed here in ATKO mice, suggesting that at least a part of amlexanox’s beneficial effects might be due to increased AMPK activity via TBK1 inhibition. However, unlike the ATKO mice, amlexanox treatment improves catecholamine sensitivity in adipose tissue (Mowers et al., 2013), while reducing insulin resistance and adipose tissue inflammation (Reilly et al., 2013). We reported that IKKε knockout increases thermogenesis and energy expenditure, while reducing adipose inflammation (Chiang et al., 2009). It is likely that IKKε is responsible for adipocyte catecholamine resistance via phosphorylation of PDE3B (Mowers et al., 2013), and must be inhibited to improve glucose homeostasis and inflammation. However, IKKε has no effect on AMPK phosphorylation. During HFD-feeding, expression of both Ikbke and Tbk1 are up-regulated by inflammation. While IKKε phosphorylates and activates PDE3B to induce catecholamine resistance, TBK1 inhibits AMPK activity to reduce catabolism via this pathway. The combination of IKKε and TBK1 activation thus decreases energy expenditure to exaggerate energy storage and obesity. Thus, we propose that the beneficial metabolic effects of amlexanox require inhibition of both IKKε and TBK1.
Previous studies showed that activation of AMPK by energy stresses, such as caloric restriction, reduces adipose inflammation and improves glucose metabolism (Canto and Auwerx, 2011; Larson-Meyer et al., 2006). We show here that loss of TBK1 disrupts the anti-inflammatory effects of AMPK activation. As TBK1 induces NIK degradation to attenuate NFκB activation, we conclude that TBK1 mediates the anti-inflammatory functions of AMPK via negatively regulating NFκB activation.
Taken together, these data have introduced a new player in the control of energy homeostasis under conditions of both over- and under-nutrition. Upon its activation, TBK1 can repress respiration by inhibiting AMPK activity, perhaps explaining in part why energy expenditure is reduced in states of obesity. At the same time, TBK1 can attenuate inflammation after it is induced by obesity, maintaining the inflammatory state at a low-grade level. Finally, TBK1 plays a crucial role in a feedback loop, both limiting the activity of AMPK under conditions, such as caloric restriction, in which it is activated, but also mediating the anti-inflammatory effects of this important energy sensor. While many questions remain, these insights open a new line of investigation concerning how TBK1 modulates energy homeostasis.
STAR Methods
Key resources table
| Reagents | Source | Identifier |
|---|---|---|
| Chemicals and Recombinant Peptides | ||
| Recombination Mouse TNFα | Thermo Scientific | PMC3014 |
| Amlexanox | Abcam | ab142825 |
| AICAR | Cell Signaling Technology | 9944 |
| Compound C | Calbiochem | 171260 |
| SBI-0206965 | Sigma-Aldrich | SML1540 |
| 5Z-7-Oxozeaenol | Sigma-Aldrich | O9890 |
| MG132 | Sigma-Aldrich | M8699 |
| Lipopolysaccharide | Sigma-Aldrich | L4391 |
| Protease inhibitor tablet | Sigma-Aldrich | 11873580001 |
| 2-[C14]-deoxyglucose | PerkinElmer | NEC720A250UC |
| ULK1 active kinase | Signal Chem | U01-11G-10 |
| Antibodies | ||
| TBK1 antibody | Cell Signaling Technology | 3013 |
| Phospho-TBK1 Ser172 antibody | Cell Signaling Technology | 5483 |
| AMPKα antibody | Cell Signaling Technology | 2532 |
| Phospho-AMPKα Thr172 antibody | Cell Signaling Technology | 2535 |
| p100/p52 antibody | Cell Signaling Technology | 4882 |
| Phosphor-IKKα/β antibody | Cell Signaling Technology | 2078 |
| p105 antibody | Cell Signaling Technology | 4717 |
| Phosphor-p105 Ser933 antibody | Cell Signaling Technology | 4806 |
| ULK1 antibody | Cell Signaling Technology | 8054 |
| NIK antibody | Cell Signaling Technology | 4994 |
| ACC antibody | Cell Signaling Technology | 3662 |
| Phospho-ACC Ser79 antibody | Cell Signaling Technology | 3661 |
| MAPK (Erk1/2) antibody | Cell Signaling Technology | 9102 |
| β-tubulin antibody | Cell Signaling Technology | 2146 |
| Myc antibody | Santa Cruz Biotechnology | sc-40 |
| Flag antibody | Sigma-Aldrich | F1804 |
| Oligonucleotides | ||
| Primers for Q-PCR, see Table S2 | This paper | N/A |
| Critical Commercial Assays | ||
| Ultra-sensitive mouse insulin ELISA kit | Crystal Chem | 90080 |
| Triglycerides liquid reagents | Pointe Scientific | T7532 |
| NEFA-HR(2) assay | Wako | 434-91795 434-91995 |
| Cyclic AMP XP assay kit | Cell Signaling Technology | 4339 |
| Free glycerol determination kit | Sigma-Aldrich | F6428 |
| Extracellular oxygen consumption assay | Abcam | ab197243 |
| NAD/NADH assay kit | Abcam | ab65348 |
| AMP-Glo assay | Promega | V6930 |
| ADP-Glo assay | Promega | V5011 |
| ENLITEN(R) ATP assay | Promega | PAFF2000 |
| Recombinant DNA | ||
| pcDNA-Flag-TBK1 | Gift from Kate Fitzgerald | N/A |
| pcDNA-HA-AMPKα1 | Gift from Ken Inoki | N/A |
| pcDNA-HA-AMPKα2 | Gift from Ken Inoki | N/A |
| pcDNA-Flag-AMPKβ1 | Gift from Ken Inoki | N/A |
| pcDNA-Myc-AMPKγ1 | Gift from Ken Inoki | N/A |
| pLentiCRISPR-v2-NIK-gRNA | Gift from Michael Karin | N/A |
| Experimental Models: Cell Lines | ||
| TBK1 WT/KO MEFs | Gift from Kate Fitzgerald | N/A |
| AMPKα1/α2 WT/KO MEFs | Gift from Ken Inoki | N/A |
| Immortalized ULK1 WT MEFs | Sigma-Aldrich | 14050804-1VL |
| Immortalized ULK1 KO MEFs | Sigma-Aldrich | 14050807-1VL |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tbk1fl/fl | Gift from Kate Fitzgerald | N/A |
| Mouse: Prkaa1fl/fl, Prkaa2fl/fl | Gift from Ken Inoki | N/A |
| Mouse: Ulk1fl/fl | Jackson Laboratory | 017976 |
| Mouse: Adiponectin-cre | Jackson Laboratory | 028020 |
| Mouse: C57BL/6J | Jackson Laboratory | 000664 |
| Software | ||
| ImageJ | NIH | N/A |
| GraphPad Prism 6 | GraphPad Software | N/A |
| Other | ||
| High-Fat Diet (45% kcal% fat) | Research Diet, Inc. | D12451 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alan Saltiel (asaltiel@ucsd.edu).
Experimental Model and Subject Details
Animals
Tbk1fl/fl mice were bred with adiponectin-cre mice to generate adipocyte-specific TBK1 knockout mice. Adipocyte-specific AMPKα1/α2 double knockout mice were generated by breeding Prkaa1fl/fl mice and Prkaa2fl/fl mice to adiponectin-cre mice. Adipocyte-specific ULK1 knockout mice were created by breeding Ulk1fl/fl mice to adiponectin-cre mice. All the mice have C57BL/6J background. Only male mice were used for experiments. We fed mice with normal chow diet or high fat diet consisting of 45% of calories from fat (D12451 Research Diets Inc.) for 12–16 weeks, starting at 6–8 weeks of age. During metabolic studies, ear tag numbers were used to identify animals. Flox and knockout mice are littermates and cage mates. Researchers performing test and collecting data were blinded during experiments. Animals in each cohort were produced from 20 breeding pairs to minimize the birthdate range. Mice were housed in a specific pathogen-free facility with a 12-h light, 12-h dark cycle, and given free access to food and water, except for fasting period. All animal use was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California-San Diego. For metabolic study, mice are subject to CLAMs indirect calorimetry and dual energy X-ray absorptionmetry (DEXA) scanning at ACP phenotyping core of UCSD. Rectal temperature was measured by Model 4600 Thermometer (Alpha Technics). Serum insulin was measured with Ultra-Sensitive Mouse Insulin ELISA kit (Crystal Chem).
Cells
3T3-L1 adipocytes
Murine 3T3-L1 preadipocytes were grown to 2 days after confluence in Dulbecco’s Modified Eagle’s Media (DMEM) with 10% newborn calf serum (NCS). 0.5mM 3-isobutyl-methylxanthine, 1μM dexamethasone, and 1μg/ml insulin (MDI) cocktail were used to induce preadipocytes differentiation in DMEM containing 10% fetal bovine serum (FBS) for the first 3 days. Cells were cultured in media containing insulin for another 3 days. After 48hrs, the media was replaced by DMEM with 10% FBS. Finally, differentiation was completed in the media containing 10% FBS (Zhao and Stephens, 2013). For serum deprivation, media was change to DMEM containing 0.2% BSA or low-glucose DMEM containing 0.2% BSA. Only cultures in which >90% of cells displayed adipocyte morphology were used.
Differentiation of primary preadipocytes
Inguinal adipose tissues of 4–6 weeks old male mice were minced and digested with 1mg/ml type II collagenase for 25 min at 37°C with gentle agitation. Cells were washed, filtered through 100 μm filter. After centrifugation, SVF was plated on 10cm collagen-coated cell culture plate. After 2 rounds of culture, Preadipocytes were grown to confluence and induced to differentiate with 0.5mM 3-isobutyl-methylxanthine, 1μM dexamethasone, 1μg/ml insulin (MDI) cocktail and 1μM rosiglitazone (Cayman).
Immortalization of TBK1 knockout MEFs
To immortalize cells, they were transduced with retroviruses encoding a temperature-sensitive (tsa58) mutant A438V of large T antigen. After antibiotic selection with 100μg/ml G418 at the permissive temperature of 33°C for two weeks, cells were trypsinized and transferred to new culture dishes. Cells were moved to 37°C incubation 48hrs before experiments to eliminate expression of SV40 large T antigen.
Generation of NIK knockout MEFs
Immortalized WT or TBK1 knockout MEFs were transduced with viral supernatant to express gRNA/Cas9 (gRNA: 5′-GGCAAAATGGCCCGTGTGTGC-3′). Supernatants were centrifuged to remove cell debris, and supplemented with Polybrene (8μg/ml) before adding to cells. After antibiotic selection with 2.5μg/ml puromycin for two weeks, cells were trypsinized and transferred to new culture dishes.
Reconstitution of TBK1 in TBK1 knockout MEFs
Immortalized TBK1 knockout MEFs were transduced with viral supernatants to express GFP or TBK1. Supernatants were centrifuged to remove cell debris, and supplemented with Polybrene (8μg/ml) before adding to cells. 48hrs after infection, expression of GFP and TBK1 was examined by Microscopy and Western blot.
Method Details
Gene expression analysis
Tissues were homogenized in TRIzol Reagent (Life Technologies). RNA was isolated with PureLink RNA mini kit (Life Technologies). 1μg of purified RNA was used for reverse transcription-PCR to generate cDNA using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystem). ΔΔCt real time PCR with Power SYBR Green PCR Master Mix (Life Technologies) and QuantStudio 5 Real-Time PCR System were used to analyze cDNA. Gapdh was used as endogenous control.
Transfection
1μg DNA for each plasmid was used to transfect HEK239T cells with Lipofectamine 3000 (Life Technologies), according the manufacturer’s instruction. 24hrs later, cells were harvested and subject to Western blot analysis.
Immunoprecipitation and lambda protein phosphatase treatment
Cells were harvested in IP buffer containing 100mM Tris (pH 7.5), 5mM MgCl2, 130mM NaCl, 1mM EDTA, 1% Triton X-100 with freshly added protease inhibitors tablet (Roche), and phosphatase inhibitor 2 mM sodium vanadate and 5mM sodium fluoride. Immunoprecipitation was performed with anti-HA or Flag affinity gels (Biotool). Affinity gel was washed with TBS for 3 times and incubated with lambda protein phosphatase (NEB) at 30°C for 30min. After 3 times wash with PBS, protein was eluted with SDS loading buffer at 95°C.
Western blot
Tissues or cells were homogenized in a non-denaturing lysis buffer that contained 10 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 0.5% Nonidet P-40, with freshly added protease inhibitors tablet (Roche), and phosphatase inhibitor 2 mM sodium vanadate and 5mM sodium fluoride. The extract was centrifuged at 9500g for 10 min at 4 °C. Supernatants were collected and analyzed with BCA (Thermo Scientific) to quantify protein content. Proteins were resolved by SDS–PAGE and transferred to nitrocellulose membranes (Bio-Rad). Individual proteins were detected with specific antibodies and visualized on film using horseradish peroxidase-conjugated secondary antibodies (Fisher Scientific) and SuperSignal West Pico Chemilunminescent Substrate (Thermo Scientific). Bands were quantified with ImageJ (NIH).
Glucose tolerance test
Fasting blood glucose were measured after 14hrs fast, using Easy Step Blood Glucose Monitoring System. Mice were then intraperitoneally injected with D-[+]-glucose (Sigma) at a dose of 2g/kg BW for ND-fed mice or 1.2g/kg BW for HFD-fed mice. Blood glucose levels were measured at 15, 30, 45, 60, 90 and 120 min after injection.
Insulin tolerance test
Fasting blood glucose were measured after 4hrs fast, using Easy Step Blood Glucose Monitoring System. Mice were then intraperitoneally injected with insulin (Humulin R) at a dose of 1U/kg BW for ND-fed mice or 1.2U/kg BW for HFD-fed mice. Blood glucose levels were measured at 15, 30, 45, 60, 90 and 120 min after injection.
In vivo 2-[C14]-deoxyglucose uptake
Mouse was fasted for 16hrs, and then injected with 1.2g/kg glucose, 1U/kg insulin with 180μCi/kg 2-[C14]-deoxyglucose. Blood was collected at 0, 15, 30, 45 and 60min. Mice were dissected at 60min. Adipose tissues were collected, weighted and homogenized in perchloric acid. After centrifugation, supernatant was collected and neutralized. Radioactivity in lysate and serum was determined by the Beckman Coulter.
Histology and determination of adipocyte size
Tissues were dissected, and then fixed in 10% formalin. Paraffin-embedding tissues were sectioned and subject to H&E and F4/80 (AbD Serotec) IHC staining at UCSD histology core. Stained tissue was visualized with NanoZoomer Slide Scanner. Size of adipocytes was determined by ImageJ analysis of H&E stained tissues.
Mature adipocytes & SVF separation and FACS
Minced white adipose tissue was digested in RPMI with 0.5% BSA containing 1mg/ml type II collagenase for 25 min at 37 °C with gentle agitation. The cell suspension was filtered through a 100 μm cell strainer and centrifuged at 500 g for 5 min to separate floating mature adipocytes and SVF. Red blood cells in SVF were lyzed with ACK red blood cell lysis buffer for 5min. SVF was washed with RPMI with 0.5% BSA. Fc receptor was blocked with anti-CD16/CD32 antibody (eBioscience). Cells were stained with anti-F4/80-FITC (eBioscience), and analyzed by flow cytometry.
Ex vivo oxygen consumption rate measurement
Adipose tissues were dissected, weighted, quickly rinsed in PBS. Minced tissues were placed in 96-well tissue culture plate. Ex vivo oxygen consumption rate was measured with Oxygen Consumption Assay Kit (Abcam) according to the manufacturer’s instruction. 150μl pre-warmed medium and 10μl oxygen consumption reagent were added to each well. Wells were sealed with pre-warmed high sensitivity mineral oil. Fluorescence was measured at 37°C for 30min by Tecan Infinite M200 Pro.
Mitochondrial staining and confocal microscopy
Adipose tissues were dissected and fixed in 10% formalin with 7.5% sucrose. OCT-embedded tissue was sectioned and stained with 200nM Mitotracker Red (Life Technologies) for 45min at room temperature, followed by 3 washes with PBS. Fluorescence was visualized with Olympus FV1000 microscope.
In vitro kinase assay
Immunoprecipitated Flag-TBK1 WT or Flag-TBK1 KD was subject to in vitro kinase assay with 40ng/ul ULK1 active kinase (SignalChem), kinase assay buffer III (SignalChem) and 100μM ATP (SignalChem). After 15min incubation in 30°C water bath, reaction was stopped by adding SDS loading buffer and heating at 95°C. Phospho-TBK1 Ser172 antibody (Cell Signaling) was used to detect phosphorylated TBK1.
NAD/NADH ratio measurement
Tissue NAD/NADH ratio was measured with NAD/NADH Assay kit (abcam). Tissues were dissected, rinsed in PBS, and homogenized in NAD/NADH extraction buffer from the kit. After centrifugation, supernatant was collected. NAD/NADH ratio was measured according to the manufacturer’s instruction.
cAMP measurement
Tissue cAMP levels were determined using the cAMP XP Assay kit (Cell Signaling). Tissues were dissected, rinsed in PBS, and homogenized in cell lysis buffer on ice. After centrifugation, supernatant was collected. 50μl sample was used to determine cAMP level according to the manufacturer’s instruction. cAMP level was normalized to protein content.
ATP, ADP, AMP measurement
Tissues were homogenized in 2% trichloroacetic acid (TCA) buffer. After centrifugation, supernatant was collected and neutralized with pH8.0 Tris-HCl. ATP was measured with ENLITEN ATP Assay System (Promega). ADP was measured with ADP-Glo Kinase Assay (Promega). AMP was measured with AMP-Glo Assay (Promega), according to the manufacturer’s instructions.
Ex vivo lipolysis measurement
Adipose tissues were dissected, weighted and quickly rinsed with PBS. Minced tissues were cultured in 12-well plate in the absence or presence of CL-316,243 (10μM) for 2hrs. Glycerol in supernatant was measured with Free Glycerol Determination kit (Sigma).
Mass spectrometry
In gel digestion followed by LC-MS/MS was performed by the Biomolecular and Proteomics Mass Spectrometry Facility at University of California-San Diego. Peptides were resolved on a nano-C18 column and sprayed into Orbitrap mass spectrometer (Thermo fusion lumos). Peak Studio 8 was used to search against human protein database (Uniprot).
Site-direct mutagenesis
HA-AMPKα1 SS/AA and HA-AMPKα1 SS/DD plasmids were generated using Kapa Hifi DNA polymerase with 0.5ng HA-AMPKα1 WT as template DNA in 50ul reaction. Template DNA was digested by DpnI (NEB). Stellar competent cell (Takara) was used for transformation and plasmids amplification. Plasmid was purified with Wizard Plus SV Minipreps DNA purification system (Promega). Mutation was confirmed by DNA sequencing (Eton Bioscience).
Quantification and Statistical Analysis
All data of animal study are shown as mean ± s.e.m., while data from in vitro studies are shown as mean ± s.d.. Replicates are indicated in figure legends. N represents the number of experimental replicates. F-test was performed to determine the equality of variance. When comparing two groups, statistical analysis was performed using a two-tailed Student’s t-test, except when the f-test suggested that variances are statistically different. For analysis of more than two groups, we used analysis of variance (ANOVA) to determine equality of variance. Comparisons between groups were performed with Tukey-Krammer post-hoc analysis (Reilly et al., 2013). For all tests, P<0.05 was considered statistically significant.
Supplementary Material
Highlights.
TBK1 operates at intersection of energy expenditure and inflammation.
TBK1 deficiency attenuates HFD-induced obesity, but exaggerates inflammation.
TBK1 represses energy expenditure by phosphorylating and inhibiting AMPK.
TBK1 attenuates NFκB activation and mediates the anti-inflammatory effect of AMPK.
Acknowledgments
We thank members of the Saltiel lab for helpful discussions. We thank Dr. Kate Fitzgerald for sharing TBK1 floxed mice. We thank Dr. Ken Inoki for sharing AMPKα1 and α2 floxed mice, as well as AMPKα, β, γ expressing plasmids. We thank Drs. Michael Karin and Jerry Wong for providing the NIK CRISPR/Cas9 KO plasmid. We thank Dr. Majid Ghassemian and Biomolecular and Proteomics Mass Spectrometry Facility for the mass spectrometry study. We thank Marilyn Hardee and Animal Phenotyping Core for support of CLAMs indirect calorimetry and DEXA studies. We thank the UCSD histology core for tissue sectioning and staining. This work was supported by grants from NIH (P30DK06349, R01DK100319, R01DK60597 and R01DK60591) to A.R.S.
Footnotes
Author Contributions
P.Z. and A.R.S. conceived the project. P.Z. designed the study and performed all the experiments. K.W. assisted with most of the experiments. X.S. contributed to Fig. 1C, 1H, S1E, S1K, 2E, 2F, S2I, S2J, 5A, 5B, 5C, S5C, S5D, S7C. S.M.R. generated ATKO mice. M.U. performed preliminary experiments. Z.L. contributed to Fig. 2E, 3E, 3F, 6E, 6F. Y.S. contributed to Fig. 5C. P.Z. and A.R.S. wrote the manuscript.
Declaration of Interest
A.R.S. is a named inventor on several patent applications that include amlexanox. The other authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, Lowell BB. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science. 2002;297:843–845. doi: 10.1126/science.1073160. [DOI] [PubMed] [Google Scholar]
- Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology (Bethesda) 2011;26:214–224. doi: 10.1152/physiol.00010.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014;5:e1102. doi: 10.1038/cddis.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma JT, Zhou J, Qi N, Westcott D, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell. 2009;138:961–975. doi: 10.1016/j.cell.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MS, Kim YJ, Kwon EY, Ryoo JY, Kim SR, Jung UJ. High-fat diet decreases energy expenditure and expression of genes controlling lipid metabolism, mitochondrial function and skeletal system development in the adipose tissue, along with increased expression of extracellular matrix remodelling- and inflammation-related genes. Br J Nutr. 2015;113:867–877. doi: 10.1017/S0007114515000100. [DOI] [PubMed] [Google Scholar]
- Clement JF, Meloche S, Servant MJ. The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 2008;18:889–899. doi: 10.1038/cr.2008.273. [DOI] [PubMed] [Google Scholar]
- Coleman DL. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14:141–148. doi: 10.1007/BF00429772. [DOI] [PubMed] [Google Scholar]
- Daval M, Foufelle F, Ferre P. Functions of AMP-activated protein kinase in adipose tissue. J Physiol. 2006;574:55–62. doi: 10.1113/jphysiol.2006.111484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhase M, Kim SY, Lee H, Naiki-Ito A, Chen Y, Ahn ER, Murata K, Kim SJ, Lautsch N, Kobayashi KS, et al. TANK-binding kinase 1 (TBK1) controls cell survival through PAI-2/serpinB2 and transglutaminase 2. Proc Natl Acad Sci U S A. 2012;109:E177– 186. doi: 10.1073/pnas.1119296109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Haro L, Garcia-Gimeno MA, Neumann D, Beullens M, Bollen M, Sanz P. The PP1-R6 protein phosphatase holoenzyme is involved in the glucose-induced dephosphorylation and inactivation of AMP-activated protein kinase, a key regulator of insulin secretion, in MIN6 beta cells. FASEB J. 2010;24:5080–5091. doi: 10.1096/fj.10-166306. [DOI] [PubMed] [Google Scholar]
- Gauthier MS, O’Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, Gokce N, Apovian C, Ruderman N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun. 2011;404:382–387. doi: 10.1016/j.bbrc.2010.11.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KD, Heymsfield SB, Kemnitz JW, Klein S, Schoeller DA, Speakman JR. Energy balance and its components: implications for body weight regulation. Am J Clin Nutr. 2012;95:989–994. doi: 10.3945/ajcn.112.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Ross FA, Gowans GJ, Tibarewal P, Leslie NR, Hardie DG. Phosphorylation by Akt within the ST loop of AMPK-alpha1 down-regulates its activation in tumour cells. Biochem J. 2014;459:275–287. doi: 10.1042/BJ20131344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JO, Wyatt HR, Peters JC. Energy balance and obesity. Circulation. 2012;126:126–132. doi: 10.1161/CIRCULATIONAHA.111.087213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A. 1994;91:4854– 4858. doi: 10.1073/pnas.91.11.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Huang BP, Lin CH, Chen HM, Lin JT, Cheng YF, Kao SH. AMPK activation inhibits expression of proinflammatory mediators through downregulation of PI3K/p38 MAPK and NF-kappaB signaling in murine macrophages. DNA Cell Biol. 2015;34:133–141. doi: 10.1089/dna.2014.2630. [DOI] [PubMed] [Google Scholar]
- Jeffery E, Berry R, Church CD, Yu S, Shook BA, Horsley V, Rosen ED, Rodeheffer MS. Characterization of Cre recombinase models for the study of adipose tissue. Adipocyte. 2014;3:206–211. doi: 10.4161/adip.29674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Xiao Y, Chang JH, Yu J, Hu H, Starr R, Brittain GC, Chang M, Cheng X, Sun SC. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat Immunol. 2012;13:1101–1109. doi: 10.1038/ni.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, Ohtsuka-Kowatari N, Kumagai K, Sakamoto K, Kobayashi M, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem. 2006;281:26602–26614. doi: 10.1074/jbc.M601284200. [DOI] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazak L, Chouchani ET, Jedrychowski MP, Erickson BK, Shinoda K, Cohen P, Vetrivelan R, Lu GZ, Laznik-Bogoslavski D, Hasenfuss SC, et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell. 2015;163:643–655. doi: 10.1016/j.cell.2015.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazak L, Chouchani ET, Lu GZ, Jedrychowski MP, Bare CJ, Mina AI, Kumari M, Zhang S, Vuckovic I, Laznik-Bogoslavski D, et al. Genetic Depletion of Adipocyte Creatine Metabolism Inhibits Diet-Induced Thermogenesis and Drives Obesity. Cell Metabolism. 2017 doi: 10.1016/j.cmet.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson-Meyer DE, Heilbronn LK, Redman LM, Newcomer BR, Frisard MI, Anton S, Smith SR, Alfonso A, Ravussin E. Effect of calorie restriction with or without exercise on insulin sensitivity, beta-cell function, fat cell size, and ectopic lipid in overweight subjects. Diabetes Care. 2006;29:1337–1344. doi: 10.2337/dc05-2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med. 1995;332:621–628. doi: 10.1056/NEJM199503093321001. [DOI] [PubMed] [Google Scholar]
- Lohman AW, Leskov IL, Butcher JT, Johnstone SR, Stokes TA, Begandt D, DeLalio LJ, Best AK, Penuela S, Leitinger N, et al. Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat Commun. 2015;6:7965. doi: 10.1038/ncomms8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U S A. 2012;109:9378–9383. doi: 10.1073/pnas.1121552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CK, Heilbronn LK, de Jonge L, DeLany JP, Volaufova J, Anton SD, Redman LM, Smith SR, Ravussin E. Effect of calorie restriction on resting metabolic rate and spontaneous physical activity. Obesity (Silver Spring) 2007;15:2964–2973. doi: 10.1038/oby.2007.354. [DOI] [PubMed] [Google Scholar]
- Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem. 2006;281:18933–18941. doi: 10.1074/jbc.M512831200. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottillo EP, Desjardins EM, Crane JD, Smith BK, Green AE, Ducommun S, Henriksen TI, Rebalka IA, Razi A, Sakamoto K, et al. Lack of Adipocyte AMPK Exacerbates Insulin Resistance and Hepatic Steatosis through Brown and Beige Adipose Tissue Function. Cell Metab. 2016;24:118–129. doi: 10.1016/j.cmet.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowers J, Uhm M, Reilly SM, Simon J, Leto D, Chiang SH, Chang L, Saltiel AR. Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKepsilon and TBK1. Elife. 2013;2:e01119. doi: 10.7554/eLife.01119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- Oral EA, Reilly SM, Gomez AV, Meral R, Butz L, Ajluni N, Chenevert TL, Korytnaya E, Neidert AH, Hench R, et al. Inhibition of IKKvarepsilon and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 2017;26:157–170. e157. doi: 10.1016/j.cmet.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos P, Lima L, Tovar S, Gonzalez-Touceda D, Dieguez C, Garcia MC. Divergent responses to thermogenic stimuli in BAT and subcutaneous adipose tissue from interleukin 18 and interleukin 18 receptor 1-deficient mice. Sci Rep. 2015;5:17977. doi: 10.1038/srep17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829– 839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Rasmussen LG, Larsen TM, Mortensen PK, Due A, Astrup A. Effect on 24-h energy expenditure of a moderate-fat diet high in monounsaturated fatty acids compared with that of a low-fat, carbohydrate-rich diet: a 6-mo controlled dietary intervention trial. Am J Clin Nutr. 2007;85:1014–1022. doi: 10.1093/ajcn/85.4.1014. [DOI] [PubMed] [Google Scholar]
- Ravussin E, Lillioja S, Knowler WC, Christin L, Freymond D, Abbott WG, Boyce V, Howard BV, Bogardus C. Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med. 1988;318:467–472. doi: 10.1056/NEJM198802253180802. [DOI] [PubMed] [Google Scholar]
- Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013;19:313–321. doi: 10.1038/nm.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. 2017 doi: 10.1038/nrendo.2017.90. [DOI] [PubMed] [Google Scholar]
- Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol. 2006;574:33–39. doi: 10.1113/jphysiol.2006.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SB, Leibel RL. Excess energy intake and low energy expenditure as predictors of obesity. Int J Obes Relat Metab Disord. 1998;22:385–386. doi: 10.1038/sj.ijo.0800640. [DOI] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 2011;89:667–676. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnurr M, Then F, Galambos P, Scholz C, Siegmund B, Endres S, Eigler A. Extracellular ATP and TNF-alpha synergize in the activation and maturation of human dendritic cells. J Immunol. 2000;165:4704–4709. doi: 10.4049/jimmunol.165.8.4704. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329– 3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, Saltiel AR, Inoki K. Inhibition of AMPK catabolic action by GSK3. Mol Cell. 2013;50:407–419. doi: 10.1016/j.molcel.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–295. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Z, Root-McCaig J, Castellani L, Kemp BE, Steinberg GR, Wright DC. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity (Silver Spring) 2014;22:730–738. doi: 10.1002/oby.20605. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ, Gauthier MS, Hess DT, Apovian CM, Cacicedo JM, Gokce N, Farb M, Valentine RJ, Ruderman NB. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. 2012;53:792–801. doi: 10.1194/jlr.P022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnegar B, Yamazaki S, He JQ, Cheng G. Control of canonical NF-kappaB activation through the NIK-IKK complex pathway. Proc Natl Acad Sci U S A. 2008;105:3503–3508. doi: 10.1073/pnas.0707959105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Stephens JM. STAT1, NF-kappaB and ERKs play a role in the induction of lipocalin-2 expression in adipocytes. Mol Metab. 2013;2:161–170. doi: 10.1016/j.molmet.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.