

Abstract
Cysteine thiols are among the most reactive functional groups in proteins, and their pairing in disulfide linkages is a common post-translational modification in proteins entering the secretory pathway. This modest amino acid alteration, the mere removal of a pair of hydrogen atoms from juxtaposed cysteine residues, contrasts with the substantial changes that characterize most other post-translational reactions. However, the wide variety of proteins that contain disulfides, the profound impact of cross-linking on the behavior of the protein polymer, the numerous and diverse players in intracellular pathways for disulfide formation, and the distinct biological settings in which disulfide bond formation can take place belie the simplicity of the process. Here we lay the groundwork for appreciating the mechanisms and consequences of disulfide bond formation in vivo by reviewing chemical principles underlying cysteine pairing and oxidation. We then show how enzymes tune redox-active cofactors and recruit oxidants to improve the specificity and efficiency of disulfide formation. Finally, we discuss disulfide bond formation in a cellular context and identify important principles that contribute to productive thiol oxidation in complex, crowded, dynamic environments.
Graphical abstract

1. INTRODUCTION
Cysteine thiols are among the most reactive functional groups in proteins, and their pairing in disulfide linkages is a common post-translational modification in proteins entering the secretory pathway. This modest amino acid alteration, the mere removal of a pair of hydrogen atoms from juxtaposed cysteine residues, contrasts with the substantial changes that characterize most other post-translational reactions. However, the wide variety of proteins that contain disulfides, the profound impact of cross-linking on the behavior of the protein polymer, the numerous and diverse players in intracellular pathways for disulfide formation, and the distinct biological settings in which disulfide bond formation can take place belie the simplicity of the process. Here we lay the groundwork for appreciating the mechanisms and consequences of disulfide bond formation in vivo by reviewing chemical principles underlying cysteine pairing and oxidation. We then show how enzymes tune redox-active cofactors and recruit oxidants to improve the specificity and efficiency of disulfide formation. Finally, we discuss disulfide bond formation in a cellular context and identify important principles that contribute to productive thiol oxidation in complex, crowded, dynamic environments.
2. WHICH PROTEINS CONTAIN DISULFIDES
Almost 40% of human protein-encoding genes are predicted to have either a signal sequence for targeting to the secretory pathway and/or at least one transmembrane segment (http://www.proteinatlas.org/humanproteome/secretome). Many of these proteins, which are either secreted or remain associated with the cell plasma membrane or the intracellular endomembrane system, are modified by disulfide bonding. Disulfide bonding is not restricted to proteins of a certain size, type, or function (Figure 1). Indeed, disulfides appear by the handful in small protein toxins containing less than 50 amino acids and by the hundreds, or even thousands, in the large proteins that make up certain biomaterials.
Figure 1.

Examples of proteins and protein complexes cross-linked by disulfide bonds. Protein backbones are represented as ribbon traces, and the Cβ and sulfur (yellow) atoms of cysteine side chains are shown as spheres. In protein complexes, different polypeptides are shown in distinct colors. PDB codes are as follows: conotoxin, 2LXG; theta-defensin, 5INZ; antifreeze protein, 1EZG; Izumo, 5JK9; ribonuclease A, 3MZR; laminin fragment, 4AQS; insulin, 2BN3; C4b-binding protein oligomerization domain, 4b0f; Kremen1-LRP6-Dickkopf complex, 5FWS.
Systems to introduce disulfide bonds into eukaryotic proteins have evolved in the endoplasmic reticulum (ER), Golgi apparatus, and mitochondria, so proteins that reside in or pass through these compartments have correspondingly evolved to contain cysteines in positions suitable for pairing. In bacteria, disulfide-generating catalysts, and thus disulfide bonded proteins, are found in the inner membrane and periplasm of Gram-negative bacteria1 and on the membrane of certain Gram-positive bacteria.2 Cytosolic proteins, whether in prokaryotes or eukaryotes, tend not to contain disulfide bonds that contribute crucially to structure or stability. However, certain thermophilic organisms provide exceptions to the generalization that cysteines in cytosolic proteins remain unpaired.3–5 In addition, a mammalian protein that may be dual-localized to the cell surface and the cytosol has been shown to contain disulfides even in its cytosolic manifestation.6 Furthermore, some viral proteins produced in cytosolic “viral factories” are disulfide bonded.7–9 Finally, cytosolic proteins may contain transient disulfide bonds, e.g., as catalytic intermediates,10 in response to chemical or thermal stress conditions,11 or as a result of cell stimulation and changes in metabolic state.12
To provide an overview of the range of protein types modified by disulfide formation, a few examples are presented. Among the smallest are the conotoxins, a diverse set of peptides or tiny proteins (10 to 50 amino acids) produced by marine Conus snails.13 Most conotoxins have between two and four disulfides, and different conotoxin types have distinct cysteine pairings, leading to enormous diversity.14 Snake venoms such as disintegrins and fasciculins are also small disulfide-rich proteins.15,16 Defensins, a class of antimicrobial peptides produced by animals and plants, have three-dimensional structures and activities that depend on disulfide formation.17,18 The above protein families, as well as growth factors, cytokines, protease inhibitors, and other small, disulfide-rich proteins or protein domains, have been analyzed structurally and classified.19 Evidently, predation and defense are frequently based on an evolved armory of disulfide-packed bioactive peptides and proteins. Nature’s arsenal inspires investigations into engineered variants with novel pharmaceutical properties.20–22
Disulfides are also a dominant structural and stabilizing element within larger proteins made up of tandem repeats of smaller, internally cross-linked units. Low density lipoprotein receptor-like repeats and epidermal growth factor-like (EGF-like) repeats are two examples of disulfide-rich modules that are linked in tandem to generate long, rod-shaped structures acting as cell-surface receptors or key components of the extracellular matrix (ECM). For example, laminin is an 800 kDa ECM protein that contains nearly 200 disulfide bonds; much of its architecture is based on distorted ladders of disulfides within tandem EGF-like repeats (Figure 1).23,24 Interestingly, protein domains can be inserted into this ladder without disrupting its form,25 suggesting that the packing of disulfides remains quite regular despite the sequencing variability among EGF-like repeats. Rigidity and regularity are supplied by disulfides in another scenario: antifreeze proteins. A few classes of highly active insect antifreeze protein consist of regular repeats of disulfide bonded loops, with the disulfides running through the protein cores (Figure 1). This orderly structural motif positions a precise array of threonine hydroxyl groups on the ice-binding face.26,27
A number of fascinating, disulfide-rich biomaterials with as yet unknown structures show particularly regular placements of cysteines in their amino acid sequences, which are likely to form repeating structural elements based on disulfide bonds (Figure 2). The pairing pattern of these cysteines is not known in all cases, but disulfides clearly contribute to the functionality of the material. One such substance is hair. During the keratinization of developing fibers within the mammalian hair follicle, an intense oxidative transformation immobilizes keratin intermediate filaments within a matrix of disulfide-rich proteins.28 The main contributors to this matrix are the high and ultra-high sulfur keratin-associated proteins (KRTAP), with cysteine contents up to 41%.28–30 The amino acid sequence of a representative KRTAP, in which the (CCX3)n pattern is evident, is shown in Figure 2. Another cysteine-rich biomaterial is minicollagen, the protein that reinforces the walls of the venomous capsules of jellyfish, hydra, and corals.31,32 The capsule walls require reinforcement because these nematocysts propel a harpoon into prey with enormous acceleration forces driven by pressures of >2000 psi.33 Protein segments with the amino acid repeat pattern (CX3)n flank the Gly-X-Y repeats of minicollagens and contribute to formation of a resilient disulfide-bonded network. Surprisingly, the N- and C-terminal motifs have identical cysteine spacings (Figure 2) but show different disulfide connectivities.34–36 A third cysteine-patterned structural material is the cysteine-rich eggshell membrane protein (CREMP), a major component of the proteinaceous fibers underlying the calcified shells of birds and oviparous reptiles.37–39 The defining feature of CREMPs is repetition of a module with the pattern (CX4CX5CX8CX6/11)n, which can extend for thousands of amino acids.37
Figure 2.

Examples of diverse disulfide-rich proteins incorporated into biomaterials. The human keratin-associated protein has an ultra-high sulfur content, with cysteine constituting 37% of the residues in the amino acid sequence. In the cnidarian mini-collagens, the six-Cys motifs amino and carboxy terminal to the short collagen triple helical region (green) form disulfide-bridged networks to reinforce the pressurized nematocyst. CREMP proteins, with cysteine contents of ~11%, feature a highly repetitive and long sequence of disulfide-linked modules, eight of which are shown here.
Despite the many disulfide-rich proteins described above, disulfide cross-links are often found more sparingly in other proteins. In cases where disulfides are not the dominant structural element, they may occur within surface loops,40 link surface loops to one another or to secondary structures,41 or bridge secondary structure elements that are part of the core fold.42,43 Examples that illustrate how disulfides can enhance rather than dominate folds are urokinase-type plasminogen activator receptor (uPAR) and phospholipase A2 (Figure 3). In uPAR, disulfide bonds affix loops to the β-strands in the three copies of the β-sheet fold that constitute the receptor. In phospholipase A2, disulfides bridge core secondary structure elements within the fold. Most disulfides in uPAR and phospholipase A2 are at least partially surface-exposed and may therefore be accessible to disulfide catalysts or other oxidants even late in the folding process. In other structures, such as the influenza virus neuraminidase and many proteins containing immunoglobulin folds, disulfides linking core secondary structure elements are entirely buried.42,43 This fact suggests that these disulfides are introduced prior to tertiary structure formation, while access by enzymes or small-molecule oxidants is still possible, or that the folded structure fluctuates sufficiently to permit exposure to oxidative agents.44
Figure 3.

Two proteins demonstrating different ways in which disulfides can decorate a fold. Disulfides link surface loops to the termini of core β-strands in uPAR. Disulfides link core helices to one another and affix a β-hairpin to the helical core in phospholipase A2. PDB codes are UPAR, 2FD6; phospholipase A2, 1C74.
In addition to stabilizing domains within proteins, disulfides are also used to build larger, intermolecular assemblies. One example is the heptameric oligomerization domain of the C4b binding protein, an inhibitor of the complement system, in which neighboring subunits in the ring are disulfide bonded to one another (Figure 1).45 Another, classic example of disulfide-mediated protein quaternary structure assembly is antibodies, in which disulfides bridge the subunits of the H2L2 heterotetramer and also link tetramers, and certain accessory proteins, into higher order structures such as IgA dimers and IgM pentamers.46,47 Another physiologically important example of intermolecular disulfide-mediated cross-linking is network formation of mucins, the proteins that form the protective mucus layers coating exposed epithelia such as in the gastrointestinal tract. Mucins are giant, heavily O-glycosylated proteins that, when properly assembled into a disulfide-bridged covalent network,48 form a physical barrier preventing the penetration of microorganisms into the epithelial cell layer.49 Mucins contain more than 200 cysteines and undergo a complex, multi-stage assembly process. An important step in this process is disulfide-mediated trimerization in the Golgi apparatus, in which mucins arriving as dimers from the ER are recruited into a hexameric array.50,51
3. HOW DISULFIDES STABILIZE PROTEINS
A number of issues must be considered before making generalizations regarding the effect of disulfide bonds on protein stability. Protein thermodynamic stability is the difference in free energy between the folded and unfolded states of a protein. Classically, disulfides are considered to stabilize proteins by decreasing the configurational entropy, and thus raising the free energy, of the unfolded state: two amino acids distant from one another in the primary structure of the protein but held in covalent association in three-dimensional space will dramatically lessen the number of conformations accessible to the unfolded protein. For a protein that has evolved to contain a disulfide between residues that are closely positioned in the folded state, there will be, to a first approximation, no accompanying decrease in entropy of the folded state. However, it has been observed that engineered disulfides are most stabilizing if they are introduced into regions of relatively high mobility in the native protein.52,53 While this finding might seem to suggest that disulfides are paradoxically stabilizing when they decrease the configurational entropy of the folded state, the implication may be instead that introducing cysteines into regions that can relax to allow good disulfide stereochemistry may be less deleterious than mutation of regions that are tightly packed and thus less accommodating. In both cases, the greater effect on unfolded state entropy may be the actual stabilizing factor, but this stabilization may be undermined by disrupting favorable interactions in well-packed regions of the folded state. Understanding the effects of disulfide bonds on protein structures is important, as disulfides can be engineered into proteins to improve stability,54 a desirable feature for protein pharmaceuticals. Engineered disulfides can also improve the functional properties of macromolecular reagents, such as the increased activity seen for certain luciferase mutants with added disulfides.55 Disulfide engineering has further applications in the mechanistic study of protein conformational changes and their relationship to protein function.56–58
Most natural disulfides have evolved to favor folded states of proteins, but disulfides can in principle also promote unfolded or partially folded states, or affect the kinetics of folding and assembly pathways. A disulfide has been proposed to stabilize an aggregation-prone mis-folded form of mutated γD-crystallin, contributing to cataract disease.59 Remarkably, a study of non-synonymous codons in a related eye-lens protein, γB-crystallin, revealed that the effect of mRNA sequence on translation and folding rates can in turn influence cysteine oxidation states.60 The order of cysteine pairing has been shown to contribute to formation of the active, metastable state of a serpin family protease inhibitor.61 Though claims for non-native disulfide formation during protein folding in vivo have been made,62 there is little evidence that non-native disulfides are required intermediates along most protein folding pathways. Instead, disulfides likely evolve primarily under the constraints of native-state functionality. The exception that proves the rule is the case of “pro regions” containing cysteine residues that form temporary, non-native disulfide bonds with other cysteines in the protein during folding.63,64 Such disulfides are transient intermediates in isomerization reactions that promote escape from other, longer-lived non-native disulfide pairings that delay native folding. The evolution of cysteine-containing pro regions thus demonstrates not the importance of non-native disulfides but rather that minimizing the extent and duration of non-native disulfides is beneficial.
Interesting cases of specific disulfide rearrangements during the folding and assembly of proteins have been discovered in mechanisms to regulate the acquisition of function. One example is the E. coli lipopolysaccharide export complex, in which the major pore subunit undergoes a disulfide rearrangement and becomes functional only when triggered by the presence of a partner protein.65 Disulfide rearrangements are also involved in the conversion from the receptor-binding form to the membrane-fusion active state of certain retrovirus surface proteins.66,67 As a final example, a reduction or isomerization event is thought to activate integrins on the cell surface for ligand binding.68,69
4. DETECTING DISULFIDES IN PROTEINS
A number of strategies have been used to demonstrate the presence of disulfide bonds within proteins. A change in the electrophoretic migration of a protein under reducing vs. non-reducing conditions is a common, straightforward, and high-throughput method,62,70 but for certain proteins can be challenging or inconclusive.71 Typically, disulfide cross-links will decrease the hydrodynamic radii of polypeptides and thus increase the migration rates of proteins in gels,72,73 but there are exceptions to this generalization, and the presence of disulfides may also decrease the migration rate in the gel.37 Such exceptions may arise if disulfides decrease binding of the ionic detergents that provide the negative charge by which proteins are driven to the positive pole.74 Disulfides may also enforce a highly elongated shape that can collapse to a faster-migrating form upon reduction.
If the electrophoretic mobility change upon reduction is insufficient or difficult to interpret, disulfides can be detected by reduction followed by modification using large alkylating groups.75 For example, any unpaired cysteines in a protein are first alkylated with a small thiol-labeling reagent such as N-ethylmaleimide (NEM; 125 Da). Disulfides are then reduced and modified with a large thiol-labeling reagent such as polyethylene glycol (PEG) (typical range 2 to 5 kD) appended with a maleimide group. The shift in migration on a gel due to two copies of this large modification per disulfide reports on the number of disulfides that had been present prior to reduction. Alternatively, free thiols can be directly labeled by PEG, and a lack of modification at certain cysteines taken to indicate their protection in disulfide bonds, with the caveat that other explanations for lack of PEG modification must be considered. It should be noted that a PEG modification of 5 kD results in retardation on the gel comparable to about 15 kD of protein.76 Another common reagent used to modify cysteines is 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS), which, with a molecular mass of 536 Da, causes a more modest retardation of the protein in the gel for each thiol group labeled.
Other methods are also available to detect disulfides. Disulfides can be identified from protein crystal structures77 or using NMR-based methods.78 Protein cysteines initially present in thiol form can be distinguished from cysteines that become reactive only upon reduction by differential labeling with small alkylating agents, which are then distinguished by mass spectrometry.79 Liquid chromatography tandem mass spectrometry (LC-MS/MS) enables both disulfide detection and mapping of cysteine connectivity.73 The presence and connectivity of disulfides can also be predicted based on the context of the cysteines in the amino acid sequence.80,81 Lastly, disulfides can be inferred by bioinformatics studies examining the presence and conservation of encoded cysteines in genomic sequence data.82–84
5. HOW DISULFIDES FORM
Disulfide bond formation requires oxidation of paired cysteine residues using either stand-alone, small-molecule oxidants or cofactors that are incorporated into enzyme active sites. These reagents will be described in turn below. We begin by presenting “disulfide exchange,” a common stratagem for forming a particular disulfide. In this process, no net disulfide formation occurs, but disulfides can be generated in new positions by sacrificing them at other loci.
5.1. Thiol-disulfide Exchange Reactions
A simple depiction of a thiol-disulfide exchange reaction is shown in equation 1:
| (1) |
This reaction can be followed, according to the same principle, by a second thiol-disulfide exchange reaction as shown in equation 2.
| (2) |
These two reactions occurring in series accomplish dithiol-disulfide exchange, in which the new disulfide (RD-S-S-RA) involves two cysteines (A and D) that did not participate in the original disulfide (RB-S-S-RC). A number of fundamental issues affect the kinetics and thermodynamics of such thiol-disulfide exchange reactions and are thus relevant to biological pathways that swap disulfide bonds, as described in subsequent sections.
Early model studies of thiol/disulfide exchange reactions demonstrated a first-order dependence on thiolate anion and disulfide85,86 and were corroborated by a series of papers showing the SN2 character of the reaction.87–89 The attacking nucleophile is almost exclusively the thiolate anion; it is some 1010-fold more reactive than the protonated thiol.87 For this reason, rapidly lowering the pH of the solution is a favored strategy for preserving the status of thiols and disulfides and preventing rearrangements prior to analysis of disulfide connectivity.88 The pH dependence of the observed second order rate constant for the exchange reaction (kobs) is related to the limiting rate constant at high pH values (k) as follows:
| (3) |
While this relationship clearly identifies the importance of the pK of the nucleophile in dictating the extent to which the thiol is ionized (and therefore reactive in disulfide exchange), a series of Bronsted analyses using a range of thiols of comparable structure showed that the intrinsic nucleophilicity of the thiolates decreases with decreasing pK values.89,90 Thus one cannot assume, for example, that an exceptionally low pK value translates to a correspondingly strong nucleophile at pH 7.5.
Theoretical studies using a range of models and solvent systems have considered whether a trisulfide (∂-S-S-S∂-) species represents a local minimum along the reaction coordinate for disulfide exchange.91–95 While the outcome of these calculations depends on the model system chosen, these studies support the expectation that exchange is optimal with in-line attack of the thiolate and with rather modest change in the charge of the central sulfur atom along the reaction coordinate.91–95
5.2. Oxidants and Oxidative Chemistry
5.2.1. Molecular Oxygen
For the net generation of disulfide bonds, other mechanisms besides thiol-disulfide exchange must come into play. Molecular oxygen is the ultimate oxidant in many pathways for disulfide bond generation. However, the direct reaction between triplet dioxygen and singlet thiol (equation 4)
| (4) |
is formally spin-forbidden and kinetically sluggish.96 Autoxidation in aqueous solutions is greatly accelerated by traces of redox-active transition metal ions, most notably copper and iron, which are persistent contaminants of solution components and glass and plastic surfaces. Metal chelating reagents such as EDTA strongly suppress, but do not totally eliminate, the background thiol oxidation in aerobic solutions. The best understood system, the copper-catalyzed oxidation of cysteine, is of considerable kinetic complexity.97–101 Oxidation proceeds via cysteine chelates and generates hydrogen peroxide at neutral pH values. Further thiol oxidation can then occur as described in section 5.2.2 below.
Given the precedents for such metal-catalyzed thiol oxidation in the chemical literature, and considering the many metal-dependent oxidoreductases in oxidative transformations, the dearth of well-documented metalloenzymes catalyzing disulfide formation is striking. The earlier literature contained a number of reports of both iron- and copper-dependent sulfhydryl oxidases, but none of them have been unequivocally confirmed.102–104 Indeed, reinvestigation of sulfhydryl oxidases from both categories have established that their apparent metal content was adventitious, and that thiol oxidation is catalyzed instead by a flavin redox center.102,103 Thus, while bona fide metal-dependent sulfhydryl oxidases capable of sustained turnovers would be expected to exist, they have yet to be definitively described.
5.2.2. Hydrogen Peroxide
The reaction between hydrogen peroxide and thiols has received sustained attention because of its importance in the context of oxidative stress and signaling in biology.105 Oxidation by hydrogen peroxide comprises two spin-allowed steps:
| (5) |
| (6) |
Sulfenic acid formation (equation 5) involves nucleophilic attack by the thiolate and is slow for simple low molecular weight thiols (with second order rate constants of about 25 M−1s−1)106,107 but can be greatly accelerated enzymatically. Subsequent disulfide bond generation (equation 6) is then inherently more rapid.
5.2.3. Dehydroascorbate
Dehydroascorbate (DHA) is reduced by reduced glutathione generating ascorbate (AA) and disulfides108,109 with the stoichiometry:
| (7) |
The reaction likely involves a thiohemiketal intermediate at C2 that can be resolved using a second nucleophilic attack (Figure 4).109–111 DHA has been described as a direct oxidant of a range of thiols, including unfolded reduced proteins and protein disulfide isomerase.112
Figure 4.
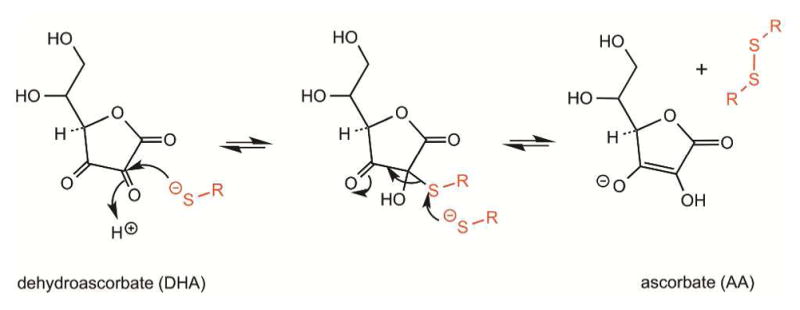
Oxidation of thiols by dehydroascorbate. Generation of a thiohemiacetal intermediate is followed by capture of the adduct by a second thiolate species to yield ascorbate and the disulfide, RS-SR.
5.2.4. Quinones
Certain quinones serve as electron carriers in biological systems,113 whereas others are found as toxic components of environmental pollutants.114,115 Quinones can form thiol adducts, catalyze the generation of reactive oxygen species, and oxidize thiols to disulfides (Figure 5).116–118 The major systems for disulfide bond formation in the periplasm of bacteria rely on ubiquinone cofactors, and the chemistry of ubiquinone reactivity with cysteines has been described most thoroughly in these systems (see sections 6.3.1 and 6.3.2). Reduction of the quinone (form 1) is initiated by formation of a Michael adduct (form 2) followed by resolution using a second thiolate to yield the hydroquinone and the corresponding disulfide (form 4; Figure 5).117,118
Figure 5.
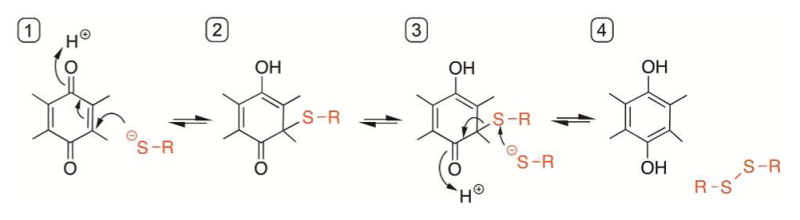
Mechanism of disulfide bond formation by 1,4-quinones. Attack of a thiolate on quinone (1) forms the Michael adduct (2) which can be resolved as in (3) to generate the reduced cofactor and the corresponding disulfide, RS-SR.
5.2.5. Flavins
Free oxidized flavins are minimally reactive towards thiols under physiological conditions, but they contribute to catalysis of disulfide bond formation as tightly bound prosthetic groups in enzymes. While flavin-linked enzymes will be discussed more fully later, the key chemistry of flavin-mediated disulfide bond generation is summarized here (Figure 6). Precedents for these steps come from early mechanistic studies of the flavin-dependent thiol/disulfide oxidoreductases dihydrolipoamide dehydrogenase (DLD) and glutathione reductase (GR),119–121 and parallels with the mechanism for quinone oxidation of thiols can be seen. Among the mechanistic commonalities are the thiolate-to-flavin charge-transfer complexes that precede the nucleophilic attack on the flavin depicted in Figure 6, form 1. Transitory C4a adducts (Figure 6, form 2) are intermediates in the net reduction of the flavin prosthetic group in other flavin-linked disulfide oxidoreductases122–125 and are likely to form in sulfhydryl oxidases as well. The corresponding adduct is then resolved by attack from a second thiolate species (Figure 6, form 3) leading to the generation of the dihydroflavin species (Figure 6, form 4) and liberation of the corresponding disulfide (R-S-S-R).122,123,126 In canonical sulfhydryl oxidases the reduced flavin is reoxidized by molecular oxygen via one-electron chemistry.127,128 All sulfhydryl oxidases release substantial to stoichiometric levels of hydrogen peroxide;129–131 however, in some cases a minor proportion of this peroxide comes from the superoxide ion that subsequently dismutes after release from the active site.132 While the nominal electron acceptor for the sulfhydryl oxidases is molecular oxygen, other electron acceptors may function in vitro or may substitute for oxygen in particular biological contexts.131,133,134
Figure 6.
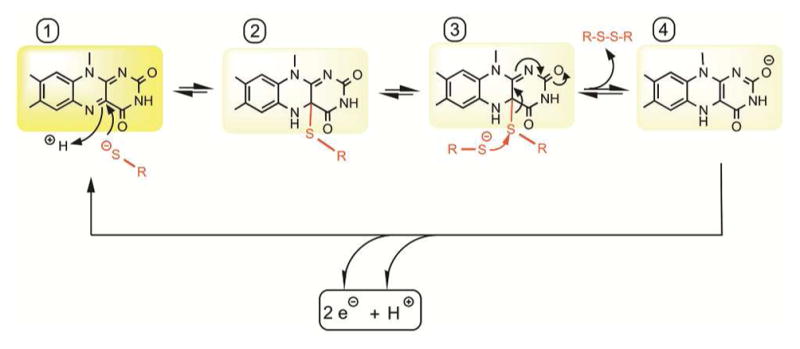
Mechanism of disulfide formation by flavin cofactors. The isoalloxazine ring system (1) can be attacked by a thiolate nucleophile at the C4a position to yield a thiol-flavin adduct (2). Resolution of this species with a second thiolate (3) leads to the generation of the disulfide. The resulting dihydroflavin (4) can be oxidized in two one-electron steps by molecular oxygen or by other oxidants.
6. HOW ENZYMES CONTRIBUTE TO THIOL OXIDATION REACTIONS
Here we revisit thiol-disulfide exchange reactions (section 5.1) and cofactors (sections 5.2.4 and 5.2.5) in the context of enzyme active sites. We will discuss how enzyme reactive groups are positioned to facilitate catalysis of disulfide formation and how other enzyme features promote substrate specificity.
6.1. Protein Disulfide Isomerase (PDI) and Other Thiol-disulfide Oxidoreductases
PDI family enzymes engage in thiol-disulfide rearrangements for disulfide introduction in proteins, and many principles can be gleaned from an examination of their active sites.
6.1.1. Geometry and Chemistry of Thiol-disulfide Exchange in the Context of PDI Active Sites
The PDI family is a large set of proteins (more than 20 in humans) that catalyze disulfide formation, isomerization, and in certain cases reduction of substrate proteins in the ER.135–136 PDI family enzymes contain domains with the same fold as thioredoxins,137,138 which are proteins with disulfide reductase activity.139 In many enzymes of the PDI family, multiple thioredoxin-fold (trx) domains are linked in tandem, and some PDI enzymes contain other domain types as well. PDI itself contains four trx domains, the first and last of which contain redox-active CXXC motifs. The domain organization of PDI is referred to as a-b-b′-a′, with “a” designating redox-active domains and “b” redox-inactive domains. Structural differences between members of the PDI family that could lead to functional specialization have been reviewed.138,140 As PDI enzymes use thiol/disulfide exchange for catalysis, they must fulfill the fundamental principles outlined above in the introduction to thiol/disulfide exchange reactions (section 5.1). Additional features may also be present to further enhance PDI-mediated reactions.
As in thioredoxin, the PDI active site contains a disulfide formed by the two cysteines in a CXXC motif, typically with the sequence CGHC. We will focus here on geometrical and chemical requirements for the catalysis of disulfide bond formation by PDI proteins using this motif. First, however, it is important to note that the presence of a CXXC motif in a thioredoxin-fold domain of a PDI family protein is not evidence of redox activity. S. cerevisiae Eps1p contains a domain with a CXXC motif in the appropriate primary structural context, but the structure of the enzyme shows the disulfide to be buried and unreactive, and the adjacent proline residue, while maintained, is found in the trans rather than the cis configuration.141
Most fundamentally, PDI and other enzymes that undergo thiol-disulfide exchange with substrate proteins should conform to the steric requirements of the reaction.142 In-line attack in the context of structured globular proteins would predict alignment of the catalytic disulfide axis as depicted schematically in Figure 7A. This arrangement allows an approaching nucleophilic thiolate from the substrate to form what is known as a “mixed” disulfide (e.g., a disulfide between a substrate and an enzyme or between two redox-active sites within a single protein or protein complex) without the need for substantial conformational rearrangement of the disulfide-containing peptide loop in the PDI protein.104,143 In contrast, an enzyme disulfide with an axis parallel to the protein surface would not readily engage in mixed disulfide formation (Figure 7B). PDI catalytic domains, like other members of the thioredoxin superfamily, position the redox-active CXXC disulfide at the amino terminus of a long surface helix in an orientation consistent with geometrical predictions (Figure 8). The sulfur atom of the N-terminal cysteine of the CXXC motif (S1 in Figure 8), also known as the interchange cysteine, is solvent accessible, whereas its redox partner (S2) is typically buried within the protein.
Figure 7.
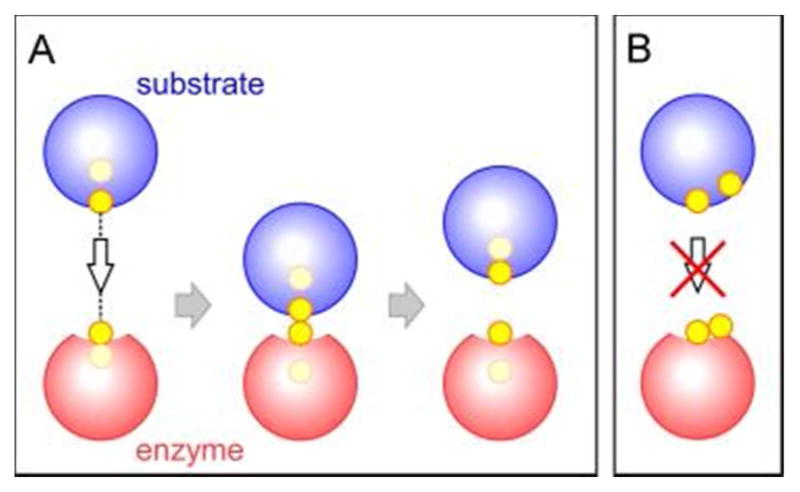
(A) Two proteins with cysteine sulfurs oriented as shown are able to engage in an in-line SN2 reaction, form a mixed disulfide, and complete a thiol-disulfide exchange reaction. (B) The orientation of cysteine sulfurs shown is incompatible with thiol-disulfide exchange.
Figure 8.
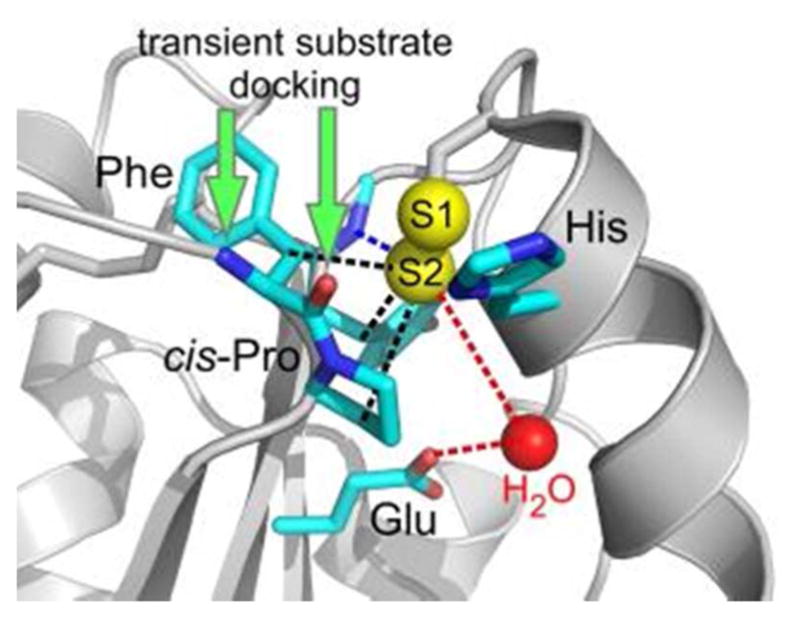
Representative active site of a PDI family protein. Amino acids commonly observed in PDIfamily active sites are shown in stick representation, with red indicating oxygen atoms and blue nitrogen. The Phe side chain is replaced by Tyr in some cases. Potential interactions of the largely buried S2 sulfur of the CXXC motif are indicated by dashed lines (black—hydrophobic; blue—hydrogen bonding; red— proton transfer). Green arrows indicate sites for hydrogen bonding interactions by the incoming substrate backbone. Red sphere is water. PDB code is 3ED3 (yeast Mpd1p).
Additional features contribute to the success of PDI family proteins in catalyzing thiol-disulfide exchange reactions. One such feature is the positioning of nearby functional groups to promote mixed disulfide formation. The second of the CXXC cysteines (S2) is released as the mixed disulfide between PDI and substrate forms (Figure 7A), and the resulting thiolate would be nestled 3 to 4 A from the three side-chain Cβ atoms of a conserved cis-proline on a neighboring loop and two nearby aromatic residues (Figure 8), an apparently unfavorable environment for development of negative charge. Even deeper within the protein, however, is a conserved glutamic acid in position to perform either a direct or a water-mediated proton transfer to the thiolate. An additional feature that may aid formation of a thiolate involving sulfur S2 is the proximity of a backbone N-H group at the carboxy terminus of the β-strand containing the buried acidic residue (Figure 8).
The PDI active site must not only be suitable for attack by a substrate thiolate to form the mixed disulfide between enzyme and substrate, it must also enable attack of this mixed disulfide by a second substrate thiolate and release of the substrate with a newly formed disulfide. Therefore, the mixed disulfide must be accessible for this second SN2 reaction. Structural information is starting to become available for intermediates in PDI thiol-disulfide exchange reactions. Stabilizing such intermediates typically requires mutation of cysteines that could potentially attack the mixed disulfides, so part of the chemical environment is sacrificed for the sake of illuminating the functional groups that remain. A few examples of mixed disulfides involving PDI family proteins stabilized by mutagenesis in this manner have been crystallized, including the PDI family protein ERp57 in association with tapasin,144 which is a component of the peptide-loading system for the class I major histocompatibility complex. In addition, structures are available for the PDI proteins P5 and ERp46 linked to a peptide derived from peroxiredoxin Prx4 (Figure 9),145 and for ERp44 linked to Prx4.146 These structures demonstrate how substrate peptides engaging in mixed disulfides with PDI proteins form extended hydrogen bonding interactions with the enzyme loop containing the cis-proline. A basic side chain from the neighboring loop may also make interactions with a substrate backbone carbonyl (Figure 9). Similar principles are observed for analogous complexes involving thioredoxins, except that the backbone of the neighboring loop, rather than a side chain, participates in hydrogen bonding to the substrate backbone.147,148 These interactions keep the substrate backbone low and tight against the enzyme surface. With the substrate main chain held to one side of the active site, the substrate cysteine is fully exposed for a second in-line attack (Figure 9).
Figure 9.
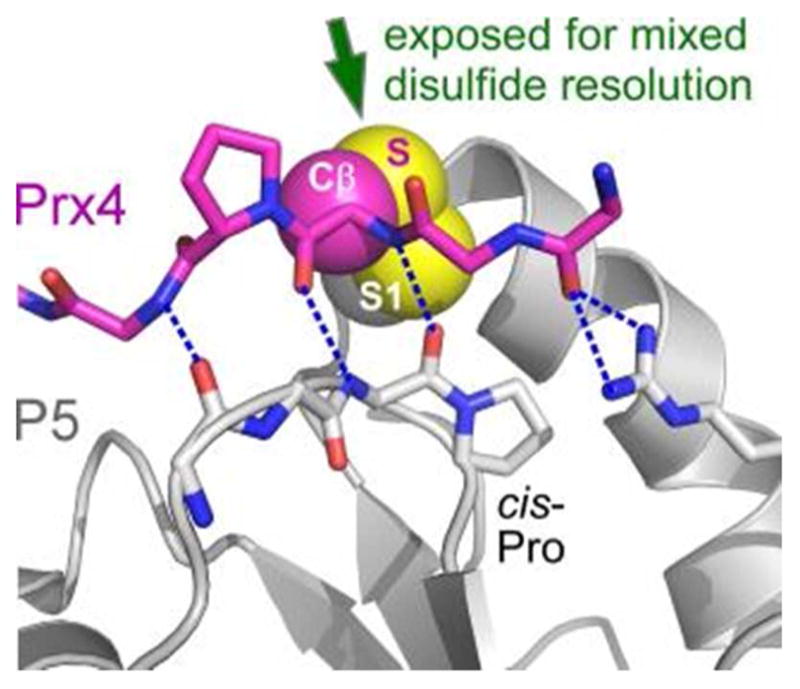
Model for substrate interaction with a PDI protein. The P5 protein (gene name PDIA6) is in gray, and the backbone (including a proline side chain) of a peptide from peroxiredoxin Prx4 is in magenta. The cysteine side chain atoms of Prx4 are labeled Cβ and S. Blue dashed lines are hydrogen bonds. Red in stick representations indicates oxygen atoms, blue nitrogens. The S1 sulfur of P5 is labeled. PDB code is 3W8J.
The final step in the electron transfer reaction that should be promoted by PDI is formation of the disulfide in the substrate and release of the reduced enzyme. The formation of a thiolate at S1 in the enzyme may be promoted by the location of the cysteine at the amino terminus of the active-site helix, i.e., at the positive end of the helix dipole. In addition, the histidine in the CGHC sequence, the presence of which raises the redox potential of the motif,149 may help stabilize the S1 thiolate.
6.1.2. Oxidation vs. Isomerization
The above description of features that promote catalysis by PDI focused on substrate oxidation. Disulfide isomerization (Figure 10) is another activity catalyzed by PDI enzymes.150 Like oxidation, isomerization occurs by formation of a mixed disulfide. The difference is that reduced PDI engages a substrate disulfide to initiate isomerization, rather than oxidized PDI acting on free substrate cysteines to pair them. The isomerization reaction may be promoted by the inherent reactivity of a disulfide between erroneously paired cysteines in a mis-folded substrate protein. The PDI-substrate mixed disulfide can then be resolved by nucleophilic attack from another substrate cysteine, forming a new substrate disulfide and releasing PDI again in the reduced state. It should be noted that the isomerization reaction is indistinguishable mechanistically from an oxidation reaction once the mixed disulfide has formed. Whether a PDI family enzyme will carry out oxidation or isomerization to promote native disulfide bond formation depends on the state of the cysteines in the substrate and the availability of the reduced form of the PDI enzyme. The presence of reduced PDI will depend on its redox potential and on various kinetic considerations, discussed in section 7.5 below.
Figure 10.
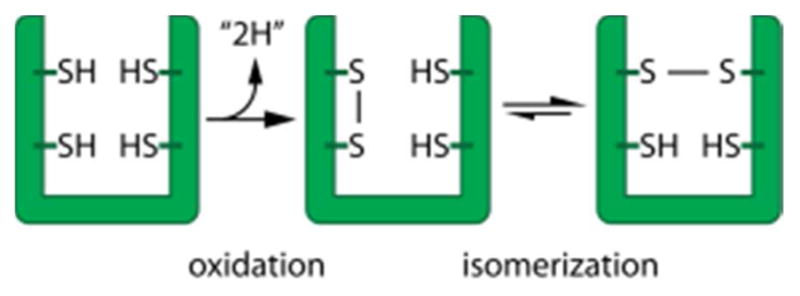
Oxidative protein folding comprises two conceptual steps: oxidation and isomerization of mispaired disulfides. These steps are schematically depicted here for the introduction and rearrangement of a single disulfide bond.
Interestingly, mutagenesis can be used to uncouple oxidation and isomerization in PDI proteins, as amino acid residues in the vicinity of the PDI active site selectively influence the isomerization vs. oxidation reactions.151 Using mutants that can oxidize but not isomerize, it was shown that the essential activity of PDI in yeast is oxidation and not isomerization.152
6.1.3. Ligand Binding and Chaperone Activity of PDI
In addition to performing catalytic activities based on thiol-disulfide exchange, PDI and some of its family members also possess ligand binding capabilities that contribute to substrate specificity and promote interaction between the PDI redox-active sites and target cysteines. Both redox-active and -inactive domains of PDI proteins participate in substrate recognition and alignment.153–155 Particularly notable is a large patch of hydrophobic surface on the b′ domain facing the interior of the U-shaped PDI structure (Figure 11).156,157 This patch is a principal docking site for exposed hydrophobic residues in unfolded or mis-folded substrates.153,156,158 Similar hydrophobic clefts or patches are found in other PDI family proteins with different domain composition.159 Recently, sophisticated models have been put forth for redox-dependent substrate binding and for how substrate interaction with non-catalytic domains influences catalytic activity at the redox centers.160–162 For example, the discovery that binding of certain small molecules to the PDI hydrophobic patch interferes with reduction of the protein substrate insulin but enhances reduction of a fluorescent GSSG analog suggested that engaging the hydrophobic site triggers an allosteric switch enhancing PDI reductase activity.162 Small-molecule binding allowed the two effects to be dissociated, but physiological protein substrates likely fulfill both roles, first binding the PDI hydrophobic patch and then benefitting from augmented catalytic activity at the PDI redox-active sites.
Figure 11.
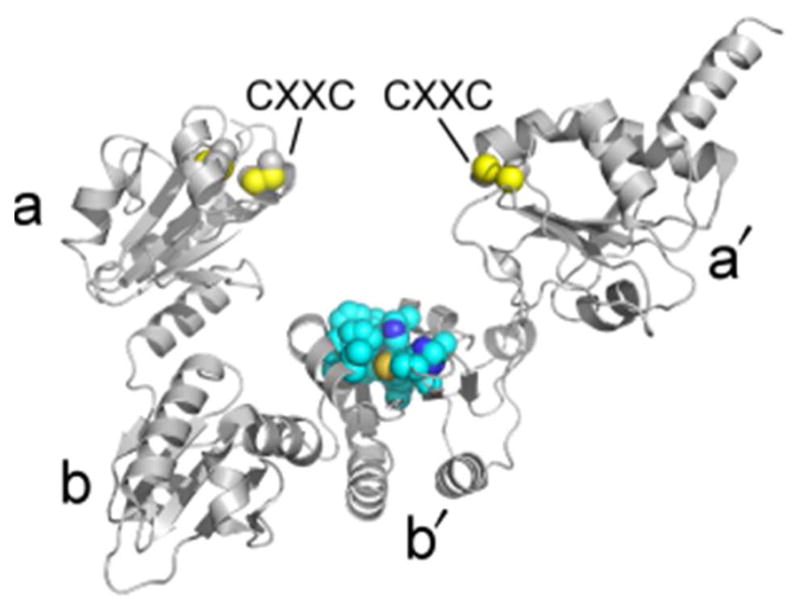
Structure of PDI. The image shows S. cerevisiae PDI. Thioredoxin fold domains are labeled according to conventional nomenclature (a-b-b′-a′). Cysteine side chains are shown as spheres with yellow sulfurs. A set of exposed hydrophobic residues in the b′ domain that may constitute a binding site for folding or misfolded proteins is shown in space-filling representation (carbons in cyan). PDB code is 2B5E.
Attempts have been made to produce small-molecule catalysts that mimic the activity of PDI in promoting native disulfide bond formation and activity of substrate proteins.163,164 Effective molecules of this type would make cheap and readily removable reagents for industrial production of disulfide bonded proteins, which include antibodies and hormones for clinical use. For basic science, success in matching the activity of PDI with a small molecule might indicate which features of PDI are key for its activity. A recent effort at producing such a reagent took into consideration not only the cysteine pK values and reduction potential of the disulfide in the designed molecules but also aimed to incorporate the capability of binding hydrophobic sites in target proteins.165 A series of dithiols decorated with various hydrophobic groups was produced, and increasing ability to refold the model substrate RNase A was observed with increasing hydrophobic bulk, until the solubility of the reagent became limiting. However, the best small-molecule catalysts did not match the improvement in folding achieved by PDI itself, suggesting that the broad binding surfaces offered by protein catalysts and the cooperation between substrate binding and redox activities described above are important for catalysis of proper disulfide bond formation in substrate proteins. Given the highly evolved and coordinated activities of PDI, it is not surprising that small molecule oxidants do not achieve equal success in oxidative protein folding.
6.1.4. Mia40, a Thiol-Disulfide Oxidoreductase with a Different Scaffold
Mia40 is an enzyme that performs thiol-disulfide exchange reactions in the mitochondrial intermembrane space (IMS) to oxidize substrates and is a rare thiol thiol-disulfide oxidoreductase not based on the trx fold. Instead, Mia40 structures from yeast and human show a helix-turn-helix motif stapled by two disulfides and an amino-terminal flexible region housing the redox-active CPC motif (Figure 12).166,167 Disulfides within CXC motifs have been shown to be relatively unstable,168 but the CPC disulfide is the most stable of the CXC disulfides, perhaps because the restricted degrees of freedom in the backbone due to the presence of the proline limit the entropic gain upon reducing the disulfide. A redox potential of approximately −200 mV has been reported for the Mia40 CPC motif,167 compared to a value of about −165 mV for PDI and −167 for a peptide with the sequence CGC.142,163 Like the active site of PDI, the CPC motif of Mia40 is solvent exposed, and the disulfide is available for attack along the preferred axis (Figure 12). Reduced substrates of Mia40 bind to a shallow hydrophobic depression on the protein surface.166,167,169 The rate of mixed disulfide formation then appears to depend on the presence of hydrophobic residues in the substrate immediately adjacent to the attacking cysteine thiolate.170 The ultimate resolution of the mixed disulfide and formation of a new substrate disulfide depends on attack by a second substrate cysteine. It has been noted that Mia40 mixed disulfides with substrate proteins are longer-lived than the typically transient encounters made by PDI.171
Figure 12.
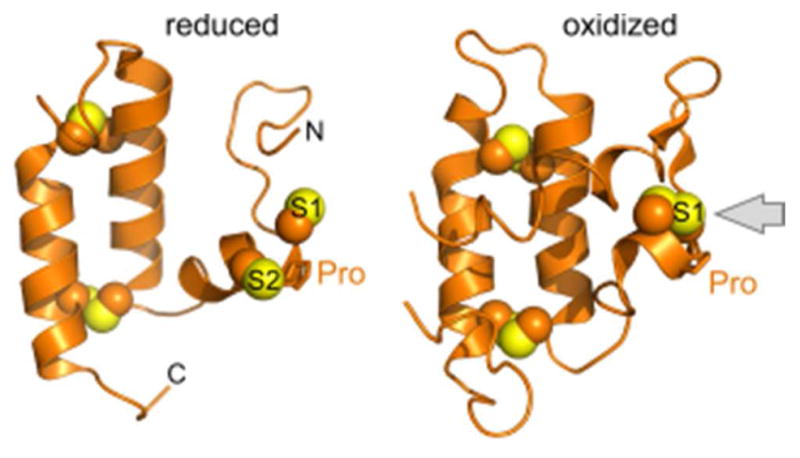
Structures of the Mia40 oxidoreductase. Sulfurs in the CPC motif are labeled S1 and S2. Gray arrow in the oxidized structure illustrates solvent accessibility of the redox-active disulfide. PDB codes are 2K3J (reduced; H. sapiens Mia40 solution NMR structure) and 2ZXT (oxidized; S. cerevisiae Mia40 crystallized as a fusion with maltose binding protein (not shown)).
6.2. Peroxiredoxins/Glutathione Peroxidases
Peroxiredoxins and glutathione peroxidases catalyze the reduction of peroxides to water, and a byproduct of this reaction is formation of a disulfide bond in the enzyme active site. Peroxidases located in cellular compartments in which oxidative protein folding occurs couple active-site regeneration to a second catalytic activity: the formation of disulfide bonds in substrate proteins.
6.2.1. Peroxiredoxin
Peroxiredoxins are a widespread protein family studied for their potential roles as antioxidants in cancer, neurodegeneration, and inflammatory disease. Many of these enzymes also function to sense and transduce redox signals borne by H2O2.172 Here we will focus on the role of peroxiredoxin as a catalyst of disulfide generation for oxidative protein folding. The peroxiredoxin relevant to disulfide formation is Prx4 of the ER,173 as most other peroxiredoxins are found in locations, i.e., the cytosol, where disulfide bond formation is not integral to protein biosynthesis. Prx4 is a ring-shaped homodecamer, and active-sites are formed between subunit pairs. The peroxidatic cysteine of Prx4 reacts very rapidly (~ 2 × 107 M−1s−1) with H2O2,174 the end of the helix containing the newly sulfenylated cysteine unfolds, and another enzyme thiolate from the neighboring subunit attacks the sulfenylated cysteine to form a disulfide (Figure 13).175 Reduction of this disulfide by thiol-disulfide exchange with a PDI family enzyme allows the oxidizing equivalents gained by reducing hydrogen peroxide to be funneled toward oxidative protein folding.145,176–178
Figure 13.

Prx4 peroxidase cycle. Isolated segments of two neighboring Prx4 subunits are shown (purple and orange). Spheres are the side chains of cysteines participating in catalytic disulfide formation, with sulfurs colored yellow and Cβ atoms colored according to the parent chain. Based on PDB codes 3TJG and 3TJF.
6.2.2. Glutathione Peroxidase
Gpx7 and Gpx8 are the glutathione peroxidase family members located in the ER. Glutathione peroxidases are thioredoxin fold superfamily proteins,179 and though some glutathione peroxidases contain selenocysteine as the active-site residue, Gpx7 and Gpx8 do not. These two enzymes are better described as PDI peroxidases, as their primary reductant appears to be PDI rather than glutathione.180–181 Gpx8 has been reported as an important detoxifier of hydrogen peroxide produced by sulfhydryl oxidase activity in the endoplasmic reticulum.182 Studies are ongoing to identify the physiological functions and physiologically relevant mechanisms of these peroxidases,183 as well as the respective roles of Gpx enzymes and Prx4.175,182
6.3. Quinone-containing Enzymes
Seminal studies on oxidative protein folding in the bacterial periplasm led to the recognition of two classes of polytopic membrane protein that utilize quinones as cofactors of enzyme-mediated oxidation of thiols (Figure 14).82,184–188 These classes are represented by the enzymes DsbB and homologs of vitamin K epoxide reductase (VKOR). Bacteria that form disulfide bonds in proteins exported from the cytosol are partitioned into those containing DsbB and those with a VKOR homolog.82 DsbB and VKOR share a similar arrangement of transmembrane helices and active-site chemistry, but their polypeptide chain termini are found at different positions in the helical bundle. These observations led to the suggestion, backed by a phylogenetic study, that DsbB and VKOR are related by circular permutation from a common ancestor.189 Disulfide bond formation in both the DsbB and VKOR involves a stepwise reduction of an enzyme-bound 1,4-quinone species according to the mechanism shown in Figure 5.
Figure 14.
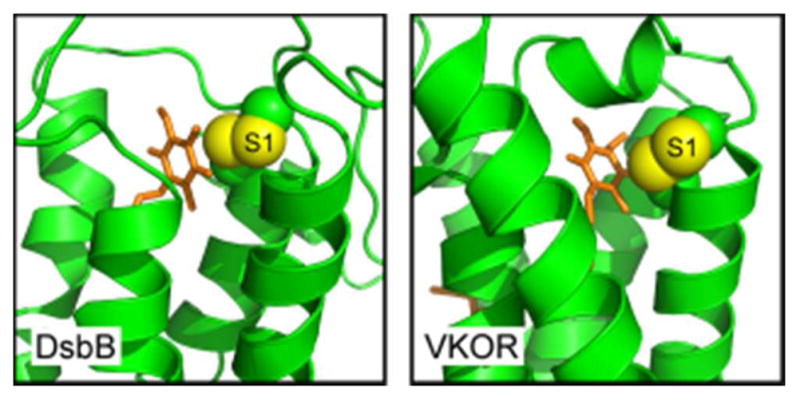
Active-site regions of quinone-containing disulfide catalysts. Quinone-proximal disulfides are shown as spheres with sulfur atoms in yellow and Cβ atoms in green. The quinone (orange sticks) is buried in the core of the helical bundle in each enzyme, and the amino-terminal cysteine of the quinone-proximal disulfide (S1) is exposed to in-line nucleophilic attack. PDB codes are 2ZUP and 3KP9.
6.3.1. DsbB
The DsbB protein from the periplasm of E. coli is the best understood mechanistically of the quinone-containing catalysts of disulfide bond formation. A series of disulfide exchange reactions transmit reducing equivalents to the quinone cofactor and ultimately to the respiratory chain (Figure 15A). The immediate reductant of DsbB is DsbA,184,185 a thioredoxin-fold protein190 that collects reducing equivalents from nascent chains undergoing oxidative protein folding in the periplasm. Mutagenesis, crystallographic and pre-steady state kinetic approaches have provided key mechanistic insight into the DsbA-DsbB system.191–195 The first observable intermediate when reduced DsbA is mixed with oxidized DsbB is a thiolate-quinone charge-transfer intermediate.194,196,197 This thiolate-to-quinone interaction is stabilized both by the charge-transfer interaction per se and via an ionic interaction with the side chain of a nearby arginine. This complex is resolved by disulfide exchange reactions that lead to rate-limiting release of DsbA and formation of the reduced quinone cofactor.196
Figure 15.

(A) Schematic representation of the DsbA-DsbB system showing the key charge-transfer intermediate observed when reduced DsbA is mixed with oxidized DsbB. Reduction of the quinone is rate-limiting and is coupled to the release of oxidized DsbA. The quinone cofactor is shown as a hexagon, in which blue represents a charge-transfer state. (B) Thermodynamics along the reaction coordinate for the events depicted in panel A. The redox potential for DsbA would appear insufficiently reducing to efficiently transfer electrons to the first disulfide in DsbB. (C) While the net conversion of reactants to products is thermodynamically unfavorable, the mixed disulfide intermediate between them can be highly populated (black line) or destabilized (grey line). Selective stabilization of mixed disulfide intermediates may allow the formation of products if those products are in turn depleted by other favorable reactions, such as electron transfer to quinone or oxygen.
Analysis of the pathway whereby reducing equivalents are relayed from DsbA to the DsbB quinone highlights a striking (> −120 mV) thermodynamic mismatch between the reduction potential of the DsbA active-site di-cysteine motif and its target disulfide within DsbB (Figure 15B). However, while the net reduction of the first disulfide in DsbB would be unfavorable by a factor >104, a mixed disulfide intermediate between them could be quite stable. This point is illustrated schematically in Figure 15C: the stability of mixed disulfides is not constrained by the energy differences between reactants and products.198 Hence a series of such disulfide exchanges in DsbA-DsbB releases the cofactor-proximal thiolate to react with the strongly oxidizing quinone center.199 This dynamic disulfide relay allows efficient redox coupling between a modest reductant, DsbA, and a distal electron-deficient center without having to traverse the formal redox barriers depicted in Figure 15B. Given the prevalence of disulfide relays in the enzymes of oxidative protein folding, this principle is likely to be exploited in other instances involving apparently mismatched redox partners.198
6.3.2. VKOR
The VKOR enzyme family is named for its function of reducing vitamin K epoxide generated during enzymatic carboxylation of glutamic acid residues in factors involved in blood coagulation, bone development, and other physiological processes. In mammals, reduced PDI generated by oxidative protein folding can provide reducing equivalents to VKOR for reduction of vitamin K1 and K2 and their corresponding epoxides.200–204 Notably, vitamin K-dependent enzymes are not restricted to mammals, suggesting that mechanisms for vitamin K reduction are needed in other organisms. For example, Conus snails contain a vitamin K-dependent carboxylase,205 which makes post-translational modifications to a class of disulfide-poor toxins,206,207 a distinct set from the disulfide-rich toxins described in section 2 above. VKOR-like sequences can be identified in various marine invertebrates including Conus bullatus, and, though a diverse and rapidly evolving set of PDI proteins has been identified in Conus snails,208 whether the activities of PDIs and VKOR-like enzymes are linked in these organisms is not known. Notably, many VKOR homologs reduce only quinones (ubiquinone or menaquinone) and do not use vitamin K epoxide as a substrate. Despite the differences in electron acceptors, various VKOR family enzymes likely share structural and certain mechanistic properties.209,210
The VKOR oxidoreductases are also mechanistically similar to the DsbB enzymes; like DsbB, they feature catalytic disulfides in redox communication with a quinone cofactor embedded in a transmembrane domain.211,212 Some VKOR enzymes accept electrons from DsbA-like proteins.213 In other VKOR-like enzymes, a trx domain is fused carboxy-terminally to the transmembrane domain (Figure 16),214 from where it presumably substitutes for DsbA. Structures of the Synechococcus VKOR enzyme revealed the nature of the association of the transmembrane and trx domains and suggested how electrons accepted from protein clients are transferred to the quinone-proximal cysteines (Figure 15).183,194 This interdomain transfer is mediated by a loop in the transmembrane domain bearing two cysteines and sandwiched between the main redox-active sites. A set of mutations were made in the VKOR electron transfer pathway to trap various mixed-disulfide intermediates and illuminate the conformational changes necessary to obtain them.196 One major intramolecular electron-transfer intermediate was recently proposed as the binding target of the anticoagulant warfarin.215
Figure 16.

Schematic representation of internal electron transfer events in a bacterial VKOR homolog. The quinone cofactor is shown as a hexagon, in which blue represents a charge-transfer state. It is not known to what extent the electron-transfer steps are concerted in VKOR enzymes, i.e., whether the electron-transfer loop is simultaneously disulfide bonded to both a trx domain active-site cysteine and the partner of the charge-transfer cysteine in the transmembrane domain. For comparison with Figure 14, the trx domain is shown to the left of the transmembrane domain, but it should be noted that the trx domain is fused carboxy terminally to the transmembrane domain.
6.4. Flavin-containing Enzymes
6.4.1. General Principles and Commonalities
All sulfhydryl oxidases described to date have been shown to be flavin-linked enzymes catalyzing the net reaction:
| (8) |
Three broad categories have been identified: the Ero1 enzymes,216–218 the Erv-fold proteins including members of the QSOX family, and enzymes based on the pyridine nucleotide disulfide oxidoreductase scaffold.219–223 Within these categories, evolution of associated domains and quaternary structure assembly modes has produced further diversity in structure (Figure 17). Despite their many differences, sulfhydryl oxidases share the unifying feature of a catalytic disulfide bond formed from closely spaced cysteines (separated by two or four residues) in redox contact with the isoalloxazine ring of the flavin adenine dinucleotide (FAD) prosthetic group, similar to the positioning of a redox-active disulfide close to the quinone in DsbB and VKOR. Four representative structures of flavin-containing sulfhydryl oxidases, emphasizing this proximity, are shown in Figure 18. In addition to the funneling of reducing equivalents through a protein disulfide dedicated to direct communication with a bound cofactor, another similarity between flavin- and quinone-containing catalysts of disulfide bond formation is the presence of a second enzyme disulfide that mediates electron transfer between substrate thiols and the FAD-proximal disulfide. Such “shuttle” disulfides may help accommodate bulky incoming thiol-containing substrates. They also may allow substrate specificity to evolve partially independently of the strict geometric requirements of the active site. Lastly, shuttle disulfides may minimize undesirable changes in active-site polarity accompanying substrate docking.
Figure 17.

Gallery of sulfhydryl oxidase flavoenzyme structures. Protein subunits are purple and green, FAD is orange, disulfide bond sulfurs are yellow. Ero1, Saccharomyces cerevisiae Ero1; QSOX1, Rattus norvegicus Quiescin Sulfhydryl Oxidase 1; QSOX (T. brucei), Trypanosoma brucei Quiescin Sulfhydryl Oxidase; ALR, Homo sapiens Augmenter of Liver Regeneration; AcMNPV, Autographa californica multicapsid nucleopolyhedrovirus; ASFV, African swine fever virus; AfGliT, Aspergillus fumigatus Gliotoxin Sulfhydryl Oxidase; APMV, Acanthamoeba polyphaga mimivirus. With the exception of APMV R596, dimer structures are viewed down the two-fold axis. PDB codes are Ero1, 1RP4; QSOX1, 4P2L; QSOX (T. brucei), 3QCP; ALR, 1OQC; AcMNV Ac92, 3QZY; ASFV pB119L, 3GWL; AfGliT, 4NTC; APMV R596, 3GWN.
Figure 18.
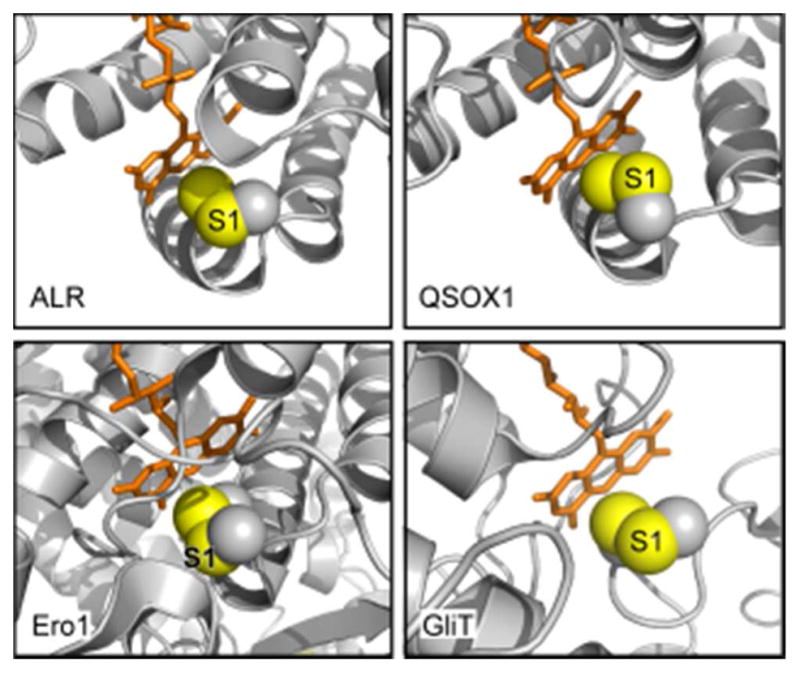
Active-site regions of flavin-dependent sulfhydryl oxidases. Flavin-proximal disulfides are shown as spheres with sulfur atoms in yellow and Cβ atoms in gray. The sulfur in each enzyme targeted for nucleophilic attack by an incoming thiolate during catalysis (the interchange sulfur) is labeled S1. The relative solvent exposure of the S1 sulfurs and accessibility of the disulfide to in-line attack is evident in ALR, QSOX1, and GliT. Only in Ero1 is the FAD-proximal disulfide buried and the S1 cysteine apparently inaccessible. PDB files are ALR, 1OQC; QSOX1, 3LLI; Ero1, 1RP4; GliT, 4NTC.
6.4.2. Erv/ALR Enzymes
The designation Erv/ALR arises from co-identification of this enzyme family in yeast and in mammals. A yeast enzyme was named Essential for Respiration and Viability 1 (Erv1) due to the impact of mutations on mitochondrial function,224,225 and a mammalian homolog was called Augmenter of Liver Regeneration for its activity in promoting hepatocyte proliferation during development or upon partial hepatectomy.226 These proteins were found to be sulfhydryl oxidase flavoenzymes.227 Fungi, animals, and plants have an Erv/ALR family enzyme localized to the mitochondrial IMS, where it contributes to disulfide bonding of IMS proteins with the aid of Mia40 (section 6.1.4). S. cerevisiae has an additional Erv/ALR paralog localized to the endoplasmic reticulum.228–229
Erv/ALR enzymes conform to the principles common to flavin-based sulfhydryl oxidases by juxtaposing a CXXC motif with the flavin isoalloxazine (Figure 18, ALR). They are compact dimers consisting of little more than the few helices encasing the FAD (Figure 17, ALR). The two FAD-binding subunits are packed with C2 symmetry, and the active sites are exposed on opposite ends of the assembly. Electron delivery from substrate to the FAD-proximal cysteines is mediated by a shuttle disulfide tethered to the amino- or carboxy-terminus of the enzyme and interacting with the opposite subunit in the dimer (Figure 19).230–232 On the other end of the internal electron-transport pathway, many Erv/ALR enzymes use oxygen as a preferred electron acceptor, but others reduce cytochrome c more rapidly than they reduce oxygen.233 A preference for cytochrome c may arise when a two-electron reduction of one of the FAD molecules bound in the Erv/ALR enzyme dimer is followed by comproportionation with the second FAD, leading to two one-electron reduced cofactors. Each active site can then perform a one-electron reduction of cytochrome c.231,233–237 In this reaction, the Erv/ALR enzyme is technically not functioning as a sulfhydryl oxidase, but because of the close evolutionary and structural relationship between Erv/ALR enzymes that utilize oxygen efficiently and those that do not, they are typically classified and discussed together.
Figure 19.
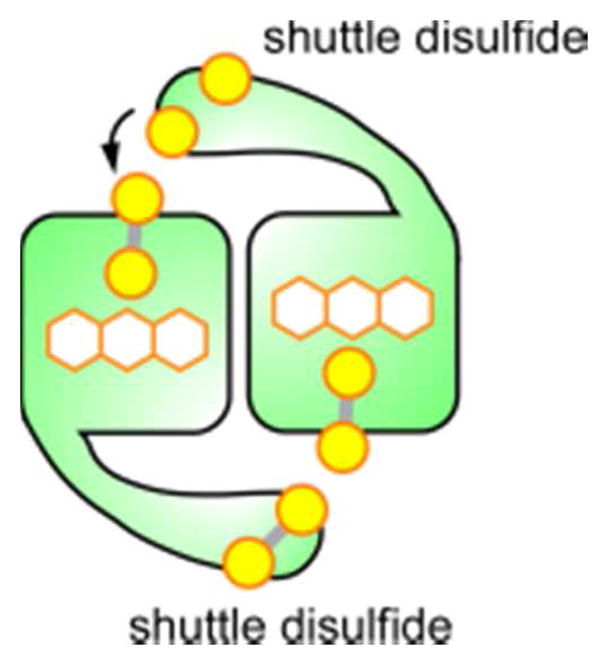
Schematic of an Erv/ALR dimeric enzyme with shuttle disulfides delivering electrons, derived from substrate oxidation, to the opposite subunit in the dimer.
In addition to the Erv/ALR orthologs localized to mitochondria or the ER, enzymes from this family are encoded by viruses. These enzymes appear to be expressed in the cytosol of virus-infected cells or in viral factories,238 which are organelle-like loci established within the cell cytosol to coordinate massive levels of DNA replication and virion self-assembly.239 Large, double-stranded DNA (dsDNA) viruses encode Erv/ALR family sulfhydryl oxidases as part of their core gene set, but the extent to which the oxidases have diversified in these viruses is extraordinary. Specifically, the tertiary and quaternary structural contexts in which the Erv/ALR module appears is highly variable.240 The canonical dimerization mode seen in cellular Erv/ALR enzymes is found in the mimivirus sulfhydryl oxidase R596, but the two subunits associate in completely different manners in the African swine fever virus and baculovirus enyzmes (Figure 20). The viral Erv/ALR enzymes thus provide a counterexample to the generalization that quaternary structures usually change by accretion of symmetries rather than by abandoning evolved interfaces and acquiring new packing modes.241 Furthermore, virus Erv/ALR sulfhydryl oxidase modules can exist either as stand-alone domains or embedded within divergent sequence contexts. It remains to be determined whether the cysteines in the regions outside the Erv/ALR domain serve as electron shuttles to the FAD-proximal active-site cysteines, or whether other viral proteins partner with them in an extended electron-transfer pathway. The Erv/ALR sulfhydryl oxidase from poxviruses functions together with a small partner protein,242 but the relationship of these two proteins in terms of electron transfer is not clear. Structural information is not yet available on the poxvirus disulfide formation enzymes, but sequence analysis suggests that they will demonstrate even further structural diversity than that already observed among the viral disulfide catalysts.243
Figure 20.

Incorporation of the Erv/ALR module into viral sulfhydryl oxidase dimers. Different dimerization modes are used in each case, and the module is embedded in different tertiary structural contexts. ASFV, African swine fever virus; AcMNPV, Autographa californica multicapsid nucleopolyhedrovirus; APMV, Acanthamoeba polyphaga mimivirus. PDB codes are ASFW pB119L, 3GWL; AcMNPV Ac92, 3QZY; APMV R596, 3GWN. For comparison, the structure of the mitochondrial enzyme ALR is also shown.
6.4.3. QSOX
The quiescin sulfhydryl oxidase, or QSOX, enzyme family is a cousin of the Erv/ALR enzymes. QSOX presumably arose from the canonical Erv/ALR dimer by gene duplication, fusion, and degradation of one of the copies of the fold. As a result of such a history, QSOX contains a single-chain pseudo-dimer with a catalytically active Erv-like sulfhydryl oxidase domain, supported by packing against a vestigial Erv domain lacking a catalytic center and flavin cofactor.244 This module was further fused to a segment resembling either the first (in plants and protists) or the first two (in animals) domains of PDI. QSOX has two redox-active CXXC motifs, one in the PDI-like module and the other adjacent to the FAD in the ERV/ALR module.245,246
The QSOX PDI-like module assumes the function that PDI plays in multi-protein disulfide relays in the ER, such that QSOX inserts disulfide bonds directly into reduced proteins without the mediation of PDI itself.247 This arrangement resembles the fusion of a trx domain to the quinone-binding domain in VKOR enzymes, except that the interaction between the two redox-active CXXC motifs of QSOX is direct (Figure 21), rather than bridged by a shuttle disulfide (electron-transfer loop) as in VKOR (Figure 15). Although QSOX contains a third, conserved CXXC motif near its carboxy terminus, this motif does not appear to mediate internal electron-transfer events in the main catalytic cycle,246,248 and the reason for its conservation is unknown. Mechanistic and structural studies support a minimal model for QSOX catalysis as depicted in Figure 22.198,248–252
Figure 21.
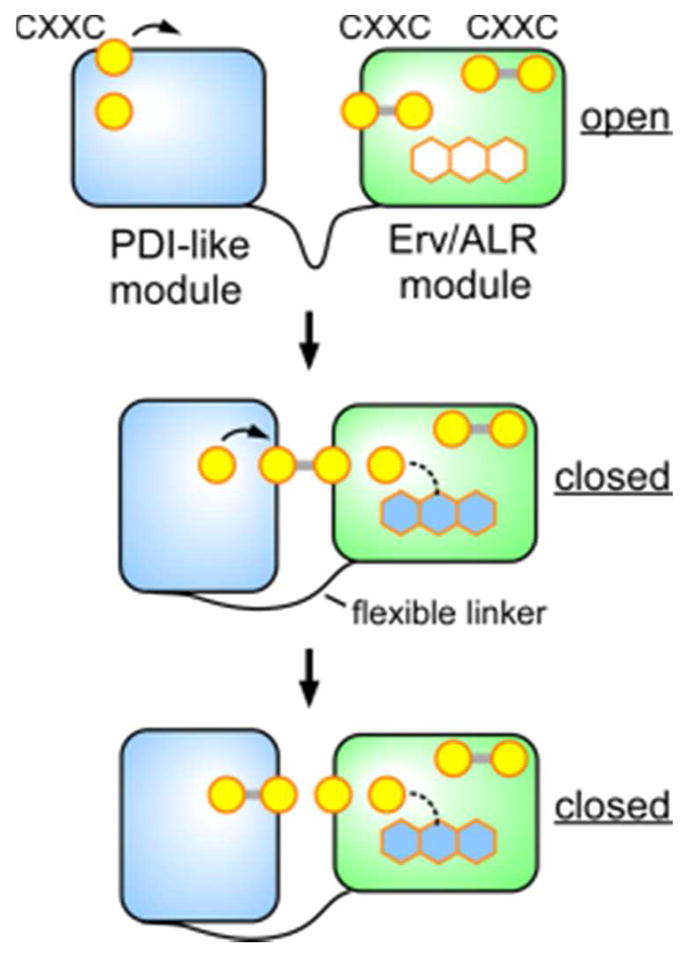
Schematic representation of conformational flexibility and internal electron transfer events in QSOX. The FAD cofactor is represented by hexagons, with blue representing a charge-transfer state. The rightmost (i.e., most carboxy-terminal) CXXC disulfide does not appear to participate in electron transfer from substrate to the FAD.
Figure 22.

QSOX catalytic cycle. This model depicts turnover involving two-electron reduced forms of the enzyme for simplicity. Additional pathways involving four-electron reduced enzyme forms may contribute at high concentration of reducing substrates and/or low oxygen tensions. Curved lines represent motion. Step 1 is reduction of QSOX by substrate protein. Step 2 is the rapid formation of an interdomain disulfide. Step 3 is a rate limiting step involving reduction of the flavin cofactor. Step 4 regenerates the oxidized enzyme and occurs from either the closed or the open (shown here) state.
Since the QSOX PDI-like module accepts electrons from substrate and then transfers them directly to the ERV/ALR active site (Figure 21), large-scale conformational changes appear to be required. A structure of trypanosome QSOX was determined with the redox-active sites far apart from one another and the PDI-domain CXXC exposed at the protein surface, appropriate for substrate interaction.248 In addition, QSOX variants containing an inter-module disulfide were readily prepared, showing how a flexible linker between the modules enables a structural transition to juxtapose and bury the redox-active sites for interdomain electron transfer.248 Two observations indicated that the closed conformational state of QSOX is not an artifact of cysteine mutagenesis. First, the wild-type rat QSOX1 enzyme containing all native cysteines was crystallized in a closed conformation in which the four redoxactive cysteine residues were aligned for in-line attack (Figure 23).252 Second, a single-molecule fluorescence resonance energy transfer (FRET) experiment on trypanosome QSOX showed that a fraction (about 5% under the conditions of the experiment) of the enzyme population assumes a closed conformation in the absence of substrate.250
Figure 23.
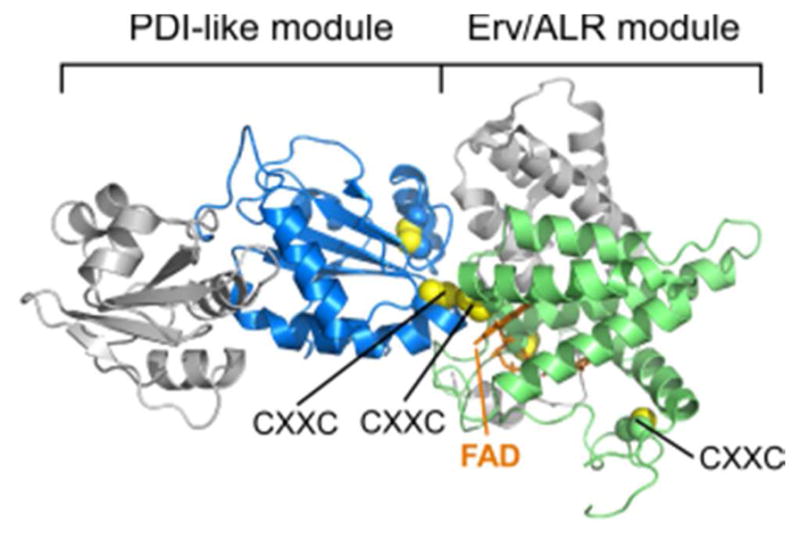
Structure of rat QSOX with juxtaposed redox-active disulfides. The protein is oriented to correspond roughly to Figure 20. The flexible linker is behind the domains. Disulfides are shown with yellow sulfur atoms. The three CXXC motifs and the FAD cofactor are labeled. Unpaired cysteines are not shown. Disulfides not in CXXC motifs are displayed but not labeled. The rightmost CXXC motif is distant from the redox-active centers. PDB code is 4P2L.
Notably, the amount of closed conformer increased substantially when substrate was added and FRET was monitored under steady-state turnover conditions. A population of charge-transfer intermediate was also seen in stopped-flow experiments monitoring FAD absorbance upon substrate addition.249 Mechanistic modeling of the single-molecule FRET results suggested that the charge-transfer species accumulating under steady-state turnover conditions does not contain the mixed disulfide. Instead, the charge-transfer species has a reduced pair of FAD-proximal cysteines, a disulfide in the PDI domain active site, and is in the closed conformation.250
An appraisal of substrate scope in vitro showed that QSOX is a facile oxidant of thiols located in areas of conformational flexibility in proteins irrespective of their amino acid sequence, isoelectric point, or overall protein size.130,247,253,254 In contrast, well-folded proteins with pairs of surface thiols are poor to negligible substrates of the enzyme.253,254 The initial reduction of QSOX (step 1; Figure 22) by a model substrate, reduced RNase, is second-order with no evidence of kinetic saturation.198,254 In addition, no obvious binding site for thiol substrates is evident from the crystal structures of QSOX family members.248,252 Together, these experiments suggest an enzyme whose targets are likely governed by substrate availability and conformational flexibility. Interestingly, QSOX has evolved to serve in physiological environments different from those in which homologs of its component domains operate. PDI family proteins function in the ER, and Erv/ALR enzymes are known for their important role in the mitochondrial intermembrane space. QSOX, on the other hand, is localized to the Golgi apparatus and secreted from cells.255,256 Physiological substrates in these contexts have yet to be discovered, but unlike proteins entering the ER or mitochondria, proteins in the late secretory pathway and extracellular environment are expected to be largely folded. Therefore, any required flexibility in natural QSOX substrates is likely to comprise only part of the protein. Alternatively, the preference for flexible model substrates may not reflect the capabilities of QSOX on its natural substrates. In any case, based on its localization, QSOX may be involved in protein assembly or re-organization rather than protein folding per se.
6.4.4. Ero1
Ero1 is a sulfhydryl oxidase found in the ER of animals, plants, fungi, and protists.257–259 Ero1 enzymes consist of a single, large, helical domain decorated with disulfide-bonded loops.77,260 Ero1 shows some underlying similarities with the Erv/ALR family, despite differences in size and quaternary structure (Figure 17). For example, the FAD cofactor is encased in a helical fold in both families and assumes a similar U-shaped structure. Like many Erv/ALR enzymes, Ero1 has a shuttle disulfide that delivers electrons to the FAD-proximal disulfide. However, some striking differences are also observed between Ero1 and Erv/ALR modules. One is that the FAD-proximal disulfide of Ero1 enzymes, as seen in crystal structures, is not exposed for in-line attack by either the shuttle motif or an exogenous cysteine (Figure 18). This situation, seen in yeast and mammalian Ero1 structures, 77,260 differs from that of PDI as discussed above (section 6.1.1), as well as from Erv/ALR enzymes. In Ero1, the FAD is buried more deeply within the fold than in Erv/ALR, and surface loops and pieces of secondary structure obscure access to the FAD-proximal disulfide. Though structures of yeast Ero1 captured with different conformations of the loop containing the shuttle disulfide suggest mobility in this region (Figure 24),77 neither of the observed conformations achieves the alignment of the relevant cysteine residues for an SN2 reaction to generate a mixed disulfide as seen for QSOX.252 More extensive rearrangement of the Ero1 structure appears to be necessary to allow formation of an electron relay between shuttle cysteines and the FAD-proximal disulfide, but how exactly this rearrangement occurs and the nature of the resulting structure remain to be determined.
Figure 24.

Structure of yeast Ero1 displaying different conformations of the loop containing the shuttle disulfide. Only the flexible loop containing the shuttle disulfide is shown for the “out” conformation (magenta). The FAD is orange. The FAD-proximal cysteine is obscured by the S1 sulfur in this view, such that the thiolate nucleophile would be expected to approach S1 from the direction of the reader. PDB codes are 2RP4 and 1RQ1.
The shielding of the active-site disulfide in Ero1 suggests the potential for regulation of Ero1 by controlled conformational changes. Indeed, reduction of regulatory disulfides in Ero1 and the presumed conformational changes that ensue have been shown to increase the catalytic activity of the enzyme.73,260–264 There is some distance between regulatory disulfides and the catalytic center, so changes in structure or flexibility upon reduction may propagate toward the active site. Reduced PDI, and in particular over-reduced PDI bearing a reduced CX6C motif near the main, CXXC disulfide,265 are potent activators of yeast Ero1.266 Ero1 appears to mediate is own de-activation by re-oxidizing the regulatory disulfides.261,266 No other sulfhydryl oxidase has been found to have such a complex and highly tuned regulatory mechanism.
6.4.5. Thioredoxin Reductase-like Enzymes
A number of secreted fungal flavin-linked sulfhydryl oxidases267–270 are efficient catalysts of the oxidation of glutathione:267,268
| (9) |
Cysteine, 2-mercaptoethanol, and dithiothreitol (DTT) are significantly poorer substrates of these enzymes.267–269 In the cases examined, these fungal sulfhydryl oxidases have weak to negligible activity towards unfolded RNase thiols.267,269,271 The physiological roles of these abundant secreted enzymes are currently unknown.
Sequence analyses revealed that these microbial glutathione oxidases are members of the pyridine nucleotide disulfide oxidoreductase superfamily, which includes glutathione and thioredoxin reductases.268,272–274 Thus the microbial sulfhydryl oxidases have appropriated the reductase framework, retaining the flavin and proximal disulfide, while eliminating a functional pyridine nucleotide binding site to allow them to use molecular oxygen as the terminal electron acceptor for substrate oxidation. For illustration, the flow of reducing equivalents in glutathione reductase and oxidase are compared here (equation 10a and 10b, respectively):
| (10a) |
| (10b) |
Phylogenetic analyses identified additional clades of specialized bacterial and fungal flavin-linked sulfhydryl oxidases that generate disulfide bonds in a number of virulence factors, antibiotics, and anticancer compounds (Figure 25).275–277 For example, the enzyme GliT catalyzes the last step in the biosynthesis of gliotoxin in the opportunistic human pathogen Aspergillus fumigatus by generating a disulfide bridge that is essential for toxicity.276–278 Holomycin is one of a series of dithiolopyrrolone broad-spectrum antibiotics produced by Actinomycetes and proteobacteria.279 In Streptomyces clavuligerus, the intramolecular disulfide in this antibiotic is generated by the dedicated oxidase, HlmI.275 A third specialized disulfide-generating enzyme, DepH, from Chromobacterium violaceum, introduces the unusual transannular cross-link in FK228 (Romidepsin) (Figure 25). While DepH was originally thought to be an NADP+ dependent oxidoreductase,280 a more recent study showed that the enzyme does not contain a functional pyridine nucleotide binding site.277 Hence the catalytic mechanism of DepH is likely to be comparable to the other members of the thioredoxin reductase-fold sulfhydryl oxidases described above.
Figure 25.
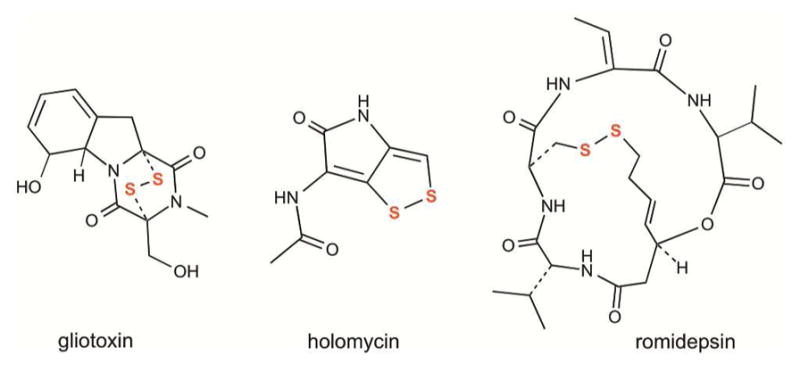
Compounds with disulfide bonds introduced by fungal and bacterial sulfhydryl oxidases related to the thioredoxin reductase enzyme family. Gliotoxin is a epipolythiodioxopiperazine virulence factor in Aspergillus. Holomycin is a broad-spectrum antibiotic from Streptomyces. Romidepsin (FK228) is a depsipeptide anticancer agent from Chromobacterium species used against T-cell lymphomas.
7. OXIDATIVE FOLDING IN BIOLOGICAL SYSTEMS
Now that the activities and mechanisms of the various enzyme families contributing to oxidative folding in biological systems have been presented, the interplay between these enzymes and their integration into their cell biological context can be discussed.
7.1. General Principles
Classical questions that have been considered regarding the biosynthesis of disulfide bonded proteins include 1) when in the process of translation/folding does a given disulfide get introduced, 2) does the vectorial nature of protein biosynthesis affect the order and kinetics of cysteine pairing, 3) what is the enzyme that introduces the disulfide, and 4) what role does non-native cysteine pairing play in oxidative protein folding and how is it resolved? In relation to the first two questions, the order of folding vs. disulfide bond formation has been studied for model proteins.281,282 A particularly creative approach was the use of single-molecule force measurements to distinguish the timing of protein folding from disulfide bond formation.283 These studies suggested that interactions of nascent proteins with PDI may delay disulfide formation until proper folding can occur, which is likely to promote native disulfide pairings. Nevertheless, due to the large number of substrates folding oxidatively and the potential parallelism of the contributing catalysts in vivo, obtaining general answers to questions regarding the order and mechanism of formation of native disulfide bonds is challenging. Apportioning the relative importance of each possible route to disulfide output in locales where multiple pathways are operating simultaneously may not be practical. Instead, it is valuable to outline general principles.
It is likely that the timing of folding vs. disulfide bonding and the requirement for isomerization following oxidation will be determined by the properties of the substrate protein to a greater extent than by the capabilities of the enzymes involved. In particular, if a protein folds sufficiently to bring correct cysteine pairs into proximity for preferred disulfide bond formation upon encounter with enzyme, or contains few competing cysteines, then pairing will likely be accurate. If, in contrast, the substrate protein arrives as a largely unfolded polymer with many cysteines available to resolve the mixed disulfide with an oxidizing enzyme, then the products will be a heterogeneous mixture of disulfide isomers. As the substrate set differs among the various organelles that promote cysteine oxidation, the balance and importance of oxidants vs. isomerases is also expected to be organelle-specific. Particularly interesting issues that are also organelle-specific are how electron flow into environments that support net protein oxidation is utilized and controlled and how electrons liberated by oxidation of folding proteins are fed into other cell metabolic pathways. In the subsequent sections we will therefore discuss key features of oxidative folding processes grouped by cellular locale.
7.2. Bacterial Periplasm
One conundrum that arose in the context of catalysts of disulfide formation in the bacterial periplasm is how reductive and oxidative pathways can co-exist in the same compartment without “short circuiting.” Disulfide bonds are produced in the E. coli periplasm by the pathway consisting of the soluble protein DsbA, the membrane-embedded DsbB, and quinones as electron acceptors. The periplasm is supplied with reducing equivalents by the soluble protein DsbC and the membrane-embedded DsbD, driven by thioredoxin, thioredoxin reductase, and NADPH in the cytosol. The activities of DsbA and DsbC, which, respectively, generate and isomerize disulfide bonds, would be undermined if DsbA could efficiently oxidize DsbC or DsbD, or if DsbC could reduce DsbA or DsbB. The solution to this potential short circuiting problem is kinetic isolation of the oxidative and reductive pathways. This isolation is accomplished in part by a simple stratagem: DsbB is prevented from oxidizing DsbC by steric seclusion of DsbC active sites through dimer formation.284,285 In fact, with the solution of the DsbB structure, it was realized that DsbC fails to engage DsbB due to a steric clash of one subunit of the DsbC dimer with the membrane were the second subunit properly positioned to transfer electrons to DsbB.193 DsbD, in turn, is prevented from reducing DsbA efficiently, most likely due to more minor steric incompatibility between regions surrounding the redox-active sites of DsbA and DsbD.286 Genetics studies investigating the phenotypes of null mutants of various Dsb pathway members, individually or in combination, led the investigators to argue that this kinetic isolation model is an over-simplification of actual events in the bacterial periplasm.287 Although the kinetic isolation may not be complete, or Dsb components may partially function off their canonical pathways, the elegant principles revealed by the initial studies of the E. coli Dsb system are nevertheless illustrative and likely also to be substantially correct.
Another principle that can be showcased with the Dsb system is how to use electrons derived from cysteine oxidation in a metabolically efficient manner. Whereas electrons accepted by sulfhydryl oxidases can be transferred, via bound cofactors, to molecular oxygen, this direct route appears to waste reducing potential. In contrast, reduction of mobile quinones by DsbB allows electrons to enter the respiratory chain where they can be additionally leveraged toward the generation of an electrochemical gradient and ATP synthesis before ultimately being delivered to oxygen.288
7.3 Mitochondrial Intermembrane Space
With the characterization of the disulfide bond formation pathway in the mitochondrial IMS, conceptual parallels were discovered with the Dsb system in the bacterial periplasm. Electrons received by DsbB are fed into the bacterial electron transport chain through lipid-soluble quinones, whereas electrons received by ALR enzymes in mammalian mitochondria are fed into the mitochondrial electron transport chain through cytochrome c.233–235 In both cases, these pathways may minimize the accumulation of reactive oxygen species within the mitochondrial intermembrane space as well as using electrons productively.233
There are some significant differences, however, between reduction of quinones by DsbB and reduction of cytochrome c by ALR. As opposed to the two-electron transfer from DsbB to quinones, ALR performs successive one-electron transfer reactions to two molecules of cytochrome c per disulfide formed. Interestingly, clues to single electron transfer by ALR were detected in studies of the purified enzyme: the two electrons received by ALR during formation of one substrate disulfide were seen to produce large amounts of neutral semiquinone, suggesting comproportionation between the two FAD molecules bound by the ALR dimer.233 The isoalloxazine moieties are separated by about 17 A edge-to-edge, which is slightly longer than that expected for unbridged tunneling distances in proteins289 but is still expected to lead to electron transfer rates consistent with the relatively low overall turnover number of the enzyme.233 Though a detailed analysis of the features of Erv/ALR enzymes that support formation of the semiquinone and promote reduction of cytochrome c vs. molecular oxygen has not been done, the preference for cytochrome c is apparently not universal for the mitochondrial enzymes, since Arabidopsis thaliana Erv1 does not resemble mammalian ALR in this regard.290
In addition to electron acceptors, other major issues have arisen in the context of the mitochondrial disulfide formation pathway: protein substrate specificity, the role of glutathione, and the relationship between cytosolic and IMS redox potentials. In eukaryotes all proteins residing in the mitochondrial intermembrane space (IMS) are encoded in the nucleus, produced in the cytosol, and enter mitochondria in a reduced form through a translocase of the outer membrane (TOM) complex. Upon entry into the IMS, these proteins encounter Mia40 and may become oxidized.291,292 Initially, Mia40 was thought to recognize substrates based on a 9-residue amphipathic helix containing a cysteine that engages in mixed disulfide formation with the redox active CPC motif in Mia40.293 However, in vitro and in vivo experiments showed that a number of kinetically-competent reducing substrates lack these sequence features (e.g., ATP23, Ccs1, DRE2, Erv1/ALR and SOD1).294–298 Indeed, comprehensive studies using mutated IMS protein sequences,171,299,300 and arrays of ARP23 peptides296 showed that Mia40 has a much broader specificity than originally envisaged. This conclusion is reinforced by the finding that the Mia40/ALR system is an efficient oxidant of three reduced proteins (RNase, lysozyme, and riboflavin binding protein) that lack the amphipathic helix motif mentioned earlier and are naturally oxidatively folded within the ER.301 The catalytic promiscuity of the Mia40/ALR system suggests that proteins entering the IMS en route to their final mitochondrial destinations may be protected from extraneous disulfide bond formation by sequestration via translocases and chaperones.302–305 Alternatively, misincorporated disulfides might be removed by IMS-resident reductases.170,294,306,307
Although the Mia40/ERV1 pathway is widely distributed in eukaryotes, a number of protists, including the human pathogens Toxoplasma gondii, Plasmodium falciparum, and the Trypanosomatids lack a detectable Mia40.237,308,309 There appears to be no compensating ability of the corresponding cognate protist Erv1 enzymes to oxidize reduced client proteins directly. Currently, the precise mode by which disulfides are introduced in the IMS of these organisms, and whether there is a factor that compensates for the missing Mia40, remains unclear.
The comparative lack of specificity in the initial oxidation step of IMS protein substrates, together with the significant complexity of disulfide connectivity in some of the products of the Mia40/ALR pathway, suggest the existence of an enzyme with isomerase activity to address mispairings that occur during oxidative protein folding in the IMS.296,297,299,310 It should be noted that Mia40 itself is an unlikely candidate for this isomerase. Mia40 showed weak to insignificant isomerase activity towards the model substrate RNase and very poor activity towards the IMS resident protein Cox17.142,299 Although reduced glutathione can catalyze the resolution of mispaired disulfides in Cox19,170 the ability of glutathione to correct mispairings in substrates with more complicated disulfide patterns in the IMS remains to be established. In any case, glutathione is certainly available to the IMS, as it readily traverses the mitochondrial outer membrane.306
7.4. Disulfide Bond Formation in Cytoplasmic Assembly of dsDNA Viruses
Among the most intriguing but poorly understood pathways involving sulfhydryl oxidases are those encoded by viruses. The genomes of large, dsDNA viruses, such as poxviruses (e.g., vaccinia virus) and mimivirus, contain hundreds or even thousands of open reading frames. These viruses assemble in viral factories established in the cytosol of infected cells, but they rely on processes considered to be characteristic of specialized, membrane-bounded compartments in eukaryotic cells. As an adaptation to their site of replication, the viruses encode their own versions of enzymes that are compartmentalized in cells. For example, mimivirus encodes a set of enzymes involved in glycan formation,311–313 allowing for the biosynthesis of surface glycoproteins independently of the host secretory pathway. In addition, as noted above (section 6.4.2), large, dsDNA viruses encode sulfhydryl oxidases. These enzymes are presumably necessary because the cytosolic virus assembly process cannot exploit the activities of cellular sulfhydryl oxidases encapsulated in the ER, mitochondria, or Golgi apparatus. Furthermore, the viral sulfhydryl oxidases can evolve independently of their cellular counterparts and thus become well-suited to the specific needs of assembling virions. The substantial architectural diversity of the viral enzymes (Figure 17) supports the notion that they have evolved features unique to each virus and its structural and biochemical requirements for disulfide bond formation.
The vaccinia virus sulfhydryl oxidase (E10R) functions on a pathway together with a glutaredoxin (G4L) and an additional small protein not apparently belonging to any class of known redox-active proteins (A2.5L).238,242,314 E10R and its partners are essential for virion morphogenesis,315,316 are associated with virion membranes,315 and have been detected by mass spectrometry proteomics in purified virions.317–319 The targets of this pathway appear to be viral membrane-embedded proteins involved in viral maturation and membrane fusion during infection.238,320 These proteins are topologically oriented such that their disulfide bonded ectodomains face the cytosol during virion assembly.238 Many glutathionylated proteins were found in a proteomics study of vaccinia virions,321 but the significance of this finding is not yet clear.
A similar pathway as that described for the vaccinia sulfhydryl oxidase may function in African swine fever virus (ASFV), sole member of a large dsDNA virus family distinct from poxviruses.322 In ASFV, the sulfhydryl oxidase was found to associate with another viral protein of unknown structure,322 perhaps paralleling the function of A2.5L in poxviruses. Also analogous to poxviruses, viral membrane proteins are the targets of disulfide bond formation in ASFV.322
Sulfhydryl oxidases have been identified in other viruses,323,324 but they have not been associated with particular pathways or viral targets. Mimiviruses encode two Erv/ALR family sulfhydryl oxidases and a number of proteins with thioredoxin-fold domains, as well as a probable glutaredoxin. A better understanding of the assembly and maturation of the virus, as well as the mechanisms by which it reprograms the cell cytosol, will require elucidating the roles of these enzymes.
In addition to the recognizable fold families corresponding to sulfhydryl oxidases and thioredoxin-like oxidoreductases, additional, novel proteins with roles in disulfide formation may be present in viruses. A protein with unknown structure but containing CXXC motifs may contribute to disulfide bond formation during herpesvirus assembly and maturation.325
7.5. Disulfide Bond Formation in the Endoplasmic Reticulum
The numerous pathways involved in generating disulfide bonds in the ER of higher eukaryotes distinguishes oxidative protein folding from other forms of posttranslational modification. The activity of PDI as a catalyst of oxidative protein folding in the ER has been recognized since the 1960s,326 but recent years have brought increasing appreciation for the complexity and multiplicity of players contributing to the redox environment of the early secretory pathway. Four enzyme classes that contribute disulfides to the ER folding environment have been identified: Ero1, VKOR, peroxiredoxin 4, and the PDI peroxidases gpx7 and gpx8. These enzymes partner with numerous PDI family enzymes, which can undergo thiol/disulfide exchange reactions among themselves in addition to oxidizing and isomerizing client proteins.327,328 Together, the whole system is exposed to varying levels of glutathione, hydrogen peroxide, DHA, and perhaps other small-molecule redox-active factors. In addition, changes in ER luminal calcium concentration may affect the localization, partnerships, and activities of redox-active ER enymes.329
Though specific interactions and electron-transfer events can be studied in vitro and identified in vivo by, for example, trapping and identifying inter-protein mixed disulfides, oxidative protein folding in the ER may deviate from the model of a strict assembly line. A distinct perspective can be obtained by considering the flux of reducing equivalents within the ER (Figure 26). In terms of inputs, electrons flow into the ER principally with cysteines on nascent polypeptide chains and with reduced glutathione transported from the cytosol.330 Reduced protein clients are then co- or post-translationally oxidized, with the majority of the reducing equivalents passing through PDI-family proteins.331 PDIs in turn reduce enzymes that catalyze the transfer of electrons from thiol groups to molecular oxygen or hydrogen peroxide, producing, respectively, either hydrogen peroxide or water. The rate of flux of electrons through this system is one crucial parameter that can be controlled, presumably by modulating the availability and activities of enzymes that oxidize PDIs. The levels and locales of hydrogen peroxide produced may be another important parameter. These levels may be controlled by the balance of, and possibly interactions between, Ero1 and PDI peroxidases,176,182 or by as yet undetermined sources.332
Figure 26.
Schematic flow of reducing equivalents in the ER during oxidative protein folding. Electron input: reducing equivalents arrive from the cytosol as cysteine residues in translocating nascent chains and imported glutathione. Output: electrons ultimately reduce molecular oxygen. The major reductive flux in the ER pipeline is believed to involve multiple PDIs in redox communication with one another and with the glutathione redox system, and capable of transferring electrons to sulfhydryl oxidases and peroxidases, which in turn reduce terminal electron acceptors.
Initial insight into how the flux of reducing equivalents in the ER is controlled came from the finding that Ero1 has a mechanism to prevent over-oxidation of its surroundings,260–263 a phenomenon not observed for other sulfhydryl oxidases. This mechanism is based on the PDI-mediated reduction of regulatory disulfide bonds for Ero1 activation and auto-oxidation for Ero1 inactivation.73,261 More recently, evidence has been put forth that Ero1 participates in complexes that further control the input and output of electrons.264,333 It has been observed that the pool of PDI family proteins in a variety of mammalian cell types is maintained largely in reduced form.142,145,330,334–337 For comparison, oxidative folding in vitro is most efficient under relatively reducing conditions.338 In one case study, the rate of oxidative refolding of riboflavin binding protein, with nine disulfides and >34 million possible isomers, progressively increased as the fraction of oxidized PDI decreased, and the most rapid folding occurred with an amount of oxidized PDI just sufficient to account stoichiometrically for the insertion of the disulfides.247 If in vitro conditions adequately represent those in the ER, it appears that the high intraluminal ratio of PDIred/PDIox is tuned for efficient oxidative folding in vivo. Somewhat paradoxically, this large reduced PDI pool should also be efficient at activating Ero1, and thereby at promoting its own depletion. The observed steady state, with substantial reducing power available to nascent protein chains, may be achieved with aid from the cytosolic thioredoxin system.339 The concentrations of relevant ER enzymes and substrates and the reaction rates between them are also clearly central to this issue, and additional factors not yet discovered or considered may further impact ER redox potential.
Regarding measurements of PDI redox state in vivo, it is important to note that the glutathione redox system is in rapid equilibrium with the isomerases.
| (11) |
In vitro studies following the reduction of both a and a′ domains of human PDI and those of human ERp57 and yeast PDI1p show that this equilibration is half complete in less than half a second using a physiologically realistic concentration of 5 mM GSH.155,340,341 The similarities among the active sites of PDI proteins and the utility of glutathione redox buffers for measurements of isomerase redox potentials155 suggests that facile redox communication with glutathione is likely to be a common feature among these oxidoreductases. Given this kinetic facility, stringent trapping procedures are needed to accurately assess the redox states of PDI within cells.142 As a flip side to this complication, equilibration of PDI proteins with glutathione has the benefit of allowing a comparison of PDIred/PDIox ratios with the redox poise of the ER measured using roGFP probes, which also communicate with luminal glutathione. Independent investigations using such probes gave redox potentials for fibroblasts, HeLa, and pancreatic acinar cells lines of −231, −208 and −236 mV, respectively.333,342,343 Applying the Nerst equation using an average intra-luminal redox potential of −225 mV predicts that the PDIs in Table 1 would be almost completely reduced if equilibrated with the ER glutathione pool. Hence the experimental observations of reduced PDI proteins in the ER and the calculations based on an equilibrium assumption are in substantial agreement. Furthermore, shifts in the balance of reduced and oxidized PDI proteins in perturbed cells are accompanied by parallel shifts in reduced and oxidized glutathione levels.344 It should be noted, however, that not all redox-active proteins in the ER need be equilibrated with the glutathione redox buffer. Protein sublocalization may allow specific pathways for oxidation to occur against the backdrop of the reducing environment. For example, the membrane-bound PDI family member Tmx1 appears to preferentially target substrates also anchored to the ER membrane.345 Reminiscent of the Dsb system in bacteria, various points of steric compatibility and incompatibility are likely to open and close routes by which reducing equivalents flow from nascent protein chains and glutathione, via PDI proteins, to sulfhydryl oxidases and PDI peroxidases.
Table 1.
Redox potentials and expected reduced/oxidized ratios of PDI proteins in the ER.
| Protein | Redox Potential | Red/Ox at −225 mV (assuming equilibrium) |
|---|---|---|
| PDI | −160 to −176 mV | ~ 90/1 |
| ERp57 | −155 to −167 mV | ~ 130/1 |
| ERp72 | −155 mV | ~ 195/1 |
| P5 | −146 mV | ~ 360/1 |
| ERp46 | −161 mV | ~ 120/1 |
| TMX3 | −157 mV | ~ 160/1 |
To balance the emphasis on sulfhydryl oxidases, other potential oxidants should also be considered for their potential effects on ER redox balance. DHA (section 5.2.3) serves as an oxidant of the reduced catalytic centers of PDI110,346, leading to the suggestion that this reaction would support oxidative folding in the ER of higher eukaryotes.347 A study of the kinetics of oxidation of reduced PDI and GSH by DHA yielded second-order rate constants of 12.5 ± 1.9 and 0.39 ± 0.01 M−1s−1 respectively at pH of 7.3.112 In contrast, DHA was observed to oxidize reduced bovine trypsin pancreatic inhibitor (BPTI) and RNase A with maximal second-order rate constants of 163 ± 33 and 104 ± 9 M−1s−1 respectively.112 DHA is thus a more efficient oxidant of unfolded reduced proteins compared to PDI; indeed, its utility was recognized decades ago by Anfinsen and Straub during their seminal work on the refolding of reduced RNase.348,349 Though DHA could have pleiotropic effects within the ER by reacting with unfolded reduced proteins, the extent to which DHA contributes to the net oxidizing flux in the mammalian ER is not currently known.350,351 An ascorbate oxidizing activity has been described on the cytosolic side of rat liver ER membranes, together with a transport system enabling dehydroascorbate to access the lumen, suggesting that a contribution is in principle possible.351,352
7.6. Quality Control at the ER/Golgi Boundary
The error-prone process of oxidation, folding, and assembly of large, complex proteins suggests the need for quality control. Not covered in this review are the multiple branches of the ER stress response that bolster ER capabilities when excessive folding failures are perceived.353 Instead, we will focus on the PDI family protein ERp44, which cycles between the ER and Golgi apparatus retrieving proteins that can be engaged through exposed cysteines and that may therefore lack their full complement of disulfides. ERp44 gives wayward subunits of protein complexes repeated chances to meet their partners and is also responsible for ER localization of some proteins lacking classical retention signals.
In contrast to many other PDI proteins, which have a redox-active di-cysteine motif as an active site, ERp44 has only a single active-site cysteine. This cysteine can be sterically shielded by the carboxy-terminal tail of ERp44, which contains a sequence recognized by the KDEL receptor responsible for retrograde transport of proteins back to the ER. Both the redox-active cysteine and the carboxy-terminal tail become exposed in a pH-dependent manner. ERp44 thereby takes advantage of the pH gradient along the secretory pathway to toggle its activity between 1) a state that forms disulfide bonds with substrate and is retrotranslocated and 2) a state that is released from the KDEL receptor, liberated from its cargo, and shielded from redox reactions.
ERp44 was first identified as a factor associated with human Ero1α354,355 and then shown to modulate the assembly of adiponectin complexes and IgM.356,357 ERp44 retains certain other ER redox-active proteins, such as Ero1 and peroxiredoxin IV, in the ER as well.358 Structural studies have provided information both on the conformational changes that underlie the toggling of ERp44 activity,359,360 as well as on the mechanism of engagement of peroxiredoxin IV.146 Some remaining questions include the mechanism for disulfide formation between ERp44 and its clients undergoing oligomeric assembly. Does ERp44 displace glutathione from protein mixed disulfides? Or is the disulfide between ERp44 and its client formed from two free thiols by the action of a sulfhydryl oxidase or other enzyme?
7.7. Disulfide Bond Formation in the Late Secretory Pathway
As described in the previous sections, the ER is the standard site for introduction of disulfide bonds into proteins in the secretory pathway, and retrieval and quality control mechanisms acting at the ER/Golgi interface reinforce the importance of the ER in this regard. Nevertheless, a unique family of sulfhydryl oxidase is localized beyond this border. The QSOX enzymes are found in the Golgi apparatus or secreted from cells, suggesting either that they function on a class of substrates distinct from the clientele for ER oxidation or, perhaps reflecting a need to reoxidize disulfides originally formed by the canonical system in the ER.
A great deal is known biochemically and structurally about QSOX, but less is known about its physiological function. The enzyme is present in a variety of body fluids102,219,361–364 and to varying degrees in blood serum, depending on the organism,365 its developmental state,366 and its disease state.367 The QSOX enzymes are localized to the Golgi apparatus in many mammalian cell types.256 Specifically in confluent fibroblasts, QSOX expression is upregulated, and the enzyme is secreted from the cells.255 In cell culture, the activity of secreted QSOX is essential for assembly of a normal and functional extracellular matrix,256 though the precise targets of QSOX activity are not yet known.
One question that arises in considering a late-secretory pathway sulfhydryl oxidase is to what extent the enzyme contributes to oxidation of thiols in the ER in transit to the Golgi and extracellular environment.368 One possible mechanism to dampen QSOX activity in the ER would be pH-dependent activity optimized for the lower pH of the Golgi. However, purified QSOX exhibits a pH optimum for activity fully consistent with the ER environment.246,249 A study of the QSOX conformational ensemble during turnover suggests another speculative but simple mechanism for controlling enzyme activity. A fraction (approximately 7%) of oxidized QSOX was observed to be in a conformation that buries the two redox-active sites against one another and secludes them from the surrounding solution.250 This observation indicates that the equilibrium between a “closed” and inactive state and an “open” and active state of QSOX only slightly favors the latter. Any hypothetical interaction made between an ER protein and QSOX as it transits that compartment, as long as the interaction is specific for the closed form, would be sufficient to tip the balance toward catalytic inactivity. Identifying such an interaction is likely to be challenging, as it would be expected to be weak and transient. Direct identification of the conformational state of QSOX in the ER is likely to be even more challenging, as undetectable levels of the enzyme accumulate there.256
Though QSOX is the only catalyst of de novo disulfide bond formation known to be efficiently secreted from mammalian cells, other enzymes typically localized to the ER have been reported to exist and function extracellularly under certain circumstances. For example, upon injury, platelets and endothelial cells release PDI.369,370 This PDI population then activates factors that initiate thrombus formation.371–373 Due to its role in strokes, PDI thus becomes a target for specific inhibition of its activity extracellularly.374 Additionally, the PDI family protein Agr2 has been found in mucus secreted from cells of the gastrointestinal tract and appears to be important for mucus structure and protective function.375,376 In certain cases, extracellular localization of ER proteins has been associated with disease, such as the correlation of ECM-associated ERp57 with renal fibrosis.377 The secretion mechanisms by which ER proteins arrive in the extracellular environment are a topic of ongoing investigation.378
8. CONCLUSIONS AND PERSPECTIVES
While the oxidation of vicinal thiols to form a disulfide bond is the most chemically conservative of post-translational modifications, disulfide bonds can profoundly impact the stability, localization, oligomerization, and function of the recipient protein. Other cross-linking chemistries have been identified in proteins in nature, but disulfide bonding is by far the most widespread. The ready reversibility of the modification also contributes to its versatility. Indeed, facile thiol-disulfide exchange reactions underpin the mechanisms of enzymes that generate and rearrange disulfide bonds in client peptides and proteins. Importantly, disulfide bond formation in various intracellular compartments is integrated into cellular metabolism, since electrons derived from disulfide bonding are either delivered productively into other pathways or generate reactive oxygen species that are deactivated using dedicated cellular resources. Historically, the study of disulfide bond formation was a subfield of the investigation of ER function. With time, however, disulfide bond formation was identified and characterized in numerous organelles and physiological contexts. Though based on common underlying chemical principles, the formation of disulfides occurs through fascinating variations on the central themes and contributes to a diversity of wondrous biological structures and phenomena.
Acknowledgments
The authors acknowledge funding from the European Research Council under the European Union’s Seventh Framework Programme, grant number 310649 (to D.F.) and the National Institutes of Health grant number GM 26643 (to C.T.).
Biographies
Colin Thorpe
Colin Thorpe obtained a Chemistry Degree from the University of Cambridge in 1969. His Ph.D. work, at the University of Kent at Canterbury, investigated how enteric bacteria can stereoselectively reduce impermeant cobalt(III) complexes. These studies led to a postdoctoral position in redox enzymology at the University of Michigan with Charles H. Williams and to a sustained interest in the mechanism and physiological role of flavoproteins. Thorpe joined the Department of Chemistry and Biochemistry at the University of Delaware in 1978 where he is now a Willis F. Harrington professor. A chance observation in 1995 led his laboratory to discover a new family of flavin-linked disulfide-generating enzymes. The Thorpe group currently applies a range of chemical, spectroscopic and mechanistic tools to study disulfide bond formation and isomerization in diverse systems and cellular locales.
Deborah Fass
Deborah Fass studied biochemistry at Harvard University and obtained a Ph.D. degree in Structural Biology from the Massachusetts Institute of Technology in 1997. She became interested in disulfide bond formation through the study of viral envelope proteins and contemplation of the process by which they fold oxidatively in the endoplasmic reticulum. In 1998, Fass joined the department of Structural Biology at the Weizmann Institute of Science, where she now holds the Fred and Andrea Fallek professorial chair. The Fass laboratory has determined the structures of a number of sulfhydryl oxidases and contributed to the study of their mechanisms.
References
- 1.Kadokura H, Katzen F, Beckwith J. Protein Disulfide Bond Formation in Prokaryotes. Annu Rev Biochem. 2003;72:111–135. doi: 10.1146/annurev.biochem.72.121801.161459. [DOI] [PubMed] [Google Scholar]
- 2.Reardon-Robinson ME, Ton-That H. Disulfide-Bond-Forming Pathways in Gram- Positive Bacteria. J Bacteriol. 2015;198:746–754. doi: 10.1128/JB.00769-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toth EA, Worby C, Dixon JE, Goedken ER, Marqusee S, Yeates TO. The Crystal Structure of an Adenylosuccinate Lyase from Pyrobaculum aerophilum Reveals an Intracellular Protein with Three Disulfide Bonds. J Mol Biol. 2000;301:433–450. doi: 10.1006/jmbi.2000.3970. [DOI] [PubMed] [Google Scholar]
- 4.Beeby M, O’Connor BD, Ryttersgaard C, Boutz DR, Perry LJ, Yeates TO. The Genomics of Disulfide Bonding and Protein Stabilization in Thermophiles. PLoS Biol. 2005;3:e309. doi: 10.1371/journal.pbio.0030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jorda J, Yeates TO. Widespread Disulfide Bonding in Proteins from Thermophilic Archaea. Archaea. 2011;2011:409156. doi: 10.1155/2011/409156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao YJ, Zhang HM, Lam CM, Hao Q, Lee HC. Cytosolic CD38 Protein Forms Intact Disulfides and Is Active in Elevating Intracellular Cyclic ADP-Ribose. J Biol Chem. 2011;268:22170–22177. doi: 10.1074/jbc.M111.228379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Locker JK, Griffiths G. An Unconventional Role for Cytoplasmic Disulfide Bonds in Vaccinia Virus Proteins. J Cell Biol. 1999;144:267–279. doi: 10.1083/jcb.144.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bisht H, Brown E, Moss B. Kinetics and Intracellular Location of Intramolecular Disulfide Bond Formation Mediated by the Cytoplasmic Redox System Encoded by Vaccinia Virus. Virol. 2010;398:187–193. doi: 10.1016/j.virol.2009.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su HP, Garman SC, Allison TJ, Fogg C, Moss B, Garboczi DN. The 1.51-Angstrom Structure of the Poxvirus L1 Protein, a Target of Potent Neutralizing Antibodies. Proc Natl Acad Sci USA. 2005;102:4240–4245. doi: 10.1073/pnas.0501103102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seo MS, Kang SW, Kim K, Baines IC, Lee TH, Rhee SG. Identification of a New Type of Mammalian Peroxiredoxin that Forms an Intramolecular Disulfide as a Reaction Intermediate. J Biol Chem. 2000;275:20346–20354. doi: 10.1074/jbc.M001943200. [DOI] [PubMed] [Google Scholar]
- 11.Voth W, Schick M, Gates S, Li S, Vilardi F, Gostimskaya I, Southworth DR, Schwappach B, Jakob U. The Protein Targeting Factor Get3 Functions as ATP-Independent Chaperone Under Oxidative Stress. Mol Cell. 2014;56:116–127. doi: 10.1016/j.molcel.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible Oxidation and Inactivation of the Tumor Suppressor PTEN in Cells Stimulated with Peptide Growth Factors. Proc Natl Acad Sci USA. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulaj G, Olivera BM. Folding of Conotoxins: Formation of the Native Disulfide Bridges During Chemical Synthesis and Biosynthesis of Conus Peptides. Antioxid Redox Signal. 2008;10:141–155. doi: 10.1089/ars.2007.1856. [DOI] [PubMed] [Google Scholar]
- 14.Akondi KB, Muttenthaler M, Dutertre S, Kaas Q, Craik DJ, Lewis RJ, Alewood PF. Discovery, Synthesis, and Structure-Activity Relationships of Conotoxins. Chem Rev. 2014;114:5815–5847. doi: 10.1021/cr400401e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calvete JJ, Moreno-Murciano MP, Theakston RD, Kisiel DG, Marcinkiewicz C. Snake Venom Disintegrins: Novel Dimeric Disintegrins and Structural Diversification by Disulphide Bond Engineering. Biochem J. 2003;372:725–734. doi: 10.1042/BJ20021739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.le Du MH, Marchot P, Bougis P, Fontecilla-Camps JC. 1.9-A Resolution Structure of Fasciculin 1, an Anti-Acetylcholinesterase Toxin from Green Mamba Snake Venom. J Biol Chem. 1992;267:22122–22130. doi: 10.2210/pdb1fas/pdb. [DOI] [PubMed] [Google Scholar]
- 17.Mattar EH, Almehdar HA, Yacoub HA, Uversky VN, Redwan EM. Antimicrobial Potentials and Structural Disorder of Human and Animal Defensins. Cytokine Growth Factor Rev. 2016;28:95–111. doi: 10.1016/j.cytogfr.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Shen M, Zhang N, Wang S, Xu Y, Chen S, Chen F, Yang K, He T, Wang A. Reduction Impairs the Antibacterial Activity but Benefits the LPS Neutralization Activity of Human Enteric Defensin 5. Sci Rep. 2016;6:22875. doi: 10.1038/srep22875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheek S, Krishna SS, Grishin NV. Structural Classification of Small, Disulfide-Rich Protein Domains. J Mol Biol. 2006;359:215–237. doi: 10.1016/j.jmb.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 20.Schroeder CI, Swedberg JE, Craik DJ. Recent Progress Towards Pharmaceutical Applications of Disulfide-Rich Cyclic Peptides. Curr Protein Pept Sci. 2013;14:532–542. doi: 10.2174/13892037113149990069. [DOI] [PubMed] [Google Scholar]
- 21.Cemazar M, Kwon S, Mahatmanto T, Ravipati AS, Craik DJ. Discover and Applications of Disulfide-Rich Cyclic Peptides. Curr Top Med Chem. 2012;12:1534–1545. doi: 10.2174/156802612802652484. [DOI] [PubMed] [Google Scholar]
- 22.Sharma H, Nagaraj R. Human β-Defensin 4 with Non-Native Disulfide Bridges Exhibit Antimicrobial Activity. PLoS One. 2015;10:e0119525. doi: 10.1371/journal.pone.0119525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stetefeld J, Mayer U, Timpl R, Huber T. Crystal Structure of Three Consecutive Laminin-Type Epidermal Growth Factor-Like (LE) Modules of Laminin γ1 Chain Harboring the Nidogen Binding Site. J Mol Biol. 1996;257:644–657. doi: 10.1006/jmbi.1996.0191. [DOI] [PubMed] [Google Scholar]
- 24.Carafoli F, Hussain SA, Hohenester E. Crystal Structures of the Network-Forming Short-Arm Tips of the Laminin β1 and γ1 Chains. PLoS One. 2012;7:e42473. doi: 10.1371/journal.pone.0042473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pulido D, Briggs DC, Hua J, Hohenester E. Crystallographic Analysis of the Laminin β2 Short Arm Reveals How the LF Domain is Inserted into a Regular Array of LE Domains. Matrix Biol. 2017;57–58:204–212. doi: 10.1016/j.matbio.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liou YC, Tocilj A, Davies PL, Jia Z. Mimicry of Ice Structure by Surface Hydroxyls and Water of a β-Helix Antifreeze Protein. Nature. 2000;406:322–324. doi: 10.1038/35018604. [DOI] [PubMed] [Google Scholar]
- 27.Graether SP, Kuiper MJ, Gagne SM, Walker VK, Jia Z, Sykes BD, Davies PL. β-Helix Structure and Ice-Binding Properties of a Hyperactive Antifreeze Protein from an Insect. Nature. 2000;406:325–328. doi: 10.1038/35018610. [DOI] [PubMed] [Google Scholar]
- 28.Rogers MA, Langbein L, Praetzel-Wunder S, Winter H, Schweizer J. Human Hair Keratin- Associated Proteins (KAPs) Int Rev Cytol. 2006;251:209–263. doi: 10.1016/S0074-7696(06)51006-X. [DOI] [PubMed] [Google Scholar]
- 29.Gillespie JM, Marshall RC. A Comparison of the Proteins of Normal and Trichothiodystrophic Human Hair. J Investig Dermatol. 1983;80:195–202. doi: 10.1111/1523-1747.ep12534032. [DOI] [PubMed] [Google Scholar]
- 30.Shimomura Y, Ito M. Human Hair Keratin-Associated Proteins. J Investig Dermatol Symp Proc. 2005;10:230–233. doi: 10.1111/j.1087-0024.2005.10112.x. [DOI] [PubMed] [Google Scholar]
- 31.Kurz EM, Holstein TW, Petri BM, Engel J, David CN. Mini-Collagens in Hydra Nematocytes. J Cell Biol. 1991;115:1159–1169. doi: 10.1083/jcb.115.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozbek S, Pertz O, Schwager M, Lustig A, Holstein T, Engel J. Structure/Function Relationships in the Minicollagen of Hydra Nematocysts. J Biol Chem. 2002;277:49200–49204. doi: 10.1074/jbc.M209401200. [DOI] [PubMed] [Google Scholar]
- 33.Nuchter T, Benoit M, Engel U, Ozbek S, Holstein TW. Nanosecond-Scale Kinetics of Nematocyst Discharge. Curr Biol. 2006;16:R316–R318. doi: 10.1016/j.cub.2006.03.089. [DOI] [PubMed] [Google Scholar]
- 34.Meier S, Jensen PR, Adamczyk P, Bachinger HP, Holstein TW, Engel J, Ozbek S, Grzesiek S. Sequence-Structure and Structure-Function Analysis in Cysteine-Rich Domains Forming the Ultrastable Nematocyst Wall. J Mol Biol. 2007;368:718–728. doi: 10.1016/j.jmb.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 35.Milbradt AG, Boulegue C, Moroder L, Renner C. The Two Cysteine-Rich Head Domains of Minicollagen from Hydra Nematocysts Differ in Their Cystine Framework and Overall Fold Despite an Identical Cysteine Sequence Pattern. J Mol Biol. 2005;354:591–600. doi: 10.1016/j.jmb.2005.09.080. [DOI] [PubMed] [Google Scholar]
- 36.Tursch A, Mercadante D, Tennigkeit J, Grater F, Ozbek S. Minicollagen Cysteine-Rich Domains Encode Distinct Modes of Polymerization to Form Stable Nematocyst Capsules. Sci Rep. 2016;6:25709. doi: 10.1038/srep25709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kodali VK, Gannon SA, Paramasivam S, Raje S, Polenova T, Thorpe C. A Novel Disulfide-Rich Protein Motif from Avian Eggshell Membranes. PloS One. 2011;6:e18187. doi: 10.1371/journal.pone.0018187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cordeiro CM, Hincke MT. Quantitative Proteomics Analysis of Eggshell Membrane Proteins During Chick Embryonic Development. J Proteomics. 2016;130:11–25. doi: 10.1016/j.jprot.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 39.Du J, Hincke MT, Rose-Martel M, Hennequet-Antier C, Brionne A, Cogburn LA, Nys Y, Gautron J. Identifying Specific Proteins Involved in Eggshell Membrane Formation Using Gene Expression Analysis and Bioinformatics. BMC Genomics. 2015;16:792. doi: 10.1186/s12864-015-2013-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duffy FJ, Devocelle M, Croucher DR, Shields DC. Computational Survey of Peptides Derived from Disulfide-Bonded Protein Loops That May Serve as Mediators of Protein-Protein Interactions. BMC Bioinformatics. 2014;15:305. doi: 10.1186/1471-2105-15-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rader AJ, Anderson G, Isin B, Khorana HG, Bahar I, Klein-Seetharaman J. Identification of Core Amino Acids Stabilizing Rhodopsin. Proc Natl Acad Sci USA. 2004;101:7246–7251. doi: 10.1073/pnas.0401429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varghese JN, Colman PM. Three-Dimensional Structure of the Neuraminidase of Influenza Virus A/Tokyo/3/67 at 2.2 A Resolution. J Mol Biol. 1991;221:473–486. doi: 10.1016/0022-2836(91)80068-6. [DOI] [PubMed] [Google Scholar]
- 43.Bork P, Holm L, Sander C. The Immunoglobulin Fold. Structural Classification, Sequence Patterns and Common Core. J Mol Biol. 1994;242:309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- 44.Gross G, Gallopin M, Vandame M, Couprie J, Stura E, Zinn-Justin S, Drevet P. Conformational Exchange is Critical for the Productivity of an Oxidative Folding Intermediate With Buried Free Cysteines. J Mol Biol. 2010;403:299–312. doi: 10.1016/j.jmb.2010.07.048. [DOI] [PubMed] [Google Scholar]
- 45.Hofmeyer T, Schmelz S, Degiacomi MT, Dal Peraro M, Daneschdar M, Scrima A, van den Heuvel J, Heinz DW, Kolmar H. Arranged Sevenfold: Structural Insights into the C-Terminal Oligomerization Domain of Human C4b-Binding Protein. J Mol Biol. 2013;425:1302–1317. doi: 10.1016/j.jmb.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 46.Biewenga J. Structure of IgA: Facts and Gaps in Our Data on Disulfide Bonds. Adv Exp Med Biol. 1995;371A:575–579. doi: 10.1007/978-1-4615-1941-6_121. [DOI] [PubMed] [Google Scholar]
- 47.Davis AC, Shulman MJ. IgM—Molecular Requirements for Its Assembly and Function. Immunol Today. 1989;10:118–122. 127–128. doi: 10.1016/0167-5699(89)90244-2. [DOI] [PubMed] [Google Scholar]
- 48.Perez-Vilar J, Hill RL. The Structure and Assembly of Secreted Mucins. J Biol Chem. 1999;274:31751–31754. doi: 10.1074/jbc.274.45.31751. [DOI] [PubMed] [Google Scholar]
- 49.Johansson ME, Larsson JM, Hansson GC. The Two Mucus Layers of Colon Are Organized by the MUC2 Mucin, Whereas the Outer Layer is a Legislator of Host-Microbial Interactions. Proc Natl Acad Sci USA. 2011;108(suppl 1):4659–4665. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Godl K, Johansson ME, Lidell ME, Morgelin M, Karlsson H, Olson FJ, Gum JR, Jr, Kim YS, Hansson GC. The N Terminus of the MUC2 Mucin Forms Trimers That Are Held Together Within a Trypsin-Resistant Core Fragment. J Biol Chem. 2002;277:47248–47256. doi: 10.1074/jbc.M208483200. [DOI] [PubMed] [Google Scholar]
- 51.Ambort D, Johansson ME, Gustafsson JK, Ermund A, Hansson GC. Perspectives on Mucus Properties and Formation—Lessons from the Biochemical World. Cold Spring Harb Perspct Med. 2012;2:a014159. doi: 10.1101/cshperspect.a014159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dani VS, Ramakrishnan C, Varadarajan R. MODIP Revisited: Re-evaluation and Refinement of an Automated Procedure for Modeling of Disulfide Bonds in Proteins. Protein Eng. 2003;16:187–193. doi: 10.1093/proeng/gzg024. [DOI] [PubMed] [Google Scholar]
- 53.Craig DB, Dombkowski AA. Disulfide by Design 2. 0: a Web-Based Tool for Disulfide Engineering in Proteins. BMC Bioinformatics. 2013;14:346. doi: 10.1186/1471-2105-14-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wijma HJ, Floor RJ, Jekel PA, Baker D, Marrink SJ, Janssen DB. Computationally Designed Libraries for Rapid Enzyme Stabilization. Prot Eng Des Sel. 2014;27:49–58. doi: 10.1093/protein/gzt061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naderi M, Moosavi-Movahedi AA, Hosseinkhani S, Nazari M, Bohlooli M, Hong J, Hadi- Alijanvand H, Sheibani N. Implication of Disulfide Bridge Induced Thermal Reversibility, Structural and Functional Stability for Luciferase. Protein Pept Lett. 2015;22:23–30. doi: 10.2174/0929866521666140827112816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kemble GW, Bodian DL, Rose J, Wilson IA, White JM. Intermonomer Disulfide Bonds Impair the Fusion Activity of Influenza Virus Hemagglutinin. J Virol. 1992;66:4940–4950. doi: 10.1128/jvi.66.8.4940-4950.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohashi T, Erickson HP. Domain Unfolding Plays a Role in Superfibronectin Formation. J Biol Chem. 2005;280:39143–39151. doi: 10.1074/jbc.M509082200. [DOI] [PubMed] [Google Scholar]
- 58.Lu C, Shimaoka M, Ferzly M, Oxvig C, Takagi J, Springer TA. An Isolated, Surface- Expressed I domain of the Integrin αLβ2 Is Sufficient for Strong Adhesive Function When Locked in the Open Conformation With a Disulfide Bond. Proc Natl Acad Sci USA. 2001;98:2387–2392. doi: 10.1073/pnas.041606398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Serebryany E, Woodard JC, Adkar BV, Shabab M, King JA, Shakhnovich EI. An Internal Disulfide Locks a Misfolded Aggregation-Prone Intermediate in Cataract-Linked Mutants of Human γD-Crystallin. J Biol Chem. 2016;291:19172–19183. doi: 10.1074/jbc.M116.735977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buhr F, Jha S, Thommen M, Mittelstaet J, Kutz F, Schwalbe H, Rodnina MV, Komar AA. Synonymous Codons Direct Cotranslational Folding Toward Different Protein Conformations. Mol Cell. 2016;61:341–351. doi: 10.1016/j.molcel.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chandrasekhar K, Ke H, Wang N, Goodwin T, Gierasch LM, Gershenson A, Hebert DN. Cellular Folding Pathway of a Metastable Serpin. Proc Natl Acad Sci USA. 2016;113:6484–6489. doi: 10.1073/pnas.1603386113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jansens A, van Duijn E, Braakman I. Coordinated Nonvectorial Folding in a Newly Synthesized Multidomain Protein. Science. 2002;298:2401–2403. doi: 10.1126/science.1078376. [DOI] [PubMed] [Google Scholar]
- 63.Weissman JS, Kim PS. The pro region of BPTI Facilitates Folding. Cell. 1992;71:841–851. doi: 10.1016/0092-8674(92)90559-u. [DOI] [PubMed] [Google Scholar]
- 64.Beer HD, Wohlfahrt G, Schmid RD, McCarthy JE. The Folding and Activity of the Extracellular Lipase of Rhizopus oryzae Are Modulated by a Prosequece. Biochem J. 1996;319:351–359. doi: 10.1042/bj3190351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chng SS, Xue M, Garner RA, Kadokura H, Boyd D, Beckwith J, Kahne D. Disulfide Rearrangement Triggered by Translocon Assembly Controls Lipopolysaccharide Export. Science. 2012;337:1665–1668. doi: 10.1126/science.1227215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wallin M, Ekstrom M, Garoff H. Isomerizatoin of the Intersubunit Disulphide-Bond in Env Controls Retrovirus Fusion. EMBO J. 2004;23:54–65. doi: 10.1038/sj.emboj.7600012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li K, Zhang S, Kronqvist M, Wallin M, Ekstrom M, Derse D, Garoff H. Intersubunit Disulfide Isomerization Controls Membrane Fusion of Human T-Cell Leukemia virus Env. J Virol. 2008;82:7135–7143. doi: 10.1128/JVI.00448-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Neill S, Robinson A, Deering A, Ryan M, Fitzgerald DJ, Moran N. The Platelet Integrin αIIbβ3 Has an Endogenous Thiol Isomerase Activity. J Biol Chem. 2000;275:36984–36990. doi: 10.1074/jbc.M003279200. [DOI] [PubMed] [Google Scholar]
- 69.Zhu G, Zhang Q, Reddy EC, Carrim N, Chen Y, Xu XR, Xu M, Wang Y, Hou Y, Ma L, Li Y, Rui M, Petruzziello-Pellegrini TN, Lavalle C, Stratton TW, Lei X, Adili R, Chen P, Zhu C, Wilkins JA, Hynes RO, Freedman J, Ni H. The Integrin PSI Domain Has an Endogenous Thiol Isomerase Function and Is a Novel Target for Antiplatelet Therapy. Blood. 2017;129:1840–1854. doi: 10.1182/blood-2016-07-729400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leichert LI, Jakob U. Global Methods to Monitor the Thiol-Disulfide State of Proteins in Vivo. Antioxid Redox Signal. 2006;8:763–772. doi: 10.1089/ars.2006.8.763. [DOI] [PubMed] [Google Scholar]
- 71.Wang S, Park S, Kodali VK, Han J, Yip T, Chen Z, Davidson NO, Kaufman RJ. Identification of Protein Disulfide Isomerase 1 as a Key Isomerase for Disulfide Bond Formation in Apolipoprotein B100. Mol Biol Cell. 2015;26:594–604. doi: 10.1091/mbc.E14-08-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Braakman I, Hoover-Litty H, Wagner KR, Helenius A. Folding of Influenza Hemaggluinin in the Endoplasmic Reticulum. J Cell Biol. 1991;114:401–411. doi: 10.1083/jcb.114.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heldman N, Vonshak O, Sevier CS, Vitu E, Mehlman T, Fass D. Steps in Reductive Activation of the Disulfide-Generating Enzyme Ero1p. Protein Sci. 2010;19:1863–1876. doi: 10.1002/pro.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pitt-Rivers R, Impiombato FS. The Binding of Sodium Dodecyl Sulphate to Various Proteins. Biochem J. 1968;109:825–830. doi: 10.1042/bj1090825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Winther JR, Thorpe C. Quantification of Thiols and Disulfides. Biochim Biophys Acta. 2014;1840:838–846. doi: 10.1016/j.bbagen.2013.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilkinson B, Xiao R, Gilbert HF. A Structural Disulfide of Yeast Protein-Disulfide Isomerase Destabilizes the Active Site Disulfide of the N-Terminal Thioredoxin Domain. J Biol Chem. 2005;280:11483–11487. doi: 10.1074/jbc.M414203200. [DOI] [PubMed] [Google Scholar]
- 77.Gross E, Kastner DB, Kaiser CA, Fass D. Structure of Ero1p, Source of Disulfide Bonds for Oxidative Protein Folding in the Cell. Cell. 2004;117:601–610. doi: 10.1016/s0092-8674(04)00418-0. [DOI] [PubMed] [Google Scholar]
- 78.Poppe L, Hui JO, Liguitti J, Murray JK, Schnier PD. PADLOC: a Powerful Tool to Assign Disulfide Bond Connectivities in Peptides and Proteins by NMR Spectroscopy. Anal Chem. 2012;84:262–266. doi: 10.1021/ac203078x. [DOI] [PubMed] [Google Scholar]
- 79.Chambers JE, Tavender TJ, Oka OB, Warwood S, Knight D, Bulleid NJ. The Reduction Potential of the Active Site Disulfides of Human Protein Disulfide Isomerase Limits Oxidation of the Enzyme by Ero1α. J Biol Chem. 2010;285:29200–29207. doi: 10.1074/jbc.M110.156596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ferre F, Clote P. DiANNA: a Web Server for Disulfide Connectivity Prediction. Nucleic Acids Res. 2005;33:W230–W223. doi: 10.1093/nar/gki412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yaseen A, Li Y. Dinosolve: a Protein Disulfide Bonding Prediction Server Using Context-Based Features to Enhance Prediction Accuracy. BMC Bioinformatics. 2013;14(Suppl 13):S9. doi: 10.1186/1471-2105-14-S13-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dutton RJ, Boyd D, Berkmen M, Beckwith J. Bacterial Species Exhibit Diversity in Their Mechanisms and Capacity for Protein Disulfide Bond Formation. Proc Natl Acad Sci USA. 2008;105:11933–11938. doi: 10.1073/pnas.0804621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wong JW, Ho SY, Hogg PJ. Disulfide Bond Acquisition Through Eukaryotic Protein Evolution. Mol Biol Evol. 2011;28:327–334. doi: 10.1093/molbev/msq194. [DOI] [PubMed] [Google Scholar]
- 84.Raimondi D, Orlando G, Vranken WF. An Evolutionary View on Disulfide Bond Connectivities Prediction Using Phylogenetic Trees and a Simple Cysteine Mutation Model. PLoS One. 2015;10:e0131792. doi: 10.1371/journal.pone.0131792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fava A, Iliceto A, Camera E. Kinetics of the Thiol-Disulfide Exchange. J Am Chem Soc. 1957;79:833–838. [Google Scholar]
- 86.Senatore L, Ciuffari E, Fava A, Levita G. Nucleophilic Substitution at Sulfur - Effect of Nucleophile and Leaving Group Basicity as Probe of Bond Formation and Breaking. J Am Chem Soc. 1973;95:2918–2922. [Google Scholar]
- 87.Bednar RA. Reactivity and pH Dependence of Thiol Conjugation to N-ethylmaleimide: Detection of a Conformational Change in Chalcone Isomerase. Biochem. 1990;29:3684–3690. doi: 10.1021/bi00467a014. [DOI] [PubMed] [Google Scholar]
- 88.Appenzeller-Herzog C, Riemer J, Zito E, Chin KT, Ron D, Spiess M, Ellgaard L. Disulphide Production by Ero1α-PDI Relay is Rapid and Effectively Regulated. EMBO J. 2010;29:3318–3329. doi: 10.1038/emboj.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shaked Z, Szajewski RP, Whitesides GM. Rates of Thiol-Disulfide Interchange Reactions Involving Proteins and Kinetic Measurements of Thiol Pka Values. Biochem. 1980;19:4156–4166. doi: 10.1021/bi00559a004. [DOI] [PubMed] [Google Scholar]
- 90.Bulaj G, Kortemme T, Goldenberg DP. Ionization-Reactivity Relationships for Cysteine Thiols in Polypeptides. Biochem. 1998;37:8965–8972. doi: 10.1021/bi973101r. [DOI] [PubMed] [Google Scholar]
- 91.Bachrach SM, Mulhearn DC. Nucleophilic Substitution at Sulfur: S(N)2 or Addition- Elimination? J Phys Chem. 1996;100:3535–3540. [Google Scholar]
- 92.Bachrach SM, Woody JT, Mulhearn DC. Effect of Ring Strain on the Thiolate-Disulfide Exchange. A Computational Study. J Org Chem. 2002;67:8983–8990. doi: 10.1021/jo026223k. [DOI] [PubMed] [Google Scholar]
- 93.Hayes JM, Bachrach SM. Effect of Micro and Bulk Solvation on the Mechanism of Nucleophilic Substitution at Sulfur in Disulfides. J Phys Chem A. 2003;107:7952–7961. [Google Scholar]
- 94.Fernandes PA, Ramos MJ. Theoretical Insights into the Mechanism for Thiol/Disulfide Exchange. Chem. 2004;10:257–266. doi: 10.1002/chem.200305343. [DOI] [PubMed] [Google Scholar]
- 95.Bach RD, Dmitrenko O, Thorpe C. Mechanism of Thiolate-Disulfide Interchange Reactions in Biochemistry. J Org Chem. 2008;73:12–21. doi: 10.1021/jo702051f. [DOI] [PubMed] [Google Scholar]
- 96.Miller DM, Buettner GR, Aust SD. Transition Metals as Catalysts of “Autoxidation” Reactions. Free Radic Biol Med. 1990;8:95–108. doi: 10.1016/0891-5849(90)90148-c. [DOI] [PubMed] [Google Scholar]
- 97.Cavallini D, Demarco C, Dupre S, Rotilio G. Copper Catalyzed Oxidation of Cysteine to Cystine. Arch Biochem Biophys. 1969;130:354–361. doi: 10.1016/0003-9861(69)90044-7. [DOI] [PubMed] [Google Scholar]
- 98.Hanaki A, Kamide H. The Copper-Catalyzed Autoxidation of Cysteine - the Amount of Hydrogen-Peroxide Produced under Various Conditions and the Stoichiometry of the Reaction. Bull Chem Soc Jpn. 1983;56:2065–2068. [Google Scholar]
- 99.Ehrenberg L, Harmsringdahl M, Fedorcsak I, Granath F. Kinetics of the Copper-Catalyzed and Iron-Catalyzed Oxidation of Cysteine by Dioxygen. Acta Chem Scand. 1989;43:177–187. [Google Scholar]
- 100.Kachur AV, Koch CJ, Biaglow JE. Mechanism of Copper-Catalyzed Autoxidation of Cysteine. Free Radic Res. 1999;31:23–34. doi: 10.1080/10715769900300571. [DOI] [PubMed] [Google Scholar]
- 101.Munday R, Munday CM, Winterbourn CC. Inhibition of Copper-Catalyzed Cysteine Oxidation by Nanomolar Concentrations of Iron Salts. Free Radic Biol Med. 2004;36:757–764. doi: 10.1016/j.freeradbiomed.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 102.Jaje J, Wolcott HN, Fadugba O, Cripps D, Yang AJ, Mather IH, Thorpe C. A Flavin- Dependent Sulfhydryl Oxidase in Bovine Milk. Biochem. 2007;46:13031–13040. doi: 10.1021/bi7016975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brohawn SG, Rudik I, Thorpe C. Avian Sulfhydryl Oxidase is Not a Metalloenzyme: Adventitious Binding of Divalent Metal Ions to the Enzyme. Biochem. 2003;42:11074–11082. doi: 10.1021/bi0301385. [DOI] [PubMed] [Google Scholar]
- 104.Kodali VK, Thorpe C. Oxidative Protein Folding and the Quiescin-Sulfhydryl Oxidase Family of Flavoproteins. Antioxid Redox Signal. 2010;13:1217–1230. doi: 10.1089/ars.2010.3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brewer TF, Garcia FJ, Onak CS, Carroll KS, Chang CJ. Chemical Approaches to Discovery and Study of Sources and Targets of Hydrogen Peroxide Redox Signaling Through NADPH Oxidase Proteins. Annu Rev Biochem. 2015;84:765–790. doi: 10.1146/annurev-biochem-060614-034018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barton JP, Packer JE, Sims RJ. Kinetics of Reaction of Hydrogen-Peroxide with Cysteine and Cysteamine. J Chem Soc Perkin Trans 2. 1973:1547–1549. [Google Scholar]
- 107.Winterbourn CC, Metodiewa D. Reactivity of Biologically Important Thiol Compounds with Superoxide and Hydrogen Peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 108.Hopkins FG, Morgan EJ. Some Relations Between Ascorbic Acid and Glutathione. Biochem J. 1936;30:1446–1462. doi: 10.1042/bj0301446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Winkler BS, Orselli SM, Rex TS. The Redox Couple between Glutathione and Ascorbic-Acid - a Chemical and Physiological Perspective. Free Radic Biol Med. 1994;17:333–349. doi: 10.1016/0891-5849(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 110.Washburn MP, Wells WW. The Catalytic Mechanism of the Glutathione-Dependent Dehydroascorbate Reductase Activity of Thioltransferase (Glutaredoxin) Biochem. 1999;38:268–274. doi: 10.1021/bi980480v. [DOI] [PubMed] [Google Scholar]
- 111.Moutiez M, Quemeneur E, Sergheraert C, Lucas V, Tartar A, Davioud-Charvet E. Glutathione-Dependent Activities of Trypanosoma cruzi p52 Makes It a New Member of the Thiol:Disulphide Oxidoreductase Family. Biochem J. 1997;322:43–48. doi: 10.1042/bj3220043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saaranen MJ, Karala AR, Lappi AK, Ruddock LW. The Role of Dehydroascorbate in Disulfide Bond Formation. Antioxid Redox Signal. 2010;12:15–25. doi: 10.1089/ars.2009.2674. [DOI] [PubMed] [Google Scholar]
- 113.Wang Y, Hekimi S. Understanding Ubiquinone. Trends Cell Biol. 2016;26:367–378. doi: 10.1016/j.tcb.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 114.Liebeke M, Pother DC, van Duy N, Albrecht D, Becher D, Hochgrafe F, Lalk M, Hecker M, Antelmann H. Depletion of Thiol-Containing Proteins in Response to Quinones in Bacillus subtilis. Mol Microbiol. 2008;69:1513–1529. doi: 10.1111/j.1365-2958.2008.06382.x. [DOI] [PubMed] [Google Scholar]
- 115.Kumagai Y, Koide S, Taguchi K, Endo A, Nakai Y, Yoshikawa T, Shimojo N. Oxidation of Proximal Protein Sulfhydryls by Phenanthraquinone, a Component of Diesel Exhaust. Chem Res Toxicol. 2002;15:483–498. doi: 10.1021/tx0100993. [DOI] [PubMed] [Google Scholar]
- 116.O’Brien PJ. Molecular Mechanisms of Quinone Cytotoxicity. Chem Biol Interact. 1991;80:1–41. doi: 10.1016/0009-2797(91)90029-7. [DOI] [PubMed] [Google Scholar]
- 117.Brunmark A, Cadenas E. Redox and Addition Chemistry of Quinoid Compounds and Its Biological Implications. Free Radic Biol Med. 1989;7:435–77. doi: 10.1016/0891-5849(89)90126-3. [DOI] [PubMed] [Google Scholar]
- 118.Finley KT. The Addition and Substitution Chemistry of Quinones. In: Patai S, editor. The Chemistry of Quinoid Compounds. London: Wiley; 1974. pp. 877–1144. [Google Scholar]
- 119.Williams CH., Jr . Lipoamide Dehydrogenase, Glutathione Reductase, Thioredoxin Reductase, and Mercuric Ion Reductase-A Family of Flavoenzyme Transhydrogenases. In: Muller F, editor. Chemistry and Biochemistry of Flavoenzymes. Chemistry and Biochemistry of Flavoenzymes. CRC Press; 1992. pp. 121–211. [Google Scholar]
- 120.Argyrou A, Blanchard JS. Flavoprotein Disulfide Reductases: Advances in Chemistry and Function. Prog Nuc Acid Res Mol Biol. 2004;78:89–142. doi: 10.1016/S0079-6603(04)78003-4. [DOI] [PubMed] [Google Scholar]
- 121.Miller SM. Flavoprotein Ddisulfide Reductases and Structurally Related Flavoprotein Thiol/Disulfide-Linked Oxidoreductases. In: Hille R, Miller SM, Palfey BA, editors. Handbook of Flavoproteins. Berlin: De Gruyter; 2013. pp. 165–201. [Google Scholar]
- 122.Thorpe C, Williams CH., Jr Spectral Evidence for a Flavin Adduct in a Monoalkylated Derivative of Pig Heart Lipoamide Dehydrogenase. J Biol Chem. 1976;251:7726–7728. [PubMed] [Google Scholar]
- 123.O’Donnell ME, Williams CH., Jr Reconstitution of Escherichia coli Thioredoxin Reductase with 1-Deaza-FAD: Evidence for 1-Deaza-FAD-C4a Adduct Linked to the Ionization of an Active Site Base. J Biol Chem. 1984;259:2243–2251. [PubMed] [Google Scholar]
- 124.Sahlman L, Lambeir AM, Lindskog S. Rapid-Scan Stopped-Flow Studies of the pH-Dependence of the Reaction between Mercuric Reductase and NADPH. Eur J Biochem. 1986;156:479–488. doi: 10.1111/j.1432-1033.1986.tb09606.x. [DOI] [PubMed] [Google Scholar]
- 125.Miller SM, Massey V, Ballou D, Williams CH, Jr, Distefano MD, Moore MJ, Walsh CT. Use of a Site-Directed Triple Mutant to Trap Intermediates: Demonstration That the Flavin C(4a)-Thiol Adduct Reduced Flavin are Kinetically Competent Intermediates in Mercuric Ion Reductase. Biochem. 1990;29:2831–2841. doi: 10.1021/bi00463a028. [DOI] [PubMed] [Google Scholar]
- 126.Dmitrenko O, Thorpe C. A Computational Analysis of the Interaction Between Flavin and Thiol(ate) Groups. Implications for Flavoenzyme Catalysis. J Sulfur Chem. 2008;29:415–424. [Google Scholar]
- 127.Baron R, Riley C, Chenprakhon P, Thotsaporn K, Winter RT, Alfieri A, Forneris F, van Berkel WJ, Chaiyen P, Fraaije MW, Mattevi A, McCammon JA. Multiple Pathways Guide Oxygen Diffusion into Flavoenzyme Active Sites. Proc Natl Acad Sci USA. 2009;106:10603–10608. doi: 10.1073/pnas.0903809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chaiyen P, Fraaije MW, Mattevi A. The Enigmatic Reaction of Flavins With Oxygen. Trends Biochem Sci. 2012;37:373–380. doi: 10.1016/j.tibs.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 129.Ostrowski MC, Kistler WS. Properties of a Flavoprotein Sulfhydryl Oxidase from Rat Seminal Vesicle Secretion. Biochem. 1980;19:2639–2645. doi: 10.1021/bi00553a016. [DOI] [PubMed] [Google Scholar]
- 130.Hoober KL, Joneja B, White HB, III, Thorpe C. A Sulfhydryl Oxidase from Chicken Egg White. J Biol Chem. 1996;271:30510–30516. doi: 10.1074/jbc.271.48.30510. [DOI] [PubMed] [Google Scholar]
- 131.Gross E, Sevier CS, Heldman N, Vitu E, Bentzur M, Kaiser CA, Thorpe C, Fass D. Generating Disulfides Enzymatically: Reaction Products and Electron Acceptors of the Endoplasmic Reticulum Thiol Oxidase Ero1p. Proc Natl Acad Sci USA. 2006;103:299–304. doi: 10.1073/pnas.0506448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Daithankar VN, Wang W, Trujillo JR, Thorpe C. Flavin-Linked Erv-Family Sulfhydryl Oxidases Release Superoxide Anion During Catalytic Turnover. Biochem. 2012;51:265–272. doi: 10.1021/bi201672h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Papp E, Nardai G, Mandl J, Banhegyi G, Csermely P. FAD Oxidizes the ERO1-PDI Electron Transfer Chain: the Role of Membrane Integrity. Biochem Biophys Res Commun. 2005;338:938–945. doi: 10.1016/j.bbrc.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 134.Riemer J, Fischer M, Herrmann JM. Oxidation-driven Protein Import into Mitochondria: Insights and Blind Spots. Biochim Biophys Acta. 2011;1808:981–989. doi: 10.1016/j.bbamem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 135.Appenzeller-Herzog C, Ellgaard L. The Human PDI Family: Versatility Packed Into a Single Fold. Biochim Biophys Acta. 2008;1783:535–548. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 136.Galligan JJ, Petersen DR. The Human Protein Disulfide Isomerase Gene Family. Hum Genomics. 2012;6:6. doi: 10.1186/1479-7364-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lu J, Holmgren A. The Thioredoxin Superfamily in Oxidative Protein Folding. Antioxid Redox Signal. 2014;21:457–470. doi: 10.1089/ars.2014.5849. [DOI] [PubMed] [Google Scholar]
- 138.Kozlov G, Maattanen P, Thomas DY, Gehring K. A Structural Overview of the PDI Family of Proteins. FEBS J. 2010;277:3924–3936. doi: 10.1111/j.1742-4658.2010.07793.x. [DOI] [PubMed] [Google Scholar]
- 139.Lu J, Holmgren A. The Thioredoxin Antioxidant System. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 140.Okumura M, Kadokura H, Inaba K. Structures and Functions of Protein Disulfide Isomerase Family Members Involved in Proteostasis in the Endoplasmic Reticulum. Free Radic Biol Med. 2015;83:314–322. doi: 10.1016/j.freeradbiomed.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 141.Biran S, Gat Y, Fass D. The Eps1p Protein Disulfide Isomerase Conserves Classic Thioredoxin Superfamily Amino Acid Motifs but Not Their Functional Geometries. PLoS One. 2014;9:e113431. doi: 10.1371/journal.pone.0113431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hudson DA, Gannon SA, Thorpe C. Oxidative Protein Folding: from Thiol-Disulfide Exchange Reactions to the Redox Poise of the Endoplasmic Reticulum. Free Radic Biol Med. 2015;80:171–182. doi: 10.1016/j.freeradbiomed.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Heckler EJ, Rancy PC, Kodali VK, Thorpe C. Generating Disulfides With the Quiescin- Sulfhydryl Oxidases. Biochim Biophys Acta. 2008;1783:567–577. doi: 10.1016/j.bbamcr.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dong G, Wearsch PA, Peaper DR, Cresswell P, Reinisch KM. Insights into MHC Class I Peptide Loading from the Structure of the Tapasin-ERp57 Thiol Oxidase Heterodimer. Immunity. 2009;30:21–32. doi: 10.1016/j.immuni.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sato Y, Kojima R, Okumura M, Hagiwara M, Masui S, Maegawa K, Saiki M, Horibe T, Suzuki M, Inaba K. Synergistic Cooperation of PDI Family Members in Peroxiredoxin 4-Driven Oxidative Protein Folding. Sci Rep. 2013;3:2456. doi: 10.1038/srep02456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yang K, Li DF, Liang J, Sitia R, Wang CC, Wang X. Crystal Structure of the ERp44- Peroxiredoxin 4 Complex Reveals the Molecular Mechanisms of Thiol-Mediated Protein Retention. Structure. 2016;24:1755–1765. doi: 10.1016/j.str.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 147.Maeda K, Hagglund P, Finnie C, Svensson B, Henriksen A. Structural Basis for Target Protein Recognition by the Protein Disulfide Reductase Thioredoxin. Structure. 2006;14:1701–1710. doi: 10.1016/j.str.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 148.Hwang J, Suh HW, Jeon YH, Hwang E, Nguyen LT, Yeom J, Lee SG, Lee C, Kim KJ, Kang BS, et al. The Structural Basis for the Negative Regulation of Thioredoxin by Thioredoxin- Interacting Protein. Nat Commun. 2014;5:2958. doi: 10.1038/ncomms3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lundstrom J, Krause G, Holmgren A. A Pro to His Mutation in Active Site of Thioredoxin Increases Its Disulfide-Isomerase Activity 10-Fold. New Refolding Systems for Reduced or Randomly Oxidized Ribonuclease. J Biol Chem. 1992;267:9047–9052. [PubMed] [Google Scholar]
- 150.Kersteen EA, Barrows SR, Raines RT. Catalysis of Protein Disulfide Bond Isomerization in a Homogeneous Substrate. Biochem. 2005;44:12168–12178. doi: 10.1021/bi0507985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Karala AR, Lappi AK, Ruddock LW. Modulation of an Active-Site Cysteine pKa Allows PDI to Act as a Catalyst of Both Disulfide Bond Formation and Isomerization. J Mol Biol. 2010;396:883–892. doi: 10.1016/j.jmb.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 152.Xiao R, Wilkinson B, Solovyov A, Winther JR, Holmgren A, Lundstrom-Ljung J, Gilbert HF. The Contributions of Protein Disulfide Isomerase and its Homologues to Oxidative Protein Folding in the Yeast Endoplasmic Reticulum. J Biol Chem. 2004;279:49780–49786. doi: 10.1074/jbc.M409210200. [DOI] [PubMed] [Google Scholar]
- 153.Klappa P, Ruddock LW, Darby NJ, Freedman RB. The b′ Domain Provides the Principal Peptide-Binding Site of Protein Disulfide Isomerase but All Domains Contribute to Binding of Misfolded Proteins. EMBO J. 1998;17:927–935. doi: 10.1093/emboj/17.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Funkner A, Parthier C, Schutkowski M, Zerweck J, Lilie H, Gyrych N, Fischer G, Stubbs MT, Ferrari DM. Peptide Binding by Catalytic Domains of the Protein Disulfide Isomerase-Related Protein ERp46. J Mol Biol. 2013;425:1340–1362. doi: 10.1016/j.jmb.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 155.Hatahet F, Ruddock LW. Protein Disulfide Isomerase: A Critical Evaluation of Its Function in Disulfide Bond Formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 156.Denisov AY, Maattanen P, Dabrowski C, Kozlov G, Thomas DY, Gehring K. Solution Structure of the bb′ Domains of Human Protein Disulfide Isomerase. FEBS J. 2009;276:1440–1449. doi: 10.1111/j.1742-4658.2009.06884.x. [DOI] [PubMed] [Google Scholar]
- 157.Tian G, Xiang S, Noiva R, Lennarz WJ, Schindelin H. The Crystal Structure of Yeast Protein Disulfide Isomerase Suggests Cooperativity Between Its Active Sites. Cell. 2006;124:61–73. doi: 10.1016/j.cell.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 158.Byrne LJ, Sidhu A, Wallis AK, Ruddock LW, Freedman RB, Howard MJ, Williamson RA. Mapping of the Ligand-Binding Site on the b′ Domain of Human PDI: Interaction With Peptide Ligands and the x-Linker Region. Biochem J. 2009;423:209–217. doi: 10.1042/BJ20090565. [DOI] [PubMed] [Google Scholar]
- 159.Kober FX, Koelmel W, Kuper J, Drechsler J, Mais C, Hermanns HM, Schindelin H. The Crystal Structure of the Protein-Disulfide Isomerase Family Member ERp27 Provides Insights into Its Substrate Binding Capabilities. J Biol Chem. 2013;288:2029–2039. doi: 10.1074/jbc.M112.410522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang C, Yu J, Huo L, Wang L, Feng W, Wang CC. Human Protein-Disulfide Isomerase is a Redox-Regulated Chaperone Activated by Oxidation of Domain a′. J Biol Chem. 2012;287:1139–1149. doi: 10.1074/jbc.M111.303149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yagi-Utsumi M, Satoh T, Kato K. Structural Basis of Redox-Dependent Substrate Binding of Protein Disulfide Isomerase. Sci Rep. 2015;5:13909. doi: 10.1038/srep13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bekendam RH, Bendapudi PK, Lin L, Nag PP, Pu J, Kennedy DR, Feldenzer A, Chiu J, Cook KM, Furie B, et al. A Substrate-Driven Allosteric Switch That Enhances PDI Catalytic Activity. Nat Commun. 2016;7:12579. doi: 10.1038/ncomms12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Woycechowsky KJ, Raines RT. The CXC Motif: a Functional Mimic of Protein Disulfide Isomerase. Biochem. 2003;42:5387–5394. doi: 10.1021/bi026993q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kersteen EA, Raines RT. Catalysis of Protein Folding by Protein Disulfide Isomerase and Small- Molecule Mimics. Antioxid Redox Signal. 2003;5:413–424. doi: 10.1089/152308603768295159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lukesh JC, 3rd, Andersen KA, Wallin KK, Raines RT. Organocatalysis of Oxidative Protein Folding Inspired by Protein Disulfide Isomerase. Org Biomol Chem. 2014;12:8598–8602. doi: 10.1039/c4ob01738b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kawano S, Yamano K, Naoe M, Momose T, Terao K, Nishikawa S, Watanabe N, Endo T. Structural Basis of Yeast Tim40/Mia40 as an Oxidative Translocator in the Mitochondrial Intermembrane Space. Proc Natl Acad Sci USA. 2009;106:14403–14407. doi: 10.1073/pnas.0901793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Banci L, Bertini I, Cefaro C, Ciofi-Baffoni S, Gallo A, Martinelli M, Sideris DP, Katrakili N, Tokatlidis K. MIA40 is an Oxidoreductase that Catalyzes Oxidative Protein Folding in Mitochondria. Nat Struct Mol Biol. 2009;16:198–206. doi: 10.1038/nsmb.1553. [DOI] [PubMed] [Google Scholar]
- 168.Zhang RM, Snyder GH. Dependence of Formation of Small Disulfide Loops in Two-Cysteine Peptides on the Number and Types of Intervening Amino Acids. J Biol Chem. 1989;264:18472–18479. [PubMed] [Google Scholar]
- 169.Banci L, Bertini I, Calderone V, Cefaro C, Ciofi-Baffoni S, Gallo A, Kallergi E, Lionaki E, Pozidis C, Tokatlidis K. Molecular Recognition and Substrate Mimicry Drive the Electron- Transfer Process Between MIA40 and ALR. Proc Natl Acad Sci USA. 2011;108:4811–4816. doi: 10.1073/pnas.1014542108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bien M, Longen S, Wagener N, Chwalla I, Herrmann JM, Riemer J. Mitochondrial Disulfide Bond Formation is Driven by Intersubunit Electron Transfer in Erv1 and Proofread by Glutathione. Mol Cell. 2010;37:516–528. doi: 10.1016/j.molcel.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 171.Koch JR, Schmid FX. Mia40 is Optimized for Function in Mitochondrial Oxidative Protein Folding and Import. ACS Chem Biol. 2014;9:2049–2057. doi: 10.1021/cb500408n. [DOI] [PubMed] [Google Scholar]
- 172.Perkins A, Nelson KJ, Parsonage D, Poole LB, Karplus PA. Peroxiredoxins: Guardians Against Oxidative Stress and Modulators of Peroxide Signaling. Trends Biochem Sci. 2015;40:435–445. doi: 10.1016/j.tibs.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tavender TJ, Sheppard AM, Bulleid NJ. Peroxiredoxin IV Is an Endoplasmic Reticulum- Localized Enzyme Forming Oligomeric Complexes in Human Cells. Biochem J. 2008;411:191–199. doi: 10.1042/BJ20071428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang X, Wang L, Wang X, Sun F, Wang CC. Structural Insights into the Peroxidase Activity and Inactivation of Human Peroxiredoxin 4. Biochem J. 2012;441:113–118. doi: 10.1042/BJ20110380. [DOI] [PubMed] [Google Scholar]
- 175.Cao Z, Tavender TJ, Roszak AW, Cogdell RJ, Bulleid NJ. Crystal Structure of Reduced and of Oxidized Peroxiredoxin IV Enzyme Reveals a Stable Oxidized Decamer and a Non-Disulfide- Bonded Intermediate in the Catalytic Cycle. J Biol Chem. 2011;286:42257–42266. doi: 10.1074/jbc.M111.298810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tavender TJ, Bulleid NJ. Peroxiredoxin IV Protects Cells from Oxidative Stress by Removing H2O2 Produced During Disulphide Formation. J Cell Sci. 2010;123:2672–2679. doi: 10.1242/jcs.067843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tavender TJ, Springate JJ, Bulleid NJ. Recycling Peroxiredoxin IV Provides a Novel Pathway for Disulphide Formation in the Endoplasmic Reticulum. EMBO J. 2010;29:4185–4197. doi: 10.1038/emboj.2010.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zito E, Melo EP, Yang Y, Wahlander A, Neubert TA, Ron D. Oxidative Protein Folding by an Endoplasmic Reticulum-Localized Peroxiredoxin. Mol Cell. 2010;40:787–797. doi: 10.1016/j.molcel.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Martin JL. Thioredoxin—a Fold for All Reasons. Structure. 1995;3:245–250. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 180.Nguyen VD, Saaranen MJ, Karala AR, Lappi AK, Wang L, Raykhel IB, Alanen HI, Salo KE, Wang CC, Ruddock LW. Two Endoplasmic Reticulum PDI Peroxidases Increase the Efficiency of the Use of Peroxide During Disulfide Bond Formation. J Mol Biol. 2011;406:503–515. doi: 10.1016/j.jmb.2010.12.039. [DOI] [PubMed] [Google Scholar]
- 181.Wang L, Zhang L, Niu Y, Sitia R, Wang CC. Glutathione Peroxidase 7 Utilizes Hydrogen Peroxide Generated by Ero1α to Promote Oxidative Protein Folding. Antioxid Redox Signal. 2014;20:545–556. doi: 10.1089/ars.2013.5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ramming T, Hansen HG, Nagata K, Ellgaard L, Appenzeller-Herzog C. GPx8 Peroxidase Prevents Leakage of H2O2 from the Endoplasmic Reticulu. Free Radic Biol Med. 2014;70:106–116. doi: 10.1016/j.freeradbiomed.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 183.Maiorino M, Bosello-Travain V, Cozza G, Miotto G, Roveri A, Toppo S, Zaccarin M, Ursini F. Understanding Mammalian Glutathione Peroxidase 7 in Light of Its Homologs. Free Radic Biol Med. 2015;83:352–360. doi: 10.1016/j.freeradbiomed.2015.02.017. [DOI] [PubMed] [Google Scholar]
- 184.Bardwell JCA, Mcgovern K, Beckwith J. Identification of a Protein Required for Disulfide Bond Formation In Vivo. Cell. 1991;67:581–589. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- 185.Kamitani S, Akiyama Y, Ito K. Identification and Characterization of an Escherichia coli Gene Required for the Formation of Correctly Folded Alkaline Phosphatase, a Periplasmic Enzyme. EMBO J. 1992;11:57–62. doi: 10.1002/j.1460-2075.1992.tb05027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Missiakas D, Georgopoulos C, Raina S. Identification and Characterization of the Escherichia coli Gene dsbB, Whose Product Is Involved in the Formation of Disulfide Bonds In Vivo. Proc Natl Acad Sci USA. 1993;90:7084–7088. doi: 10.1073/pnas.90.15.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Inaba K, Ito K. Structure and Mechanisms of the DsbB-DsbA Disulfide Bond Generation Machine. Biochim Biophys Acta. 2008;1783:520–529. doi: 10.1016/j.bbamcr.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 188.Hatahet F, Boyd D, Beckwith J. Disulfide Bond Formation in Prokaryotes: History, Diversity and Design. Biochim Biophys Acta. 2014;1844:1402–1414. doi: 10.1016/j.bbapap.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bevans CG, Krettler C, Reinhart C, Watzka M, Oldenburg J. Phylogeny of the Vitamin K 2,3- Epoxide Reductase (VKOR) Family and Evolutionary Relationship to the Disulfide Bond Formation Protein B (DsbB) Family. Nutrients. 2015;7:6224–6249. doi: 10.3390/nu7085281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Martin JL, Bardwell JCA, Kuriyan J. Crystal Structure of the DsbA Protein Required for Disulfide Bond Formation In Vivo. Nature. 1993;365:464–468. doi: 10.1038/365464a0. [DOI] [PubMed] [Google Scholar]
- 191.Kadokura H, Beckwith J. Four Cysteines of the Membrane Protein DsbB Act in Concert to Oxidize Its Substrate DsbA. EMBO J. 2002;21:2354–2363. doi: 10.1093/emboj/21.10.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Regeimbal J, Bardwell JC. DsbB Catalyzes Disulfide Bond Formation De Novo. J Biol Chem. 2002;277:32706–32713. doi: 10.1074/jbc.M205433200. [DOI] [PubMed] [Google Scholar]
- 193.Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. Crystal Structure of the DsbB-DsbA Complex Reveals a Mechanism of Disulfide Bond Generation. Cell. 2006;127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 194.Inaba K, Murakami S, Nakagawa A, Iida H, Kinjo M, Ito K, Suzuki M. Dynamic Nature of Disulphide Bond Formation Catalysts Revealed by Crystal Structures of DsbB. EMBO J. 2009;28:779–791. doi: 10.1038/emboj.2009.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Grauschopf U, Fritz A, Glockshuber R. Mechanism of the Electron Transfer Catalyst DsbB from Escherichia coli. EMBO J. 2003;22:3503–3513. doi: 10.1093/emboj/cdg356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tapley TL, Eichner T, Gleiter S, Ballou DP, Bardwell JC. Kinetic Characterization of the Disulfide Bond-Forming Enzyme DsbB. J Biol Chem. 2007;282:10263–10271. doi: 10.1074/jbc.M611541200. [DOI] [PubMed] [Google Scholar]
- 197.Inaba K, Takahashi YH, Ito K, Hayashi S. Critical Role of a Thiolate-Quinone Charge Transfer Complex and Its Adduct Form in De Novo Disulfide Bond Generation by DsbB. Proc Natl Acad Sci USA. 2006;103:287–292. doi: 10.1073/pnas.0507570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Israel BA, Kodali VK, Thorpe C. Going Through the Barrier: Coupled Disulfide Exchange Reactions Promote Efficient Catalysis in Quiescin Sulfhydryl Oxidase. J Biol Chem. 2014;289:5274–5284. doi: 10.1074/jbc.M113.536219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhou Y, Cierpicki T, Jimenez RH, Lukasik SM, Ellena JF, Cafiso DS, Kadokura H, Beckwith J, Bushweller JH. NMR Solution Structure of the Integral Membrane Enzyme DsbB: Functional Insights into DsbB-Catalyzed Disulfide Bond Formation. Mol Cell. 2008;31:896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jin DY, Tie JK, Stafford DW. The Conversion of Vitamin K Epoxide to Vitamin K Quinone and Vitamin K Quinone to Vitamin K Hydroquinone Uses the Same Active Site Cysteines. Biochem. 2007;46:7279–7283. doi: 10.1021/bi700527j. [DOI] [PubMed] [Google Scholar]
- 201.Westhofen P, Watzka M, Marinova M, Hass M, Kirfel G, Muller J, Bevans CG, Muller CR, Oldenburg J. Human Vitamin K 2,3-Epoxide Reductase Complex Subunit 1-Like 1 (VKORC1L1) Mediates Vitamin K-Dependent Intracellular Antioxidant Function. J Biol Chem. 2011;286:15085–15094. doi: 10.1074/jbc.M110.210971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Soute BA, Groenen-van Dooren MM, Holmgren A, Lundstrom J, Vermeer C. Stimulation of the Dithiol-Dependent Reductases in the Vitamin K Cycle by the Thioredoxin System. Biochem J. 1992;281:255–159. doi: 10.1042/bj2810255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wajih N, Hutson SM, Wallin R. Disulfide-Dependent Protein Folding is Linked to Operation of the Vitamin K Cycle in the Endoplasmic Reticulum. A protein Disulfide Isomerase-VKORC1 Redox Enzyme Complex Appears to be Responsible for Vitamin K1 2,3-Epoxide Reduction. J Biol Chem. 2007;282:2626–2635. doi: 10.1074/jbc.M608954200. [DOI] [PubMed] [Google Scholar]
- 204.Singh AK, Bhattacharyya-Pakrasi M, Pakrasi HB. Identification of an Atypical Membrane Protein Involved in the Formation of Protein Disulfide Bonds in Oxygenic Photosynthetic Organisms. J Biol Chem. 2008;283:15762–15770. doi: 10.1074/jbc.M800982200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Stanley TB, Stafford DW, Olivera BM, Bandyopadhyay PK. Identification of a Vitamin K-Dependent Carboxylase in the Venom Duct of a Conus Snail. FEBS Lett. 1997;407:85–88. doi: 10.1016/s0014-5793(97)00299-8. [DOI] [PubMed] [Google Scholar]
- 206.McIntosh JM, Olivera BM, Cruz LJ, Gray WR. γ-Carboxyglutamate in a Neuroactive Toxin. J Biol Chem. 1984;259:14343–14346. [PubMed] [Google Scholar]
- 207.Lebbe EK, Tytgat J. In the Picture: Disulfide-Poor Conopeptides, a Class of Pharmacologically Interesting Compounds. J Venom Anim Toxins Incl Trop Dis. 2016;22:30. doi: 10.1186/s40409-016-0083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Safavi-Hemami H, Li Q, Jackson RL, Song AS, Boomsma W, Bandyopadhyay PK, Gruber CW, Purcell AW, Yandell M, Olivera BM, Ellgaard L. Rapid Expansion of the Protein Disulfide Isomerase Gene Family Facilitates the Folding of Venom Peptides. Proc Natl Acad Sci USA. 2016;113:3227–3232. doi: 10.1073/pnas.1525790113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cao Z, van Lith M, Mitchell LJ, Pringle MA, Inaba K, Bulleid NJ. The Membrane Topology of Vitamin K Epoxide Reductase is Conserved Between Human Isoforms and the Bacterial Enzyme. Biochem J. 2016;473:851–858. doi: 10.1042/BJ20151223. [DOI] [PubMed] [Google Scholar]
- 210.Tie JK, Jin DY, Stafford DW. Conserved Loop Cysteines of Vitamin K Epoxide Reductase Complex Subunit 1-Like (VKORC1L1) Are Involved in Its Active Site Regeneration. J Biol Chem. 2014;289:9396–9407. doi: 10.1074/jbc.M113.534446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li WK, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. Structure of a Bacterial Homologue of Vitamin K Epoxide Reductase. Nature. 2010;463:507–512. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Liu S, Cheng W, Fowle Grider R, Shen G, Li W. Structures of an Intramembrane Vitamin K Epoxide Reductase Homolog Reveal Control Mechanisms for Electron Transfer. Nat Commun. 2014;5:3110. doi: 10.1038/ncomms4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Premkumar L, Heras B, Duprez W, Walden P, Halili M, Kurth F, Fairlie DP, Martin JL. Rv2969c, Essential for Optimal Growth in Mycobacterium tuberculosis, is a DsbA-Like Enzyme That Interacts with VKOR-Derived Peptides and Has Atypical Features of DsbA-Like Disulfide Oxidases. Acta Crystallogr D. 2013;69:1981–1994. doi: 10.1107/S0907444913017800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Goodstadt L, Ponting CP. Vitamin K Epoxide Reductase: Homology, Active Site and Catalytic Mechanism. Trends Biochem Sci. 2004;29:289–292. doi: 10.1016/j.tibs.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 215.Shen G, Cui W, Zhang H, Zhou F, Huang W, Liu Q, Yang Y, Li S, Bowman GR, Sadler JE, Gross ML, Li W. Warfarin Traps Human Vitamin K Epoxide Reductase in an Intermediate State During Electron Transfer. Nat Struct Mol Biol. 2017;24:69–76. doi: 10.1038/nsmb.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Pollard MG, Travers KJ, Weissman JS. Ero1p: a Novel and Ubiquitous Protein with an Essential Role in Oxidative Protein Folding in the Endoplasmic Reticulum. Mol Cell. 1998;1:171–182. doi: 10.1016/s1097-2765(00)80018-0. [DOI] [PubMed] [Google Scholar]
- 217.Frand AR, Kaiser CA. The ERO1 Gene of Yeast is Required for Oxidation of Protein Dithiols in the Endoplasmic Reticulum. Mol Cell. 1998;1:161–170. doi: 10.1016/s1097-2765(00)80017-9. [DOI] [PubMed] [Google Scholar]
- 218.Tu BP, Ho-Schleyer SC, Travers KJ, Weissman JS. Biochemical Basis of Oxidative Protein Folding in the Endoplasmic Reticulum. Science. 2000;290:1571–1574. doi: 10.1126/science.290.5496.1571. [DOI] [PubMed] [Google Scholar]
- 219.Hoober KL, Glynn NM, Burnside J, Coppock DL, Thorpe C. Homology Between Egg White Sulfhydryl Oxidase and Quiescin Q6 Defines a New Class of Flavin-Linked Sulfhydryl Oxidases. J Biol Chem. 1999;274:31759–31762. doi: 10.1074/jbc.274.45.31759. [DOI] [PubMed] [Google Scholar]
- 220.Lee J, Hofhaus G, Lisowsky T. Erv1p from Saccharomyces cerevisiae Is a FAD-Linked Sulfhydryl Oxidase. FEBS Lett. 2000;477:62–66. doi: 10.1016/s0014-5793(00)01767-1. [DOI] [PubMed] [Google Scholar]
- 221.Thorpe C, Hoober K, Raje S, Glynn N, Burnside J, Turi G, Coppock D. Sulfhydryl Oxidases: Emerging Catalysts of Protein Disulfide Bond Formation in Eukaryotes. Arch Biochem Biophys. 2002;405:1–12. doi: 10.1016/s0003-9861(02)00337-5. [DOI] [PubMed] [Google Scholar]
- 222.Fass D. The Erv Family of Sulfhydryl Oxidases. Biochim Biophys Acta. 2008;1783:557–566. doi: 10.1016/j.bbamcr.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 223.Sevier C. Erv2 and Quiescin Sulfhydryl Oxidases: Erv-Domain Enzymes Associated with the Secretory Pathway. Antioxid Redox Signal. 2012;16:800–808. doi: 10.1089/ars.2011.4450. [DOI] [PubMed] [Google Scholar]
- 224.Lisowsky T. Dual Function of a New Nuclear Gene for Oxidative Phosphorylation and Vegetative Growth in Yeast. Mol Gen Genet. 1992;232:58–64. doi: 10.1007/BF00299137. [DOI] [PubMed] [Google Scholar]
- 225.Lisowsky T. ERV1 is Involved in the Cell-Division Cycle and the Maintenance of Mitochondrial Genomes in Saccharomyces cerevisiae. Curr Genet. 1994;26:15–20. doi: 10.1007/BF00326299. [DOI] [PubMed] [Google Scholar]
- 226.Hagiya M, Francavilla A, Polimeno L, Ihara I, Sakai H, Seki T, Shimonishi M, Porter KA, Starzl TE. Cloning and Sequence Analysis of the Rat Augmenter of Liver Regeneration (ALR) Gene: Expression of Biologically Active Recombinant ALR and Demonstration of Tissue Distribution. Proc Natl Acad Sci USA. 1994;91:8142–8146. doi: 10.1073/pnas.91.17.8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lee J, Hofhaus G, Lisowsky T. Erv1p from Saccharomyces cerevisiae is a FAD-Linked Sulfhydryl Oxidase. FEBS Lett. 2000;477:62–66. doi: 10.1016/s0014-5793(00)01767-1. [DOI] [PubMed] [Google Scholar]
- 228.Gerber J, Muhlenhoff U, Hofhaus G, Lill R, Lisowsky T. Yeast ERV2p is the First Microsomal FAD-Linked Sulfhydryl Oxidase of the Erv1p/Alrp Protein Family. J Biol Chem. 2001;276:23486–23491. doi: 10.1074/jbc.M100134200. [DOI] [PubMed] [Google Scholar]
- 229.Sevier CS, Cuozzo JW, Vala A, Aslund F, Kaiser CA. A Flavoprotein Oxidase Defines a New Endoplasmic Reticulum Pathway for Biosynthetic Disulphide Bond Formation. Nat Cell Biol. 2001;3:874–882. doi: 10.1038/ncb1001-874. [DOI] [PubMed] [Google Scholar]
- 230.Gross E, Sevier CS, Vala A, Kaiser CA, Fass D. A New FAD-Binding Fold and Intersubunit Disulfide Shuttle in the Thiol Oxidase Erv2p. Nat Struct Biol. 2002;9:61–67. doi: 10.1038/nsb740. [DOI] [PubMed] [Google Scholar]
- 231.Daithankar VN, Farrell SR, Thorpe C. Augmenter of Liver Regeneration: Substrate Specificity of a Flavin-Dependent Oxioreductase from the Mitochondrial Intermembrane Space. Biochem. 2009;48:4828–4837. doi: 10.1021/bi900347v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Vitu E, Bentzur M, Lisowsky T, Kaiser CA, Fass D. Gain of Function in an ERV/ALR Sulfhydryl Oxidase by Molecular Engineering of the Shuttle Disulfide. J Mol Biol. 2006;362:89–101. doi: 10.1016/j.jmb.2006.06.070. [DOI] [PubMed] [Google Scholar]
- 233.Farrell SR, Thorpe C. Augmenter of Liver Regeneration: a Flavin-Dependent Sulfhydryl Oxidase With Cytochrome c Reductase Activity. Biochem. 2005;44:1532–1541. doi: 10.1021/bi0479555. [DOI] [PubMed] [Google Scholar]
- 234.Allen S, Balabanidou V, Sideris DP, Lisowsky T, Tokatlidis K. Erv1 Mediates the Mia40-dependent Protein Import Pathway and Provides a Functional Link to the Respiratory Chain by Shuttling Electrons to Cytochrome c. J Mol Biol. 2005;353:937–944. doi: 10.1016/j.jmb.2005.08.049. [DOI] [PubMed] [Google Scholar]
- 235.Bihlmaier K, Mesecke N, Terziyska N, Bien M, Hell K, Herrmann JM. The Disulfide Relay System of Mitochondria is Connected to the Respiratory Chain. J Cell Biol. 2007;179:389–395. doi: 10.1083/jcb.200707123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Dabir DV, Leverich EP, Kim SK, Tsai FD, Hirasawa M, Knaff DB, Koehler CM. A Role for Cytochrome c and Cytochrome c Peroxidase in Electron Shuttling from Erv1. EMBO J. 2007;26:4801–4811. doi: 10.1038/sj.emboj.7601909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Eckers E, Petrungaro C, Gross D, Riemer J, Hell K, Deponte M. Divergent Molecular Evolution of the Mitochondrial Sulfhydryl:Cytochrome C Oxidoreductase Erv in Opisthokonts and Parasitic Protists. J Biol Chem. 2013;288:2676–2688. doi: 10.1074/jbc.M112.420745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Senkevich TG, White CL, Koonin EV, Moss B. A Viral Member of the ERV1/ALR Protein Family Participates in a Cytoplasmic Pathway of Disulfide Bond Formation. Proc Natl Acad Sci USA. 2000;97:12068–12073. doi: 10.1073/pnas.210397997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. Virus Factories: Associations of Cell Organelles for Viral Replication and Morphogenesis. Biol Cell. 2005;97:147–172. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hakim M, Mandelbaum A, Fass D. Structure of a Baculovirus Sulfhydryl Oxidase, a Highly Divergent Member of the Erv Flavoenzyme Family. J Virol. 2011;85:9406–9413. doi: 10.1128/JVI.05149-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Levy ED, Boeri-Erba E, Robinson CV, Teichmann SA. Assembly Reflects Evolution of Protein Complexes. Nature. 2008;453:1262–1265. doi: 10.1038/nature06942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Senkevich TG, White CL, Koonin EV, Moss B. Complete Pathway for Protein Disulfide Bond Formation Encoded by Poxviruses. Proc Natl Acad Sci USA. 2002;99:6667–6672. doi: 10.1073/pnas.062163799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Hakim M, Fass D. Dimer Interface Migration in a Viral Sulfhydryl Oxidase. J Mol Biol. 2009;391:758–768. doi: 10.1016/j.jmb.2009.06.070. [DOI] [PubMed] [Google Scholar]
- 244.Alon A, Heckler EJ, Thorpe C, Fass D. QSOX Contains a Pseudo-Dimer of Functional and Degenerate Sulfhydryl Oxidase Domains. FEBS Lett. 2010;584:1521–1525. doi: 10.1016/j.febslet.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 245.Raje S, Thorpe C. Inter-domain Redox Communication in Flavoenzymes of the Quiescin/Sulfhydryl Oxidase Family: Role of a Thioredoxin Domain in Disulfide Bond Formation. Biochem. 2003;42:4560–4568. doi: 10.1021/bi030003z. [DOI] [PubMed] [Google Scholar]
- 246.Heckler EJ, Alon A, Fass D, Thorpe C. Human Quiescin-Sulfhydryl Oxidase, QSOX1: Probing Internal Redox Steps by Mutagenesis. Biochem. 2008;47:4955–4963. doi: 10.1021/bi702522q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rancy PC, Thorpe C. Oxidative Protein Folding In Vitro: a Study of the Cooperation Between Quiescin-sulfhydryl Oxidase and Protein Disulfide Isomerase. Biochem. 2008;47:12047–12056. doi: 10.1021/bi801604x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Alon A, Grossman I, Gat Y, Kodali VK, DiMaio F, Mehlman T, Haran G, Baker D, Thorpe C, Fass D. The Dynamic Disulphide Relay of Quiescin Sulfhydryl Oxidase. Nature. 2012;488:414–418. doi: 10.1038/nature11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kodali VK, Thorpe C. Quiescin Sulfhydryl Oxidase from Trypanosoma brucei: Catalytic Activity and Mechanism of a QSOX Family Member with a Single Thioredoxin Domain. Biochem. 2010;49:2075–2085. doi: 10.1021/bi902222s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Grossman I, Aviram HY, Armony G, Horovitz A, Hofmann H, Haran G, Fass D. Single- Molecule Spectroscopy Exposes Hidden States in an Enzymatic Electron Relay. Nat Commun. 2015;6:8624. doi: 10.1038/ncomms9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Hoober KL, Thorpe C. Egg White Sulfhydryl Oxidase: Kinetic Mechanism of the Catalysis of Disulfide Bond Formation. Biochem. 1999;38:3211–3217. doi: 10.1021/bi9820816. [DOI] [PubMed] [Google Scholar]
- 252.Gat Y, Vardi-Kilshtain A, Grossman I, Major DT, Fass D. Enzyme Structure Captures Four Cysteines Aligned for Disulfide Relay. Protein Sci. 2014;23:1102–1112. doi: 10.1002/pro.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hoober KL, Sheasley SS, Gilbert HF, Thorpe C. Sulfhydryl Oxidase from Egg White: a Facile Catalyst for Disulfide Bond Formation in Proteins and Peptides. J Biol Chem. 1999;274:22147–22150. doi: 10.1074/jbc.274.32.22147. [DOI] [PubMed] [Google Scholar]
- 254.Codding JA, Israel BA, Thorpe C. Protein Substrate Discrimination in the Quiescin Sulfhydryl Oxidase (QSOX) Family. Biochem. 2012;51:4226–4235. doi: 10.1021/bi300394w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Coppock D, Kopman C, Gudas J, Cina-Poppe DA. Regulation of the Quiescence-Induced Genes: Quiescin Q6, Decorin, and Ribosomal Protein S29. Biochem Biophys Res Commun. 2000;269:605–610. doi: 10.1006/bbrc.2000.2324. [DOI] [PubMed] [Google Scholar]
- 256.Ilani T, Alon A, Grossman I, Horowitz B, Kartvelishvily E, Cohen SR, Fass D. A Secreted Disulfide Catalyst Controls Extracellular Marix Composition and Function. Science. 2013;341:74–76. doi: 10.1126/science.1238279. [DOI] [PubMed] [Google Scholar]
- 257.Frand AR, Kaiser CA. The ERO1 Gene of Yeast is Required for Oxidation of Protein Dithiols in the Endoplasmic Reticulum. Mol Cell. 1998;1:161–170. doi: 10.1016/s1097-2765(00)80017-9. [DOI] [PubMed] [Google Scholar]
- 258.Pollard MG, Travers KJ, Weissman JS. Ero1p: a Novel and Ubiquitous Protein With an Essential Role in Oxidative Protein Folding in the Endoplasmic Reticulum. Mol Cell. 1998;1:171–182. doi: 10.1016/s1097-2765(00)80018-0. [DOI] [PubMed] [Google Scholar]
- 259.Araki K, Inaba K. Structure, Mechanism, and Evolution of Ero1 Family Enzymes. Antioxid Redox Signal. 2012;16:790–799. doi: 10.1089/ars.2011.4418. [DOI] [PubMed] [Google Scholar]
- 260.Inaba K, Masui S, Iida H, Vavassori S, Sitia R, Suzuki M. Crystal Structures of Human Ero1α Reveal the Mechanisms of Regulated and Targeted Oxidation of PDI. EMBO J. 2010;29:3330–3343. doi: 10.1038/emboj.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Sevier CS, Qu H, Heldman N, Gross E, Fass D, Kaiser CA. Modulation of Cellular Disulfide- Bond Formation and the ER Redox Environment by Feedback Regulation of Ero1. Cell. 2007;129:333–344. doi: 10.1016/j.cell.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 262.Appenzeller-Herzog C, Riemer J, Christensen B, Sorensen ES, Ellgaard L. A Novel Disulphide Switch Mechanism in Ero1α Balances ER Oxidation in Human Cells. EMBO J. 2008;27:2977–2987. doi: 10.1038/emboj.2008.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Baker KM, Chakravarthi S, Langton KP, Sheppard AM, Lu H, Bulleid NJ. Low Reduction Potential of Ero1α Regulatory Disulphides Ensures Tight Control of Substrate Oxidation. EMBO J. 2008;27:2988–2997. doi: 10.1038/emboj.2008.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ramming T, Okumura M, Kanemura S, Baday S, Birk J, Moes S, Spiess M, Jeno P, Berneche S, Inaba K, et al. A PDI-Catalyzed Thiol-Disulfide Switch Regulates the Production of Hydrogen Peroxide by Human Ero1. Free Radic Biol Med. 2015;83:361–372. doi: 10.1016/j.freeradbiomed.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 265.Niu Y, Zhang L, Yu J, Wang CC, Wang L. Novel Roles of the Non-Catalytic Elements of Yeast Protein-Disulfide Isomerase and Its Interplay with Endoplasmic Reticulum Oxidoreductin 1. J Biol Chem. 2016;291:8283–8294. doi: 10.1074/jbc.M115.694257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Kim S, Sideris DP, Sevier CS, Kaiser CA. Balanced Ero1 Activation and Inactivation Establishes ER Redox Homeostasis. J Cell Biol. 2012;196:713–725. doi: 10.1083/jcb.201110090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.de la Motte RS, Wagner FW. Aspergillus niger Sulfhydryl Oxidase. Biochem. 1987;26:7363–7371. doi: 10.1021/bi00397a025. [DOI] [PubMed] [Google Scholar]
- 268.Faccio G, Kruus K, Buchert J, Saloheimo M. Secreted Fungal Sulfhydryl Oxidases: Sequence Analysis and Characterisation of a Representative Flavin-Dependent Enzyme from Aspergillus oryzae. BMC Biochem. 2010;11:31. doi: 10.1186/1471-2091-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Kusakabe H, Kuninaka A, Yoshino H. Purification and Properties of a New Enzyme, Glutathione Oxidase from Penicillium sp. K-6-5. Agric Biol Chem. 1982;46:2057–2067. [Google Scholar]
- 270.Nivala O, Mattinen ML, Faccio G, Buchert J, Kruus K. Discovery of Novel Secreted Fungal Sulfhydryl Oxidases with a Plate Test Screen. Appl Microbiol Biotechnol. 2013;97:9429–9437. doi: 10.1007/s00253-013-4753-9. [DOI] [PubMed] [Google Scholar]
- 271.Janolino VG, Swaisgood HE. A Comparison of Sulfhydryl Oxidase from Bovine Milk and Aspergillus niger. Milchwissenschaft. 1992;47:143–146. [Google Scholar]
- 272.Williams CH., Jr . Lipoamide Dehydrogenase, Glutathione Reductase, Thioredoxin Reductase, and Mercuric Ion Reductase-A Family of Flavoenzyme Transhydrogenases. In: Muller F, editor. Chemistry and Biochemistry of Flavoenzymes. Chemistry and Biochemistry of Flavoenzymes. CRC Press; 1992. pp. 121–211. [Google Scholar]
- 273.Argyrou A, Blanchard JS. Flavoprotein Disulfide Reductases: Advances in Chemistry and Function. Prog Nucleic Acid Res Mol Biol. 2004;78:89–142. doi: 10.1016/S0079-6603(04)78003-4. [DOI] [PubMed] [Google Scholar]
- 274.Williams CH., Jr Mechanism and Structure of Thioredoxin Reductase from Escherichia coli. FASEB J. 1995;9:1267–1276. doi: 10.1096/fasebj.9.13.7557016. [DOI] [PubMed] [Google Scholar]
- 275.Li B, Walsh CT. Streptomyces clavuligerus HlmI Is an Intramolecular Disulfide-Forming Dithiol Oxidase in Holomycin Biosynthesis. Biochem. 2011;50:4615–4622. doi: 10.1021/bi200321c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Scharf DH, Remme N, Heinekamp T, Hortschansky P, Brakhage AA, Hertweck C. Transannular Disulfide Formation in Gliotoxin Biosynthesis and Its Role in Self-Rresistance of the Human Pathogen Aspergillus fumigatus. J Am Chem Soc. 2010;132:10136–10141. doi: 10.1021/ja103262m. [DOI] [PubMed] [Google Scholar]
- 277.Li J, Wang C, Zhang ZM, Cheng YQ, Zhou J. The Structural Basis of an NADP(+)- Independent Dithiol Oxidase in FK228 Biosynthesis. Sci Rep. 2014;4:4145. doi: 10.1038/srep04145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Schrettl M, Carberry S, Kavanagh K, Haas H, Jones GW, O’Brien J, Nolan A, Stephens J, Fenelon O, Doyle S. Self-Protection against Gliotoxin-A Component of the Gliotoxin Biosynthetic Cluster, GliT, Completely Protects Aspergillus fumigatus Against Exogenous Gliotoxin. PLoS Pathog. 2010;6:e1000952. doi: 10.1371/journal.ppat.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Li B, Wever WJ, Walsh CT, Bowers AA. Dithiolopyrrolones: Biosynthesis, Synthesis, and Activity of a Unique Class of Disulfide-Containing Antibiotics. Nat Prod Rep. 2014;31:905–923. doi: 10.1039/c3np70106a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Wang C, Wesener SR, Zhang H, Cheng YQ. An FAD-Dependent Pyridine Nucleotide- Disulfide Oxidoreductase is Involved in Disulfide Bond Formation in FK228 Anticancer Depsipeptide. Chem Biol. 2009;16:585–593. doi: 10.1016/j.chembiol.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 281.Robinson PJ, Pringle MA, Woolhead CA, Bulleid NJ. Folding of a Single Domain Protein Entering the Endoplasmic Reticulum Precedes Disulfide Formation. J Biol Chem. 2017;292:6978–6986. doi: 10.1074/jbc.M117.780742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Ellgaard L, McCaul N, Chatsisvili A, Braakman I. Co- and Post-Translational Protein Folding in the ER. Traffic. 2016;17:615–638. doi: 10.1111/tra.12392. [DOI] [PubMed] [Google Scholar]
- 283.Kosuri P, Alegre-Cebollada J, Feng J, Kaplan A, Ingles-Prieto A, Badilla CL, Stockwell BR, Sanchez-Ruiz JM, Holmgren A, Fernandez JM. Protein Folding Drives Disulfide Formation. Cell. 2012;151:794–806. doi: 10.1016/j.cell.2012.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Bader MW, Hiniker A, Regeimbal J, Goldstone D, Haebel PW, Riemer J, Metcalf P, Bardwell JC. Turning a Disulfide Isomerase into an Oxidase: DsbC Mutants That Imitate DsbA. EMBO J. 2001;20:1555–1562. doi: 10.1093/emboj/20.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Segatori L, Paukstelis PJ, Gilberg HF, Georgiou G. Engineered DsbC Chimeras Catalyze Both Protein Oxidation and Disulfide-Bond Isomerization in Escherichia coli: Reconciling Two Competing Pathways. Proc Natl Acad Sci USA. 2004;101:10018–10023. doi: 10.1073/pnas.0403003101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Rozhkova A, Stirnimann CU, Frei P, Grauschopf U, Brunisholz R, Grutter MG, Capitani G, Glockshuber R. Structural Basis and Kinetics of Inter- and Intramolecular Disulfide Exchange in the Redox Catalyst DsbD. EMBO J. 2004;23:1709–1719. doi: 10.1038/sj.emboj.7600178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Vertommen D, Depuydt M, Pan J, Leverrier P, Knoops L, Szikora JP, Messens J, Bardwell JC, Collet JF. The Disulphide Isomerase DsbC Cooperates With the Oxidase DsbA in a DsbD-Independent Manner. Mol Microbiol. 2008;67:336–349. doi: 10.1111/j.1365-2958.2007.06030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Bader M, Muse W, Ballou DP, Gassner C, Bardwell JC. Oxidative Protein Folding Is Driven by the Electron Transport System. Cell. 1999;98:217–227. doi: 10.1016/s0092-8674(00)81016-8. [DOI] [PubMed] [Google Scholar]
- 289.Page CC, Moser CC, Chen X, Dutton PL. Natural Engineering Principles of Electron Tunnelling in Biological Oxidation-Reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 290.Farver O, Vitu E, Wherland S, Fass D, Pecht I. Electron Transfer Reactivity of the Arabidopsis thaliana Sulfhydryl Oxidase AtErv1. J Biol Chem. 2009;284:2098–2105. doi: 10.1074/jbc.M806316200. [DOI] [PubMed] [Google Scholar]
- 291.Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. A Disulfide Relay System in the Intermembrane Space of Mitochondria that Mediates Protein Import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 292.Naoe M, Ohwa Y, Ishikawa D, Ohshima C, Nishikawa S, Yamamoto H, Endo T. Identification of Tim40 that Mediates Protein Sorting to the Mitochondrial Intermembrane Space. J Biol Chem. 2004;279:47815–47821. doi: 10.1074/jbc.M410272200. [DOI] [PubMed] [Google Scholar]
- 293.Sideris DP, Petrakis N, Katrakili N, Mikropoulou D, Gallo A, Ciofi-Baffoni S, Banci L, Bertini I, Tokatlidis K. A novel intermembrane space-targeting signal docks cysteines onto Mia40 during mitochondrial oxidative folding. Journal of Cell Biology. 2009;187(7):1007–22. doi: 10.1083/jcb.200905134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Kojer K, Peleh V, Calabrese G, Herrmann JM, Riemer J. Kinetic Control by Limiting Glutaredoxin Amounts Enables Thiol Oxidation in the Reducing Mitochondrial Intermembrane Space. Mol Biol Cell. 2015;26:195–204. doi: 10.1091/mbc.E14-10-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Herrmann JM, Kohl R. Catch Me if You Can! Oxidative Protein Trapping in the Intermembrane Space of Mitochondria. J Cell Biol. 2007;176:559–563. doi: 10.1083/jcb.200611060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Weckbecker D, Longen S, Riemer J, Herrmann JM. Atp23 Biogenesis Reveals a Chaperone- Like Folding Activity of Mia40 in the IMS of Mitochondria. EMBO J. 2012;31:4348–4358. doi: 10.1038/emboj.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Chatzi A, Tokatlidis K. The Mitochondrial Intermembrane Space: a Hub for Oxidative Folding Linked to Protein Biogenesis. Antioxid Redox Signal. 2013;19:54–62. doi: 10.1089/ars.2012.4855. [DOI] [PubMed] [Google Scholar]
- 298.Longen S, Woellhaf MW, Petrungaro C, Riemer J, Herrmann JM. The Disulfide Relay of the Intermembrane Space Oxidizes the Ribosomal Subunit mrp10 on its Transit into the Mitochondrial Matrix. Dev Cell. 2014;28:30–42. doi: 10.1016/j.devcel.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 299.Koch JR, Schmid FX. Mia40 Combines Thiol Oxidase and Disulfide Isomerase Activity to Efficiently Catalyze Oxidative Folding in Mitochondria. J Mol Biol. 2014;426:4087–4098. doi: 10.1016/j.jmb.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 300.Koch JR, Schmid FX. Mia40 Targets Cysteines in a Hydrophobic Environment to Direct Oxidative Protein Folding in the Mitochondria. Nat Commun. 2014;5:3041. doi: 10.1038/ncomms4041. [DOI] [PubMed] [Google Scholar]
- 301.Hudson DA, Thorpe C. Mia40 is a Facile Oxidant of Unfolded Reduced Proteins but Shows Minimal Isomerase Activity. Arch Biochem Biophys. 2015;579:1–7. doi: 10.1016/j.abb.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Fischer M, Riemer J. The Mitochondrial Disulfide Relay System: Roles in Oxidative Protein Folding and Beyond. Int J Cell Biol. 2013;2013:742923. doi: 10.1155/2013/742923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Wenz LS, Opalinski L, Wiedemann N, Becker T. Cooperation of Protein Machineries in Mitochondrial Protein Sorting. Biochim Biophys Acta. 2015;1853:1119–1129. doi: 10.1016/j.bbamcr.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 304.Dudek J, Rehling P, van der Laan M. Mitochondrial Protein Import: Common Principles and Physiological Networks. Biochim Biophys Acta. 2013;1833:274–285. doi: 10.1016/j.bbamcr.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 305.Stojanovski D, Bohnert M, Pfanner N, van der Laan M. Mechanisms of Protein Sorting in Mitochondria. Cold Spring Harb Perspect Biol. 2012;4:a011320. doi: 10.1101/cshperspect.a011320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Kojer K, Riemer J. Balancing Oxidative Protein Folding: The Influences of Reducing Pathways on Disulfide Bond Formation. Biochim Biophys Acta. 2014;1844:1383–1390. doi: 10.1016/j.bbapap.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 307.Vogtle FN, Burkhart JM, Rao S, Gerbeth C, Hinrichs J, Martinou JC, Chacinska A, Sickmann A, Zahedi RP, Meisinger C. Intermembrane Space Proteome of Yeast Mitochondria. Mol Cell Proteomics. 2012;11:1840–1852. doi: 10.1074/mcp.M112.021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Allen JW, Ferguson SJ, Ginger ML. Distinctive Biochemistry in the Trypanosome Mitochondrial Intermembrane Space Suggests a Model for Stepwise Evolution of the MIA Pathway for Import of Cysteine-Rich Proteins. FEBS Lett. 2008;582:2817–2825. doi: 10.1016/j.febslet.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 309.Basu S, Leonard JC, Desai N, Mavridou DA, Tang KH, Goddard AD, Ginger ML, Lukes J, Allen JW. Divergence of Erv1-Associated Mitochondrial Import and Export Pathways in Trypanosomes and Anaerobic Protists. Eukaryot Cell. 2013;12:343–355. doi: 10.1128/EC.00304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Deponte M, Hell K. Disulfide Bond Formation in the Intermembrane Space of Mitochondria. J Biochem. 2009;146:599–608. doi: 10.1093/jb/mvp133. [DOI] [PubMed] [Google Scholar]
- 311.Piacente F, Marin M, Molinaro A, De Castro C, Seltzer V, Salis A, Damonte G, Bernardi C, Claverie JM, Abergel C, et al. Giant DNA Virus Mimivirus Encodes Pathway for Biosynthesis of Unusual Sugar 4-Amino-4,6-dideoxy-D-glucose (Viosamine) J Biol Chem. 2012;287:2009–3018. doi: 10.1074/jbc.M111.314559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Rommel AJ, Hulsmeier AJ, Jurt S, Hennet T. Giant Mimivirus R707 Encodes a Glycogenin Paralogue Polymerizing Glucose Through α- and β-Glycosidic Linkages. Biochem J. 2016;473:3451–3462. doi: 10.1042/BCJ20160280. [DOI] [PubMed] [Google Scholar]
- 313.Hulsmeier AJ, Hennet T. O-Linked Glycosylation in Acanthamoeba polyphaga Mimivirus. Glycobiol. 2014;24:703–714. doi: 10.1093/glycob/cwu034. [DOI] [PubMed] [Google Scholar]
- 314.White CL, Senkevich TG, Moss B. Vaccinia Virus G4L Glutaredoxin Is an Essential Intermediate of a Cytoplasmic Disulfide Bond Pathway Required for Virion Assembly. J Virol. 2002;76:467–472. doi: 10.1128/JVI.76.2.467-472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Senkevich TG, Weisberg AS, Moss B. Vaccinia Virus E10R Protein is Associated With the Membranes of Intracellular Mature Virions and Has a Role in Morphogenesis. Virol. 2000;278:24–252. doi: 10.1006/viro.2000.0656. [DOI] [PubMed] [Google Scholar]
- 316.Senkevich TG, White CL, Weisberg AS, Granek JA, Wolffe EJ, Koonin EV, Moss B. Expression of the Vaccinia Virus A2.5L Redox Protein is Required for Virion Morphogenesis. Virol. 2002;300:296–303. doi: 10.1006/viro.2002.1608. [DOI] [PubMed] [Google Scholar]
- 317.Yoder JD, Chen TS, Gagnier CR, Vemulapalli S, Maier CS, Hruby DE. Pox Proteomics: Mass Spectrometry Analysis and Identification of Vaccinia Virion Proteins. Virol J. 2006;3:10. doi: 10.1186/1743-422X-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Chung CS, Chen CH, Ho MY, Huang CY, Liao CL, Chang W. Vaccinia Virus Proteome: Identification of Proteins in Vaccinia Virus Intracellular Mature Virion Particles. J Virol. 2006;80:2127–2140. doi: 10.1128/JVI.80.5.2127-2140.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Resch W, Hixson KK, Moore RJ, Lipton MS, Moss B. Protein Composition of the Vaccinia Virus Mature Virion. Virol. 2007;358:233–247. doi: 10.1016/j.virol.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 320.Ojeda S, Senkevich TG, Moss B. Entry of Vaccinia Virus and Cell-Cell Fusion Require a Highly Conserved Cysteine-Rich Membrane Protein Encoded by the A16L Gene. J Virol. 2006;80:51–61. doi: 10.1128/JVI.80.1.51-61.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ngo T, Mirzakhanyan Y, Moussatche N, Gershon PD. Protein Primary Structure of the Vaccinia Virion at Increased Resolution. J Virol. 2016;90:9905–9919. doi: 10.1128/JVI.01042-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Rodriguez I, Redrejo-Rodriguez M, Rodriguez JM, Alejo A, Salas J, Salas ML. African Swine Fever Virus pB119L Protein is a Flavin Adenine Dinucleotide-Linked Sulfhydryl Oxidase. J Virol. 2006;80:3157–3166. doi: 10.1128/JVI.80.7.3157-3166.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Long CM, Rohrmann GF, Merrill GF. The Conserved Baculovirus Protein p33 (Ac92) is a Flavin Adenine Dinucleotide-Linked Sulfhydryl Oxidase. Virol. 2009;388:231–235. doi: 10.1016/j.virol.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 324.Hakim M, Ezerina D, Alon A, Vonshak O, Fass D. Exploring ORFan Domains in Giant Viruses: Structure of Mimivirus Sulfhydryl Oxidase R596. PLoS One. 2012;7:e50649. doi: 10.1371/journal.pone.0050649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Albright BS, Kosinski A, Szczepaniak R, Cook EA, Stow ND, Conway JF, Weller SK. The Putative Herpes Simplex Virus 1 Chaperone Protein UL32 Modulates Disulfide Bond Formation During Infection. J Virol. 2015;89:443–453. doi: 10.1128/JVI.01913-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Goldberger RF, Epstein CJ, Anfinsen CB. Acceleration of Reactivation of Reduced Bovine Pancreatic Ribonuclease by a Microsomal System from Rat Liver. J Biol Chem. 1963;238:628–635. [PubMed] [Google Scholar]
- 327.Oka OB, Yeoh HY, Bulleid NJ. Thiol-Disulfide Exchange Between the PDI Family of Oxidoreductases Negates the Requirement for an Oxidase or Reductase for Each Enzyme. Biochem J. 2015;469:279–288. doi: 10.1042/BJ20141423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Araki K, Iemura S, Kamiya Y, Ron D, Kato K, Natsume T, Nagata K. Ero1-α and PDIs Constitute a Hierarchical Electron Transfer Network of Endoplasmic Reticulum Oxidoreductases. J Cell Biol. 2013;202:861–874. doi: 10.1083/jcb.201303027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Avezov E, Konno T, Zyryanova A, Chen W, Laine R, Crespillo-Casado A, Melo EP, Ushioda R, Nagata K, Kaminski CF. Retarded PDI Diffusion and a Reductive Shift in Poise of the Calcium Depleted Endoplasmic Reticulum. BMC Biol. 2015;13:2. doi: 10.1186/s12915-014-0112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Jessop CE, Bulleid NJ. Glutathione Directly Reduces an Oxidoreductase in the Endoplasmic Reticulum of Mammalian Cells. J Biol Chem. 2004;279:55341–55347. doi: 10.1074/jbc.M411409200. [DOI] [PubMed] [Google Scholar]
- 331.Braakman I, Bulleid NJ. Protein Folding and Modification in the Mammalian Endoplasmic Reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 332.Konno T, Pinho Melo E, Lopes C, Mehmeti I, Lenzen S, Ron D, Avezov E. ERO1-Independent Production of H2O2 Within the Endoplasmic Reticulum Fuels Prdx4-Mediate Oxidative Protein Folding. J Cell Biol. 2015;211:253–259. doi: 10.1083/jcb.201506123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Zhang L, Niu Y, Zhu L, Fang J, Wang X, Wang L, Wang CC. Different Interaction Modes for Protein-Disulfide Isomerase (PDI) as an Efficient Regulator and a Specific Substrate of Endoplasmic Reticulum Oxidoreductin-1α (Ero1α) J Biol Chem. 2014;289:31188–31199. doi: 10.1074/jbc.M114.602961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Birk J, Meyer M, Aller I, Hansen HG, Odermatt A, Dick TP, Meyer AJ, Appenzeller-Herzog C. Endoplasmic Reticulum: Reduced and Oxidized Glutathione Revisited. J Cell Sci. 2013;126:1604–1617. doi: 10.1242/jcs.117218. [DOI] [PubMed] [Google Scholar]
- 335.Toldo S, Boccellino M, Rinaldi B, Seropian IM, Mezzaroma E, Severino A, Quagliuolo L, Van Tassell BW, Marfella R, Paolisso G, et al. Altered Oxido-Reductive State in the Diabetic Heart: Loss of Cardioprotection Due to Protein Disulfide Isomerase. Mol Med. 2011;17:1012–1021. doi: 10.2119/molmed.2011.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Appenzeller-Herzog C, Ellgaard L. In Vivo Reduction-Oxidation State of Protein Disulfide Isomerase: the Two Active Sites Independently Occur in the Reduced and Oxidized Forms. Antioxid Redox Signal. 2008;10:55–64. doi: 10.1089/ars.2007.1837. [DOI] [PubMed] [Google Scholar]
- 337.Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R. Manipulation of Oxidative Protein Folding and PDI Redox State in Mammalian Cells. EMBO J. 2001;20:6288–6296. doi: 10.1093/emboj/20.22.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Lyles MM, Gilbert HF. Catalysis of the Oxidative Folding of Ribonuclease-a by Protein Disulfide Isomerase - Pre-Steady-State Kinetics and the Utilization of the Oxidizing Equivalents of the Isomerase. Biochem. 1991;30:619–625. doi: 10.1021/bi00217a005. [DOI] [PubMed] [Google Scholar]
- 339.Poet GJ, Oka OB, van Lith M, Cao Z, Robinson PJ, Pringle MA, Arner ES, Bulleid NJ. Cytosolic Thioredoxin Reductase 1 Is Required for Correct Disulfide Formation in the ER. EMBO J. 2017;36:693–702. doi: 10.15252/embj.201695336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Darby NJ, Creighton TE. Characterization of the Active Site Cysteine Residues of the Thioredoxin-like Domains of Protein Disulfide Isomerase. Biochem. 1995;34:16770–16780. doi: 10.1021/bi00051a027. [DOI] [PubMed] [Google Scholar]
- 341.Lappi AK, Ruddock LW. Reexamination of the Role of Interplay Between Glutathione and Protein Disulfide Isomerase. J Mol Biol. 2011;409:238–249. doi: 10.1016/j.jmb.2011.03.024. [DOI] [PubMed] [Google Scholar]
- 342.van Lith M, Tiwari S, Pediani J, Milligan G, Bulleid NJ. Real-Time Monitoring of Redox Changes in the Mammalian Endoplasmic Reticulum. J Cell Sci. 2011;124:2349–2356. doi: 10.1242/jcs.085530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Avezov E, Cross BC, Kaminski Schierle GS, Winters M, Harding HP, Melo EP, Kaminski CF, Ron D. Lifetime Imaging of a Fluorescent Protein Sensor Reveals Surprising Stability of ER Thiol Redox. J Cell Biol. 2013;201:337–349. doi: 10.1083/jcb.201211155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Stocki P, Chapman DC, Beach LA, Williams DB. Depletion of Cyclophilins B and C Leads to Dysregulation of Endoplasmic Reticulum Redox Homeostasis. J Biol Chem. 2014;289:23086–23096. doi: 10.1074/jbc.M114.570911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Pisoni GB, Ruddock LW, Bulleid N, Molinari M. Division of Labor Among Oxidoreductases: TMX1 Preferentially Acts on Transmembrane Polypeptides. Mol Biol Cell. 2015;26:3390–3400. doi: 10.1091/mbc.E15-05-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Wells WW, Xu DP, Yang YF, Rocque PA. Mammalian Thioltransferase (Glutaredoxin) and Protein Disulfide Isomerase Have Dehydroascorbate Reductase-Activity. J Biol Chem. 1990;265:15361–15364. [PubMed] [Google Scholar]
- 347.Wells WW, Xu DP. Dehydroascorbate Reduction. J Bioenerg Biomembr. 1994;26:369–77. doi: 10.1007/BF00762777. [DOI] [PubMed] [Google Scholar]
- 348.Givol D, Anfinsen CB, Goldberger RF. Oxidation and Disulfide Interchange in Reactivation of Reduced Ribonuclease. J Biol Chem. 1964;239:3114–3116. [PubMed] [Google Scholar]
- 349.Venetianer P, Straub FB. Mechanism of Action of Ribonuclease-Reactivating Enzyme. Biochim Biophys Acta. 1964;89:189–190. doi: 10.1016/0926-6569(64)90123-3. [DOI] [PubMed] [Google Scholar]
- 350.Margittai E, Banhegyi G. Oxidative Folding in the Endoplasmic Reticulum: Towards a Multiple Oxidant Hypothesis? FEBS Lett. 2010;584:2995–2998. doi: 10.1016/j.febslet.2010.05.055. [DOI] [PubMed] [Google Scholar]
- 351.Szarka A, Stadler K, Jenei V, Margittai E, Csala M, Jakus J, Mandl J, Banhegyi G. Ascorbyl Free Radical and Dehydroascorbate Formation in Rat Liver Endoplasmic Reticulum. J Bioenerg Biomembr. 2002;34:317–323. doi: 10.1023/a:1020212720330. [DOI] [PubMed] [Google Scholar]
- 352.Banhegyi G, Marcolongo P, Puskas F, Fulceri R, Mandl J, Benedetti A. Dehydroascorbate and Ascorbate Transport in Rat Liver Microsomal Vesicles. J Biol Chem. 1998;273:2758–2762. doi: 10.1074/jbc.273.5.2758. [DOI] [PubMed] [Google Scholar]
- 353.Walter P, Ron D. The Unfolded Protein Response: from Stress Pathway to Homeostatic Regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 354.Anelli T, Alessio M, Mezghrani A, Simmen T, Talamo F, Bachi A, Sitia R. ERp44: a Novel Endoplasmic Reticulum Folding Assistant of the Thioredoxin Family. EMBO J. 2002;21:835–844. doi: 10.1093/emboj/21.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Anelli T, Alessio M, Bachi A, Bergamelli L, Bertoli G, Camerini S, Mezghrani A, Ruffato E, Simmen T, Sitia R. Thiol-Mediated Protein Retention in the Endoplasmic Reticulum: the Role of ERp44. EMBO J. 2003;22:5015–5022. doi: 10.1093/emboj/cdg491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Wang ZV, Schraw TD, Kim JY, Khan T, Rajala MW, Follenzi A, Scherer PE. Secretion of the Adipocyte-Specific Secretory Protein Adiponectin Critically Depends on Thiol-Mediated Protein Retention. Mol Cell Biol. 2007;27:3716–3731. doi: 10.1128/MCB.00931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Anelli T, Ceppi S, Bergamelli L, Cortini M, Masciarelli S, Valetti C, Sitia R. Sequential Steps and Checkpoints in the Early Exocytic Compartment During Secretory IgM Biogenesis. EMBO J. 2007;26:4177–4188. doi: 10.1038/sj.emboj.7601844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Kakihana T, Araki K, Vavassori S, Iemura S, Cortini M, Fagioli C, Natsume T, Sitis R, Nagata K. Dynamic Regulation of Ero1α and Peroxiredoxin 4 Localization in the Secretory Pathway. J Biol Chem. 2013;288:29586–29594. doi: 10.1074/jbc.M113.467845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Wang L, Wang L, Vavassori S, Li S, Ke H, Anelli T, Degano M, Ronzoni R, Sitia R, Sun F, et al. Crystal Structure of Human ERp44 Shows a Dynamic Functional Modulation by Its Carboxy-Terminal Tail. EMBO Rep. 2008;8:642–647. doi: 10.1038/embor.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Watanabe S, Harayama M, Kanemura S, Sitia R, Inaba K. Structural Basis of pH-Dependent Client Binding by ERp44, a Key Regulator of Protein Secretion at the ER-Golgi Interface. Proc Natl Acad Sci USA. 2017;114:E3224–E3232. doi: 10.1073/pnas.1621426114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Raje S, Glynn N, Thorpe C. A Continuous Fluorescence Assay for Sulfhydryl Oxidase. Anal Biochem. 2002;307:266–272. doi: 10.1016/s0003-2697(02)00050-7. [DOI] [PubMed] [Google Scholar]
- 362.Benayoun B, Esnard–Feve A, Castella S, Courty Y, Esnard F. Rat Seminal Vesicle FAD-Dependent Sulfhydryl Oxidase. Biochemical Characterization and Molecular Cloning of a Member of the New Sulfhydryl Oxidase/Quiescin Q6 Gene Family. J Biol Chem. 2001;276:13830–13837. doi: 10.1074/jbc.M010933200. [DOI] [PubMed] [Google Scholar]
- 363.Chang TSK, Zirkin BR. Distribution of Sulfhydryl Oxidase Activity in the Rat and Hamster Male Reproductive Tract. Biol Reprod. 1978;17:745–748. doi: 10.1095/biolreprod18.5.745. [DOI] [PubMed] [Google Scholar]
- 364.Ostrowski MC, Kistler WS. Properties of a Flavoprotein Sulfhydryl Oxidase from Rat Seminal Vesicle Secretion. Biochem. 1980;19:2639–2645. doi: 10.1021/bi00553a016. [DOI] [PubMed] [Google Scholar]
- 365.Israel BA, Jiang L, Gannon SA, Thorpe C. Disulfide Bond Generation in Mammalian Blood Serum: Detection and Purification of Quiescin-Sulfhydryl Oxidase. Free Radic Biol Med. 2014;69:129–135. doi: 10.1016/j.freeradbiomed.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Zanata SM, Luvizon AC, Batista DF, Ikegami CM, Pedrosa FO, Souza EM, Chaves DF, Caron LF, Pelizzari JV, Laurindo FR, et al. High Levels of Active Quiescin Q6 Sulfhydryl Oxidase Are Selectively Present in Fetal Serum. Redox Rep. 2005;10:319–323. doi: 10.1179/135100005X83699. [DOI] [PubMed] [Google Scholar]
- 367.Antwi K, Hostetter G, Demeure MJ, Katchman BA, Decker GA, Ruiz Y, Sielaff TD, Koep LJ, Lake DF. Analysis of the Plasma Peptidome from Pancreas Cancer Patients Connects a Peptide in Plasma to Overexpression of the Parent Protein in Tumors. J Proteome Res. 2009;8:4722–4731. doi: 10.1021/pr900414f. [DOI] [PubMed] [Google Scholar]
- 368.Rutkevich LA, Williams DB. Vitamin K Epoxide Reductase Contributes to Protein Disulfide Formation and Redox Homeostasis Within the Endoplasmic Reticulum. Mol Biol Cell. 2012;23:2017–2027. doi: 10.1091/mbc.E12-02-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Chen K, Detwiler TC, Essex DW. Characterization of Protein Disulphide Isomerase Released from Activated Platelets. Br J Haematol. 1995;90:425–431. doi: 10.1111/j.1365-2141.1995.tb05169.x. [DOI] [PubMed] [Google Scholar]
- 370.Flaumenhaft R, Furie B. Vascular Thiol Isomerases. Blood. 2016;128:893–901. doi: 10.1182/blood-2016-04-636456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Cho J, Furie BC, Coughlin SR, Furie B. A Critical Role for Extracellular Protein Disulfide Isomerase During Thrombus Formation in Mice. J Clin Invest. 2008;118:1123–1131. doi: 10.1172/JCI34134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Kim K, Hahm E, Li J, Holbrook LM, Sasikumar P, Stanley RG, Ushio-Fukai M, Gibbins JM, Cho J. Platelet Protein Disulfide Isomerase Is Required for Thrombus Formation but Not for Hemostasis in Mice. Blood. 2013;122:1052–1061. doi: 10.1182/blood-2013-03-492504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Bowley SR, Fang C, Merrill-Skoloff G, Furie BC, Furie B. Protein Disulfide Isomerase Secretion Following Vascular Injury Initiates a Regulatory Pathway for Thrombus Formation. Nat Commun. 2017;8:14151. doi: 10.1038/ncomms14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Bekendam RH, Flaumenhaft R. Inhibition of Protein Disulfide Isomerase in Thrombosis. Basic Clin Pharmacol Toxicol. 2016;119:42–48. doi: 10.1111/bcpt.12573. [DOI] [PubMed] [Google Scholar]
- 375.Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, Killeen N, Erle DJ. The Protein Disulfide Isomerase AGR2 Is Essential for Production of Intestinal Mucus. Proc Natl Acad Sci USA. 2009;106:6950–6955. doi: 10.1073/pnas.0808722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Bergstrom JH, Berg KA, Rodriguez-Pineiro AM, Stecher B, Johansson ME, Hansson GC. AGR2, an Endoplasmic Reticulum Protein, is Secreted into the Gastrointestinal Mucus. PLoS One. 2014;9:e104186. doi: 10.1371/journal.pone.0104186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Dihazi H, Dihazi GH, Bibi A, Eltoweissy M, Mueller CA, Asif AR, Rubel D, Vasko R, Mueller GA. Secretion of ERP57 is Important for Extracellular Matrix Accumulation and Progression of Renal Fibrosis, and Is and Early Sign of Disease Onset. J Cell Sci. 2013;126:3649–3663. doi: 10.1242/jcs.125088. [DOI] [PubMed] [Google Scholar]
- 378.Araujo TL, Zeidler JD, Oliveira PV, Dias MH, Armelin HA, Laurindo FR. Protein Disulfide Isomerase Externalization in Endothelial Cells Follows Classical and Unconventional Routes. Free Radic Biol Med. 2017;103:199–208. doi: 10.1016/j.freeradbiomed.2016.12.021. [DOI] [PubMed] [Google Scholar]