

Abstract
Particulate methane monooxygenase (pMMO) is one of the few enzymes that can activate methane. The metal content of this enzyme has been highly controversial, with suggestions of a dinuclear Fe site or mono-, di-, or trinuclear Cu sites. Crystal structures have shown a mono- or dinuclear Cu site, but the resolution was low and the geometry of the dinuclear site unusual. We have employed quantum refinement (crystallographic refinement enhanced with quantum-mechanical calculations) to improve the structure of the active site. We compared a number of different mono- and dinuclear geometries, in some cases enhanced with more protein ligands or one or two water molecules, to determine which structure fits the two sets of crystallographic raw data best. In all cases, the best results were obtained with mononuclear Cu sites, occasionally with an extra water molecule. Thus we conclude that there is no crystallographic support for a dinuclear Cu site in pMMO.
Keywords: copper, density functional theory, particulate methane monooxygenase, quantum refinement
Graphical abstract
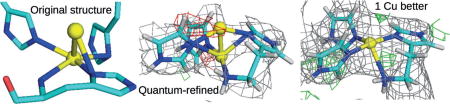
Two crystal structures of particulate methane monooxygenase (pMMO) were studied with quantum refinement. For the putative active site, several different mono- and dinuclear geometries, in some cases with more protein ligands or one or two water molecules, were compared to determine which structure fits the crystallographic raw data best. The results indicate that there is no crystallographic support for a dinuclear Cu site in pMMO.
Methane is one of the most difficult organic substrates to activate, with a C–H bond dissociation energy of 435 kJ mol−1.[1] In biology, there are only two groups of enzymes that can perform this reaction. The first one is composed of the soluble methane monooxygenases,[2] which contain a dinuclear Fe active site, similar to that of the ribonucleotide reductases. This is a well-characterised group of enzymes, and their reaction mechanism is understood in some detail.[2] The second group contains the particulate (i.e., membrane-bound) methane monooxygenases (pMMO), which are the predominant enzymes in methanotrophic bacteria, except under copper-limiting conditions.[1, 3] The pMMOs have been more difficult to characterise, and the metal content has been highly controversial, with suggestions of dinuclear Fe centres as well as mono-, di-, and trinuclear Cu sites.[1, 4–6]
In 2005, the first crystal structure of a pMMO was published (2.8 Å resolution), showing a heterotrimeric α3β3γ3 structure with a dinuclear and a mononuclear Cu site in the soluble periplasmic parts of the PmoB subunit and a monomeric Zn site in the PmoC subunit within the membrane (these three metal sites will be called A, B, and C in the following; Figure 1).[7] Site A was suggested to be the active site and involves three histidine residues, which are conserved in all pMMO sequences, except for those from one phylum of bacteria.[1] All three residues interact with the metals via their side-chain imidazole group, but one of them is the first residue of PmoB and the N-terminal N atom also coordinates to one of the metals. This gives a binding site similar to the histidine brace in the lytic polysaccharide monooxygenase enzymes,[8] although that site involves only two His ligands and binds only one Cu atom. In 2011, a crystal structure was reported at a slightly improved resolution (2.68 Å).[9] It also showed Cu ions in site A, but it was modelled as mononuclear in two of the protomers and dinuclear in one protomer. In addition, it showed a Zn ion in site C (the Zn ions in these structures derive from the crystallization buffer). Three additional crystal structures of pMMO have been published, showing only a single Cu ion in site A.[10]
Figure 1.

a) Trimeric structure of the pMMO (3RGB)[9] with metal sites A–C indicated. Cu ions in yellow, Zn ions in red; PmoA, PmoB, and PmoC subunits in magenta, green, and cyan, respectively. Geometry of Cu site A in protomers 1 (b) and 2 (c), involving three histidine residues and the amino terminal group. Cu ions are shown as yellow balls whereas C, N, and O atoms are shown as cyan, blue, and red sticks, respectively.
Unfortunately, the crystal structures did not settle the location and composition of the pMMO active site. Computational studies have supported site A and have indicated that both mono- and dinuclear Cu sites are reactive enough to oxidise CH4.[3, 11] On the other hand, Chan and co-workers have argued that the active site instead consists of three Cu ions, not observed in the crystal structure, but coordinated by residues from PmoA and PmoC.[5] Other groups have instead suggested that site C, with its conserved ligands, is a dinuclear Fe active site, similar to that in the soluble methane monooxygenases.[6]
A prime problem with the mechanistic studies of pMMO is the poor resolution of the crystal structures, giving metal sites with unclear composition and unusual geometries (Figures 1b, c). These issues may be addressed by using computational methods. Quantum refinement is an approach to supplement crystallographic refinement with quantum mechanical (QM) calculations for a small but interesting part of the protein.[12] It is standard crystallographic refinement in which the empirical potential, used to ensure chemically reasonable bond lengths and angles in the structure, is replaced by more accurate QM calculations. Quantum refinement has been shown to locally improve crystal structures and has been used to determine protonation and oxidation states of metal sites.[13, 14] Herein, we applied quantum refinement to two crystal structures of pMMO[9] with the aim of improving the structure of the Cu site A and determining the number of copper ions observed crystallographically.
We started by performing quantum refinement of site Ain the first protomer of the 3RGB structure[9] with a dinuclear Cu site. This gave a topology similar to that in the starting crystal structure (Figure 1b), but with a more reasonable geometry. The two Cu ions are on top of each other, with all three imidazole groups directed towards the Cu ions. One of the Cu ions is coordinated by all four N atoms while the other binds to only one or two N atoms (Figure 2). The distance to the N-terminal N atom is typically longer than those to the imidazole groups. The exact distances depend on the oxidation states of the two Cu ions, as is shown in Table S1 and discussed in the Supporting Information.
Figure 2.

QM-refined structure of site A in protomer 1 of the 3RGB structure, modelled with two Cu ions in the fully reduced state, including electron-density maps[9] (2mFo−DFc map, contoured at 1.0 σ (gray), and the mFo−DFc maps, contoured at +3.0 σ (green) and −3.0 σ (red)). Atoms coloured as in Figure 1 (and H atoms as white sticks).
The electron-density maps in Figure 2 show that the dinuclear site fits the crystallographic raw data[9] rather poorly. In particular, there is negative difference density around the upper Cu ion, indicating that it is not correctly modelled or that it does not have full occupancy. This was also quantified by the real-space difference density Z-score (RSZD),[15] which is 1.9–2.1 (C2W0 in Table 1). Still, the structures are appreciably better than the starting structure (RSZD=4.7).
Table 1.
RSZD and strain energies (ΔEstr, kJ mol−1) for the various QM-refined structures in the three protomers 1–3.
| PDB[a] | Structure[b] | State[c] | RSZD | ΔEstr | ||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | |||
| 3RGB | C2W0 | Red | 2.1 | 2.8 | 2.0 | 179 | 195 | 176 |
| MV | 1.9 | 2.9 | 2.6 | 224 | 178 | 160 | ||
| Ox | 2.1 | 2.9 | 2.8 | 300 | 247 | 224 | ||
| Y2W2 | Red | 3.2 | 2.2 | 3.0 | 153 | 144 | 160 | |
| MV | 1.5 | 1.9 | 3.2 | 149 | 110 | 104 | ||
| Ox | 1.5 | 2.1 | 2.9 | 158 | 126 | 104 | ||
| Y2W0 | Red | 2.7 | 2.5 | 1.8 | 220 | 226 | 227 | |
| MV | 1.3 | 2.7 | 2.1 | 170 | 136 | 124 | ||
| Ox | 1.3 | 2.8 | 2.2 | 214 | 179 | 167 | ||
| C1W0 | Red | 1.7 | 1.1 | 1.5 | 85 | 62 | 76 | |
| Ox | 1.6 | 1.7 | 1.6 | 75 | 47 | 37 | ||
| C1W1 | Red | 1.7 | 90 | |||||
| Ox | 1.5 | 2.4 | 2.0 | 64 | 53 | 52 | ||
| 3RFR | C2W0 | Red | 2.7 | 311 | ||||
| MV | 1.6 | 292 | ||||||
| Ox | 1.9 | 357 | ||||||
| C1W0 | Red | 1.4 | 0.4 | 0.9 | 88 | 101 | 89 | |
| Ox | 1.3 | 0.4 | 0.9 | 32 | 47 | 37 | ||
| C1W1 | Red | 2.6 | 82 | |||||
| Ox | 2.3 | 1.3 | 1.0 | 42 | 68 | 58 | ||
PDB code.
Number of Cu ions (C2 or C1; Y2 represents planar structures, see Figure 3) and number of water molecules, W0–W2.
Oxidation state of the Cu ion(s), fully reduced (Red), oxidised (Ox), or mixed valence (MV; CuI+CuII).
The strain energy (i.e., the difference between the QM energies of the QM regions when optimised in vacuum or in the crystal),[12] is a measure of how well the metal site fits into the crystal structure. This value is high for all three structures (179–300 kJ mol−1Table 1). Thus QM refinement can improve the structure of site A in pMMO, giving reasonable geometries for the ligands, but the results indicate that the site is not well modelled by two Cu ions. In particular, there are not enough ligands for the higher oxidation states.
Therefore, we tried to enhance the site with two water molecules. After many attempts, we were able to obtain some reasonable structures, and the best are shown in Table 1 (Y2W2). The RSZD scores improved for the higher oxidation states (1.5), and the strain energies were also appreciably better (149–159 kJ mol−1). However, the maps in Figure 3a show that there is no electron density around the water molecules. Consequently, we also optimised the corresponding structures without the water molecules (Y2W0 in Table 1 and Figure 3b, that is, with the Cu ions in the same plane and not on top of each other as in Figure 2). They gave slightly better RSZD scores (down to 1.3), but somewhat higher strain energies (170–220 kJ mol−1).
Figure 3.

QM-refined structures of site A in protomer 1 of the 3RGB structure with a) two or b) no extra water molecules (MV state). Atom colours and electron-density maps[9] are the same as in Figure 2.
We also tried to enhance the QM system with potentially coordinating nearby groups, as suggested by some QM studies.[16] However, as detailed in the Supporting Information, only the carbonyl group of His-137 was found to coordinate, and such structures reproduced the crystallographic raw data quite poorly.
Finally, we instead tried to model site A with a single Cu ion (C1W0 in Table 1). This gave chemically more reasonable structures, and the electron density maps in Figure 4a show that this state fits the crystallographic raw data[9] almost as well as the best dinuclear structures with RSZD=1.6–1.7. Moreover, the strain energies were much lower (75–85 kJ mol−1), reflecting a minimal distortion by the protein.
Figure 4.

QM-refined structures of site A in protomer 1 of the 3RGB structure, modelled with a single Cu ion a) without or b) with an extra water molecule (oxidised state). Atom colours and electron-density maps[9] are the same as in Figure 2.
There remained some positive difference density on top of the Cu ion (green mesh in Figure 4a), which corresponds to the additional Cu ion in the original structure. Yet, it is too close to the Cu ion, and its density is too low for it to be another metal. Therefore, we tried to model it as a water molecule instead (C1W1 in Table 1), which gave reasonable structures for the oxidised state. The electron density maps in Figure 4b show that this state fits the crystallographic raw data[9] even better than the structure without the water molecule, with a RSZD score of 1.5 and a strain energy of only 64 kJ mol−1.
We also studied site A in the other two protomers of the 3RGB structure, which have somewhat different structures of site A to the original structure, as can be seen in Figure 1c. Quantum refinement also gave a more planar structure (Figure 5a; see the Supporting Information for details). However, the same types of structures could also be found in these two protomers (Figures 5b, c), and the results in Table 1 show that a mononuclear site still gives the best RSZD scores and strain energies in both protomers.
Figure 5.

QM-refined structures of site A in protomer 2 of the 3RGB structure with a) two Cu ions, b) two Cu ions and two water molecules, or c) one Cu ion in the reduced state. Atom colours and electron-density maps[9] are the same as in Figure 2.
Finally, we considered also the 3RFR crystal structure,[9] which contains a dinuclear site A only in protomer 1. Again, the results in Table 1 show that the best results were obtained with a mononuclear site in all three protomers (see the Supporting Information for details).
Thus quantum refinement of two crystal structures of pMMO gives quite conclusive results for the nature of the Cu site A: The mononuclear models always yield lower strain energies than the dinuclear models (37–85 kJ mol−1 compared to 104–300 kJ mol−1). Therefore, the dinuclear structures are chemically not reasonable even after quantum refinement.
Likewise, the RSZD score is also lowest for the mononuclear sites in the 3RFR structure and protomers 2 and 3 of 3RGB (by 0.3–0.8). However, for protomer 1 in 3RGB, the lowest score was actually obtained for the structure in Figure 3b, but only marginally (1.3 compared to 1.5 for the best mononuclear site; however, as discussed in the Supporting Information, this depends on how the score is calculated).
To further investigate the catalytic relevance of a mononuclear copper site, we studied a putative reaction mechanism of pMMO based on the mononuclear site in Figure 4. Following the studies by Yoshizawa and Shiota,[11] who studied both dinuclear and mononuclear Cu sites, but used site B (Figure 1) as the mononuclear site (see the Supporting Information for an additional discussion), we started from a CuIII–oxo species. This complex can bind methane and extract a hydrogen atom with a barrier of only 22 kJ mol−1giving an intermediate with a methyl radical and an OH group (Figure 6). Finally, the latter rebounds to the radical, forming methanol with a barrier of 53 kJ mol−1. The net reaction is strongly exothermic. The first half of the process takes place on the triplet spin surface, but at the intermediate, it shifts over to singlet spin, as was also observed in the previous study.[11] These results show that a mononuclear site can oxidise methane and that the activation barriers are actually lower than those obtained in a previous study (79 and 107 kJ mol−1 for the mono- and dinuclear sites, respectively).[11]
Figure 6.

Putative mechanism for the reaction of the mononuclear site with methane. Reaction energies in kJ mol−1 are shown at the top whereas the lower part shows structures of the various states, with key distances indicated in Å. All states were studied both in the singlet and triplet states, indicated by superscript 1 and 3, respectively.
Consequently, we have found no conclusive evidence that the crystal structures of pMMO involve a dinuclear Cu site A. Instead, a mononuclear site fits the crystallographic raw data at least as well. Moreover, it gives chemically much more reasonable structures, which are similar in both crystals and all protomers. This finding, if applicable to functional pMMO in the cell, has strong implications for its possible reaction mechanisms.
Experimental Section
Structures were obtained with quantum refinement[12, 13] using the 3RGB and 3RFR crystal structures at 2.8 and 2.68 Å resolution, respectively.[9] QM calculations were performed with the TPSS[17] method and the def2-SV(P)[18] basis set. QM regions are shown in Figures 2–5.
Supplementary Material
Acknowledgments
This investigation has been supported by grants from the Swedish research council project 2014-5540 (U.R.), from COST through Action CM1305 (U.R.), by NIH grant GM118035 (A.C.R.), and by a scholarship to L.C. from the China Scholarship Council. The computations were performed on computer resources provided by the Swedish National Infrastructure for Computing (SNIC) at Lunarc at Lund University and HPC2N at Umeå University.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201708977.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Lili Cao, Department of Theoretical Chemistry, Lund University, Chemical Centre, P.O. Box 124, 22100 Lund (Sweden).
Octav Caldararu, Department of Theoretical Chemistry, Lund University, Chemical Centre, P.O. Box 124, 22100 Lund (Sweden).
Prof. Amy C. Rosenzweig, Departments of Molecular Biosciences and of Chemistry, Northwestern University, Evanston, IL 60208 (USA)
Prof. Ulf Ryde, Department of Theoretical Chemistry, Lund University, Chemical Centre, P.O. Box 124, 22100 Lund (Sweden)
References
- 1.Culpepper MA, Rosenzweig AC. Crit. Rev. Biochem. Mol. Biol. 2012;47:483–492. doi: 10.3109/10409238.2012.697865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sazinsky MH, Lippard SJ. Methane Monooxygenase: Functionalizing Methane at Iron and Copper. 2015 doi: 10.1007/978-3-319-12415-5_6. [DOI] [PubMed] [Google Scholar]
- 3.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Chem. Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lieberman RL, Shrestha DB, Doan PE, Hoffman BM, Stemmler TL, Rosenzweig AC. Proc. Natl. Acad. Sci. USA. 2003;100:3820–3825. doi: 10.1073/pnas.0536703100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang VC-C, Maji S, Chen PP-Y, Lee HK, Yu SS-F, Chan SI. Chem. Rev. 2017;117:8574–8621. doi: 10.1021/acs.chemrev.6b00624. [DOI] [PubMed] [Google Scholar]
- 6.Martinho M, Choi DW, DiSpirito AA, Antholine WE, Semrau JD, Münck E. J. Am. Chem. Soc. 2007;129:15783–15785. doi: 10.1021/ja077682b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lieberman RL, Rosenzweig AC. Nature. 2005;434:177–182. doi: 10.1038/nature03311. [DOI] [PubMed] [Google Scholar]
- 8.Walton PH, Davies GJ. Curr. Opin. Chem. Biol. 2016;31:195–207. doi: 10.1016/j.cbpa.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Smith SM, Rawat S, Telser J, Hoffman BM, Stemmler TL, Rosenzweig AC. Biochemistry. 2011;269:26344–26348. doi: 10.1021/bi200801z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sirajuddin S, Barupala D, Helling S, Marcus K, Stemmler TL, Rosenzweig AC. J. Biol. Chem. 2014;289:21782–21794. doi: 10.1074/jbc.M114.581363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshizawa K, Shiota Y. J. Am. Chem. Soc. 2006;128:9873–9881. doi: 10.1021/ja061604r. [DOI] [PubMed] [Google Scholar]
- 12.Ryde U, Olsen L, Nilsson K. J. Comput. Chem. 2002;23:1058–1070. doi: 10.1002/jcc.10093. [DOI] [PubMed] [Google Scholar]
- 13.Ryde U. Dalton Trans. 2007:607–625. doi: 10.1039/b614448a. [DOI] [PubMed] [Google Scholar]
- 14.Nilsson K, Ryde U. J. Inorg. Biochem. 2004;98:1539–1546. doi: 10.1016/j.jinorgbio.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Tickle IJ. Acta Crystallogr. Sect. D. 2012;68:454–467. doi: 10.1107/S0907444911035918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiota Y, Yoshizawa K. Inorg. Chem. 2009;48:838–845. doi: 10.1021/ic8003933. [DOI] [PubMed] [Google Scholar]
- 17.Tao J, Perdew JP, Staroverov VN, Scuseria GE. Phys. Rev. Lett. 2003;91:146401. doi: 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- 18.Schäfer A, Horn H, Ahlrichs R. J. Chem. Phys. 1992;97:2571–2577. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.