

Abstract
Understanding gene-gene interaction and its causal relationship to protein-protein interaction is a viable route for understanding drug action at the genetic level, which is largely hindered by inability to robustly map gene regulatory networks. Here, we use biological prior knowledge of family-to-family gene interactions available in the KEGG database to reveal individual gene-to-gene interaction networks that underlie the gene expression profiles of 2 cell line data sets, sensitive and resistive to neoadjuvant docetaxel breast anticancer drug. Comparison of the topology of the 2 networks revealed that the resistant network is highly connected with 2 large domains of connectivity: one in which the RAF1 and MAP2K2 genes form hubs of connectivity and another in which the RAS gene is highly connected. On the contrary, the sensitive network is highly disrupted with a lower degree of connectivity. We investigated the interactions of the neoadjuvant docetaxel drug with the protein chains encoded by gene-gene interactions that underlie the disruption of the sensitive network topology using protein-protein and drug-protein docking techniques. We found that the sensitive network is likely to be disrupted by interaction of the neoadjuvant docetaxel drug with the DAXX and FGR1 proteins, which is consistent with the observed accumulation of cytoplasmic DAXX and overexpression of FGR1 precursors in cancer cell lines. This indicates that the DAXX and FGR1 proteins could be potential targets for the neoadjuvant docetaxel drug. The work, therefore, provides a new route for understanding the effect of the drug mode of action from the viewpoint of the change in the topology of gene-gene regulatory networks and provides a new avenue for bridging the gap between gene-gene interactions and protein-protein interactions which could have deep implications on mainstream drug development protocols.
Keywords: Gene regulatory network, KEGG database, network comparison, drug mode of action, neoadjuvant docetaxel drug, breast anticancer
Introduction
Recent advances in high-throughput DNA microarray technologies have fostered a growing interest in genomics,1 proteomics,2 and drug discovery,3 offering a platform for a deeper understanding of gene-gene interaction and shedding light on previously uncharted avenues for understanding protein-protein and drug-protein interactions. However, a framework for understanding the causal relationship between gene-gene interactions and protein-protein interactions (PPIs) is yet to be developed. In fact, a growing number of studies have indicated that gene expression profiles could be functionally related to the protein expression levels.4 For example, direct interactions between proteins were found to be directly linked to their gene-gene expression profiles.5 Such interactions form a network that underlies the causal relationship between gene-gene interactions and PPIs. Learning gene regulatory networks (GRNs) from gene expression profiles could, therefore, shed light on the underlying PPI networks which could open new avenues for posterior cellular systems investigations.
Using GRNs as a route for mapping the underlying PPI networks has been the focus of a growing number of recent investigations. In a recent study,6 it was indicated that integration between gene expressions and PPI networks could lead to prioritizing and ranking the genes most likely to be associated with breast and lung cancers. Moreover, experimental studies that complementarily use inferred GRNs and PPI networks have been able to suggest missing gene-gene interactions that does not initially show up in the inferred GRNs.7
Currently, different approaches are being pursued to learn gene-gene regulatory networks. Many of these approaches focus on establishing methodologies for statistically unveiling correlated pairs of genes.4,5,8-10 An inherent assumption in these approaches is that if 2 genes show statistical correlation, then it is likely that they influence each other at the cellular level. In fact, coregulation relationships between genes were shown to be related to similarity in their expression profiles.4 Machine learning of graphical models is usually used to unveil correlations between multiple genes, inferring gene-gene regulatory networks. Typical machine learning models that are widely used comprise Boolean networks,4 Bayesian networks,8 dynamic Bayesian networks,9 and dependency networks.10 Preference of a particular graphical model over another is largely dependent on the problem under investigation. However, learning of gene-gene regulatory networks from gene expression data, that are usually sparse, often suffers from overfitting problems, leading to many pairs of genes showing correlation by chance even though they are not biologically related.4 This severely restricts the applicability and utility of machine learning of graphical models techniques in the context of gene-gene regulatory networks.
Interestingly, incorporation of biological prior knowledge in machine learning of graphical models has shown the prospects of efficient learning of gene-gene regulatory networks such as to overcome problems incurred by sparse gene expression data.11–14 In this context, incorporation of biological prior knowledge amounts to supplementing the graphical model technique with available information about gene expression data using cell signaling pathways relevant to the problem under investigation. This leads to restricting the variable space to lower dimensions and thereby circumventing the overfitting problems incurred in dealing with sparse gene expression profile data. Information about cell signaling pathways is available from many biological knowledge databases, such as KEGG,15,16 CPDB,17 REACTOME,18 GOLD.db,19 and PROTEIN LOUNGE.20 However, one shortcoming of the signaling pathways available, for example, from KEGG,15 is that they are usually represented in terms of family-to-family connections between genes rather than individual gene-to-gene connections. For example, the KEGG database shows that the MAPK kinase signaling pathway involves interaction between the fibroblast growth factor (FGF) family (22 genes) and the FGF receptor (FGFR) family (4 genes) without any reference to which individual genes are responsible for this interaction (Figure 1). To the best of our knowledge, there is no existing methodology for unveiling the specific gene-to-gene connections between gene families that underlie the generic connections represented in KEGG signaling diagrams/pathways. This definitely restricts the usability of the biological knowledge expressed in KEGG pathways for learning gene-gene regulatory networks.
Figure 1.
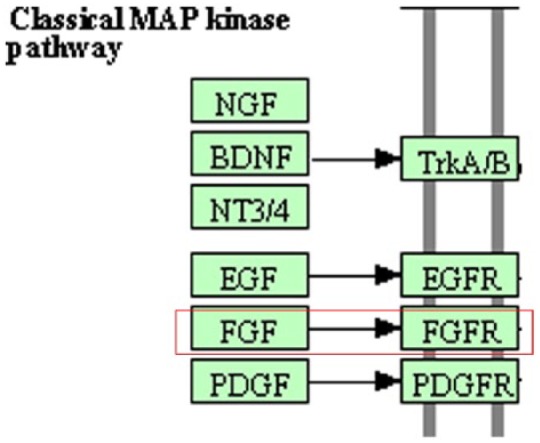
KEGG database illustration of the MAP kinase pathway. Family-to-family interactions are indicated, eg, the FGF (22 genes) and FGFR (4 genes) families, without indication of their gene-to-gene interaction makeup. FGF indicates fibroblast growth factor; FGFR, fibroblast growth factor receptor.
Hence, the purpose of this work is 2-fold: first, to show that using family-to-family biological knowledge available from KEGG pathways along with gene expression profiles available from microarray experiments could be used to unveil individual gene-gene interactions whereof gene-gene regulatory networks could be robustly constructed and second, to use the gene-gene regulatory networks to infer the drug mode of action at the PPI level. To achieve this, we use microarray gene-gene expression profiles from 2 breast cancer cell line samples: resistant and sensitive to neoadjuvant docetaxel drug. We conduct 3 stages of investigation. First, we incorporate prior biological knowledge (family-to-family gene interactions) from the MAPK signaling pathway available from KEGG knowledge database in machine learning and feature selection graphical models to construct gene-gene regulatory networks for both samples. The reason behind choosing MAPK signaling pathway is that because we found that the gene expression profiles in our study are enriched with genes from MAPK signaling pathway (240 genes). Second, we conduct network-network comparison of the resistant and sensitive gene-gene regulatory networks to identify individual gene-gene interactions that were affected by the drug, that is, disappeared from the resistive sample. The affected gene-gene interactions are then mapped to the corresponding encoded protein chains. Third, we identify potential drug-protein interactions that underlie affected gene-gene interactions. To achieve this, we investigate the likelihood of interactions of expressed protein chains and the possibility of disruption of these interactions by the drug using protein-protein and drug-protein docking molecular modeling protocols.
Methodology
Inference of gene-gene regulatory network
Affymetrix breast cancer cell line data that were subjected to the neoadjuvant docetaxel anticancer drug were retrieved from a previous study.21 The data comprise 2 sets of samples: 14 samples that were found to be resistant to the anticancer drug and 10 samples that were found to be sensitive to it. To get meaningful gene expression data sets from the cell lines, the data (24 samples) were normalized using the Robust Multichip Average (RMA) algorithm.6 After normalizations, the genes in the 2 data sets were annotated by their probe-IDs. The probe-IDs were obtained from the hgu95av2.db database available in Bioconductor database, which corresponds to the microarray chips used in the original study.21 The 2 annotated data sets were then mapped to KEGG database using the KEGG.db package available in Bioconductor. This showed that many genes in the 2 data sets are annotated to MAPK signaling pathway (Table 1). The MAPK signaling pathway was therefore used as a prior biological knowledge to guide inference of gene-gene regulatory network as detailed in the following sections.
Table 1.
The total number of genes that were found to be matched between the resistive and sensitive data sets and MAPK signaling pathway in KEGG knowledgebase database.
| Data set type | Genes vs samples |
|---|---|
| Resistant data set | 209 genes, 14 samples |
| Sensitive data set | 209 genes, 10 samples |
The genes were found by mapping probe-IDs to KEGG database using KEGG database (KEGG.db).
Inference of gene-gene regulatory networks under Akaike information criterion-lasso restraints
We modeled the gene-gene regulatory network for each data set using Bayesian graphical models in which the graph represents the probabilistic conditional dependence between the graph vertices, genes in our case. Learning Bayesian graphical models can be achieved by assigning to each vertex (X) a number of parents (pa) and computing the corresponding conditional distributions P as follows7:
In this context, for each gene (X), in the data sets (resistive and sensitive; Table 1), we search for a subset of (causals) parents (xi) that best predict that gene using a linear regression model of the form:
One way to find the best set of parents is to use a score function such as Akaike information criterion (AIC).22 For a given number of parents (n), the AIC uses the residual sum of squares as the likelihood estimate that these parents are good predictors of a particular gene and incorporates a complexity penalty parameter (2p) that increases with the number of parents thereby discouraging overfitting:
In fact, the AIC was shown to give more reliable results compared with alternative methods such as leave-one-out crossvalidation method (LOOCV) especially in gene expression data sets as well as in big data sets.11 The AIC score function in that form focuses on finding best-fit parents, which does not necessarily incur concurrent best estimate of the regressive parameters (βs), namely, model selection. To alleviate this concern, the lasso estimate23,24 was used alongside the AIC such as to ensure that the sum of the absolute values of the model regressive parameters (βs) is below a prespecified threshold parameter (s) using the following penalty function:
Optimization of gene-gene regulatory networks using KEGG prior knowledge restraints and feature ranking
Systematic optimization of the value of the (s) parameter could be prohibitive due to the large dimensionality of the search space defined by the model regressive parameters (βs). To circumvent this problem, we use prior knowledge of gene-gene interactions to guide the optimization process such that all possible subsets of parents for any particular gene are restricted to only those genes that are known to interact with that gene. To achieve this, we used prior knowledge of family-to-family gene connectivity shown in the MAPK signaling pathway, available from the KEGG database. We restricted all possible subsets of parents of any particular gene to the family of genes that show connectivity with the family of that gene. For example, the MAPK signaling pathway (Figure 1) shows that the FGF family of genes has a direct connection with the FGFR family, which indicates that the search space for predicting any of the genes in the FGFR family should be restricted only to the genes in the FGF family. A notable advantage of this procedure is that individual gene-gene connectivity that was not initially visible in the family-to-family connections in the MAPK signaling pathway becomes visible. After restricting the search space for each gene using prior knowledge from MAPK signaling pathway, the univariate filtering feature selection method was used to order the search space for each gene. The filtering feature selection method is a ranking method that incrementally orders all subset of predictors according to correlation coefficients from the highest to the lowest, and therefore it is usually called feature ranking.21
We then test for a particular value of (s) to choose the best subset of parents using the AIC score function under lasso restraint, validating the results using LOOCV. The lowest correlated parent is then iteratively removed (Algorithm 1). Using the feature ranking method with AIC score function, the lasso estimate allows, therefore, to examine different values of (s) for all possible parents of a particular gene. This procedure was found to allow for a more relaxed (s) parameter (Table 2) compared with that using only the lasso estimate described above.
Table 2.
Comparison between the combined ranking method with AIC and the lasso estimate vs the lasso estimate.
| Data set | Corr.AIC.Lasso | Lasso estimate |
|---|---|---|
| Resistive samples | 0.33 | 0.4 |
| Sensitive samples | 0.30 | 0.4 |
Abbreviation: AIC, Akaike information criterion.
The combination method gives a better result than using the lasso estimate only.
| Algorithm 1: THE SKELETON OF HOW THE LASSO ESTIMATE WORKS WHEN EMBEDDED WITH AIC SCORE FUNCTION AND FEATURE SELECTION RANKING METHOD WHILE THE SEARCH SPACE IS RESTRICTED BY MAPK-KEGG SIGNALING PATHWAY. |
|---|
|
for
i = 1 to
length(Genes) do
Y = GENE[i] Features=MAPK.kegg.prior(Y, GENES[–i]) PR = OrderFeatures(Y, fiiter.rank(Features)) for j = 1 to length(PR) do SP = Seareh,SpaceFromLassoPath(Y, PR) return BestFeatures = mini[(AIC(SP))] return FinalError = LOOCV(BestFeatures) PR= PR[,–j] end for return BestFeatures(Y, min(FinalError)) end for |
GRN comparison and identifying potential drug-protein interactions
Network comparison and gene-to-protein mapping
The gene-gene regulatory networks for the 2 data sets, resistive and sensitive to the neoadjuvant docetaxel drug, were compared based on their pairwise gene-gene connectivity. The gene-gene interactions that disappeared from the resistive network were retained and are referred to as “disappeared connections” throughout. Mapping of the genes corresponding to the disappeared connections to the corresponding encoded protein chains was performed through the Human Gene Database (http://www.genecards.org/) and the corresponding structures of protein chains were obtained from the Protein Data Bank (http://www.rcsb.org/). This leads to mapping each gene to several PDB IDs that represent the same encoded protein, yet in different conformations.
Examining interaction between protein pairs corresponding to disappeared gene-gene connections: protein-protein docking
To investigate the physical reasons behind gene-gene correlation corresponding to disappeared connections, pairwise interactions of the protein chains encoded by these genes were investigated. Investigation of PPIs was performed using the ClusPro 2.0 protein-protein docking Web server (http://cluspro.bu.edu/).25 The ClusPro server was recently reported to outperform the best human predictor groups, to select the top-ranked models of PPI complexes and to reliably generate high-quality structures of these complexes from the structures of separately crystallized proteins in the absence of biological information.26 As the mode of interaction of the protein chains is largely unknown a priori, the balanced mode for computing the interaction scores was used in ClusPro. Each PPI mode is represented by ClusPro as a cluster of structure pairs that are structurally similar (root-mean-square deviation < 1.5). The number of structures of each cluster is indicative of the breadth of the free energy valley of the PPI and is used by the ClusPro server to rank different PPI modes. The scores of the top 10 interaction modes were retained.
Examining interaction of neoadjuvant docetaxel drug with protein pairs: drug-protein docking
Blind docking27,28 of the neoadjuvant docetaxel drug was then performed against all of the protein pairs that were retained from protein-protein docking using the AutoDock 4.1 suite of programs.29 Computation of the potential energy grids required by AutoDock at appropriate resolution is quite demanding for large proteins in terms of memory storage and CPU time; therefore, the space around each protein was partitioned using a grid that encompasses the whole protein and extends outward by 40% of its extent in each direction. A grid cell extent of 64 Å was used throughout. For each grid cell, the grid potentials were computed at 0.5-Å resolution for the docking calculations. To alleviate the possibility of missing binding sites that lie across neighboring grid cells, 2 overlapping grids were used that are shifted by 20 Å in each direction. We note that partitioning the space around the protein into grid cells that are treated independently by AutoDock serves 2 purposes: first, it circumvents the prohibitive computational cost of performing blind docking to the whole of each protein in a single run; second, because the same grid cell extent is used throughout, this setup maintains a consistent ratio of the number of docking simulations to protein size, which varies across different protein chains. In all of the docking simulations, the protein was kept rigid and fixed in space, whereas the peptide was placed at a random initial position and orientation. In the drug docking, a search in the space surrounding the protein was performed using the Lamarckian genetic algorithm (GA-LS)30 with no restriction on the drug conformation. For every arbitrary starting position of the drug, 20 hybrid GA-LS docking runs were performed using a population size of 200, a maximum number of energy evaluations of 3 000 000, a maximum number of generations of 27 000, and 300 iterations of local search. The structures of the docked drug were stored and subsequently checked for overlap with the interface regions of each protein pairs. Drug structures that overlap with at least 3 residues of the interface of neighboring proteins, using a 3.0-Å distance threshold between the CA atoms, were selected and the drug-protein complex with the lowest binding free energy was retained in each case.
Figure 2 summarizes the aforementioned methodology steps that underline the approach used in this work.
Figure 2.

Flowcharts for the main steps involved in the 2 stages underlying the approach used in this work for identifying potential protein targets for neoadjuvant docetaxel anticancer drug.
Results and Discussion
Incorporating KEGG prior knowledge and construction of gene-gene regulatory networks
As described in the “Methodology” section, we incorporated prior biological knowledge (gene family-to-family interactions) from the MAPK signaling pathway that we extracted from KEGG knowledgebase into the machine learning and feature selection graphical model to construct gene-gene regulatory networks from the resistive and sensitive data sets (Table 1). The key point is that a gene can be involved in hundreds of biological functions or pathways and it is not a priori known which pathways or gene set annotations are relevant in a given context. Incorporation of prior knowledge allowed for a statistically robust estimation of pairwise correlations between individual genes annotated in the MAPK signaling pathway. These correlations were used for the construction of the gene-gene interaction network for both sensitive and resistive samples (Figure 3A and B).
Figure 3.

Gene-gene regulatory networks constructed from the gene expression profiles for (A) neoadjuvant docetaxel resistive samples and (B) neoadjuvant docetaxel sensitive samples after incorporation of biological prior knowledge from the MAPK signaling pathway. Active and inactive genes are highlighted in gray and white, respectively. The domains of connectivity are delineated by a dashed line in each network and genes with vertex connectivity greater than 4 are highlighted in blue.
Network comparison: resistive vs sensitive
Comparison of the resistant and sensitive gene-gene interaction networks (Figure 3A and B) reveals that the resistant network is highly connected with distinct domains of connectivity, delineated by dashed lines in Figure 3. These domains comprise one large domain in which the RAF1 and MAP2K2 genes are pivotal for its subdomain connection, a domain in which the RAS gene is highly connected and other smaller domains that are sparsely populated. Although the resistant network shows 178 edges, only 52 edges exist in the sensitive network where the network domains, compared with the resistive network, are highly disrupted with lower degree of connectivity. Clearly, the change in the network topology embodies the effect of the docetaxel drug; however, the mechanism underlying this change is largely unknown. After all, it is the interactions between the myriad of proteins encoded by the network genes that cause gene-gene correlations and it is the drug interactions with these proteins that cause cessation of gene-gene correlations. To unveil such a mechanism, we studied these interactions at the molecular level.
Assessment of the docetaxel drug potential to disrupt the network
One way to account for such change in the gene-gene network topology is to assess the extent of disruption incurred by the drug to the interaction of the proteins encoded by the network genes. Such disruption, if any, could be expansive such as to prevail the network or could be localized with a large-scale domino effect (second-order effect) throughout the network. Both scenarios could lead to network disruption; for the purpose of this work, we focused on the former. We focused on the affected network edges (pairs of genes), that is, disappeared from the resistive sample, 126 edges. We mapped each pair of genes to known pairs of protein chain structures, as outlined in the “Methodology” section and performed protein-protein docking simulations, 354 simulations, to delineate their favorable modes of interaction using the ClusPro server.25 This leads to identification of 289 protein pairs that showed favorable modes of interactions. Favorable PPIs in ClusPro are determined by the cluster size of the docked structure clusters; this criterion has been shown to outperform human predictor groups to reliably generate high-quality structures of these complexes from the structures of separately crystallized proteins in the absence of biological information.26 To determine whether the drug is able to disrupt these favorable protein-protein modes of interaction, we performed blind drug-protein docking to each protein, 42 191 simulations, and inspected 3 criteria: (1) favorable drug-protein thermodynamic interaction, (2) drug interaction with the protein-protein interface region is thermodynamically more favorable than other regions on the protein surface, and (3) the drug-protein interaction takes place such that the drug overlaps at least 3 residues in the hotspot regions—residues essential for PPI31,32—in the protein-protein interface. These 3 criteria are tailored to target 3 competitive molecular levels of interactions in the thermodynamic domain such as to rule out the integrity of PPI in the presence of the drug. In a previous work, we used the same strategy for identifying potential lead structures that can disrupt virus assembly.33 Imposing these criteria leads to identification of 34 PPIs, namely, 34 gene-gene interactions that are potentially disrupted by the docetaxel drug (Figure 4A). Interestingly, most of the proteins (and thereby the corresponding genes) that satisfy the above criteria show a high degree of connectivity (Figure 4B) are spread throughout the network (Figure 4C). This indicates that the docetaxel drug disrupts the network by concurrent attacks that target different proteins that exist in different domains of connectivity. To further understand the cellular mechanisms that are possibly disrupted by the docetaxel drug, we need to inspect the specific gene-gene connections that are most likely affected by the drug.
Figure 4.

(A) Drug-protein binding energies (in kJ mol−1) that underlie the disappearance of gene-gene interactions from the resistive sample network compared with the sensitive sample network. (A) Proteins targeted by the drug are indicated in parentheses and (B) their connectivity indices in the resistive sample network. (C) Targeted genes are superposed in black on the resistive sample network.
Docetaxel mode of action: DAXX-mediated interaction
The drug-protein interactions that underlie the disruption of the gene-gene connections were ordered in terms of the corresponding drug-protein binding energy as shown in Figure 3A. Of these interactions, drug interaction with the DAXX-gene–encoded protein (that disrupts DAXX-FAS interaction) is the most thermodynamically favorable interaction (Figure 4A) and the drug interaction with the FGR1-encoded protein (that disrupts FGFR1-FGF13 interaction) corresponds to the highest vertex connectivity (Figure 4B). The molecular details of theses interactions are shown in Figure 5. The molecular interactions shown in Figure 5 show faithful complementarity between interacting protein surfaces and clear overlap between the drug and the hotspots on the target proteins, providing clues for the thermodynamic stability of the PPI and highlighting the drug potential to disrupt these interactions.
Figure 5.

Structural details of protein-protein interactions (left column; yellow and green), hotspot regions (blue) on the protein-protein interface and interaction of the neoadjuvant docetaxel drug (red) with the hotspot regions (right column) for (A) DAXX-FAS interaction and (B) FGFR1-FGF13 interaction.
In agreement with our results, highlighting the DAXX-FAS connection as a potential drug target, the DAXX protein, the protein encoded by the DAXX gene, has been reported to bind specifically to the FAS death domain.34 Moreover, accumulation of more cytoplasmic DAXX in cancer cell lines, for example, due to interaction with a drug, is believed to participate in cellular apoptosis (programmed cell death)35 and thereby providing routes for anticancer drug development.
Our results also suggest that drug FGR1 interaction has the potential to disrupt FGFR1-FGF13 interaction. Overexpression of FGFs has been reported in prostate cancer malignancies such that increased FGF signaling activates multiple signal transduction pathways, all of which play a role in prostate cancer progression.36 This agrees with the high vertex connectivity that FGR1 shows in the resistive network (Figure 4B) and indicates that docetaxel interaction with FGR1 probably breaks the chain of reaction throughout these signaling transduction pathways (disrupting FGR1 connections in the sensitive sample).
Conclusions
We used biological prior knowledge of family-to-family gene interactions available in the KEGG database to reveal individual gene-to-gene interaction networks that underlie the gene expression profiles of 2 cell line data sets: sensitive and resistive to neoadjuvant docetaxel breast anticancer drug. Using machine learning of graphical models to infer the topology of the gene-gene interaction networks revealed that incorporation of biological prior knowledge allows for a more robust regression model optimization compared with mainstream optimization techniques that are statistically driven by AIC penalization and lasso estimation of regression parameters.
Interestingly, comparison of the topology of the 2 networks reveals that the resistant network is highly connected with 2 large domains of connectivity: one in which the RAF1 and MAP2K2 genes form hubs of connectivity and another in which the RAS gene is highly connected. On the contrary, the sensitive network was found to be highly disrupted with a lower degree of connectivity. To unveil the physical reasons behind such change in network topology, we investigated the interactions of the neoadjuvant docetaxel drug with the protein chains encoded by the gene-gene connectivities that disappeared from the resistive sample. We performed protein-protein docking simulations of the encoded protein chains, to delineate their thermodynamically favorable modes of interaction using the ClusPro server.25 The potential of the neoadjuvant docetaxel drug to disrupt these thermodynamically favorable modes of interaction was further investigated by estimation of the drug-protein interaction free energy at the chain-chain interface using the AutoDock program.29 We found that the sensitive network is likely disrupted by interaction of the neoadjuvant docetaxel drug with the DAXX and FGR1 proteins which is consistent with the observed accumulation of cytoplasmic DAXX35 and overexpression FGR1 precursors in cancer cell lines.36 This indicates that he DAXX and FGR1 proteins could be potential targets for the neoadjuvant docetaxel drug.
The work, therefore, provides a new route for understanding the effect of the drug mode of action from the viewpoint of the change in the topology of gene-gene regulatory networks and provides a new avenue for bridging the gap between gene-gene interactions and PPIs which could have deep implications on mainstream drug development protocols.
Footnotes
Author Contributions: The paper is equity contributed by authors. The first author has conducted machine learning and graphical construction through mentioned algorithms and prior to that all preprocessing steps for cell files have been done by the first author. The second author has contributed to protein mapping and docking analysis. The second author has also proofed read the paper thoroughly. The paper has been written equally by authors.
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by King Abdulaziz City for Science and Technology (KACST)—Project Number: LGP-35-120.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1. Molla M, Waddell M, Page D, Shavlik J. Using machine learning to design and interpret gene-expression microarrays. AI Mag. 2004;25:23–44. [Google Scholar]
- 2. Huang JX, Mehrens D, Wiese R, et al. High-throughput genomic and proteomic analysis using microarray technology. Clin Chem. 2001;47:1912–1916. [PubMed] [Google Scholar]
- 3. Iskar M, Zeller G, Zhao XM, van Noort V, Bork P. Drug discovery in the age of systems biology: the rise of computational approaches for data integration. Curr Opin Biotechnol. 2012;23:609–616. [DOI] [PubMed] [Google Scholar]
- 4. Markowetz F, Spang R. Inferring cellular networks—a review. BMC Bioinformatics. 2007;8:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Webb E, Westhead D. The transcriptional regulation of protein complexes: a cross-species perspective. Genomics. 2009;94:369–376. [DOI] [PubMed] [Google Scholar]
- 6. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geiger D, Heckerman D. Learning Gaussian networks. UAI. 1994:235–243. [Google Scholar]
- 8. Heckerman D, Meek C, Cooper G, Holmes D, Jain L. A Bayesian Approach to Causal Discovery: Innovations in Machine Learning. Berlin, Germany; Heidelberg, Germany: Springer; 2006. [Google Scholar]
- 9. Murphy K, Mian S. Modelling gene expression data using dynamic Bayesian networks. Technical Report, Computer Science Division, University of California, Berkeley, CA; 1999. [Google Scholar]
- 10. Heckerman D, Chickering DM, Meek C, Rounthwaite R, Kadie C. Dependency networks for inference, collaborative filtering, and data visualization. J Machine Learning Res. 2000;1:49–75. [Google Scholar]
- 11. Aloraini AAM. Extending the Graphical Representation of Four KEGG Pathways for a Better Understanding of Prostate Cancer Using Machine Learning of Graphical models. York, UK: University of York; 2011. [Google Scholar]
- 12. Aloraini A, Cussens J, Birnie R. Extending prostate cancer KEGG pathways using machine learning of graphical models. Syst Inform World Net. 2010;10:56–67. [Google Scholar]
- 13. Aloraini A, Cussens J, Birnie R. Extending KEGG pathways for a better understanding of prostate cancer using graphical models. Paper presented at: Proceedings of the 3rd International Workshop on Machine Learning in Systems Biology (MLSB); Ljubljana, Slovenia; September 5-6, 2009. [Google Scholar]
- 14. Aloraini A. A directed acyclic graphical approach and ensemble feature selection for a better drug development strategy using partial knowledge from KEGG signalling pathways. Paper presented at: 2014 13th International Conference on Machine Learning and Applications; Detroit, MI; December 3-5, 2014:620–624. [Google Scholar]
- 15. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gold LS, Slone TH, Manley NB, et al. The carcinogenic potency database: analyses of 4000 chronic animal cancer experiments published in the general literature and by the U.S. National Cancer Institute/National Toxicology Program. Environ Health Perspect. 1991;96:11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fabregat A, Sidiropoulos K, Garapati P, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2016;44:D481–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hackl H, Maurer M, Mlecnik B, et al. GOLD.db: genomics of lipid-associated disorders database. BMC Genomics. 2004;5:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Besaw ME. Protein lounge. JMLA. 2013;101:164. [Google Scholar]
- 21. Chang JC, Wooten EC, Tsimelzon A, et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 2003;362:362–369. [DOI] [PubMed] [Google Scholar]
- 22. Dietterich TG. Approximate statistical tests for comparing supervised classification learning algorithms. Neural Comput. 1998;10:1895–1923. [DOI] [PubMed] [Google Scholar]
- 23. Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc B Met. 1996;58:267–288. [Google Scholar]
- 24. Tibshirani R. Regression shrinkage and selection via the lasso: a retrospective. J R Stat Soc B. 2011;73:273–282. [Google Scholar]
- 25. Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. [DOI] [PubMed] [Google Scholar]
- 26. Kozakov D, Hall DR, Beglov D, et al. Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13-19. Proteins. 2010;78:3124–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hetenyi C, van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002;11:1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hetényi C, van der Spoel D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006;580:1447–1450. [DOI] [PubMed] [Google Scholar]
- 29. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garrett MM, David SG, Robert SH, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 31. Bowman AL, Nikolovska-Coleska Z, Zhong H, Wang S, Carlson HA. Small molecule inhibitors of the MDM2-p53 interaction discovered by ensemble-based receptor models. J Am Chem Soc. 2007;129:12809–12814. [DOI] [PubMed] [Google Scholar]
- 32. Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. [DOI] [PubMed] [Google Scholar]
- 33. ElSawy KM, Twarock R, Verma CS, Caves LSD. Peptide inhibitors of viral assembly: a novel route to broad-spectrum antivirals. J Chem Inf Model. 2012;52:770–776. [DOI] [PubMed] [Google Scholar]
- 34. Yang X, Khosravi-Far R, Chang HY, Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997;89:1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mo YY, Yu Y, Ee PL, Beck WT. Overexpression of a dominant-negative mutant Ubc9 is associated with increased sensitivity to anticancer drugs. Cancer Res. 2004;64:2793–2798. [DOI] [PubMed] [Google Scholar]
- 36. Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr Relat Cancer. 2004;11:709–724. [DOI] [PubMed] [Google Scholar]