

Abstract
Dental infection is risk for preterm birth (PTB) through unclear mechanisms. We established a dental infection-induced PTB mouse model, in which Porphyromonas gingivalis (P.g.) induced PTB by 2 days. We analysed pathogenic factors contributing to PTB and their effects on trophoblasts in vitro. TNF-α, IL-8, and COX-2 were upregulated in P.g.-infected placenta. Galectin-3 (Gal-3), an immune regulator, was significantly upregulated in placenta, amniotic fluid, and serum. In vitro, P.g.-lipopolysaccharide (P.g.-LPS) increased TNF-α and Gal-3 in trophoblasts via NF-κB/MAPK signalling. Gal-3 inhibition significantly downregulated P.g.-LPS-induced TNF-α production. TNF-α upregulated Gal-3. Gal-3 also increased cytokines and Gal-3 through NF-κB/MAPK signalling. Moreover, Gal-3 suppressed CD-66a expression at the maternal-foetal interface. Co-stimulation with Gal-3 and P.g.-LPS upregulated cytokine levels, while Gal-3 plus Aggregatibacter actinomycetemcomitans (A.a.)- or Escherichia coli (E. coli)-LPS treatment downregulated them, indicating the critical role of Gal-3 especially in P.g. dental infection-induced PTB. P.g.-dental infection induced PTB, which was associated with Gal-3-dependent cytokine production. New therapies and/or diagnostic systems targeting Gal-3 may reduce PTB.
Introduction
Preterm birth (PTB) is a leading cause of infant morbidity and mortality in new-borns. PTB is characterized as delivery at <37 weeks of gestation1. The frequency of PTB is approximately 5% in Japan and several European countries, 12–13% in the United States, and 18% in some African countries2. PTB is associated with 75% of perinatal mortality and >50% of the long-term morbidity, such as cerebral palsy, bronchopulmonary dysplasia, intraventricular haemorrhage, and necrotizing enterocolitis3. Maternal or fetal indications account for 30% of PTB cases (so-called indicated PTB), while spontaneous PTB with or without premature preterm rupture of the membranes accounts for 25% or 45% of PTB cases, respectively1. Causes of spontaneous PTB include infection, inflammation, vascular disease, and uterine over-distention. Infections and infection-driven inflammatory responses are considered to be the leading causes of spontaneous PTB4. It is well accepted that various stimulatory factors such as proinflammatory cytokines, chemokines, and prostaglandins trigger the onset of labour near the end of pregnancy by inducing cervical ripening and myometrium activation. These stimulatory factors can be induced by intrauterine infection and then lead to PTB5. It is reported that intrauterine infection increases the production of prostaglandins, TNF-α, IL-6, and IL-8, which leads to more frequent and intense uterine contractions with consequential PTB6,7.
Periodontal disease is a chronic infectious disease, mainly caused by gram-negative periodontal pathogens, which can enter the blood circulation and disseminate throughout the entire body. Periodontitis is considered a risk factor of systemic diseases such as cardiovascular disease and diabetes mellitus8,9. In 1996, the results from a case-control study performed by Offenbacher et al. suggested that maternal periodontal disease was associated with a 7.9-fold increased risk for PTB/low birth weight10. In pregnant women with a diagnosis of threatened premature labour, periodontal pathogens were detected both in the periodontal pocket and in the amniotic fluid11. In addition, the presence of periodontal pathogens in the placenta was confirmed in cases of chorioamnionitis where PTB was also indicated12. However, the underlining mechanisms of periodontitis-induced PTB remain unclear.
Several experimental studies have revealed that Porphyromonas gingivalis (P.g.), a main periodontal pathogen, can invade the placental-fetal barrier to induce pregnancy complications in rodents and rabbit animal models13–15. However, the bacteria-delivery systems used, such as intravenous or peritoneal injection, the subcutaneous chamber model, and oral-gavage are quite different from the natural infection route used by P.g. to infect patients. Recently, we established a dental infection-induced PTB mouse model that showed long-term low-grade inflammation, which ideally imitates the clinical condition of pregnant women with periodontitis. In the model, P.g. dental infection induced PTB (by an average of 2 days). P.g. originating from a dental infection was detected in placental tissue by immunohistochemistry and PCR. Moreover, in P.g.-infected placenta, upregulation of inflammatory mediators such as TNF-α and COX-2 (inducible synthetase of prostaglandins) was observed, which can promote the initiation of labour16.
In the present study, our aim was to clarify PTB-related molecules that can differentiate PTB from term birth (TB), using our established animal model. We identified an interesting immune-regulatory molecule, Galectin-3 (Gal-3), which was upregulated in P.g.-infected placental tissue compared control placentas. Moreover, the underlining mechanisms, in which Gal-3 induces PTB, were studied a using human trophoblast cell line.
Results
Dentally Applied P.g. Induced Defects in Mouse Placental Tissue through Inflammatory Mediators
We confirmed that histological damage occurred in P.g.-infected placental tissue, as we reported previously16. In normal placentas of gd15, no regressive changes were observed, and the decidua was firmly attached to the uterus. However, in P.g.-infected placental tissue, the amniotic membrane was degenerated, and necrosis was seen in the labyrinth and decidua (Fig. 1A). COX-2, Gal-3, and TNF-α mRNA expressions were significantly upregulated in P.g.-infected placental tissue (*p < 0.05) (Fig. 1B and C).
Figure 1.

Dentally applied P.g. induced tissue damages and upregulation of PTB related cytokines in mouse placental tissues. (A) Representative histological findings of gd15-placental tissue of the NC group and the P.g.-infected group by H&E staining (n = 6, each group). The amniotic membrane degenerated, and trophoblasts and endothelial cells were necrotic in the P.g.-infected group. Scale bars; 100 μm. (B) Representative COX-2, Gal-3, IL-8, and TNF-α mRNA expression data in placental tissues in the NC and P.g.-infected groups. GAPDH expression was detected as an internal control. (C) Quantitative real time PCR analysis of COX-2, Gal-3, IL-8, and TNF-α. *p < 0.05 significant difference between NC and P.g.-infected group.
Dentally Applied P.g. Increased Gal-3 Expression in the Placenta, Amniotic Fluid, and Maternal Serum
Because the expression of Gal-3 mRNA, an important immune regulatory molecule, was prominently upregulated in P.g.-infected placental tissue (Fig. 1B,C), Gal-3 immunoexpression was examined to confirm its production in the placenta. Interestingly, ubiquitous Gal-3 staining was detected in only P.g.-infected placenta of 15gd mice. Gal-3 was expressed not only in inflammatory macrophages, but also in trophoblasts (Fig. 2A).
Figure 2.

Gal-3 expression in the P.g.-infected group was upregulated. (A) Gal-3 immunohistochemistry in gd15-placental tissue of the NC group and P.g.-infected group. In addition to macrophages, trophoblasts strongly expressed Gal-3. Scale bars, 100 μm. ELISA analysis of the Gal-3 concentration in amniotic fluid (B) and maternal serum (C) from the NC group and P.g.-infected group (n = 6, each). Data are presented as the mean ± SD. **P < 0.01; Student’s t-test.
Because intensive Gal-3 production in P.g.-infected placenta may induce elevated Gal-3 levels in the amniotic fluid and maternal serum, we examined Gal-3 levels in both locations. As expected, Gal-3 concentrations in the amniotic fluid (Fig. 2B) and maternal serum (Fig. 2C) were significantly increased in the P.g.-infected group, compared to those in the NC group (**P < 0.01).
Gal-3 Plays an Important Role in the Inflammatory Activity of P.g.-LPS
It is well known that Gal-3 is one of important products from macrophages with LPS stimulation and is a key immunoregulator to produce inflammatory cytokines17.
P.g.-LPS upregulated Gal-3 secretion from trophoblasts (HTR-8 cells: a; p < 0.01) at day 3 (Fig. 3A) as well as macrophages (THP-1 cells: p < 0.01) at day 1, 2 (Supplement Fig. 1A) and endothelial cells (Huh-1 cells: p < 0.01) at day 3 (Supplement Fig. 1B). At 3 days after P.g.-LPS stimulation, TNF-α significantly secreted from HTR-8 cells (a; P < 0.01; Fig. 3B). To examine the effect of TNF-α produced by P.g.-LPS stimulation on Gal-3 production, HTR-8 cells were incubated with rhTNF-α and Gal-3 secretion was examined in the culture medium. Significantly increased Gal-3 production was detected after 3 days rhTNF-α-stimulation (a; p < 0.01, Fig. 3A).
Figure 3.

Gal-3 played an important role in the Inflammatory activity of P.g.-LPS. (A) Gal-3 levels in culture media were analysed by ELISA. P.g.-LPS (1 µg/ml) significantly induced Gal-3 in trophoblasts (HTR-8 cells) at 3 days. HTR-8 cells incubated with rhTNF-α (10ng/ml) also significantly produced Gal-3 at 3 days after incubation. (B) TNF-α levels produced from HTR-8 cells with P.g.-LPS (1 μg/ml) were measured. After 3 days, TNF-α production was significantly upregulated. (C,D) HTR-8 cells were pre-treated with or without CAPE (1 μg/ml) for 4 h, pre-exposed to dicumarol/SB203580/U0126 (10 μM) for 30 min, and then grown with or without P.g.-LPS (1 µg/ml) or rhTNF-α (10ng/ml) for 3 days. The Gal-3 or TNF-α concentration in the culture media was analysed by ELISA. CAPE, and U0126 inhibited Gal-3 production induced both by P.g.-LPS and rhTNF-α (C). All inhibitors used were significantly reduced TNF-α secretion from HTR-8 cells at 3 days after LPS-stimulation. Data are presented as the mean ± SD. a and b: significant difference (P < 0.01, p < 0.05) between untreated control, c: significant difference (P < 0.01) between P.g.-LPS/rhTNF-α stimulated and control group; 1-way ANOVA. (E,F) HTR-8 cells were treated with P.g.-LPS (1 μg/ml) with or without a Gal-3 blocking antibody (clone M3/38) or rat control IgG for 3 Days. Gal-3 antibody significantly reduced P.g.-LPS induced TNF-α production (E). COX-2, IL-8, and TNF-α mRNA-expression levels were examined. GAPDH expression was detected as an internal control (F). Data are presented as the mean ± SD. **P < 0.01; 1-way ANOVA. The experiments were performed at least 3 times with similar results.
To clarify the signaling pathways relating to Gal-3 production, inhibitors were used. CAPE (an NF-κB inhibitor) and U0126 (an ERK inhibitor) suppressed P.g.-LPS and rhTNF-α induced upregulation of Gal-3 secretion (c; p < 0.01, Fig. 3C). Inhibition of NF-κB signalling significantly reduced constitutive secretion of Gal-3 in HTR-8 cells (c; p < 0.01, Fig. 3C).
All examined NF-κB and MAPK signalling inhibitors completely downregulated P.g.-LPS induced TNF-α production (c; p < 0.01, Fig. 3D). Moreover, the inflammatory activity of P.g.-LPS was restricted by treatment with a Gal-3 blocking antibody. P.g.-LPS-induced TNF-α secretion by HTR-8 cells was significantly reduced to control level after Gal-3 antibody treatment (**P < 0.01, Fig. 3E). Gal-3, IL-8, and TNF-α gene-expression levels were down-regulated by Gal-3 blocking antibody but not COX-2 (Fig. 3F).
Gal-3 Activated the JNK and NF-κB Pathways, Playing an Important Role in the Inflammatory Activity of P.g.-LPS
rhGal-3 treatment activated the phosphorylation of JNK, c-Jun, ERK and p65 within 5 min. p-JNK and pERK peaked at 5 min and lasted till 15 min. On the other hand, p-c-JUN and p-p65 gradually upregulated in 60 min (Fig. 4A). Similarly, rhGal-3 induced Gal-3 mRNA expression in HTR-8 cells (Fig. 4B). rhGal-3 also significantly upregulated the mRNA expression of inflammatory mediators such as COX-2, IL-8, and TNF-α (Fig. 4B).
Figure 4.

Gal-3 activated the JNK and NF-κB pathways and played an important role in the inflammatory activity of P.g.-LPS. (A) Trophoblasts (HTR-8 cells) were stimulated with rhGal-3 (0.5 μg/ml), and cell lysates were examined by immunoblot analysis. Phosphorylation of JNK, c-Jun, ERK and p65 was evident. (B) HTR-8 cells were stimulated by different concentrations of rhGal-3 (0, 0.5, 2.5 μg/ml) for 48 h. The mRNA-expression levels of COX-2, Gal-3, IL-8, and TNF-α were analysed by PCR and real time PCR. The expression of cytokines were markedly upregulated. (C) TNF-α production from rhGal-3 (1 μg/ml)-stimulated HTR-8 with or without CAPE (1 μg/ml) for 4 h, pre-exposed to dicumarol/U0126 (10 μM) for 30 min were measured by ELISA after 2 days. Data are presented as the mean ± SD. *P < 0.05; **P < 0.01; 1-way ANOVA. The experiments were performed at least 3 times with similar results.
To clarify the signaling pathways relating Gal-3-induced TNF-α production, inhibitors were used. Inhibition of NF-κB, ERK and JNK signaling pathway significantly reduced Gal-3-induced TNF-α secretion (Fig. 4C).
Gal-3 Inhibition Using a Blocking Antibody or Gene Knockdown Increased CD66a Expression
CD66a is an adhesion molecule that is normally expressed in trophoblasts, functioning as an important adhesion mediator at the maternal-foetal interface18 and a Gal-3 receptor on neutrophils19. Both rhGal-3 and P.g.-LPS treatment decreased CD66a mRNA expression, and a Gal-3 blocking antibody rescued CD66a downregulation induced by P.g.-LPS (Fig. 5A). Moreover, siRNA-mediated Gal-3 knockdown caused CD66a upregulation at both the mRNA (Fig. 5B) and protein levels (Fig. 5C).
Figure 5.

Gal-3 inhibition via a blocking antibody or gene knockdown increased CD66a expression in trophoblasts. (A) Trophoblasts (HTR-8 cells) were stimulated for 48 h with rhGal-3, P.g.-LPS, or a Gal-3 blocking antibody together with P.g.-LPS. CD66a mRNA expression was examined after exposure to P.g.-LPS and rhGal-3 with or without the Gal-3 antibody. (B,C) siRNA-mediated Gal-3 knockdown was performed for 48 h. CD66a expression at both the mRNA and protein levels. GAPDH or β-actin expression was detected as an internal control. The experiments were performed at least 3 times with similar results.
Gal-3 could not Bind to P.g.-LPS and Both Molecules Synergistically Upregulated the Expression of Inflammatory Mediators
Gal-3 can bind LPS and might inhibit LPS-induced proinflammatory responses20. Here, a binding assay was performed to examine the relationship between LPS and Gal-3. Gal-3 could bind to LPS from A.a. and E. coli; however, P.g.-LPS and Gal-3 did not bind each other (Fig. 6A). Co-stimulation of rhGal-3 and P.g.-LPS upregulated COX-2, and TNF-α mRNA expression, compared with their levels of P.g.-LPS treatment alone (Fig. 6B, supplement Fig. 2A). In contrast, co-stimulation of rhGal-3 and E. coli-LPS significantly downregulated TNF-α, IL-8 and COX-2 versus LPS treatment alone. Co-stimulation of rhGal-3 and A.a.-LPS also showed a tendency to downregulate these molecules (Fig. 6B, Supplement Fig. 2A).
Figure 6.
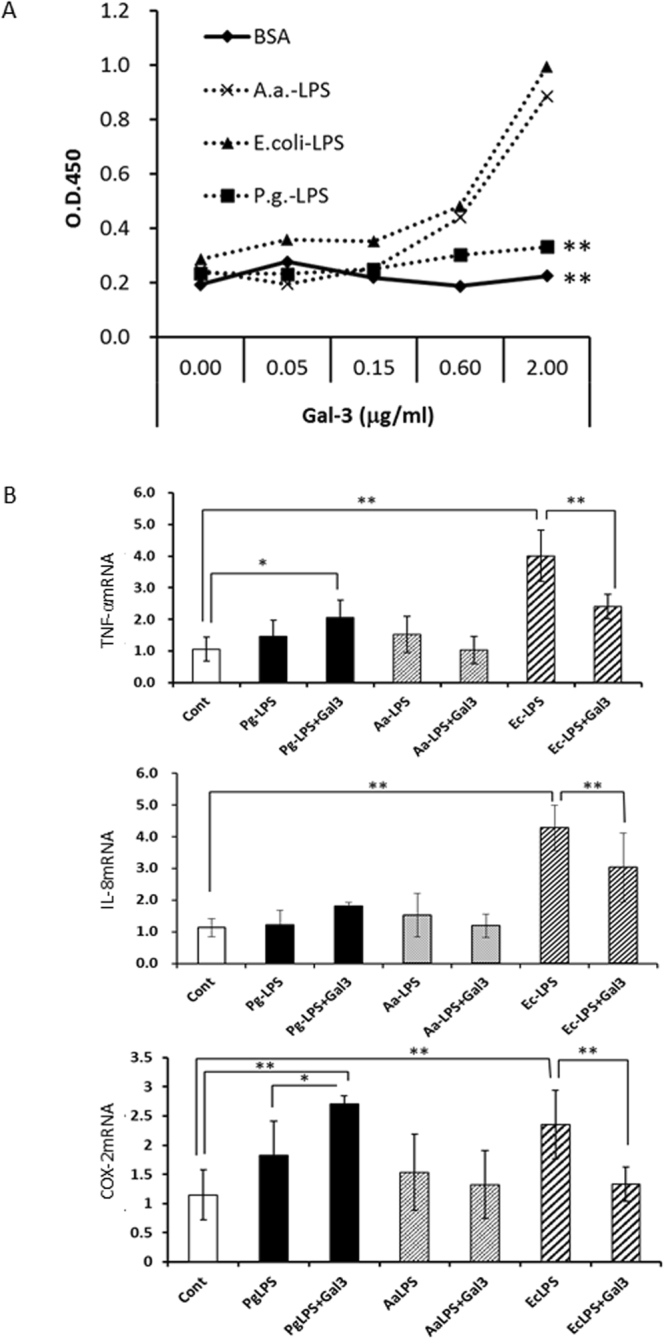
Gal-3 did not bind to P.g.-LPS, and both molecules synergistically upregulated the expression of inflammatory mediators. (A) Studying the binding affinity of Gal-3 for LPS from A.a., E. coli, or P.g., as described in the Material and Methods section. P.g.-LPS did not bind Gal-3. (B) Trophoblasts (HTR-8 cells) were stimulated for 48 h with LPS (100 ng/ml) from P.g., A.a., or E. coli with or without rhGal-3 (0.5 µg/ml). Synergistical upregulation of cytokine mRNA expression was evident by P.g- LPS. but not by A.a. or E. coli-LPS using the real time PCR analysis. GAPDH was used as an internal control. Data are presented as the mean ± SD. *P < 0.05; **P < 0.01; 1-way ANOVA. Experiments were performed at least three with similar results.
Discussion
In this study, we used a mouse model of P.g.-dental infection. P.g., which is a main causative pathogen of chronic and periapical periodontitis, is infected from the pulp chamber because it requires anaerobic growth conditions21. The pulp chamber provides ideal anaerobic conditions and is the natural infecting way of periapical periodontitis. Previously, we demonstrated that P.g. remained alive inside the pulp chamber, reproduced long enough to induce periapical periodontitis, and maintained upregulated LPS levels in the serum22. Moreover, we confirmed the establishment of chronic, low-grade systemic inflammation, including upregulated TNF-α, IL-17, IL-6, and IL-1β serum levels at 6-weeks post-P.g. infection, the time of mating16. These data indicated that the mouse model successfully mimicked the clinical conditions of pregnant women with chronic dental infection. We also reported previously that P.g.-infected mice showed PTB by 2 days, on average16, which was comparable with that observed by direct intrauterine injection of E. coli (104 CFU)23.
Interestingly, Gal-3, TNF-α and COX-2 immunoexpression dramatically increased in P.g.-infected placenta in this study. The Gal-3 level in the amniotic fluid and serum was also significantly upregulated in P.g.-infected mice (Fig. 2). Gal-3, a beta-galactoside-binding lectin, plays both pro-inflammatory and anti-inflammatory roles in the inflammation process24. Extracellular Gal-3 contributes to neutrophil adhesion, macrophage migration, and endocytosis, indicating the pro-inflammatory roles of Gal-3. Therefore, it is possible that extracellular Gal-3 may play an important role in the onset of PTB. However, to our knowledge, the relationship between Gal-3 and PTB has not yet been elucidated.
It is well accepted that bacterial LPS contributes to pathogenesis of PTB through TLR425. LPS is a main virulence factor of P.g., and elevated LPS levels in the sera of P.g.-infected mice were observed in a previous study22. We reported that TNF-α, IL-8 and COX-2 expression, which relate to onset of PTB, were secreted from HTR-8 cells with P.g.-LPS stimulation16. Ren et al. also demonstrated that P.g.-LPS promoted IL-8 secretion from HTR-8 cells26.
P.g.-LPS stimulation increased Gal-3 expression in placental cells (trophoblasts) by activating the NF-κB and MAPK signaling pathways (Fig. 3A,C). Moreover, exogenously applied rhGal-3 or rhTNF-α, which were produced by LPS-stimulated trophoblasts, also increased Gal-3 production. It was previously reported that extracellular Gal-3 modulated the production of cytokines such as IL-1β27, IL-528, and IL-829 not only in immune cells, but also in another cell types30. In the present study, Gal-3 was also responsible for upregulating proinflammatory mediators, such as COX-2, IL-8, and TNF-α. Gal-3 and TNF-α, which were produced by P.g.-LPS stimulation in P.g.-infected placenta, induced additional Gal-3-production, finally leading to excessive cytokine expression. It was suggested that in placenta of P.g.-odontogenic infected animals, harmful cycle in cytokine through Gal-3 may be established. Moreover, inhibition of Gal-3 by Gal-3 neutralizing antibody significantly suppressed P.g.-LPS induced TNF-α production, resulting in reduction of IL-8 and COX-2. We consider that the regulation of Gal-3 in placenta may be a possible preventive strategy of PTB.
Our previous report showed that immunohistochemically P.g. detected in the placental cells (trophoblasts) of P.g.- dental infection mouse. In vitro study showed that HTR-8 cells with P.g.-infection increased secretion of COX-2, TNF-α, and IL-816. Several reports indicated that infection of trophoblasts by P.g. induced cytokine expression26,31 and apoptosis32,33 through MAP kinase signalling34. It is indicating that P.g.-infection to trophoblasts also has impacts on pathogenesis of PTB caused by periodontitis.
In contrast, it was also reported that Gal-3 shows an anti-inflammatory effect by binding to LPS. Mey et al.35 reported that Gal-3 binds to LPS from gram-negative bacteria, and Li et al. showed the inhibitory effect of Gal-3 in E. coli-LPS-induced inflammation20. We examined the binding affinity between Gal-3 and various types of LPS. Here, we found that Gal-3 bound to LPS from E. coli and A.a., but not P.g.-LPS, in agreement with the report by Kato et al.36. LPS consists of a hydrophobic lipid embedded in the outer leaflet of the outer membrane, a core oligosaccharide (OS), and the O-polysaccharide side chain composed of several repeating units37. It was reported that heptose residues in core OS of LPS provided the attachment point with various types of LPS. Using heptose mutation, Quattroni et al. found that heptose in the core OS in Neisseria meningitidis was necessary for the Gal-3 binding to LPS38. The LPS core OS molecules of E. coli and A.a. contain heptose residues39, whereas P.g.-LPS lacks heptose residues37,40. Co-stimulation of rhGal-3 and E. coli-LPS or A.a.-LPS significantly downregulated cytokine levels versus LPS treatment alone. However, since the anti-inflammatory effect of Gal-3 is limited in early stage of LPS stimulation, the prolonged and excessive production of Gal-3 finally may induce proinflammatory effect. On the other hand, co-stimulation with rhGal-3 and a low dose of P.g.-LPS revealed a synergistic effect on the upregulation of inflammatory mediators. Since P.g.-LPS can synergistically induce intense cytokine production in early stage of placental inflammation, in which abundant Gal-3 present, even P.g with low virulence LPS than E. coli, can be a risk for PTB.
Our investigation showed that Gal-3 inhibited CD66a expression. CD66a is an adhesion molecule of the carcinoembryonic antigen family. Bamberger et al.18 reported that CD66a was strongly expressed by intermediate trophoblasts, endometrial epithelial cells, and endothelial cells at the maternal-fetal interface throughout pregnancy. Thus, CD66a may play an important role in adhesive trophoblast-endometrial and trophoblast-endothelial cell interactions. CD66a downregulation by Gal-3 may induce cell adhesion breakdown at the maternal-fetal interface and contribute to the initiation of placental abruption from the uterus.
Moreover, we clarified that Gal-3 levels in the amniotic fluid and maternal serum of P.g.-infected mice were significantly higher than those of NC mice, indicating that the Gal-3 level may be potent predictive biomarker for PTB. Predictive biomarkers for PTB are urgently needed to reduce PTB caused by maternal infection/inflammation during the gestation period. Several inflammatory biomarkers present in amniotic fluid have been proposed as predictors of intra-amniotic inflammation and preterm labour. IL-6 has shown the best sensitivity for predicting infection in patients with preterm labour7,41. Recently, the combined use of IL-6 and proteomic biomarkers was recommended42. However, their clinical use is still limited. As mentioned above, we demonstrated significant Gal-3 elevation in the amniotic fluid of P.g. dentally infected mice. Gal-3 in the amniotic fluid may be an important predictive marker for PTB. However, examination of the amniotic fluid is invasive testing that causes an increased risk of abortion. Non-invasive diagnostic testing is key for screening pregnant individuals. Maternal serum is easily collected without threatening the pregnancy. In this study, we also detected Gal-3 upregulation in the maternal serum. This is the first investigation of the association of serum Gal-3 upregulation with PTB. In human serum, Gal-3 is constitutively detected with mean serum levels of approximately 5 ng/ml in healthy adults43. Increase serum Gal-3 is associated with various pathological conditions, such as immune responses44 and malignancy24. Demmert et al.45 demonstrated that the Gal-3 level was significantly higher in cord blood of small-for-gestational-age infants. However, there is still no report showing a relationship between maternal serum Gal-3 levels and PTB. In the present study, we detected significant upregulation of the serum Gal-3 level at 15gd in P.g. dentally infected mice with PTB. Serum Gal-3 may serve as a novel predictive biomarker for PTB, although further clinical studies are needed to clarify the usefulness of Gal-3 upregulation in the amniotic fluid and/or serum as a predictive marker of PTB.
Based on our results, we considered possible mechanisms responsible for P.g. dental infection-induced PTB through Gal-3 (Fig. 7). Dental P.g. infection increases inflammatory cytokines in the serum and induces P.g.-LPS translocation in the placenta through the blood circulation. In the placenta, P.g.-LPS upregulated inflammatory mediators such as TNF-α, IL-8, and COX-2 directly or indirectly through Gal-3 production. Upregulated TNF-α can also induce Gal-3 production, which may help establish the excessive production of pro-inflammatory mediators through Gal-3. Gal-3 inhibits CD66a expression, which results in the breakdown of cell adhesion and induces placental abruption. In addition, the regulation of inflammatory mediators induces cervical ripening and activation of the myometrium. Taken together, the findings indicate that Gal-3 plays an important role in PTB induced by dental infection with P.g. Thus, dental treatments to prevent and/or eliminate P.g. infection should be meticulously performed throughout the world to reduce the PTB rate. Gal-3 should be considered as one of the PTB biomarkers in the amniotic fluid and/or serum, and should be considered as a possible new target for preventing PTB.
Figure 7.
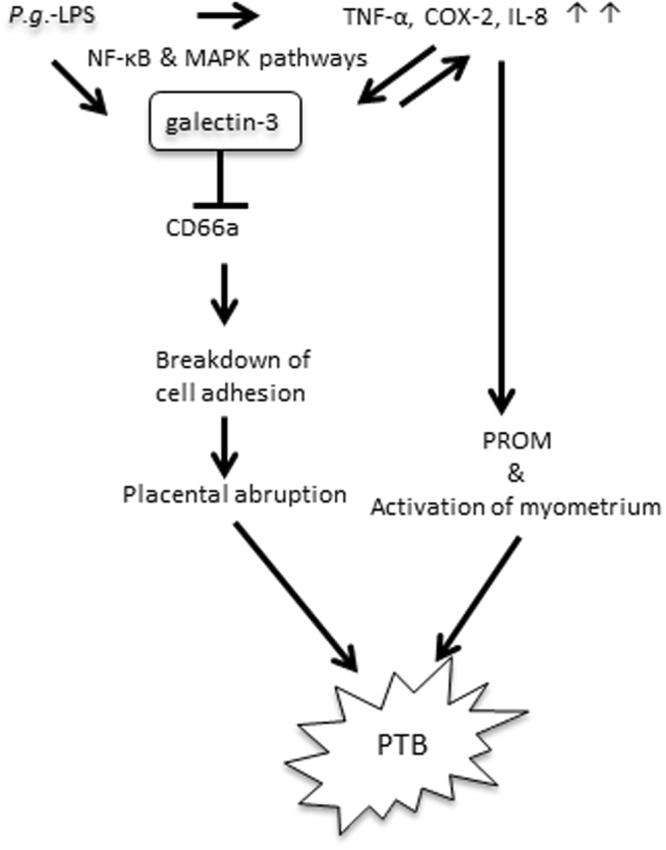
The possible mechanisms responsible for P.g. dental infection induced PTB through Gal-3. Dental infection of P.g. increases P.g.-LPS and inflammatory cytokines in serum. In placenta, P.g.-LPS activates NF-κB and MAPK pathways and induces production of TNF-α, IL-8 and COX-2 that lead to premature rupture of membrane (PROM) and activation of myometrium. Moreover, P.g.-LPS and cytokines produced by P.g.-LPS stimulation upregulate Gal-3. The produced Gal-3 may also contribute to excessive production of cytokines. Eventually, excessively produced cytokines may result in PTB. Alternatively, P.g.-LPS induced Gal-3 may inhibit CD66a. Breakdown of cell adhesion at maternal-infant interface leads to placental abruption and PTB.
Materials and Methods
Animal Model
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Hiroshima University Animal Research Committee and AVMA Guidelines on Euthanasia. The protocol (Supplementary Fig. 3) described below was approved by the Committee on the Ethics of Animal Experiments of the Hiroshima University (Permit Number: A09-89). Eight-week-old female C57Bl/6J mice (Charles River Japan, Inc., Yokohama, Japan) were housed in a specific-pathogen free facility in 12-h light-dark cycles with access to water and food ad libitum, and health monitoring was conducted every day. Mice were randomly divided into 2 groups with or without P.g. infection and designated as the P.g.-infected group or the non-infected control (NC) group, respectively. P.g. dental infection was performed as described previously16,21. Briefly, the pulp chambers of the upper first molars on both sides were opened with a #1/2 round burr. After removing the coronal pulp, a small cotton swab containing 108 colony-forming units (CFU) of P.g. (W83 strain) was inserted into the pulp chamber and sealed with Caviton (GC Co., Tokyo, Japan). At 6 weeks post-P.g. infection, mating was started. Placental tissues, amniotic fluid, and serum were harvested at 15 gestational day (gd).
Histology
Placental tissues were fixed in 10% neutral buffered formalin for 24 h. Samples were processed, embedded in paraffin, and serial sections of 4.5 μm in thickness were obtained for hematoxylin and eosin (HE) staining and immunohistochemistry, which was performed as described previously16. Staining was visualized using the DAB Peroxidase (HRP) Substrate Kit (Dako Japan, Tokyo, Japan) to produce a brown reaction product. Immunohistochemistry were performed using high polymer method (Histofine Max-PO; Nichirei Biosciences). A primary Gal-3 antibody (BioLegend, Inc., San Diego, CA, USA; 1: 500) was used. Specificity was ascertained by substituting PBS/serum for the antibody.
Cell Lines and Cell Culture
HTR-8/Svneo cells (human trophoblast cell line) were kindly provided by Professor Charles H. Graham (Queen’s University, Canada). The cells were grown in RPMI 1640 media (Nissui Pharmaceutical Co., Tokyo, Japan) supplemented with 10% heat-inactivated foetal bovine serum (Invitrogen) and 100 U/ml penicillin–streptomycin (Gibco, Tokyo, Japan). For the experiments, cells were seeded at a density of 5 × 105 cells in 35-mm culture dishes. The cells were maintained at 37 °C in a normal atmosphere containing 5% CO2.
Enzyme-Linked Immunosorbent Assay (ELISA)
Mouse Gal-3 concentrations in amniotic fluids and sera were measured using the Galectin-3 DuoSet ELISA Development Kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. P.g.-LPS, which is a TLR2 and TLR4 ligand, was purchased from InvivoGen (San Diego, CA, USA). HTR-8 cells were stimulated with P.g.-LPS, recombinant human Gal-3 (rhGal-3; PeproTech, Rocky Hill, NJ, USA), or recombinant human TNF-α (rhTNF-α; R & D Systems), as indicated below, and the protein levels of Gal-3 or TNF-α in cell culture supernatants were analysed using the Human Galectin-3 Immunoassay (R & D Systems) or the Human TNF-α Immunoassay (R & D Systems). Culture supernatants from P.g.-LPS-stimulated HRT-8 cells, grown in the presence or absence of dicumarol (a JNK MAPK inhibitor; 10 μM), SB203580 (a p38 MAPK inhibitor; 10 μM), U0126 (an ERK1/2 MAPK inhibitor; 10 μM) or caffeic acid phenethyl ester (CAPE; an NF-kB inhibitor; 10 μg) (Sigma-Aldrich) were also analysed.
RNA Isolation from Placental Tissues/Cells and RT-PCR Analysis
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, K.K., Tokyo, Japan) following the manufacturer’s instructions. The RNA concentration and purity were determined using standard spectrophotometric methods. One microgram of total RNA was used for cDNA synthesis using the ReverTra Dash Kit (TOYOBO, Osaka, Japan). Total cDNA was amplified using the Go Taq Green Master Mix (Promega, Madison, WI, USA). Amplification of human or mouse COX-2, Gal-3, IL-8/mKC, and TNF-α was performed in a MyCyclerTM thermal cycler (Bio-Rad, Tokyo, Japan) for 30 cycles of denaturation for 30 s at 94 °C, annealing for 30 s at 58 °C, and extension for 1 min at 72 °C, using primers for all each target mentioned above. GAPDH expression was detected as an internal control. PCR primer sequences are listed in Supplemental Table 1. The amplification products were resolved on 1.5% agarose/TAE gels (Nacalai Tesque, Inc., Kyoto, Japan), electrophoresed at 100 mV, and visualized by ethidium-bromide staining.
Quantitative real time PT-PCR was performed in the Applied Biosystem StepOnePlusTM (Applied Biosystems, http://www.appliedbiosystems.jp) using TaqMan® Fast Advanced Master Mix (Applied Biosystems) and specific primers and probe. Specific primers and probe for mTNF-α (Forward: CCAAATGGCCTCCCTCTCAT, Reverse; GCTACAGGCTTGTCACTCGAATT, Probe; CCCAGACCCTCACACTCAGATCATCT) and hTNF-α (Forward: CCTGCCCCAATCCCTTTATT, Reverse; CCAATTCTCTTTTTGAGCCAGAA, Probe; CCCCCTCCTTCAGACACCCTCAACC) were used. Other sets of specific primers and probe for mGal-3 (Mm00478303), mCOX-2 (Mm03294838), mIL-8(Cxcl15; Mm00441263), mGAPDH (Mm03302249), hGal-3 (Hs00174774), hCOX2 (PTGS2; Hs00153133), hIL-8 (CXCL8; Hs00174103) and hGAPDH (Hs02786624) were purchased from Applied Biosystems. The reaction product was quantified with each GAPDH as the reference gene.
ELISA Analysis for Binding of Gal-3 to LPS
Experiments was performed as described previously, with slight modifications36. The 96-well ELISA plates (Costar High binding 3590, Fisher Scientific, Japan) were coated with Aggregatibacter actinomycetemcomitans-LPS (A.a.-LPS) kindly provided by Dr. Tatsuji Nishihara of Kyushu Dental College, Japan), P.g.-LPS, Escherichia coli-LPS (E. coli-LPS; Wako Pure Chemical Industries, Ltd., Ohsaka, Japan) (50 μg/ml each), or 3% bovine serum albumin (BSA; as a control) and incubated overnight at 4 °C. Plates were washed twice with ELISA buffer (Tris-buffered saline with 0.05% Tween 20, 1 mM CaCl2, and 0.1% BSA, pH 7.4), and 3% Gelatin was used for blocking at 37 °C for 1 h. rhGal-3 was added to each well at different final concentrations (0, 0.05, 0.15, 0.6, or 2 μg/ml) and incubated overnight at 4 °C. An anti-Gal-3 antibody (BioLegend, Inc.) was also added to each well, and the plates were incubated overnight at 4 °C. For detection, a horseradish peroxidase-conjugated anti-rat IgG (GE Healthcare Life Sciences) was added, and tetramethyl benzidine substrate (colour reagent A & B, 1: 1, R&D Systems) was used for visualization. After 20 min, stop solution (R&D Systems) was added. O.D.s were measured at 450 nm.
Small interfering RNA (siRNA)-mediated Gal-3 Gene Knockdown
siRNAs against human Gal-3 were purchased from Bonac Corporation (Fukuoka, Japan). The siRNA sequences are shown in Table 1. Transfection was performed using Lipofectamine RNAi Max (Invitrogen), according to the manufacturer’s recommended protocol.
Table 1.
Sequences of primer pairs.
| Gene | Forward | Reverse | Size | Accession number |
|---|---|---|---|---|
| Mouse | ||||
| COX-2 (Ptgs2) | TTCAAAAGAAGTGCTGGAAAAGGT | GATCATCTCTACCTGAGTGTCTTT | 304 | NM_011198.3 |
| mKC (IL-8 homologue) | CGCTGCTGCTGCTGGCCACCA | GGCTATGACTTCGGTTTGGGTGCAG | 164 | NM_008176.3 |
| TNF-α | ATGAGCACAGAAAGCATGATC | TACAGGCTTGTCACTCGAATT | 276 | NM_013693.2 |
| GAPDH | GCATCCTGGGCTACACTGAG | TCCACCACCCTGTTGCTGTA | 163 | NM_008084.2 |
| Human | ||||
| COX-2 (Ptgs2) | TGAGCATCTACGGTTTGCTG | TGCTTGTCTGGAACAACTGC | 158 | NM_000963.2 |
| IL-8 | TAGCAAAATTGAGGCCAAGG | GGACTTGTGGATCCTGGCTA | 204 | NM_000584.3 |
| TNF-α | AAGAATTCAAACTGGGGCCT | GGCTACATGGGAACAGCCTA | 402 | NM_000594.2 |
| GAPDH | TGAACGGGAAGCTCACTGG | TCCACCACCCTGTTGCTGTA | 307 | NM_002046.4 |
| siRNA | ||||
| siGal-3 | CACGCUUCAAUGAGAACAACA | UGUUGUUCUCAUUGAAGCGUG | ||
Western Blotting
Western blotting was performed as previously described46. Briefly, HTR-8 cell pellets (5 × 105 cells) were resuspended in ice-cold lysis buffer. Proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and electro-blotted onto a nitrocellulose membrane. After blocking the membrane in 3% milk for 30 min, immunoblotting was performed using primary antibodies (anti-phospho-JNK, anti-phospho-c-Jun, anti-phospho-ERK1/2, anti-ERK1/2, anti-phospho-NF-κB p65, anti-RelB [Cell Signaling Technology], or anti-CD66a [Epitomics, Burlingame, CA, USA]) and secondary antibodies (GE Healthcare Life Science, Tokyo, Japan), as indicated. β-actin (Sigma-Aldrich, St. Louis, MO, USA) was detected as an internal control. The results were visualized using the ECL Western Blotting Detection System (GE Healthcare, UK).
Statistical Analysis
Data are presented as the mean ± standard deviation. Statistical differences among experimental groups were evaluated by 1-way analysis of variance (ANOVA), followed by Tukey’s post-hoc test or Student’s t-test using GraphPad Prism (version 6.0c, CA, USA) with the levels of significance set at *P < 0.05 and **P < 0.01.
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information files.
Electronic supplementary material
Acknowledgements
We thank Dr. Junzou Hisatune (Department of Bacteriology, Hiroshima University) for providing the P.g. culture and Professor Tatsuji Nishihara (Kyushu Dental College) for providing A.a.-LPS. We also thank Professor Charles H. Graham (Queen’s University, Canada) for providing the HTR-8/Svneo. This work was supported by Grants-in-Aid for Scientific Research [Challenging Exploratory Research; Research Project number 26670802 to T.T., Scientific Research (C); Research Project number 16K11444 to M.M.]. The manuscript was reviewed by Editage prior to submission.
Author Contributions
A.M. performed the experiments, contributed to the project design and wrote the manuscript text. M.M. performed the experiments, contributed to the project design, wrote the manuscript text and supervised the project. H.F. and A.N. contributed to the animal experiments in Figs 1 and 2. C.C. and T.A. contributed to additional ELISA in Figs 3 and 4. S.S. and T.I. contributed to the experiments in Fig. 6. K.K. refined the project design. T.T. Supervised the project and wrote the manuscript text. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Mutsumi Miyauchi, Min Ao and Hisako Furusho contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-21072-y.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mutsumi Miyauchi, Email: mmiya@hiroshima-u.ac.jp.
Takashi Takata, Email: ttakata@hiroshima-u.ac.jp.
References
- 1.Goldenberg RL, et al. Epidemiology and causes of preterm birth. Lancet. 2008;371:75–84. doi: 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blencowe H, et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet. 2012;379:2162–72. doi: 10.1016/S0140-6736(12)60820-4. [DOI] [PubMed] [Google Scholar]
- 3.Mwaniki MK, et al. Long-term neurodevelopmental outcomes after intrauterine and neonatal insults: a systematic review. Lancet. 2012;379:445–52. doi: 10.1016/S0140-6736(11)61577-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Institute of Medicine (US) Committee on Understanding Premature Birth and Assuring Healthy Outcomes. Preterm Birth: Causes, Consequences, and Prevention. (edis Behrman R.E., Butler A.S.), (National Academies Press (US) 2007). [PubMed]
- 5.MacIntyre DA, et al. Prevention of preterm labour via the modulation of inflammatory pathways. J Matern Fetal Neonatal Med. 2012;25(Suppl 1):17–20. doi: 10.3109/14767058.2012.666114. [DOI] [PubMed] [Google Scholar]
- 6.LeBlanc MM, et al. Relationship between infection, inflammation and premature parturition in mares with experimentally induced placentitis. Equine Vet J Suppl. 2012;41:8–14. doi: 10.1111/j.2042-3306.2011.00502.x. [DOI] [PubMed] [Google Scholar]
- 7.Cappelletti M, et al. Inflammation and preterm birth. J Leukoc Biol. 2016;99(1):67–78. doi: 10.1189/jlb.3MR0615-272RR. [DOI] [PubMed] [Google Scholar]
- 8.Jeftha A, Holmes H. Periodontitis and cardiovascular disease. SADJ. 2013;68(60):62–3. [PubMed] [Google Scholar]
- 9.Xiong X, et al. Periodontal disease as a potential risk factor for the development of diabetes in women with a prior history of gestational diabetes mellitus. J Public Health Dent. 2013;73:41–9. doi: 10.1111/jphd.12004. [DOI] [PubMed] [Google Scholar]
- 10.Offenbacher S, et al. Periodontal infection as a possible risk factor for preterm low birth weight. J Periodontol. 1996;67:1103–13. doi: 10.1902/jop.1996.67.10s.1103. [DOI] [PubMed] [Google Scholar]
- 11.Leon R, et al. Detection of Porphyromonas gingivalis in the amniotic fluid in pregnant women with a diagnosis of threatened premature labor. J Periodontol. 2007;78:1249–1255. doi: 10.1902/jop.2007.060368. [DOI] [PubMed] [Google Scholar]
- 12.Katz J, et al. Localization of P. gingivalis in preterm delivery placenta. J Dent Res. 2009;88:575–8. doi: 10.1177/0022034509338032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin D, et al. Porphyromonas gingivalis infection in pregnant mice is associated with placental dissemination, an increase in the placental Th1/Th2 cytokine ratio, and fetal growth restriction. Infect Immun. 2003;71:5163–5168. doi: 10.1128/IAI.71.9.5163-5168.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belanger M, et al. Colonization of maternal and fetal tissues by Porphyromonas gingivalis is strain-dependent in a rodent animal model. Am J Obstet Gynecol. 2008;199:86e1–7. doi: 10.1016/j.ajog.2007.11.067. [DOI] [PubMed] [Google Scholar]
- 15.Polak D, et al. Mouse model of experimental periodontitis induced by Porphyromonas gingivalis/Fusobacterium nucleatum infection: bone loss and host response. In J Clin Periodontol. 2009;36:406–10. doi: 10.1111/j.1600-051X.2009.01393.x. [DOI] [PubMed] [Google Scholar]
- 16.Ao M, et al. Dental Infection of Porphyromonas gingivalis Induces Preterm Birth in Mice. PLoS One. 2015;10(8):e0137249. doi: 10.1371/journal.pone.0137249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerliani JP, et al. Expanding the universe of cytokines and pattern recognition receptors: galectins and glycans in innate immunity. J Clin Immunol. 2011;31(1):10–21. doi: 10.1007/s10875-010-9494-2. [DOI] [PubMed] [Google Scholar]
- 18.Bamberger AM, et al. The adhesion molecule CEACAM1 (CD66a, C-CAM, BGP) is specifically expressed by the extravillous intermediate trophoblast. Am J Pathol. 2000;156:1165–70. doi: 10.1016/S0002-9440(10)64985-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feuk-Lagerstedt E, et al. Identification of CD66a and CD66b as the major galectin-3 receptor candidates in human neutrophils. J Immunol. 1999;163:5592–8. [PubMed] [Google Scholar]
- 20.Li Y, et al. Galectin-3 is a negative regulator of lipopolysaccharide-mediated inflammation. J Immunol. 2008;181:2781–9. doi: 10.4049/jimmunol.181.4.2781. [DOI] [PubMed] [Google Scholar]
- 21.Pereira CV, et al. Detection and clonal analysis of anaerobic bacteria associated to endodontic-periodontal lesions. J Periodontol. 2011;82:1767–1775. doi: 10.1902/jop.2011.110063. [DOI] [PubMed] [Google Scholar]
- 22.Furusho H, et al. Dental infection of Porphyromonas gingivalis exacerbates high fat diet-induced steatohepatitis in mice. J Gastroenterol. 2013;48:1259–70. doi: 10.1007/s00535-012-0738-1. [DOI] [PubMed] [Google Scholar]
- 23.Nath CA, et al. Can sulfasalazine prevent infection-mediated pre-term birth in a murine model? Am J Reprod Immunol. 2010;63:144–9. doi: 10.1111/j.1600-0897.2009.00773.x. [DOI] [PubMed] [Google Scholar]
- 24.Dumic J, Dabelic S, Flogel M. Galectin-3: an open-ended story. In Biochim Biophys Acta. 2006;1760:616–35. doi: 10.1016/j.bbagen.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 25.Elovitz, M. A. et al. A new model for inflammation-induced preterm birth: the role of platelet-activating factor and Toll-like receptor-4. Am J Pathol. 163(5), 2103–11. 2003 Nov;pmid:14578208 (2003). [DOI] [PMC free article] [PubMed]
- 26.Ren, H., Li, Y., Jiang, H. & Du, M. Porphyromonas gingivalis induces IL-8 and IFN-gamma secretion and apoptosis in human extravillous trophoblast derived HTR8/SVneo cells via activation of ERK1/2 and p38 signaling pathways. Placenta. 45, 8–15. 10.1016/j.placenta.2016.06.010. 2016 Sep, Epub 2016 Jun 27 (2016). [DOI] [PubMed]
- 27.Jeng KC, et al. An endogenous lectin, galectin-3 (epsilon BP/Mac-2), potentiates IL-1 production by human monocytes. Immunol Lett. 1994;42:113–116. doi: 10.1016/0165-2478(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 28.Cortegano I, et al. Interaction between galectin-3 and FcgammaRII induces down-regulation of IL-5 gene: implication of the promoter sequence IL-5REIII. Glycobiology. 2000;10:237–242. doi: 10.1093/glycob/10.3.237. [DOI] [PubMed] [Google Scholar]
- 29.Nishi Y, et al. Role of galectin-3 in human pulmonary fibrosis. Allergol Int. 2007;56:57–65. doi: 10.2332/allergolint.O-06-449. [DOI] [PubMed] [Google Scholar]
- 30.Filer A, et al. Galectin 3 induces a distinctive pattern of cytokine and chemokine production in rheumatoid synovial fibroblasts via selective signalling pathways. Arthritis Rheum. 2009;60:1604–1614. doi: 10.1002/art.24574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riewe, S. D. et al. Human trophoblast responses to Porphyromonas gingivalis infection. Mol Oral Microbiol. 2010 Aug, 25(4), 252–9. 10.1111/j.2041-1014.2010.00573.x. (2010) [DOI] [PMC free article] [PubMed]
- 32.Inaba, H. et al. Identification of signaling pathways mediating cell cycle arrest and apoptosis induced by Porphyromonas gingivalis in human trophoblasts. Infect Immun. 80(8), 2847–57. 10.1128/IAI.00258-12. 2012 Aug;Epub 2012 Jun 11 (2012). [DOI] [PMC free article] [PubMed]
- 33.Inaba, H. et al. Porphyromonas gingivalis invades human trophoblasts and inhibits proliferation by inducing G1 arrest and apoptosis. Cell Microbiol. 11(10), 1517–32. 10.1111/j.1462-5822.2009.01344.x. 2009 Oct;Epub 2009 Jun 11. (2009). [DOI] [PMC free article] [PubMed]
- 34.Ren, H., Li, Y., Jiang, H. & Du, M. Interferon-Gamma and Fas Are Involved in Porphyromonas gingivalis-Induced Apoptosis of Human Extravillous Trophoblast-Derived HTR8/SVneo Cells via Extracellular Signal-Regulated Kinase 1/2 Pathway. J Periodontol. 87(11), e192–e199. Epub 2016 Jun 28. (2016). [DOI] [PubMed]
- 35.Mey A, et al. The animal lectin galectin-3 interacts with bacterial lipopolysaccharides via two independent sites. J Immunol. 1996;156:1572–1577. [PubMed] [Google Scholar]
- 36.Kato T, Uzawa A, Ishihara K. Inhibitory effect of galectin-3 on the cytokine-inducing activity of periodontopathic Aggregatibacter actinomycetemcomitans endotoxin in splenocytes derived from mice. FEMS Immunol Med Microbiol. 2009;57:40–5. doi: 10.1111/j.1574-695X.2009.00577.x. [DOI] [PubMed] [Google Scholar]
- 37.Paramonov NA, et al. Structural Analysis of the Core Region of O-Lipopolysaccharide of Porphyromonas gingivalis from Mutants Defective in O-Antigen Ligase and O-Antigen Polymerase. J Bacteriol. 2009;191:5272–82. doi: 10.1128/JB.00019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quattroni P. Galectin-3 binds Neisseria meningitidis and increases interaction with phagocytic cells. Cell Microbiol. 2012;14:1657–75. doi: 10.1111/j.1462-5822.2012.01838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakao R, et al. Enhanced biofilm formation by Escherichia coli LPS mutants defective in Hep biosynthesis. PLoS One. 2012;7(12):e51241. doi: 10.1371/journal.pone.0051241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang PL, Ohura K. Porphyromonas gingivalis lipopolysaccharide signalling in gingival fibroblasts-CD14 and Toll-like receptors. Crit Rev Oral Biol Med. 2002;13:132–42. doi: 10.1177/154411130201300204. [DOI] [PubMed] [Google Scholar]
- 41.Puchner K, et al. The implication of second-trimester amniotic fluid TNF-alpha, cytochrome C and cell death nucleosomes in the prediction of preterm labor and/or premature rupture of membranes. Arch Gynecol Obstet. 2012;285:37–43. doi: 10.1007/s00404-011-1909-7. [DOI] [PubMed] [Google Scholar]
- 42.Cobo T, et al. Intra-amniotic inflammation predicts microbial invasion of the amniotic cavity but not spontaneous preterm delivery in preterm prelabor membrane rupture. Acta Obstet Gynecol Scand. 2012;91:930–5. doi: 10.1111/j.1600-0412.2012.01427.x. [DOI] [PubMed] [Google Scholar]
- 43.Weigert J, et al. Serum galectin-3 is elevated in obesity and negatively correlates with glycosylated hemoglobin in type 2 diabetes. J Clin Endocrinol Metab. 2010;95:1404–11. doi: 10.1210/jc.2009-1619. [DOI] [PubMed] [Google Scholar]
- 44.Ohshima S, et al. Galectin 3 and its binding protein in rheumatoid arthritis. Arthritis Rheum Oct. 2003;48:2788–95. doi: 10.1002/art.11287. [DOI] [PubMed] [Google Scholar]
- 45.Demmert M, et al. Galectin-3 in cord blood of term and preterm infants. Clin Exp Immunol. 2012;167:246–51. doi: 10.1111/j.1365-2249.2011.04509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inubushi T, et al. Molecular mechanisms of the inhibitory effects of bovine lactoferrin on lipopolysaccharide-mediated osteoclastogenesis. J Biol Chem. 2012;287:23527–36. doi: 10.1074/jbc.M111.324673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article and its Supplementary Information files.