

Abstract
Inhibition of DYRK1A kinase, produced by chromosome 21 and consequently overproduced in trisomy 21 subjects, has been suggested as a therapeutic approach to treating the cognitive deficiencies observed in Down syndrome (DS). We now report the synthesis and potent DYRK1A inhibitory activities of fluoro derivatives of 3,5-di(polyhydroxyaryl)-7-azaindoles (F-DANDYs). One of these compounds (3-(4-fluorophenyl)-5-(3,4-dihydroxyphenyl)-1H-pyrrolo[2,3-b]pyridine, 5a) was selected for in vivo studies of cognitive rescuing effects in a standard mouse model of DS (Ts65Dn line). Using the Morris water maze task, Ts65Dn mice treated i.p. with 20 mg/kg of 5a performed significantly better than Ts65Dn mice treated with placebo, confirming the promnesiant effect of 5a in the trisomic mice. Overall, these results demonstrate for the first time that selective and competitive inhibition of DYRK1A kinase by the F-DANDY derivative 5a may provide a viable treatment strategy for combating the memory and learning deficiencies encountered in DS.
Introduction
Down syndrome (DS), or trisomy 21, is the most common genetically acquired form of intellectual disability1–3, occurring in approximately 1 out of 650–1000 newborns in Europe4,5 and North America6. In addition to their characteristic physical appearance, people with DS present developmental neurological delay, including a lower IQ7 and reduced learning and memory capacities3. DS, for which there is currently no cure, is caused by the presence of an extra copy of chromosome 21 leading to increased production and resulting imbalance of the proteins and enzymes encoded by this chromosome8,9. One of these enzymes is the dual-specificity tyrosine phosphorylation kinase 1a, or DYRK1A, belonging to the CGMC kinome group and which is expressed in all mammalian tissues but especially so in the developing brain10–12. DYRK1A is implicated in cell proliferation13 and neuronal development14 as well as a wide range of signaling pathways. In DS, the triplication of chromosome 21 leads to approximately 1.5-fold higher DYRK1A levels compared to the general euploid population15 and this overproduction has been linked to the cognitive deficits associated with DS16,17, and notably to imbalance of excitation/inhibition18. Through hyperphosphorylation of Tau protein19 and the resulting formation of insoluble tau aggregates and neurofibrillary tangles, DYRK1A is also involved in neurodegeneration and neuronal loss appearing in Alzheimer’s disease (AD)20,21. DYRK1A has been found to be abnormally expressed in both DS and AD22 and indeed, people with DS develop AD precociously23, the amyloid precursor protein (APP) at the origin of senile plaques also being overexpressed by chromosome 21 in DS individuals24.
A plausible therapeutic strategy for cognitive deficits associated with DS and eventually AD would thus entail controlled inhibition of the activity of cerebral DYRK1A kinase25. To this end, a variety of DYRK1A inhibitors has been developed over the past few years most of which bind to the active ATP site of the enzyme. Examples of such competitive inhibitors of DYRK1A are shown in Fig. 1. These include harmine, an alkaloid isolated from Peganum harmala26, the INDY derivatives27, the leucettine derivative L4128, as well as analogues of the naturally-occurring (aza) indolic compounds meriolin29 and lamellarin30. While the leucettine derivative L41 was shown to prevent memory impairment produced by administration of the β-amyloid peptide Aβ25–35 in rodents31, to the best of our knowledge, none of the current competitive DYRK1A inhibitors has passed the in vitro stage of investigation with respect to improvement of cognitive impairments in DS. In contrast, epigallocatechin gallate (EGCG), the major active principle of green tea, has been demonstrated to be a relatively potent allosteric inhibitor of DYRK1A12,32 and to produce cognitive enhancement in Ts65Dn mice, the most widely used mouse model for DS33.
Figure 1.

Naturally-occurring and synthetic inhibitors of DYRK1A kinase.
We recently reported that hydroxy derivatives of 3,5-diaryl-7-azaindoles (DANDYs) were potent, competitive inhibitors of DYRK1A34. The di-, tri- and tetrahydroxy diaryl azaindoles I-IV displayed in vitro inhibition of this kinase with IC50s in the 3 to 23 nM range (Fig. 2) and selectivity with respect to a panel of structurally related kinases including DYRK2 and DYRK3. Starting from the known resolved crystal structure of DYRK1A27, molecular modeling and docking studies of compounds I-IV revealed an extended network of hydrogen bonds between these heterocycles and the amino acid residues of the active site, accounting for their high inhibitory potency in vitro.
Figure 2.

Structures of the most active DYRK1A competitive inhibitors of the 3,5-diaryl-7-indole family (DANDYs)34.
Because the large number of polar hydroxy groups in compounds I-IV might interfere with brain penetration or be a source of rapid metabolization, we initiated a study whereby one or two of the hydroxy groups were systematically replaced by fluorine atoms. In this report, we describe the synthesis of fluoro analogues of compounds I-IV (F-DANDYs) and their in vitro DYRK1A inhibitory activities. Moreover, for a selected, active F-DANDY (compound 5a) administered to mice, we demonstrate, using mass spectral analysis of plasma and brain tissue, that this compound is stable in vivo and enters the brain in therapeutically relevant quantities. Finally, preliminary studies showed that this F-DANDY compound 5a significantly improved the performance of Ts65Dn mice in the Morris water maze, a standard learning and memory paradigm for rodents, but had no observable effect on wild type mice.
Results and Discussion
Chemistry
The synthetic strategy used to prepare the fluorinated or selectively O-methylated 3,5-diaryl-7-azaindoles 5a-5g was essentially identical to the one we used for the synthesis of our first series of DANDYs34. Thus, as illustrated in Fig. 3, N-phenylsulfonyl-3-iodo-5-bromo-7-azaindole 1, easily prepared from 5-bromo-7-azaindole34, was subjected to a first Suzuki-Miyaura coupling35 with 4-fluoro- or 3,4-difluorophenylboronic acid to provide good yields of the corresponding C-3-aryl 7-azaindoles 2a and 2b, respectively. No products resulting from coupling at the less reactive C-5 position were observed. Similarly, Suzuki coupling of 1 with 3-fluoro-4-methoxy- and 3,4-dimethoxyphenylboronic acid gave only the C-3 mono-coupled products 2c and 2d, respectively. Using the same reaction conditions, compounds 2a-2d then served as starting materials for the second Suzuki-Miyaura coupling, this time with 3,4-dimethoxy-, 2,4-dimethoxy or 4-benzyloxyphenylboronic acid, giving access to the 3,5-diaryl-7-azaindoles 3a-3e in mostly good to excellent yields. These conditions did not allow, however, coupling of 2d with 3,4-dibenzyloxyphenylboronic acid. This was circumvented by coupling 2d with 3,4-dibenzyloxyphenylboronic acid pinacol ester using dichlorobis(triphenylphosphine)palladium(II) and triphenylphosphine36 in dioxane as catalytic system, providing the desired product 3f in 61% yield.
Figure 3.

Chemical synthesis of the target substituted 3,5-diaryl-7-azaindole derivatives 5a-5ga.
The N-phenylsulfonyl protecting groups of compounds 3a-3f were efficiently removed by the action of aqueous NaOH in methanol, providing the 3,5-diaryl-7-azaindoles 4a-4f in yields ranging from 66% (4e) to 88% (4b). De-O-methylation of the 7-azaindole derivatives 4a-4d with excess boron tribromide in dichloromethane at room temperature then provided the first of the targeted F-DANDY analogues, the mono- and difluorinated products 5a-5d. Hydrogenolytic cleavage of the O-benzyl groups of 4e and 4f using ammonium formate and
palladium black in methanol gave access to the mixed hydroxy/methoxy F-DANDY analogues 5e and 5f, respectively, in quantitative yields. Finally, treatment of 5e with BBr3 in CH2Cl2 furnished 5g, the monofluorinated analogue of the highly active DYRK1A inhibitors 3,5-di-(4-hydroxyphenyl)-7-azaindole (IV) and 3-(4-hydroxyphenyl)-5-(3,4-dihydroxyphenyl)-7-azaindole (I).
Inhibition of DYRK1A activity in vitro
All the newly synthesized N-deprotected F-DANDY analogues were evaluated for their in vitro DYRK1A inhibitory activity using a fluorescent peptide substrate of this enzyme and UFLC (Ultra Fast Liquid Chromatography) assay as previously described34,37. We first tested the fluorinated analogues 4a-4e having only methoxy or benzyloxy groups instead of free phenolic hydroxy functions on the C-3 and C-5-phenyl rings and compared the results to our previously reported non-fluorinated methoxy derivatives MeO-I to MeO-IV.
As shown in Fig. 4, the IC50s of these compounds, fluorinated or not, were mostly in the micromolar range. In general, it appears that introduction of one or two fluorine atoms on the C-3 phenyl ring has little effect on DYRK1A inhibitory activity. Thus, replacement of one of the methoxy groups of MeO-I and MeO-II by a fluorine atom (compounds 4a and 4b) produced a 3-fold loss in activity (IC50 = 0.46 μM and 0.28 μM compared to IC50 = 1.43 μM and 0.92 μM for the fluorinated analogues, respectively). On the other hand, replacement of two methoxy groups of compound MeO-III (IC50 = 57.78 μM) by two fluorine atoms (4c, IC50 = 5.26 μM) led to a 10-fold improvement in DYRK1A inhibition. Interestingly, introduction of a supplementary fluorine atom at the C-3 position of the C-4 fluoro derivative 4b (to give 4d) led to an almost 40-fold loss of activity (IC50 = 37.36 μM for the 3,4-difluoro compound compared to 0.92 μM for the 4-fluoro analogue). This effect of an additional fluorine atom was much less evident when comparing the 3,4-difluoro derivative 4c with the 4-fluoro analogue 4a (both differing only in the position of a methoxy group with respect to 4b and 4d) which displayed only a 4-fold diminishment of inhibitory activity (4c: IC50 = 5.26 μM; 4a: IC50 = 1.43 μM). Finally, our attention turned to the benzyloxy derivatives 4e and 4f which were prepared in order to allow orthogonal protection of the phenolic hydroxy groups. While the mono-benzyloxy derivative 4e was equipotent with the dimethoxy compound 4c (IC50 = 6.09 μM and 5.26 μM, respectively), replacement of the 3′,4′-dimethoxy groups of MeO-III by dibenzyloxy groups as in 4f surprisingly led to a 3-fold superior activity though both compounds can be considered to be among the least active of our study (4f: IC50 = 18.54 μM; MeO-III: IC50 = 57.78 μM).
Figure 4.

In Vitro inhibition of DYRK1A by 3,5-diaryl-7-azaindoles 4a-4f compared to activities of previously reported DANDYs (IC50 values in μM)34.
We then went on to evaluate the DYRK1A inhibitory activities of the de-O-protected analogues of 4a-4f, that is, compounds 5a-5g. As expected from the results of our previous study, the latter phenolic derivatives proved generally to be considerably more potent inhibitors than their O-methylated counterparts (Fig. 5, IC50 values given in nM). Following the same comparative analysis as for the O-methyl derivatives of Fig. 4, replacement of the C-4 hydroxy group of compound I by a fluorine atom (compound 5a) led to a 7-fold decrease in inhibitory activity (IC50 = 3.0 nM and 20.7 nM, respectively). But this effect was much less deleterious than operating the same modification on compound II, the resulting analogue 5b displaying this time an almost 20-fold loss of activity (IC50 = 11.7 nM and 190.5 nM, respectively).
Figure 5.

In Vitro inhibition of DYRK1A by 3,5-diaryl-7-azaindoles 5a-5g compared to activities of previously reported DANDYs (IC50 values in nM)34.
Adding a fluorine atom to the C-3 positions of compounds 5a and 5b, yielding compounds 5c and 5d, led to a further loss of activity in both cases (IC50 = 56.6 nM and 231.7 nM, respectively). Interestingly, the 3-fluoro-4-methoxy derivative 5e proved to be surprisingly active despite the presence of the methoxy group (IC50 = 41.5 nM) and this activity was gratifyingly improved almost 5-fold in the de-O-methylated counterpart, compound 5g (IC50 = 9.34 nM), the most active F-DANDY of our study. Compound 5g also represents the only example wherein simple addition of a fluorine atom to an active DANDY derivative led to a significant increase in activity. Thus, 5g was 2.5-fold more active than compound IV (IC50 = 23.1 nM). Finally, another strong indication that free phenolic hydroxy groups are essential to high inhibitory activity was provided by the observation that the dimethoxy compound 5f was 40 times less active than its phenolic equivalent III (IC50 = 476 nM and 12.4 nM, respectively).
Cytotoxicity Evaluation
Because the potential cytotoxicity of the F-DANDY derivatives could account, at least in part, for their DYRK1A inhibition in cellulo, the cytotoxicities of compounds 5a and 5g, as well as of 5c and 5e, were evaluated in vitro for growth inhibition of KB cells32. As shown in Table 1, compounds 5a and 5c were essentially non-cytotoxic at a concentration of 10−6 M and only modestly cytotoxic at 10−5 M, inhibiting less than 50% of KB cell growth at the latter concentration. In contrast, compounds 5e and 5g were somewhat more cytotoxic, compound 5g displaying the highest inhibitory potency with an estimated IC50 value of 10−6 M.
Table 1.
Inhibition of KB Cell Proliferation in vitro by Selected F-DANDYs.
| Compound | % Growth inhibition 10−5 M | % Growth inhibition 10−6 M |
|---|---|---|
| 5a | 49 ± 3 | 0 ± 9 |
| 5c | 22 ± 2 | 6 ± 2 |
| 5e | 66 ± 3 | 30 ± 1 |
| 5 g | 95 ± 1 | 46 ± 1 |
Inhibition of tau phosphorylation by DYRK1A in cellulo
In order to verify that compound 5a can penetrate cells and inhibit tau phosphorylation by DYRK1A in cellulo, we compared the activity of DYRK1A in the absence or in the presence of various concentrations of 5a in human embryonic kidney cells (HEK293) transiently transfected with DYRK1A and tau. The activity of 5a was compared to the activity of 5g and harmine. Table 2 shows that all three compounds inhibited tau phosphorylation with IC50s in the 0.2 to 1 μM range. Ratios between in cellulo and in vitro inhibitory potencies were 0.6, 48 and 41 for harmine, compounds 5a and 5g respectively (Fig. 5 and Table 2). Compounds 5a and 5g showed comparable ratios indicating that the difference in cytotoxicity (5g being more cytotoxic than 5a, Table 1) was not due to a difference in cell penetration and activity in cellulo.
Table 2.
Inhibition of Tau Phosphorylation by DYRK1A by Selected DANDYs.
| Compound | IC50 for tau phosphorylation (μM) |
|---|---|
| 5a | 1 |
| 5g | 0.38 |
| Harmine | 0.19 |
Kinase Profiling of 5a and 5g
As for the DANDY derivatives I-IV34, the selectivity of F-DANDYs 5a (the least cytotoxic) and 5g (the most active) was evaluated at 5 × 10−8 M, together with harmine (at 10−6 M), on a panel of 13 kinases belonging to several families including DYRK (DYRK1A, DYRK2, DYRK3), the closely related CMGCs (CDK2, CDK5, CLK1, ERK2, GSK3β) as well as serine/threonine (AKT1, Pim1, CK1α) and tyrosine kinases (JAK3, TRKA) (Fig. 6). Neither 5a nor 5g was observed to be as selective as harmine. However, of these two F-DANDYs, 5a was considerably more selective than 5g for DYRK1A with respect to the other 12 kinases. As compared to harmine, 5a did not show any activity on CLK1 and a higher selectivity for DYRK1A among the three DYRK tested.
Figure 6.

In vitro inhibition of a panel of 13 kinases by F-DANDYs 5a and 5g (5.10−8 M) and harmine (10−6 M) (100 represents full inhibition of the enzyme).
Taken together, these results suggested that compound 5a presented the best compromise in terms of DYRK1A inhibitory activity and selectivity as well as low cytotoxicity. Compound 5a was thus selected for further study as described below.
Mass Spectral Studies on Compound 5a
Bioavailability of 5a in plasma
Before undertaking behavioral studies in Ts65Dn mice, the bioavailability of F-DANDY 5a in plasma after peripheral (intraperitoneal – i.p.) injection in mice was determined. UHPLC-MS/MS was used to detect and quantify compound 5a in plasma of 12 wild-type (WT) mice that were treated with the drug (20 mg/kg; 2.4 mg/mL) and then sacrificed at 12 successive time points. The sample preparation and analytical protocol were adapted from Bonneau et al.38,39. As shown in Fig. 7, plasma concentrations of compound 5a peaked a few minutes after injection (311.5 pg/μL at t = 4.4 min and 235.8 pg/μL at t = 5.4 min) and slowly decreased over time, reaching a plateau between 15 and 45 min. A 30 min interval was thus selected between time of injection and behavioral assessment.
Figure 7.
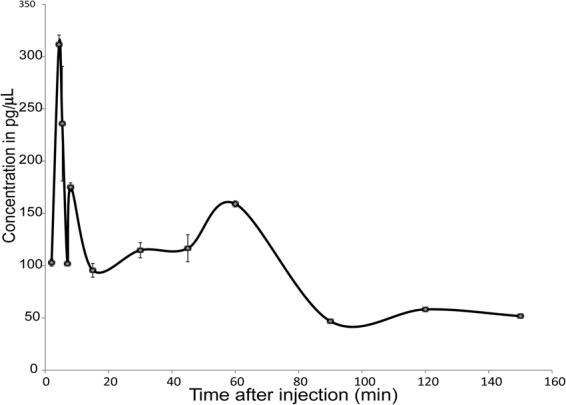
Evolution of compound 5a concentration in plasma extracts after i.p. injection in WT mice. Standard deviations are noted after 3 injections except for points at 5.4, 8, 30 and 45 min where 3 extractions of each sample were realized.
Bioavailability of 5a in brain homogenates
Using UHPLC-MS/MS as described above, we detected and quantified compound 5a in plasma and brain homogenates to assess whether it had likely crossed the blood-brain barrier. To that end, all animals were sacrificed directly after the Morris Water Maze (MWM) and plasma and brain homogenates (one hemisphere) were extracted and analyzed. Compound 5a-treated Ts65Dn and WT mice had an average plasma concentration of 0.84 ± 0.54 μM (n = 9) and 0.50 ± 0.28 μM (n = 10), respectively, with no significant difference between Ts65Dn and WT mice. Mean brain concentrations were found to be 6.67 ± 4.64 μM (n = 5) and 6.61 ± 5.35 µM (n = 4) for Ts65Dn and WT mice, respectively, assuming a vascular space of 15 µL/g of tissue40. The partition coefficients at steady state (Kp = brain concentration/plasma concentration) for Ts65Dn and WT mice were 6.43 ± 3.38 and 10.81 ± 9.55 respectively, indicating that a substantial fraction of compound 5a had crossed the blood-brain barrier. Compared to other drugs acting on the central nervous system, compound 5a showed values similar to buproprion (4.97), fluoxetine (5.23) and perphenazine (7.12)41. However, Kp values might be underestimated due to high plasma levels that could saturate efflux transport to the brain.
Behavioral Studies with Compound 5a
Ts65Dn mice, the best-characterized and most widely used mouse model of DS, are trisomic for a segment of chromosome 16 extending between genes Mrp139 and Znf295 and containing approximately 92 orthologues of human chromosome 21 genes42 including the Dyrk1a gene. Using a standard Morris Water Maze (MWM) paradigm, spatial learning was assessed in Ts65Dn mice and euploid WT littermates treated either with compound 5a (20 mg/Kg) or with vehicle (vehicle solution) every day for the two weeks preceding behavioral testing and also during the MWM protocol (mice being injected 30 min before each daily session). Mice were trained for a total of 5 days to learn a fixed spatial goal location in the maze. Figure 8 shows the learning skills of the mice assessed by their corrected integrated path length (CIPL) to reach the platform. In this paradigm, learning occurred when mice succeeded in decreasing their traveled distances after repeated training trials.
Figure 8.
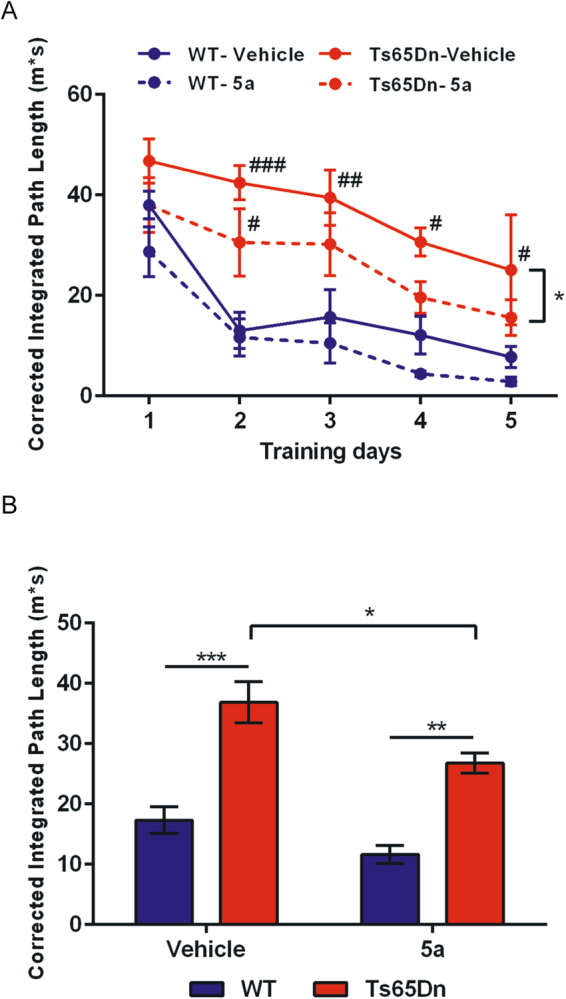
Mice were trained in a standard spatial learning protocol (MWM paradigm). (A) All mice showed a gradual improvement across training sessions. As expected, Ts65Dn mice showed a behavioral impairment (increased path lengths to reach the goal location as compared to WT mice). This deficit was partly corrected following treatment with 5a. (B) The same differences between groups were observed using pooled data over the 5 training sessions. Overall comparisons between groups: *p < 0.05; **p < .01; ***p < 0.001. Different from control (WT-Vehicle group): #p < 0.05; ##p < 0.01; ###p < 0.001.
ANOVA on CIPL with two main factors (Genotype and Treatment) and repeated measures (Training days) indicated a significant effect of repetition (F(4,52) = 16.69; p < 0.0001) underscoring that mice gradually improved their performance across training sessions. A main effect of the Genotype factor was concurrently observed (F(1,13) = 64.25; p < 0.0001) due to poorer learning performance of Ts65Dn mice as compared to WT littermates. ANOVA also underscored an effect of the Treatment factor (p < 0.005) that was explained by an overall increase of performance (i.e., decrease of path lengths) in 5a-treated animals. Importantly posthoc analysis (Bonferroni tests) confirmed a promnesiant effect of 5a in Ts65Dn (p < 0.05, see Fig. 8A) but not in WT mice (p > 0.44). Day-by-day analysis indicated that while Ts65Dn-vehicle mice were constantly impaired when compared to WT-vehicle mice, Ts65Dn-5a mice displayed only an initial delay in learning but then rapidly recovered a level of performance not statistically different from that of WT-vehicle littermates. The rescuing effect of 5a in the trisomic mice, although significant, was partial as a Genotype effect was still observed in the drug condition (WT > Ts65Dn, posthoc test, p < 0.005). A global analysis of performance with all training sessions pooled confirmed that Ts65Dn-vehicle mice were impaired in comparison to WT littermates (p < 0.001; Fig. 8B). A significant improvement of performance was observed in Ts65Dn mice following treatment with 5a (p < 0.05) but these mice remained impaired in comparison to WT-5a treated animals (p < 0.01).
Subsequently, the percentage of thigmotaxis, i.e. the propensity to swim close to the pool walls, was analyzed. Thigmotaxis refers to an unadapted navigational strategy and hence is considered as a complementary index to assess learning failures in the MWM paradigm. ANOVA with two factors (Genotype and Treatment) revealed a strong increase of thigmotaxis in Ts65Dn mice as compared to WT mice (F(1,13) = 50.1; p < 0.0001) that paralleled their spatial learning impairment (see above). Interestingly, treatment with 5a induced an overall reduction of thigmotaxis (F(1,13) = 7.8; p < 0.025), observed in WT mice (−6.5% between vehicle and 5a conditions) which was even more pronounced in Ts65Dn mice (−12.9%). The decrease of thigmotaxis in the trisomic animals might partly be explained by their better learning proficiency under the drug condition (see above).
Finally, analysis of swim speeds underscored differences between genotypes (ANOVA) with two main factors (Genotype and Treatment) and repeated measures (Training days); F(3,65) = 6.493; p < 0.001) with untreated Ts65Dn mice swimming at lower speeds when compared to the three other groups (Bonferroni posthoc tests, all ps < 0.005). Treatment with 5a restored normal swim speed in Ts65Dn mice (comparison with 5a-treated WT mice: p > 0.99).
Conclusions
The cognitive deficiencies observed in DS subjects have been linked to the over-production of cerebral DYRK1A kinase due to the extra copy of chromosome 21. Inhibition of this enzyme would seem, then, to be a viable option for the treatment of learning and memory problems encountered in DS. We have now developed a new family of potent DYRK1A inhibitors, F-DANDYs, which are fluorinated analogues of our previously described DANDY inhibitors. Our preliminary structure-activity relationship study has indicated that replacing one or two of the phenolic OH groups of the DANDY derivatives by fluoride led to some loss of activity though IC50s nevertheless remained in the nanomolar range (e.g., compounds 5a, IC50 = 20.7 nM and 5g, IC50 = 9.3 nM). Moreover, as with the DANDY derivatives, methylation of the phenolic alcohol functions led to an important loss of inhibitory potency (e.g., compounds 4a, IC50 = 1.43 µM). These results encouraged us to conduct in vivo studies on one of these F-DANDY derivatives, compound 5a. We first demonstrated, using tandem LC-MS/MS, that 5a administered i.p. to WT mice at a dose of 20 mg/kg was detectable in plasma for up to one hour at a level of approximately 100 to 150 pg/µL with a slow decrease over 2.5 hours to approximately 50 pg/µL. Using a similar mass spectral technique, compound 5a was then shown to enter the brain of both WT and Ts65Dn mice, the latter being a commonly utilized model for DS. Spatial learning evaluation of WT and Ts65Dn mice, treated with 5a (20 mg/kg i.p.), pointed to a significant improvement in performance in the Ts65Dn mice but not the WT mice, providing proof-of-concept as to the working hypothesis that inhibition of DYRK1A by compound 5a leads to improvement in cognitive abilities of Ts65Dn mice. Only partial recovery of performance of Ts65Dn was observed but it is expected that a more robust effect could be obtained by prenatal treatment with 5a43. This study is the first demonstration of a therapeutic effect of a competitive DYRK1A inhibitor in mice modeling DS. Supporting our data, a recent report underscored similar protective effect of DYRK1A inhibition in an animal model of Alzheimer’s disease31, a pathology that shares many links with DS. In summary, we have demonstrated that compound 5a is a potent, non-toxic inhibitor of DYRK1A whose metabolic stability in plasma and ability to cross the blood-brain barrier translates into cognitive rescuing effects in a mouse model of DS. While further in vivo studies will be required, compound 5a thus represents a potentially viable drug candidate for the treatment of cognitive deficits associated with DS and associated pathologies.
Methods
Synthesis of inhibitors
F-DANDY derivatives were prepared using standard synthetic procedures as described in the Supplementary Information.
Expression and purification of the recombinant DYRK1A catalytic domain (DYRK1A-ΔC)
The cDNA coding for the catalytic domain (residues 1-502) of rat DYRK1A (99.6% amino acid identity with human DYRK1A) was a kind gift of Prof. W. Becker (Aachen University, Germany). This cDNA was subcloned into pET28 plasmid and used to produce recombinant 6xHis-tagged DYRK1A catalytic domain (DYRK1A-ΔC). The pET28-DYRK1A-ΔC plasmid was transformed into E. coli BL21(DE3) cells for production and purification of the protein. Briefly, transformed bacterial cells were grown at 37 °C for 4 h in the presence of 0.5 mM isopropyl β-D-1-thiogalactopyranoside and further grown at 4 °C overnight. Cells were harvested by centrifugation and resuspended in phosphate buffered saline buffer (PBS) supplemented with protease inhibitors, 1 mg/mL lysozyme and 0.1% Triton X-100. After 30 min incubation at 4 °C, the lysate was subjected to sonication on ice and pelleted (12000 × g, 30 min). The supernatant was incubated with His-select Nickel resin (Sigma) for 2 h at 4 °C. The resin was poured into a column and washed with Tris-HCl 20 mM, pH 7.5, 10 mM imidazole. Proteins were eluted in Tris-HCl 20 mM, pH 7.5, 300 mM imidazole. Proteins were reduced with 10 mM dithiothreitol (DTT) and dialyzed overnight against Tris-HCl 20 mM, pH 7.5. Proteins were quantified with Bradford’s reagent (Bio-rad). Purity was assessed by SDS-PAGE. Proteins were kept at −80 °C.
DYRK1A inhibition assays
-
In vitro measurement of DYRK1A kinase activity was carried out using a UFLC-based approach in combination with a fluorescent peptide substrate of DYRK1A. The fluorescent peptide substrate was derived from the sequence of the human transcription factor FKHR that is known to be a physiological substrate of DYRK1A37. The peptide substrate, coupled to fluorescein by its N-terminal amino acid, had the following sequence: KISGRLSPIMTEQ (the serine residue that is phosphorylated by DYRK1A is underlined). Purified recombinant rat DYRK1A catalytic domain (DYRK1A-ΔC) was used in all enzymatic assays as previously described37. Activity assays were performed in 96-well plates, in a total volume of 50 μL consisting of kinase buffer (Tris-HCl 50 mM, pH 7.5, 10 mM DTT, 5 mM MgCl2), peptide substrates (ranging from 5 to 60 μM) and 20 ng of purified DYRK1A-ΔC. The reaction was initiated by addition of ATP (ranging from 50 to 800 μM) and the mixture was incubated for 30 min at 37 °C. Reactions were stopped by addition of 50 μL of 15% HClO4 (v/v) and 20 μL of the mixture was analyzed by UFLC (Shimadzu) on a C8 reverse-phase column (Ascentis). Mobile phases used consisted of 0.12% trifluoroacetic acid (TFA) (solvent A) and acetonitrile in 0.12% TFA. Phosphorylated and unphosphorylated peptides were separated by isocratic flow (85% solvent A/15% solvent B) at a flow rate of 1.5 mL/min. The peptides were monitored by fluorimetry (excitation at 485 nm, emission at 530 nm) and quantified by integration of the peak absorbance area. A calibration curve established with different known concentrations of peptides was used for quantification. Initial velocities and kinetic parameters were determined by fitting the data iteratively to the following equation: Vi = Vmax [SA][SB]/(KAKmB + KmB[SA] + KmA[SB] + [SA][SB]) using the Prism 5 (GraphPad) program, where Vi is the initial velocity, Vm is the maximal velocity, [SA] and [SB] are the substrate concentrations, KA is the dissociation constant of the enzyme for SA while KmA and KmB are the Michaelis-Menten constants for SA and SB44,45.
For inhibition studies, the compounds were added at different concentrations to the well prior to addition of ATP. Determination of the mode of inhibition (competitive/non-competitive) of the compounds and inhibition constant (Ki) values were obtained by carrying out assays with different concentrations of inhibitors and fitting the data to the following equations: Vi = Vmax [S]/(Km (1 + [I]/KI) + [S]) and Vi = (Vmax/(1 + [I]/KI) [S])/(Km + [S]) for competitive and non-competitive inhibition, respectively45.
In cellulo studies were performed by Cell Assay Innovations (Beverly, MA) using ClariCELL™ technology (www.cellassayinnov.com). HEK293 human embryonic kidney cells were transiently co-transfected with plasmid sequence verified vectors encoding full-length human DYRK1A and Tau and dispensed into multi-well plates. Cells were dispersed and incubated for 2 h in the presence of harmine, compounds 5a and 5g at various concentrations. Harmine was used as a control. The cells were lysed and an ELISA was performed by capturing the phosphorylated Tau substrate and detecting phosphorylation levels using an antibody to phosphor-TauT212. DYRK1A-dependent kinase activity in these assays was validated using a kinase-deficient DYRK1A[K188R].
Cell culture and proliferation assay
Cancer cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA) and were cultured according to the supplier’s instructions. Briefly, human KB epidermal carcinoma cells were grown in Dulbecco minimal essential medium (DMEM) containing 4.5 g/L glucose supplemented with 10% fetal calf serum (FCS) and 1% glutamine, 100 UI penicillin, 100 µg/mL streptomycin and 1.5 µg/mL fungizone and maintained at 37 °C in a humidified atmosphere containing 5% CO2. Cell viability was assessed using Promega CellTiter-Blue TM reagent according to the manufacturer’s instructions. Cells were seeded in 96-well plates (5 × 103 cells/well) containing 50 mL growth medium. After 24 h of culture, the cells were supplemented with 50 mL of the studied compound dissolved in DMSO (less than 0.1% in each preparation). After 72 h of incubation, 20 mL of resazurin was added for 2 h before recording fluorescence (λex = 560 nm, λem = 590 nm) using a Victor microtiter plate fluorimeter (Perkin-Elmer, USA). Results are shown in Table 1.
Kinase profiling
Kinase profiling was performed by Eurofins-CEREP, Le Bois l’Evêque, France. Compounds were tested at 5.10−8 M except for harmine which was tested at 10−6 M. All experiments were performed in duplicate and the values reported are the mean values.
Liquid chromatography/tandem mass spectrometry of compound 5a
All plasma and brain homogenate extracts were analyzed by an ultra-high performance liquid chromatography (UHPLC) system coupled to a triple-quadrupole mass spectrometer (Dionex Ultimate 3000 RSLC system coupled to a TSQ Vantage EMR, Thermo Scientific, Les Ulis, France). UHPLC separation was performed using a C18 column (HSS T3, 2.1 mm × 150 mm, 1.7 μm, Waters, Guyancourt, France) equipped with a guard column (HSS T3, 2.1 mm × 5 mm, 1.7 μm, Waters, Guyancourt, France) and maintained at 40 °C. The mobile phases were water, 0.1% formic acid (A) and acetonitrile (B). Compound 5a and the internal standard were eluted at a flow rate of 0.5 mL/min, with first 20% B for 4 min then from 20% B to 100% B in 6 min. A washing step in acetonitrile/isopropanol was added to limit carryover problems. Injection volume was 4 µL and the autosampler was maintained at 6 °C. Ionization was performed with a heated electrospray ionization source in positive mode. The source parameters were set as follows: spray voltage 3350 V, vaporizer temperature 250 °C, sheath gas (N2) pressure 55 psi, auxiliary gas pressure (N2) 25 psi, capillary temperature 350 °C. Analysis was performed under the Selected Reaction Monitoring (SRM) mode using several transitions for each compound at optimized collision energies (see Table SI2 and Figure SI1). Ion ratios were used to confirm the identity of compound 5a in plasma and brain samples. Data acquisition was performed using ThermoScientific Xcalibur 2.1 software system. All samples were analyzed in triplicate.
Two compounds, V34 (Fig. 9) and 5d, were tested as internal standard (IS). Compound V was found to be the most suitable to correct for matrix effects. Calibration curve points and quality control (QC) points were realized in spiked plasma or brain homogenate samples (Figure SI2). Spiking of compound 5a and IS was realized before extraction to correct for the extraction recovery. In plasma samples, linear curves were obtained from 1.6 to 320.3 pg/μL of compound 5a with within-run trueness in the 85–125% range and within-run precision better than 15% for all calibration points (r2 > 0.995). Samples with concentrations above the upper calibration point were diluted to be in the calibration range. Limit of quantification (LOQ) was determined as the lowest point of the calibration curve at 1.6 pg/μL. QC samples were prepared at 14.5 and 58 pg/μL to validate the between-run precision (n = 3, RSD of 13% and 5%, respectively), between-run trueness was 95 and 88% respectively. In brain homogenate samples, linear curves were obtained from 1.6 to 80 pg/mg of compound 5a with within-run trueness in the 85–125% range and within-run precision better than 15% (r2 > 0.990). Limit of quantification (LOQ) was determined as the lowest point of the calibration curve at 1.6 pg/mg. QC samples were prepared at 3.2 and 22.4 pg/mg to validate the between-run precision (n = 3, RSD of 8% and 2% respectively), between-run trueness was 129 and 93% respectively.
Figure 9.
Structure of internal standard compound V.
Animals
Male Ts65Dn (n = 8) mice and euploid wild-type (WT) littermates (n = 10) were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). As the recessive retinal degeneration 1 mutation Pde6brd1 in the original Ts65Dn mice causes blindness in homozygotes, we used an alternative strain (SN 5252) that is wild-type for Pde6b, thereby preventing retinal degeneration. Housing conditions comprised a standard cage (air-controlled system) with ad libitum food and water and a 12 h light/dark cycle. Mice received at least one week of habituation to the animal facility, followed by three days of handling (three minutes per mouse per day). Behavioral experiments were conducted following a two-week treatment with compound 5a (20 mg/kg) or vehicle by i.p. injections. Treatment was continued during the MWM protocol (mice being injected 30 min before each daily session).
All experiments were conducted in compliance with the ethical standards and animal welfare regulations of the French (Ministry of Agriculture) and European authorities (European Communities Council Directive of 24 November 1986). In vivo protocols used in the present study were also approved by our local ethics committee (Charles Darwin Committee). MC Potier has authorization n° A-75-2138 and B. Delatour has authorization n° A-75e-1741 from the Direction Départementale de la Protection des Populations de Paris (Service Protection et Santé Animales, Environnement) to perform experiments on vertebrates.
Formulation of compound 5a
Compound 5a in powder form was dissolved for injection in a mixture of DMSO, Cremophor EL (Sigma-Aldrich Chemie, Steinheim, Germany) and Proamp® sterile water for parenteral use (Laboratoire Aguettant, Lyon, France) (10:15:75) with a final concentration of 2.4 mg/mL46. Vehicle consisted of an identical composition without compound 5a. Mice received i.p. injections of compound 5a (20 mg/kg) or vehicle.
Bioavailability of compound 5a in plasma
To determine the bioavailability of compound 5a after i.p. injections, 12 WT mice were injected with 20 mg/kg of compound 5a and sacrificed at 12 successive time points. After decapitation, blood was sampled and transferred into BD Microtainer® SST tubes (Becton, Dickinson and Company, Plymouth, UK). Plasma was obtained according to the manufacturer’s protocol and stored at −80 °C.
Using an optimized Ultra Performance Liquid Chromatography with tandem mass spectrometry (UPLC-MS/MS), compound 5a was subsequently quantified in plasma. In short, proteins were precipitated using liquid/liquid extraction with ethyl acetate and supernatant was transferred into 40 µL DMSO/water/acetonitrile (12/44/44). Thereupon, UPLC separation was performed using a C18 column (HSS T3, Waters, Guyancourt, France) and a gradient mobile phase (acetonitrile and water with 0.1% formic acid), followed by triple quadrupole MS (TSQ Vantage, Thermo Scientific) with electrospray ionization. All samples were analyzed in triplicate.
Effect of compound 5a on behavioral deficit of Ts65Dn mice in the Morris water maze
To analyze the effect of compound 5a on spatial learning, a Morris Water Maze (MWM) protocol was used as previously described46. A white pool with a diameter of 150 cm was filled with water made opaque with non-toxic white paint (Acusol OP301, Rohm and Haas Company, Landskrone, Sweden) and kept at a constant temperature (20–21.5 °C). A 9-cm diameter platform was placed in the south-east (SE) quadrant of the pool, 1 cm under the water surface. The light intensity in the experimental room was kept constant at 70 Lux. Mice were habituated to the room for at least 30 min before starting each daily session. The acquisition phase comprised 5 days of training, four trials per day with an inter-trial interval of at least 20 min to minimize motor fatigue. The order of release positions in the pool was pseudo-randomly at the four cardinal points and changed every day to maximize spatial allocentric training and formation of a “cognitive map”. Each trial ended as soon as the mouse found the platform or after a maximum of 90 s, after which the animal was gently manually guided to the platform. Mice were left on the platform for 20 s before they were gently dried with a towel and individually placed in a cage with paper towels for further drying. The animals returned to their homecage after 5 min.
ANY-maze Video Tracking System (Stoelting, USA) was used to record all trials. Calculated measures included 1) thigmotaxis (percent distance traveled in the 10 cm wide peripheral annulus of the pool) indicative of unsuccessful spatial strategy to locate the platform and 2) the Corrected Integrated Pathway Length (CIPL) an unbiased measure of distance-to-platform travelled taking into account the animal’s speed and the initial distance from the goal for each starting position47.
Quantification of compound 5a in mouse brain
Immediately following last day of training in the MWM, mice were sacrificed by decapitation for maximal blood collection and the brain was immediately extracted from the skull, the cerebellum removed and the hemispheres separated and frozen at −80 °C. Plasma was obtained as described above. For UHPLC-MS/MS quantification of compound 5a in the brain, one hemisphere of each mouse brain was homogenized in methanol (J.T. Baker analyzed LC-MC reagent, Avantor Performance Materials BV, Deventer, The Netherlands), 1 mL per 100 mg brain. Subsequently, the IS was added to 400 µL of homogenate which was then centrifuged (12 min, 14000 × g, 4 °C). The supernatant was evaporated and transferred into 40 µL acetonitrile/methanol/water (2/2/1). After centrifugation (12 min, 14000 × g, 4 °C), 4 µL of supernatant was used for UHPLC-MS/MS analysis.
Electronic supplementary material
Acknowledgements
This study was part of the Therapeutics-21 project “Drug targets for improving cognitive deficits in Down syndrome”, funded by the Agence Nationale de la Recherche (ANR), by Labex Lermit (ANR grant ANR-10-LABX-33 under the program Investissements d’Avenir ANR-11-IDEX-0003-01) and by FCS Campus Saclay. This work was also supported by grants from Université Paris Diderot and Jérôme Lejeune Foundation (FJL). We also acknowledge the technical platform “Bioprofiler” for provision of high-performance liquid chromatography facilities (Unité BFA, Université Paris Diderot, CNRS UMR 8251). We thank CNPq (Brazil) for a Sandwich Fellowship (F.N.). The research leading to these results has received funding from the program “Investissements d’avenir” ANR-10-IAIHU-06.
Author Contributions
F.N. and S.G. performed the chemistry. A.D.D., B.D., C.A., J.M.D. and M.-C.P designed and performed the in vivo studies and wrote the corresponding part of the manuscript. J.D., L.C.B. and F.R.-L. designed and performed the in vitro inhibition studies on DYRK1A. J.D. and M.-C.P. designed and supervised the in cellulo studies of tau phosphorylation by DYRK1A. J.-P.L., N.H., I.S.-A. and D.T. designed and performed the mass spectral studies. K.C. and R.H.D. designed and supervised the chemistry and writing of the manuscript. All authors have given approval of the final version of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-20984-z.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Asim A, Kumar A, Muthuswamy S, Jain S, Agarwal S. Down syndrome: an insight of the disease. J. Biomed. Sci. 2015;22:2–9. doi: 10.1186/s12929-015-0138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiseman FK, Alford KA, Tybulewicz VLJ, Fisher EMC. Down syndrome-recent progress and future prospects. Human Mol. Genet. 2009;18:R75–R83. doi: 10.1093/hmg/ddp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010;9:623–633. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- 4.Khoshnood B, Greenlees R, Loane M, Dolk H. Paper 2: EUROCAT public health indicators for congenital anomalies inEurope. Birth Defects Res. A Clin. Mol. Teratol. 2011;91:S16–S22. doi: 10.1002/bdra.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bittles AH, Bower C, Hussain R, Glasson EJ. The four ages of Down syndrome. Eur. J. Public Health. 2007;17:221–225. doi: 10.1093/eurpub/ckl103. [DOI] [PubMed] [Google Scholar]
- 6.Parker SE, et al. Updated national birth prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res. A Clin. Mol. Teratol. 2010;88:1008–1016. doi: 10.1002/bdra.20735. [DOI] [PubMed] [Google Scholar]
- 7.Vicari S. Verbal short-term memory in Down’s syndrome: an articulatory loop deficit? J. Intellect. Disabil. Res. 2004;48:80–92. doi: 10.1111/j.1365-2788.2004.00478.x. [DOI] [PubMed] [Google Scholar]
- 8.Hattori M, et al. The DNA sequence of human chromosome 21. Nature. 2000;405:311–319. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- 9.Dierssen M. Down syndrome: the brain in trisomic mode. Nat. Rev. Neurosci. 2012;13:844–858. doi: 10.1038/nrn3314. [DOI] [PubMed] [Google Scholar]
- 10.Becker W, Joost HG. Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog. Nucleic Acid Res. Mol. Biol. 1999;62:1–17. doi: 10.1016/s0079-6603(08)60503-6. [DOI] [PubMed] [Google Scholar]
- 11.Hanks SK, Quinn AM. Protein kinase catalytic domain sequence database: identification of conserved features of primary structure and classification of family members. Methods Enzymol. 1991;200:38–62. doi: 10.1016/0076-6879(91)00126-H. [DOI] [PubMed] [Google Scholar]
- 12.Becker W, Sippl W. Activation, regulation, and inhibition of DYRK1A. FEBS J. 2011;278:246–256. doi: 10.1111/j.1742-4658.2010.07956.x. [DOI] [PubMed] [Google Scholar]
- 13.Ionescu A, et al. DYRK1A kinase inhibitors with emphasis on cancer. Mini Rev. Med. Chem. 2012;12:1315–1329. doi: 10.2174/13895575112091315. [DOI] [PubMed] [Google Scholar]
- 14.Tejedor FJ, Hämmerle B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2010;278:223–235. doi: 10.1111/j.1742-4658.2010.07954.x. [DOI] [PubMed] [Google Scholar]
- 15.Dowjat WK, et al. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci. Lett. 2007;413:77–81. doi: 10.1016/j.neulet.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park J, Song WJ, Chung KC. Function and regulation of DYRK1A: towards understanding Down syndrome. Cell. Mol. Life Sci. 2009;66:3235–3240. doi: 10.1007/s00018-009-0123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altafaj J, et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum. Mol. Genet. 2001;10:1915–1923. doi: 10.1093/hmg/10.18.1915. [DOI] [PubMed] [Google Scholar]
- 18.Sheppard O, et al. Altered regulation of tau phosphorylation in a mouse model of Down syndrome aging. Neurobiol. Aging. 2012;33:828.e31–828.e44. doi: 10.1016/j.neurobiolaging.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Souchet B, et al. Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol. Dis. 2014;69:65–75. doi: 10.1016/j.nbd.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 20.Wegiel J, Gong C-X, Hwang Y-W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011;278:236–245. doi: 10.1111/j.1742-4658.2010.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryoo S-R, et al. DYRK1A-mediated hyperphosphorylation of tau: a functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 2007;282:34850–34857. doi: 10.1074/jbc.M707358200. [DOI] [PubMed] [Google Scholar]
- 22.Ferrer I, Barrachina M, Puig B. Martinez de Lagran, M., Marti, E., Avila, J. & Dierssen, M. Constitutive DYRK1A is abnormally expressed in Alzheimer disease, Down yndrome, Pick disease, and related transgenic models. Neurobiol. Dis. 2005;20:392–400. doi: 10.1016/j.nbd.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 23.Ryoo S-R, et al. Dual-specificity tyrosine(γ)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 2008;104:1333–1344. doi: 10.1111/j.1471-4159.2007.05075.x. [DOI] [PubMed] [Google Scholar]
- 24.Kimura R, et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007;16:15–23. doi: 10.1093/hmg/ddl437. [DOI] [PubMed] [Google Scholar]
- 25.Smith B, Medda F, Gokhale V, Dunckley T, Hulme C. Recent advances in the design, synthesis, and biological evaluation of selective DYRK1A inhibitors: a new avenue for a disease modifying treatment of Alzheimer’s. ACS Chem. Neurosci. 2012;3:857–872. doi: 10.1021/cn300094k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adayev T, Wegiel J, Hwang YW. Harmine is an ATP-competitive inhibitor for dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) Arch. Biochem. Biophys. 2011;2:212–218. doi: 10.1016/j.abb.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogawa Y, et al. Development of a novel selective inhibitor of the Down syndrome-related kinase DYRK1A. Nature Commun. 2010;1:1–9. doi: 10.1038/ncomms1090. [DOI] [PubMed] [Google Scholar]
- 28.Tahtouh T, et al. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012;55:9312–9330. doi: 10.1021/jm301034u. [DOI] [PubMed] [Google Scholar]
- 29.Echalier A, et al. Meriolins (3-(pyrimidin-4-yl)-7-azaindoles): synthesis, kinase inhibitory activity, cellular effects, and structure of a CDK2/cyclin A/meriolin complex. J. Med. Chem. 2008;51:737–751. doi: 10.1021/jm700940h. [DOI] [PubMed] [Google Scholar]
- 30.Neagoie C, et al. Synthesis of chromeno[3,4-b]indoles as lamellarin D analogues: a novel DYRK1A inhibitor class. Eur. J. Med. Chem. 2012;49:379–396. doi: 10.1016/j.ejmech.2012.01.040. [DOI] [PubMed] [Google Scholar]
- 31.Naert G, et al. & Maurice, T. Leucettine L41, a DYRK1A-preferential DYRKs/CLKs inhibitor, prevents memory impairments and neurotoxicity induced by oligomeric Aβ25-35 peptide administration in mice. Eur. Neuropsychopharmacol. 2015;25:2170–2182. doi: 10.1016/j.euroneuro.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 32.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem. J. 2003;371:199–204. doi: 10.1042/bj20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De la Torre R, et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food. Res. 2014;58:278–288. doi: 10.1002/mnfr.201300325. [DOI] [PubMed] [Google Scholar]
- 34.Gourdain S, et al. Development of DANDYs, new 3,5-diaryl-7-azaindoles demonstrating potent DYRK1A kinase inhibitory activity. J. Med. Chem. 2013;56:9569–9585. doi: 10.1021/jm401049v. [DOI] [PubMed] [Google Scholar]
- 35.Minard, C., Palacio, C., Cariou, K. & Dodd, R. H. Selective Suzuki monocouplings with symmetrical dibromoarenes and aryl ditriflates for the one-pot synthesis of unsymmetrical triaryls. Eur. J. Org. Chem. 2942–2955 (2014).
- 36.Desage-El Murr, M., Nowaczyk, S., Le Gall, T. & Mioskowski, C. Synthesis of pulvinic acid and norbadione A analogues by Suzuki-Miyaura cross-coupling of benzylated intermediates. Eur. J. Org. Chem. 1489–1498 (2006).
- 37.Bui LC, et al. A high-performance liquid chromatography assay for Dyrk1a, a Down syndrome-associated kinase. Anal. Biochem. 2014;449:172–178. doi: 10.1016/j.ab.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 38.Bonneau N, Schmitz-Afonso I, Brunelle A, Touboul D, Champy P. Method development for quantification of the environmental neurotoxin annonacin in rat plasma by UPLC-MS/MS and application to a pharmacokinetic study. J. Chrom. B. 2015;1004:46–52. doi: 10.1016/j.jchromb.2015.09.039. [DOI] [PubMed] [Google Scholar]
- 39.Bonneau N, Schmitz-Afonso I, Brunelle A, Touboul D, Champy P. Quantification of the environmental neurotoxin annonacin in rat brain by UPLC-MS/MS. Toxicon. 2016;118:129–133. doi: 10.1016/j.toxicon.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 40.Sharp CJ, et al. Investigation into the role of P2X(3)/P2X(2/3) receptors in neuropathic pain following chronic constriction injury in the rat: an electrophysiological study. Br. J. Pharmacol. 2006;148:845–852. doi: 10.1038/sj.bjp.0706790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Summerfield SG, Zhang Y, Liu H. Examining the uptake of central nervous system drugs and candidates across the blood-brain barrier. J. Pharmacol. Exp. Ther. 2016;358:294–305. doi: 10.1124/jpet.116.232447. [DOI] [PubMed] [Google Scholar]
- 42.Reeves RH, et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 43.Nakano-Kobayashi A, et al. Prenatal neurogenesis induction therapy normalizes brain structure and function in Down syndrome mice. Proc. Natl. Acad. Sci. USA. 2017;114:10268–10273. doi: 10.1073/pnas.1704143114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woods YL, et al. The kinase DYRK phosphorylates protein-synthesis initiation factor EIF2Bepsilon at Ser539 and the microtubule-associated protein Tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem. J. 2001;355:609–615. doi: 10.1042/bj3550609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cornish-Bowden, A. Fundamentals of Enzyme Kinetics, 4th edition, Wiley–Blackwell, Weinheim (2012).
- 46.Braudeau J, et al. Specific targeting of the GABA-A receptor α5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. J. Psychopharmacol. 2011;25:1030–1042. doi: 10.1177/0269881111405366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barnes CA, Suster MS, Shen J, McNaughton BL. Multistability of cognitive maps in the hippocampus of old rats. Nature. 1997;388:272–275. doi: 10.1038/40859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.