

Abstract
The utility of metabolic synthons as the building blocks for new biomaterials is based on the early application and success of hydroxy acid based polyesters as degradable sutures and controlled drug delivery matrices. The sheer number of potential monomers derived from the metabolome (e.g., lactic acid, dihydroxyacetone, glycerol, fumarate) gives rise to almost limitless biomaterial structural possibilities, functionality, and performance characteristics, as well as opportunities for the synthesis of new polymers. This review describes recent advances in new chemistries, as well as the inventive use of traditional chemistries, toward the design and synthesis of new polymers. Specific polymeric biomaterials can be prepared for use in varied medical applications (e.g., drug delivery, tissue engineering, wound repair, etc.) through judicious selection of the monomer and backbone linkage.
Graphical Abstract
1. INTRODUCTION
To keep pace with the rapid progress in medicine, advanced biomaterials are needed to provide the unique functionality required by the new devices, tissue scaffolds, and drug delivery systems to meet patient care needs. One approach to addressing this functional demand and to maintaining the biocompatibility necessary for new polymeric biomaterials is to synthesize new polymers derived from structures within the human metabolome. The human metabolome is made up of metabolic pathways comprised of thousands of structures needed to sustain human life. The idea is to molecularly engineer specific functions into new materials that, upon implantation, function as designed, degrade in conformance to clinical needs and are eliminated from the body via their natural metabolic pathways. This review focuses on the advances over the past decade, with some historical context included, in polymeric biomaterial design and synthesis with this specific design philosophy in mind.
Specifically, we describe the use of metabolically derived synthons to prepare unique polymers and the subsequent evaluation of these biomaterials for medical applications. We do not describe the successes in derivatization of natural polymers or un-natural poly(amino acid)s. Nor do we describe the classical condensation polymerization methods (e.g., between a diol and diacid) to prepare polymers as these topics have been extensively reviewed elsewhere and/or are outside of the scope of this article. We begin with a discussion of polymers prepared from hydroxy acids, which have been known for more than three decades. We then transition to synthons that are more compositionally complex, possessing several reactive/functional groups or stereochemical centers. We highlight the use of these biomaterials to address specific challenges observed in the clinic and note the important role that composition, structure, and physical properties play in their success. An overview of topics covered in this review, including the metabolic synthons, polymerization strategies, polymer architectures and linkages, form factors, and their applications is shown in Table 1. Finally, we conclude with a summary of the findings and the outlook for the future.
Table 1.
Summary of Polymerization Strategies, Polymers, And Applications of the Metabolic Synthons Covered in This Review
| synthon | polymerization routes | polymer architectures and linkages | form factors | applications |
|---|---|---|---|---|
| hydroxy acids | polycondensation, ring-opening polymerization, microbial synthesis, divergent dendrimer synthesis, and convergent dendrimer synthesis | linear polyesters, polyester dendrimers, and hyperbranched polyesters | microparticles, nanoparticles, hydrogels, tablets, plastics, solids, and dendrimers | biodegradable sutures, controlled drug delivery, pulmonary drug delivery, biodegradable tissue engineering scaffolds, implantable glues, packaging, medical devices, and wound repair |
| dihydroxyacetone | ring-opening polymerization, transketalization, and condensation | linear polycarbonates, linear poly(carbonate-ester)s, symmetrical lipids, poly(spiro-acetal)s, and poly(carbonate-acetal)s | microparticles, nanoparticles, hydrogels, solids, tablets, powders, and thermoplastics | seroma prevention, hemostatic agents, controlled drug delivery, tissue engineering scaffolds, and functionalizable surfaces |
| glycerol | ring-opening polymerization, epoxide/CO2 coupling, divergent dendrimer synthesis, and convergent dendrimer synthesis | linear poly(1,3-carbonate)s, linear poly(1,2-carbonate)s, poly(carbonate-ester)s, polyether and polyester dendrimers, polyether and poly(ether-ester) hyperbranched polymers, and hybrid dendritic-linear esterether copolymers | films; dendritic drug carriers, hydrogels, and anionic amphiphilic dendrons | controlled drug delivery, medical devices, tumor recurrence reduction, tissue engineering, wound repair, sealants for corneal lacerations, antibacterial agents, and cartilage tissue engineering scaffolds |
| natural amino acids | N-carboxyanhydride polymerization, genetic engineering, living systems, convergent dendrimer synthesis, divergent dendrimer synthesis, and solution or solid phase synthesis using coupling reagents (DCC, EDCI, BOP, oxalyl chloride, and pentafluoroesters) | linear poly(amino acid)s, polypeptide dendrimers, and tyrosine-based polycarbonates | hydrogels, dendrimers, and nanoparticles | drug delivery, gene delivery, surgical adhesives and sealants, and cortical neural probe carriers |
| fumarate | condensation to form a bis(hydroxypropyl) fumarate diester, followed by transesterification | poly(propylene fumarate) | viscous liquids and cross-linked networks | tissue engineering scaffolds, orthopedic implants, and drug delivery |
| saccharides | enzymatic catalysis, living systems, polycondensation, click-chemical polymerization (Huisen cycloaddition), organocatalyzed ring-opening polymerization, anionic ring-opening polymerization, ring-opening metathesis polymerization, and reversible addition–fragmentation chain transfer | linear polyesters, linear polyethers, poly(glycoamidoamine) s, poly(amidosaccharide)s, sugar-derived polycarbonates, and glycopolymers | micelles, nanoparticles, microparticles, and hydrogels | drug delivery, nucleic acid delivery, wound repair, tissue engineering, interactions with natural carbohydrate receptors, protein stabilizing agents, promotion/inhibition of biofilm formation, vaccine development, biosensing, and signal transduction studies |
2. HYDROXY ACID SYNTHONS
Hydroxy acids are aliphatic organic acids bearing a hydroxyl group and a carboxylic acid on each end of the molecule. Examples of common metabolic synthon hydroxy acids and their corresponding dimers or cyclized structures (in parentheses) are glycolic acid (glycolide), lactic acid (lactide), and 6-hydroxyhexanoic acid (ε-caprolactone) (Figure 1). These weak acids (pKa ranges from ~3 to ~ 5) are soluble in aqueous solution when they contain one to two methylene units separating the acid and hydroxyl functionality, and aqueous solubility decreases with an increasing number of methylene units. The first commercially available poly(hydroxy acid), poly(glycolic acid) (PGA), was developed and reported by Frazza and Schmitt at the former American Cyanamid Company in 1971,1 followed by linear polyesters of lactic acid (LA), GA:LA copolymers, and 6-hydroxyhexanoic acid, all of which are now common and widely used biomaterials. High molecular weight polymers of these hydroxy acids, in particular PGA, were introduced as biodegradable sutures in the late 1960s and early 1970s based on their safety profile, mechanical strength, and manufacturing processability.2–5 Success in sutures spurred the continued investigation of these materials for more advanced applications, such as controlled drug delivery systems,6–8 degradable tissue engineering scaffolds,9 and implantable glues.10
Figure 1.
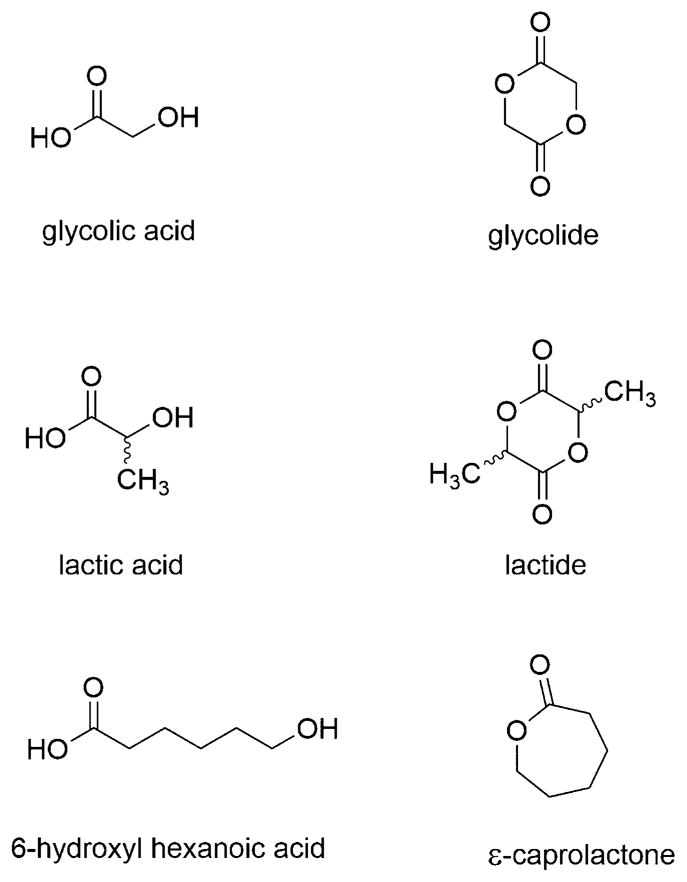
Chemical structures of common hydroxy acids and their corresponding dimers or cyclized structures used for the synthesis of polyesters.
2.1. General
2.1.1. Hydroxy Acids in Metabolism
Cellular metabolism, a series of regulated enzymatic reactions that sustain life, is dependent on three major dietary sources of energy: carbohydrates, fats, and proteins.11 The premise of using metabolites as monomer units for the synthesis of biodegradable polymers is that the body is already equipped with pathways to eliminate downstream polymer degradation products. Therefore, the likelihood of generating a local toxic environment or initiating an inflammatory response is reduced. The representation of synthons in natural metabolic pathways is the subject of most college-level biochemistry textbooks. For example, lactic acid is an end product of glycolysis. The accumulation of lactic acid in muscle under anaerobic conditions is one cause of muscle fatigue. The transport of lactic acid to the liver feeds glyconeogenesis, thereby providing additional glucose to active muscles. This production-conversion feedback is known as the Cori cycle, and allows burden sharing of new energy source production between the muscle and liver.
GA is represented in the process of photorespiration, wherein a plant consumes oxygen and releases carbon dioxide, opposite to the more recognized photosynthesis. The process is a result of a complex interaction between the enzymes in plant chloroplasts, peroxisomes, and mitochondria. While GA is not actively involved in the metabolic pathways of humans, its consumption results in its metabolism by glycolate oxidase, and it is ultimately metabolized to oxalic acid or carbon dioxide.
The building block 6-hydroxyhexanoic acid is a substrate for alcohol dehydrogenase, wherein the primary alcohol at the 6-position is converted to an aldehyde, which is further converted by aldehyde dehydrogenase to form a diacid, a process known as ω-oxidation. The resulting 1,6-diacid (adipic acid) is conjugated to coenzyme A and ultimately enters the citric acid cycle as succinic acid or is eliminated from the body as adipic acid.
2.1.2. Structure and Reactivity
The synthesis of linear polyesters from GA, LA, and 6-hydroxyhexanoic acid by direct polycondensation is complicated by the competing formation of the 6-membered cyclic dimers, glycolide and lactide (or 7-membered ε-caprolactone), and by the difficulty in removing water. The polycondensation of these hydroxy acids creates two equilibria: dehydration to form the linear polyester and depolymerization to form the ring structures. In the absence of control over these competing equilibra, only low molecular weight polyesters (low tens of thousands) are obtained. Strategies to obtain high molecular weight polyesters with molecular weights on the order of 100 kDa by direct polymerization include, but are not limited to, melt/solid polycondensation, where a low molecular weight polymer is made by direct polycondensation, and then crystallized to the solid state, and heated to ~150 °C in the presence of a catalyst to form a large molecular weight polymer (~500 kDa) with high yield.12 Careful selection and screening of catalysts13 and azeotropic removal of water are required.14 These and other direct polycondensation routes are reviewed by Gupta and Kumar with a focus on poly(lactic acid) (PLA).15 The more common synthetic route to polyesters of this type is ring-opening polymerization (ROP), which is discussed in the following section.
2.2. Linear Polyesters
Cyclic dimers of GA and LA are produced on a manufacturing scale in a continuous process through vacuum distillation of the oligomers and purified by crystallization, although the specifics of each process vary greatly among manufacturers.16 The synthesis of ε-caprolactone (ε-CL) follows a two-step process: first, the production of a strong oxidizer, peracetic acid, followed by its use to oxidize cyclohexanone to make the final monomer.
Synthesis of these linear polyesters by ROP allows good control over the molecular weight, dispersity, and end group composition. Closed dimers of both LA and GA into 6-membered lactones, and ring closing of 6-hydroxyhexanoic acid into a 7-membered lactone, afford structures that readily polymerize in both the melt and in solution (Figure 2). The most common mechanism through which these cyclic monomers are polymerized is by coordination–insertion with tin octoate as the coordination catalyst. However, both cationic and anionic polymerization are reported, with over 100 catalysts identified for the polymerization of lactide alone.15
Figure 2.

Structures of poly(glycolic acid) [PGA], poly(D,L-lactic acid) [PLA], poly(D,L-lactic acid-co-glycolic acid) [PLGA], poly(3-hydroxybutyrate) [P3-HB], and poly(ε-caprolactone) [PCL].
Microbial routes of polyester synthesis are also reported. Of particular importance for the field of biomaterials are the poly(hydroxyalkanoate)s. These polyesters are synthesized by microbes in response to stress and starvation, generally accumulating as inclusion bodies when carbon sources exceed nitrogen sources.17 Biomass can be used as feedstock for the manufacture of poly(hydroxyalkanoate)s, and the polyesters are degradable in the environment, giving these materials a designation as “green” since they are not derived from petrochemicals, and can also be produced in plants.18 Because of their favorable stress/strain, thermal, and processing characteristics, the polymer can be used in the packaging industry.19 Medical applications of poly(hydroxyalkanoate)s are also described, and these polymers are used in tissue engineering,20 drug delivery,21 and medical devices.22 Modulation of the polyester characteristics is achieved by alteration of the feedstock, as well as by genetic manipulation of the microbe. The recent application of CRISPRi to engineer the metabolic pathways associated with poly(hydroxyalkanoate) synthesis is an exciting advance with the potential to fine-tune microbial gene expression and allow the production of tailored polyesters.23
Of particular importance for these polyesters is the ability to tune their degradation rates through engineering of their molecular architecture. Rapid degradation kinetics (i.e., days) is desirable for an application like pulmonary drug delivery;24 medium degradation kinetics (i.e., weeks) is desirable for an application like an absorbable suture, and longer degradation kinetics (i.e., years) is desirable for an application like a drug-eluting stent.25 Approaches to alter degradation rates include modulation of molecular weight, dispersity, crystallinity, monomer hydrophobicity or hydrophilicity, and end group characteristics (Table 2).
Table 2.
Design Characteristics for Modulating the Rate of Hydrolysis of Polyester Biomaterials
| parameter | increase degradation kinetics | decrease degradation kinetics |
|---|---|---|
| molecular weight | lower molecular weight broad dispersity | higher molecular weight narrow dispersity |
| crystallinity | highly crystalline matrix | amorphous matrix |
| monomer content | more hydrophilic | more hydrophobic |
| end group | more hydrophilic ionized | more hydrophobic end-capped |
2.3. Polyester Dendrimers
Dendrimers are three-dimensional (3D) globular nearly monodisperse macromolecules with low dispersities (Đ), composed of a central core from which are emitted branched repeating units (generations, Gn), and peripheral terminal groups that can be further functionalized for a desired application.26–30 The size, architecture, density, and surface groups of these macromolecules can be easily tuned during their synthesis, enabling a highly versatile platform for materials and biomedical research. Dendrimers are already used for a variety of applications, including drug delivery, gene transfection, tissue engineering, and molecular imaging.29 Among various dendritic structures being investigated for biomedical applications, polyester dendrimers based on metabolically derived synthons [e.g., LA and glycerol (GL), glucose, succinic acid (SA), LA and aspartic acid (AA), which are linked via ester groups] are of interest as promising biomaterials because they are biocompatible and biodegradable.31 Currently, these specific dendrimer formulations are being evaluated for drug delivery, tissue engineering, and wound repair.31–37
Dendrimers are synthesized by either a divergent (from core to surface) or a convergent (from surface to core) route, as depicted in Figure 3. Biocompatible and biodegradable polyester dendrimers based on metabolically derived synthons are one example of this class of materials, and these dendrimers are synthesized via one of the two approaches shown in Figure 3.
Figure 3.

Pictorial representation of dendrimer synthesis by (A) divergent and (B) convergent routes.
The divergent synthesis allows the growth of the dendrimer from a multifunctional core to the periphery by reacting it with a monomer unit, which possesses one reactive site and multiple protected groups, giving the first generation (G1) dendrimer (Figure 3A). Following this reaction, the protected groups on the monomer units of the G1-dendrimer are deprotected and subsequently reacted with another reactive monomer unit to generate a growing branching structure, in a stepwise fashion. Specifically, Grinstaff et al. used this strategy to synthesize GL and LA polyester Gn-dendrimers, namely Gn-PGLLAs, starting from a tetrafunctional G0-PGLLA–OH core (Figure 4).38
Figure 4.

Chemical structures of polyester dendrimers based on metabolically derived synthons from (top) lactic acid (LA, black) and glycerol (GL, blue) and (bottom) citric acid (CA, black) and poly(ethylene glycol) (PEG, green).
Each Gn is obtained in a two-step sequence: esterification of a benzylidene acetal-protected GLLA monomer with an unprotected Gn–1-PGLLA–OH dendrimer, followed by hydrogenolysis of the protecting-groups. At each sequence, the protected-dendrimers are purified by silica gel chromatography to remove unreacted monomers and coupling reagents. Nuclear magnetic resonance (NMR) spectroscopy monitored the formation of each successive generation by following the integration of the benzylidene and methyl proton groups of protected GL and LA, respectively. Fourier transform infrared (FT-IR) spectra of the deprotected dendrimers confirmed the presence of the hydroxyls via the O–H broad stretches, at around 3400 cm−1. Molecular weights of Gn dendrimers, ranging from 0.8 to 6 kDa with narrow dispersity, are described and confirmed by matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) and size exclusion chromatography (SEC) against polystyrene standards. The glass transition temperatures (Tg) of the dendrimers are generation and compositionally dependent and range from 19 to 31 °C and from −34 to −28 °C for the protected and deprotected structures, respectively.
Namazi and Adeli also used the divergent route for the synthesis of citric acid (CA)-based polyester dendrimers starting from a CA-PEG multifunctional core (Figure 4).39 The growth of the macromolecule is achieved by activating the Gn–1 dendrimer and subsequently reacting it with CA. The material is separated from the crude mixture by precipitation in diethyl ether. Gn-PCA-PEG dendrimers are purified by dialysis, and their structures were confirmed by 1H NMR and FT-IR spectroscopies. Interestingly, these dendrimers form thermoreversible hydrogels in the presence of water and viscosity measurements reveal a sol–gel transition temperature (Tsol–gel) above 37 °C (the G1 and G2-PCA-PEG dendrimers exhibited Tsol–gel of 40–45 °C and 50–55 °C at 5 wt %, respectively). The sol–gel properties and ability to synthesize various dendrimer generations allowed for facile drug encapsulation and tunable release properties from the hydrogel network.40
Another example based on the divergent synthetic approach was reported by Yeates et al. for the synthesis of aromatic polyester dendrimers based on 3,5-dihydroxybenzoic acid (3,5-DHBA).41 The DHBA synthon can be considered as an analog of the hydroxy acid synthon possessing an aromatic backbone, although the reactivity of the phenol and corresponding carboxylic acid are different than LA, for example.
Two- and three-branched initiator cores (i.e., hydroquinone, naphthalene-2,6-diol and phloroglucinol) are first coupled to either benzyl-3,5-dihydroxybenzoic acid (Bn-3,5-DHBA) monomers, using N,N′-dicyclohexylcarbodiimide (DCC) as the activating agent, or to 3,5-dibenzyloxybenzoyl chlorides (Bn-3,5-DHBCl) to obtain G1-polyester dendrimers.42,43 Next, hydrogenolysis of the benzyl groups and their subsequent reactions with the activated monomers afforded higher generation aromatic polyester dendrimers (up to G4) in good yield. 1H and 13C NMR spectroscopies and MALDI-TOF spectrometry confirmed the structures.
While the divergent route is used to synthesize a wide variety of dendrimers, incomplete reactions during the coupling step can lead to imperfect dendrimer structures. In order to limit these undesirable side reactions, the monomer units are usually added in large excess to the reaction mixtures, thus an additional purification step is necessary to remove all unwanted byproducts and reagents before any subsequent step. These limitations have led to the optimization of the divergent route, by using monomers and reagents that can be separated from the dendrimers by simple extraction or precipitation, thus avoiding the use of time-consuming flash chromatography. Moreover, the use of complementary functional groups within the monomer structures can afford high generation dendrimers in excellent yield.
The convergent strategy is another common approach for the synthesis of dendrimers and was developed to circumvent some of the problems with using the divergent strategy (Figure 3B). Defect-free dendrimers are obtained and successfully separated from byproducts using this approach. The convergent approach enables the construction of dendrons from the periphery toward the central focal point, followed by their subsequent reaction through their focal point with the polyfunctional core to generate the dendrimers. Additionally, the purification of the dendrons is simpler after each step because their structures and molecular weights differ significantly from the starting materials and reaction byproducts. Thus, a wide panel of reaction conditions can be used to generate different dendrimer structures. Despite the advantages of the convergent approach, it is still mainly used to obtain lower generation dendrimers because the synthetic coupling yields between the focal point of the large dendron and the next building block are reduced due to steric hindrance.
Fréchet et al. reported the first convergent synthesis of polyester aromatic dendrimers based on the naturally occurring 3,5-DHBA metabolite.42,43 The dendritic starting fragments are prepared by first coupling the monomer unit, 2,2,2-trichloroethyl-3,5-dihydroxybenzoate (A), with either activated benzoic acid (BA) or Bn-3,5-DHBA (Figure 5). The trichloroethyl ester of the G1-dendron is then activated by zinc in glacial acetic acid to afford the carboxylic acid, which is subsequently coupled with A to obtain the G2-dendron. Repetition of this two-step procedure affords higher generation dendrons, which upon reaction with the 1,l,l-tris(4′-hydroxyphenyl)ethane multifunctional core (B), generated the Gn-polyester dendrimers. Deprotection of the benzyl-protected dendrimers by catalytic hydrogenolysis generated hydroxyl groups on the surface, which can potentially be functionalized for any desired application. Flash chromatography after each step provided the dendrons and dendrimers in good isolated yields and purity. Integration of the methylene proton resonance of the dendron focal point and its comparison to other resonances in the spectrum allowed the confirmation of the dendritic structures and their generation number. Moreover, coupling of the dendrons to the polyfunctional core is easily detected by the appearance of the resonances in the NMR spectrum for the core structure. Similarly, the disappearance of the benzylresonances confirmed the complete deprotection of the dendrimers. FT-IR, MALDI-MS, elemental analysis, and SEC provided additional data that support the proposed structures and their purity. The composition of the surface functional groups affects the physical properties of the dendritic materials, and thus one can tailor the surface functionalization of the dendrimer for a given property. For example, benzyl-protected polyesters of 10.746 kDa molecular weight possess a Tg of 346 K, while the Tg is 474 K for the corresponding phenolic polyester.
Figure 5.

Chemical structures of monomer units and multifunctional core used in Fréchet et al. convergent synthesis of polyester-dendrimers.42,43
Do et al. also reported the convergent synthesis of 3,5-DHBA-polyester dendrimers, similar to the type prepared by Fréchet et al., where peripheral phenolic groups are further functionalized with chromophore units, and the materials were evaluated for their optical properties.44–46
Amrein et al. described the convergent synthesis of (R)-3-hydroxy-butanoic acid (HB)- and trimesic acid (TMA)-derived polyester dendrimers, starting from the benzyl-protected dimer and tetramer of HB (A), TMA-trichloride (B), and tert-butyldiphenylsilyl ether-protected 5-hydroxymethyl-1,3-benzenedicarboxylic acid dichloride (C) (Figure 6).47 For the construction of the branching dendritic units, Bn-HB (A) is first acylated with compound C to obtain the Bn-G1 dendron. After the subsequent deprotection of benzyl-groups, activation of the carboxylic acids and their reaction with the benzyl-protected G1-dendron, the benzyl-G2 dendron is obtained. Finally, for the assembly of G1/G2 dendrimers, desilylated Bn-G1/G2-dendrons are coupled with a trifunctional HB core (D), respectively. The dendrons and dendrimers were characterized by FT-IR, 1H, and 13C NMR spectroscopies, optical rotation, MALDI-MS spectrometry, and elemental analysis. Their biodegradability, evaluated in the presence of poly(3-hydroxybutyrate) depolymerase (PHB-depolymerase) and compared to a standard substrate, the linear tetrameric HB (A; n = 2, R = H), showed that deprotected dendrimers with dimer-HB units are better candidates for the hydrolase than their respective benzyl-ones, whereas the simple dimer-HB (A; n = 1, R = H) did not decompose in the presence of the enzyme. Similar to the standard substrate, all dendrimers with tetrameric-HB units are hydrolyzed by the enzyme. In addition, other enzymes such as esterase, lipase, and protease degrade these dendrimers.
Figure 6.

Chemical structures of monomer and core units used in Amrein et al. convergent synthesis of polyester dendrimers.47
2.4. Hyperbranched Polyester Structures
Hyperbranched polymers display highly and randomly branched 3D globular architectures, which contain more defects in their structures than the previously described monodisperse and uniformly branched dendrimers. These polymers possess a branching degree (DB) (i.e., the mole fraction of fully branched monomers in the polymer structure) lower than one (generally, 0.4–0.6) and high dispersities (Đ > 3). Importantly, the advantages of dendritic structures over the linear ones, such as low molecular entanglement, high solubility, low viscosity, and highly reactive functional groups, are also exhibited in hyperbranched polymers. Moreover, hyperbranched polymers are synthesized in one-step reactions, whereas dendrimers are constructed in step-by-step sequences, which often require protection/deprotection of the monomers/dendrons before subsequent reaction with another reactive one and purification of the product after each step. Therefore, despite the irregular structures of hyperbranched polymers, their relative ease of synthesis is advantageous for applications requiring highly functionalizable globular macromolecules. Hyperbranched polymers are currently of significant interest in nanoscience and nanotechnology, as well as in biomedical research. Potential applications of these materials include their use as components for coating, blends and sensors, drug delivery, gene transfection, and bioimaging.48–51 Hyperbranched polyesters are an important subclass of hyperbranched polymers, composed of either aliphatic or aromatic monomer units, linked together by ester groups. These structures are extensively studied in the literature for their use as additives, rheology modifiers, and components for blends, coatings, and sensors.52–54 However, hyperbranched polyesters are surprisingly less explored in the biomedical field, with commercially available Boltorns Hx (x = 20, 30, and 40), based on 2,2-dimethylol propionic acid (Bis-MPA), being the most widely used hyperbranched polyesters at present.50,55 Today there are no formulations of hyperbranched polyesters, based on naturally occurring metabolites, currently in clinical use. The challenges associated with development of such a product include the reproducibility of the synthesis and control measures to ensure the quality of the hyperbranched polyester.
Hyperbranched polyesters are generally synthesized via one of two approaches: polymerization of an ABx monomer or polymerization of two types of monomers or a monomer pair, each method having advantages and disadvantages.48 The first approach can be divided into four subclasses, depending on the reaction mechanism: (1) polycondensation of ABx monomers, (2) self-condensing vinyl polymerization, (3) self-condensing ROP, and (4) proton-transfer polymerization. The second approach includes two main subclasses: (1) polymerization of A2 and Bx (x > 2) monomers and (2) in situ generation of one type of ABx intermediate due to the nonequal reactivity of specific functional groups in monomer pairs, leading to hyperbranched polyesters. A broad range of hyperbranched polyesters, specifically those based on naturally occurring metabolites, are reported via the polycondensation of ABx monomers, which was first theoretically described by Flory in the early 1950s. Therefore, this paragraph will solely focus on this type of polymerization. Flory predicted that ABx monomers, where A is a single functional group of one type and B is two or more of another type, can react with each other, in a single step, to form highly branched 3D polymer structures, free of cross-links (Figure 7).56 The polycondensation of the reactive monomers is activated by the presence of a catalyst or after activation of A and B monomer units via a thermal, chemical, or photochemical process and leads to polydisperse and irregular hyperbranched structures.
Figure 7.

Flory’s polycondensation of ABx monomers (where x ≥ 2) to form hyperbranched polymers. Adapted with permission from ref 56. Copyright 1952 American Chemical Society.
Fréchet et al. reported the first one-step synthesis of hyperbranched polyesters based on metabolically derived synthons by heating (trimethylsiloxy)benzoyl chlorides in the presence of a catalyst (Figure 8).57–59 This hyperbranched aromatic polyester is based on DHBA. This synthetic strategy afforded high mass weighted molecular weight DHBA-derived polymers (Mw = 30–65 kDa; Đ = 1.9–2.5) with irregular dendritic structures in a 60–88% yield after precipitation in methanol. Moreover, these structures possess free phenolic groups both throughout and at the periphery of the macromolecules, which can be further functionalized with various end groups to tune the physicochemical properties of the materials. The hyperbranched aromatic polyesters are soluble in most common organic solvents. 1H and 13C NMR spectroscopy confirmed their structures with determination of DB in the range of 0.55–0.60. The polymers with free phenolic groups possess a Tg of 190 °C and are stable up to 400 °C, with about 10 and 50 weight percentage (wt %) loss at 435 and 560 °C, respectively.58 These results are similar to those obtained with linear polyesters based on DHBA, demonstrating that the branching structure and the multiple functional groups at the periphery afford a minimal effect on the thermal properties of the polymers. However, further functionalization of the phenolic groups influences the thermal properties of the macromolecules, which are dependent on the nature of the side chains. For example, when flexible adipic acid chains are coupled to the phenolic hydroxyl groups, the Tg dropped to 6 °C, whereas when the phenolic hydroxyl groups are silylated with t-butyldimethyl silyl groups or esterified with acetyl chlorides, the Tg increases to 103 and 133 °C, respectively, due to the rigidity and polarity of the protecting groups.59 Additionally, the thermal decomposition temperatures (Tdecomp) of the hyperbranched aromatic polyesters also depend on the nature and composition of the side chains. Two Tdecomp are observed for the functionalized polymers, one in the range of 280–400 °C which corresponded to the wt % loss of the side chains and the second in the range of 400–545 °C, attributed to the remaining polymeric structure.59
Figure 8.

Synthesis of hyperbranched aromatic polyesters by Fréchet et al. Adapted with permission from ref 58. Copyright 1991 American Chemical Society.
Similarly, Mourey et al. reported the synthesis of DHBA-derived hyperbranched polyesters starting from 3,5-diacetoxybenzoic acid (3,5-DABA).60 The melt polymerization proceeded without any catalyst, at T < 250 °C, and afforded high Mw (30–500 kDa) amorphous polymers, with high Đ (6.3–30.2), and Tg in the range of 147–160 °C. Mild acidic hydrolysis of the dendrimer removes 89% of the acetate groups to afford a phenolic-hyperbranched aromatic polyesters with Tg around 200 °C, and allowed the measurement of DB, which equaled 0.5. Additionally, copolymerization of DABA with either a nonbranching AB or a monofunctional A monomer, proceeded successfully and afforded hyperbranched aromatic polyesters with characteristics almost similar to those of the homopolymers; however, the determination of DB was not possible by 1H NMR spectroscopy for this sample. Other variants of hyperbranched aromatic polyesters based on DHBA monomers and possessing different physicochemical properties are also described by Luebbers et al.,61 Hill et al.,62 and Ramakrishnan et al.63
Due to their highly compact and branched structures, hyperbranched aromatic polyesters often exhibit extremely low viscosities, high solubilities in most organic solvents, and interesting rheological properties as compared to the linear aromatic polyesters. However, relatively few synthetic routes are reported for the formation of these structures, and most of the generated polyesters are insoluble in aqueous solutions and/or require additional steps of modification/functionalization to control their solubility and biocompatibility,50,55 limiting their use in vivo. Additionally, their complete characterization, which is critical for understanding their physicochemical properties and for identifying potential applications, is challenging. Hyperbranched polyesters, which can be easily metabolized into degradable components due to the hydrolysis of ester backbones within their structures, are less explored.50
2.5. Hydroxy Acid Synthon Summary
Since the first synthesis of high molecular weight poly(hydroxy acid)s [e.g., poly(glycolic acid)s] and their translation to the market place as sutures for use in the clinic, these polymers continue to be widely used and investigated. However, linear poly(glycolic acid), poly(lactic acid), and poly(caproic acid) possess several limitations such as control of molecular weight, chemical modification or derivatization potential, alteration of their thermal properties, and limited solubility. Consequently, significant research efforts toward new copolymers with other metabolite or nonmetabolite synthons [e.g., glycerol and poly(ethylene glycol] and polymer architectures (e.g., linear vs dendrimer) are underway, with several notable successes reported to date. Moreover, the development of new polymerization routes, starting from a variety of functionalized monomeric structures, will allow the preparation of new biocompatible and biodegradable hyperbranched aromatic or aliphatic polyesters based on hydroxy acids and enable the understanding of how their structural features impact the thermal, mechanical, degradation, and solution properties of these materials. These advances will expand their use in a variety of biological applications such as drug delivery, bioimaging, and tissue repair.
3. DIHYDROXYACETONE SYNTHONS
Dihydroxyacetone (DHA, I, Figure 9a), a three-carbon sugar, is an important metabolite in humans, yeast, bacteria, and plants. The awareness of DHA, sometimes called “dioxyacetone,” in carbohydrate metabolism dates back to the early 1900s.64,65 One of the first proposed therapeutic applications of DHA was for the treatment of diabetes. Rabinowitch published reports on the ability of DHA to reduce the dosage of insulin needed by diabetics.64,66–68 It was not until the late 1950s, however, that DHA gained interest for its first major commercial use: sunless tanning lotions. Eva Wittgenstein noticed that when children accidentally “spit up” an orally administered solution of DHA onto their skin, pigmentation occurred.69 While it had been previously known that sugars react with primary amines to form brown pigments known as melanoidins, a reaction discovered by Maillard in 1912,70 and more mechanistically detailed by Hodge in 1953,71 it was Wittgenstein and Berry who drew a connection between the browning in foods and the browning of skin from DHA.72 They suggested that the mechanism through which DHA functions as an artificial tanning lotion is a reaction between the carbonyl group of DHA and the basic groups of amino acids in proteins present on the surface of the skin.72,73 To date, DHA-based artificial tanning lotions are FDA approved for topical use and remain a popular selling item. Although, some controversy has arisen over the unknown long-term health effects of the inhalation of spray tan products, as well as postapplication UV exposure and the generation of harmful reactive oxygen species,74–76 DHA-based artificial tanning lotions continue to be accepted as a safe alternative to UV-based tanning.77–79 A brief review on the safety concerns of DHA-based sunless tanners is covered by Pagoto.77
Figure 9.

(a) DHA dimerization mechanism.89 (b) Dissociation of DHA dimer in aqueous solutions. (c) (1) Reversible reaction of DHA with a primary amine to form a Schiff base, (2) reductive amination to a secondary amine, and (3) Heyns rearrangement. Nomenclature: I, DHA; II, DHA dimer; III, gem-diol (DHA hydrate).
DHA’s reactive ketone functionality, two α-hydroxy groups, and metabolic involvement are key reasons why this synthon is of continued interest. From a topical standpoint, DHA shows potential as a treatment for proliferative skin diseases such as psoriasis and eczema, a cosmetic solution to the masking of vitiligo, and as an ingredient for controlled release mosquito-repellent formulations.80–84 DHA has been studied for internal use in diet formulations as a method to prolong muscle endurance (oral LD50 in rats is >16,000 mg/kg) and also as a preventative for cyanide poisoning.79,85–87 In this section, we will cover macromolecules that are synthesized from DHA and their intended applications.
3.1. General
3.1.1. Dihydroxyacetone Phosphate in Metabolism
As mentioned above, the premise of using metabolites as monomer units for synthesis of biodegradable polymers is that the body is already equipped with pathways to eliminate downstream polymer degradation products. The natural form of DHA in metabolism is dihydroxyacetone phosphate (DHAP). Once DHA enters the bloodstream, it can be phosphorylated by DHA kinases and entered into a metabolic pathway if not otherwise eliminated through natural disposal of excess or foreign material.88
Glycolysis, the breakdown of glucose for energy, is the most well-known source of DHAP in the body. DHAP, as well as the structural isomer glyceraldehyde-3-phosphate, are generated during the initial “energy consumption” stages of glycolysis, which is followed by a series of reactions resulting in pyruvate and a net positive production of ATP: the cellular “energy currency.” DHAP is also an intermediate in the breakdown of other common dietary sugars including fructose, mannose, and galactose. During times of starvation, when sugar supply is low, the liver will perform gluconeogenesis, a pathway in which glucose is synthesized so that it can be released into the bloodstream and sent to necessary organs such as the brain. DHAP plays an important role in gluconeogenesis as the pathway operates much like a reverse glycolysis cycle, requiring several of the same intermediates.11
In addition to carbohydrate metabolism, DHAP is often involved in the storage of lipids. DHAP is a precursor to the glycerol backbone of triglycerides, the major method of energy storage in the body. DHAP is first generated through glycolysis before use in triacylglycerol synthesis, thus being one of the many molecules that make metabolic processes interdependent. 11
3.1.2. Structure and Reactivity
Pentose and hexose sugars, such as fructose and glucose, exist as 5- or 6-membered rings due to intramolecular nucleophilic addition reactions between their carbonyl and hydroxyl groups. As a triose, DHA cannot react intramolecularly to form an energetically favorable ring. DHA instead reacts intermolecularly to form a hemiacetal dimer, the commercially available form of DHA, as shown in Figure 9a.89 Davis observed that in aqueous solutions the dimer dissociates into ketone and gem-diol forms (Figure 9b).90 Lyophilization of a stirred aqueous solution of dimer can be used to obtain a solid form of the keto-monomer.90,91 Davis also discovered that in solution, the ketone and hydrate exist in a 4:1 ratio, respectively, at room temperature.90 Infrared and nuclear magnetic resonance studies on DHAP indicate that the ketone:hydrate ratio is temperature dependent with a trend for increasing ketone content with increasing temperature.92
A key feature of DHA is the carbonyl group, which can react with primary and secondary amines. Reactions of DHA with amino compounds are of interest in several diverse fields including biomedical materials, enzyme mechanism determination, and cosmetics.93–96 When a primary amine reacts with the ketone, an imine is formed, commonly known as a Schiff base (IV, Figure 9c). The reaction is reversible (1, Figure 9c) but can be made irreversible through reductive amination with a reducing agent such as sodium cyanoborohydride (2, Figure 9c).93,97 A third possible outcome following imine formation is a Heyn’s rearrangement (3, Figure 9c): a presumed step in a series of rearrangements and reactions leading to Maillard-type products and browning.98,99 Precise reaction mechanisms leading to melanoidins are difficult to confirm as the Maillard reaction is known to be highly dependent on the specific amine reagent, pH of solution, temperature, and various other reaction conditions.100,101
The high chemical reactivity of the carbonyl group in combination with the ability of DHA to possess multiple forms in solution limits the number of polymerization conditions and catalysts that can be applied to DHA. In this next section, we cover several synthetic procedures used to successfully synthesize polycarbonates and poly(carbonate-esters) of DHA.
3.2. Linear Polycarbonates
3.2.1. Ring-Opening Polymerization
Ring-opening polymerization (ROP) is a highly effective and widely used technique to generate polyesters, polycarbonates, polyamides, polyethers, and others.102 Fine tuning of the ratio between cyclic monomers, catalysts, and initiators affords controlled syntheses of high molecular weight polymers with low PDIs, as well as reproducible thermal and mechanical properties. Polycarbonates, in particular, are of interest in biomedical applications due to their strength, biodegradability, and versatility. For example, various polycarbonate materials are receiving considerable attention for the development of controlled drug delivery systems103–107 and regenerative medicine.108–110 ROP is among the most widely used techniques for generating polycarbonates. Song et al. and Zhang et al. recently reviewed a number of cyclic carbonates used in the ROP of aliphatic polycarbonates as well as their biomedical applications.111,112
Cyclic ketone-protected carbonate derivatives of DHA are used to synthesize homopolymers, random copolymers, and diblock copolymers via ROP with a variety of catalyst/initiator combinations. This discussion focuses on the synthesis of cyclic DHA derivatives, catalysts used in the ROP of DHA-based polymers, postpolymerization modification, polymers synthesized through these methods, and applications of the reported polymers.
3.2.2. Ketone Protection
DHA is commercially available as a dimer, which is transformed into a stable monomeric form for subsequent polymerization. The most common methods to generate DHA-based macromolecules involve protection of the reactive ketone group. Ketone protection poses two major advantages: (1) dimerization is no longer feasible, enabling a pure monomeric derivative of DHA to be obtained and (2) the likelihood of unwanted side reactions decreases, offering a greater number of plausible starting materials and catalysts.
Two cyclic ketone-protected DHA derivatives are used for ROP: 2,2-dimethoxypropylene carbonate (MeO2DHAC, Figure 10a)93 and 2,2-ethylenedioxypropane-1,3-diol carbonate (EOPDC, Figure 10b).113,114 MeO2DHAC possesses a dimethoxy-acetal protecting group, whereas EOPDC contains a cyclic acetal. The synthesis of MeO2DHAC occurs in two stages as shown in Figure 10a. In the first stage, dimethoxy-protected DHA monomer, 2,2-dimethoxy-propane-1,3-diol (MeO2DHA, Figure 10a), is synthesized by combining the DHA dimer with trimethylorthoformate and p-toluenesulfonic acid in methanol. Following treatment with sodium carbonate, the product is isolated via a number of different purification procedures from column chromatography,115 to distillation,116 or recrystallization from diethyl ether.93 We found that purification by crystallization is easily amendable to large batches, generating significant quantities of material, but also has lower overall yield and can be difficult to coax into the solid state from the oil. The second stage in MeO2DHAC synthesis is the cyclization of MeO2DHA. Three different methods are presented in the literature to prepare the cyclic carbonate. Putnam et al. reported the successful use of the common reagent triphosgene, which involves addition of the reagent to a stirring solution of MeO2DHA, pyridine, and dichloromethane at −70 °C, followed by reaction at room temperature.93 Zhuo et al. showed that the reaction could be successfully carried out entirely at room temperature.117 Putnam et al. also reported a second synthetic method that is attractive, owing to the increased simplicity in the purification procedures. The reaction utilizes an alternative organochloride, ethyl chloroformate, in tetrahydrofuran solvent with triethylamine as the organic base. Purification is accomplished through paper filtration and recrystallization in diethyl ether as opposed to aqueous/organic extraction and flash chromatography.93 Waymouth et al. demonstrated that oxidative carbonylation is also an effective method to generate the cyclic carbonate. Their method uses the palladium-based reagent (neocuproine)Pd(OAc)2 and offers increased safety compared to a phosgene-type reagent.118 In the procedure, MeO2DHA is added to a stirring solution of (neocuproine)Pd(OAc)2 and sodium dichloroisocyanuric acid in acetonitrile. The reaction occurs at 35 °C inside a vessel that is vented and pressurized with carbon monoxide, both before and after diol addition.118 All three methods are adequate synthetic techniques that generate MeO2DHAC in satisfactory yields.
Figure 10.

(a) Synthetic scheme to MeO2DHAC. I. Trimethyl orthoformate, p-toluenesulfonic acid, and methanol, followed by sodium carbonate. II. Performed through three different methods involving trisphosgene,93 ethyl chloroformate,93 or oxidative carbonylation. 121 (b) Synthetic scheme to EOPDC. I. p-toluenesulfonic acid, glycol, and benzene. II. triphosgene, pyridine, and dichloromethane.113 Figure 10b is adapted from ref 113, with permission from the Chinese Chemical Society.
Finally, Zhuo et al. introduced a cyclic-acetal protecting group to the field of DHA-based polymers as shown in compound EOPDC (Figure 10b). Both a two- and four-step process are reported for generating EOPDC.113,114 The more simplistic two-step process is described in Figure 10b. It should be noted that unlike polymers of MeO2DHAC, which are deprotected and studied in their ketone-containing form, the focus of polymers derived from EOPDC in the current literature is on their ketone-protected form.
3.2.3. Catalysts
Several catalysts are employed for the polymerization of MeO2DHAC. The first reported synthesis of p(MeO2DHAC) (Figure 11a) is described by Putnam et al. using the catalyst stannous octoate, which afforded molecular weights of approximately 8–37.5 kDa, as determined by gel permeation chromotography.93 Zhuo et al. studied variations on reaction temperature, time, and monomer:catalyst ratios to identify conditions that give molecular weights up to 138.2 kDa.117
Figure 11.

Example syntheses of protected-DHA homopolymers performed by: (a) Putnam et al.,93 (b) Guillaume et al.,119 (c) Waymouth et al.,121 and (d) Zhuo et al.114
A drawback of the method reported by Putnam et al. is its reliance on trace atmospheric moisture as a polymer initiator. Guillaume and co-workers investigated alternative catalyst systems, which would yield more control over polymer molecular weight. Guillaume et al. found that dimethoxyacetal-protected DHA homopolymers can be synthesized with molecular weights as high as 70.2 kDa via an “immortal” ROP with the zinc complex [(BDI)Zn(NTMS2)] using an alcohol initiator (Figure 11b).119 Guillaume et al. additionally tested several organocatalysts to promote the use of “green” catalytic systems where concerns over trace toxic metals in final products could be eliminated.119,120 Catalysts 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP), 4-N,N-dimethylaminopyridine (DMAP), and 1,5,7-triazabicyclo-[4.4.0]dec-5-ene (TBD) were all shown to be active in the synthesis of dimethoxy protected DHA polymers.119,120 Waymouth et al. further investigated the use of organocatalysts in ROP of MeO2DHAC by evaluating 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 1-(3,5-Bis-(trifluoromethyl)phenyl)-3-cyclohexyl-2-thiourea (TU), and (−)-sparteine as well as TBD.121 TBD is the most efficient organocatalyst reported in their study, as concluded by a monomer conversion of 95% during only 5.5 min of reaction time (Figure 11c).121 A summary of catalysts, reaction conditions, and results from syntheses of MeO2DHAC homopolymers are shown in Table 3. Copolymerizations of MeO2DHAC with poly(ethylene glycol) (PEG),122 lactide,123 ε-CL,121 and trimethylene carbonate (TMC)124 are also reported using the catalysts listed in Table 3, thus broadening the compositions and properties attained with DHA-based polymers.
Table 3.
Catalysts Used in Polymerization of MeO2DHAC
| catalyst | T (°C) | t (h) | molecular weight (kDa) | Đ | ref |
|---|---|---|---|---|---|
| metal-based catalysts | |||||
| Sn(Oct)2 | 100 | 1–2 | 8–37.5a,d | 1.35–1.5 | 93 |
| Sn(Oct)2 | 110–140 | 24 | 14.3–138.2b,e | 1.31–1.91 | 117 |
| (BDI)Zn(NTMS2) | 60, 90 | 1–2 | 3–70.2a,e | 1.12–1.79 | 119 |
| organocatalysts | |||||
| BEMP | 90 | 3 | 14–14.3a,e | 1.53, 1.66 | 119,120 |
| DMAP | 90 | 5.5 | 6.8a,e | 1.23 | 119 |
| TBD | 90 | 3 | 14.1a,e | 1.71 | 119 |
| TBD | RT | 5.5 (min) | 6.3c,e | 1.55a | 121 |
| DBU+TU | RT | 0.55, 1.17 | 7.5 and 31c,e | 1.20, 1.24a | 121 |
| (−)-sparteine + TU | RT | 7–25 | 5.8–28c,e | 1.11–1.18a | 121 |
GPC, THF, and polystyrene standards.
GPC, chloroform; no standards listed.
NMR.
Mw.
Mn.
Zhuo and co-workers first reported polymers of the cylicketone-protected DHA (Figure 11d).114 The polymer, poly-(2,2-ethylenedioxy-propane-1,3-diol carbonate) (PEOPDC), can be synthesized using two separate catalysts: stannous octoate [Sn(Oct)2] and aluminum isobutanoxide [Al(OiBu)3]. The use of stannous octoate resulted in higher molecular weights and percent yields with a reported range of 31.9–55 kDa and 79.3–92.6%, respectively.114 Copolymerizations of EOPDC with lactide,125 caprolactone,126 and 1,4-dioxane-2-one127 are also reported using stannous octoate as a catalyst.
3.2.4. Ketone Deprotection
To date, three methods are established for the deprotection of dimethoxy-acetal protected dihydroxyacetone macromolecules. Putnam et al. demonstrated that the ketone functionality could be recovered after treatment with a trifluoroacetic acid (TFA)/water solution (Figure 12a).93 Deprotection >95% is possible; however, the authors targeted 85% to maintain polymer solubility for structural characterization. 93 Putnam et al. showed that this method is also effective for diblock copolymers of DHA and PEG (Figure 12b) and that the acidic nature of the protocol does not result in degradation of the polymer backbone.122
Figure 12.

Synthetic methods of dimethoxy-acetal deprotection in various DHA-based polymers demonstrated by: (a) Putnam et al.,93 (b) Putnam et al.,122 (c) Putnam et al.,123 and (d) Waymouth et al.121 Figure 4d was reprinted from ref 121. Copyright 2012 American Chemical Society.
While polymer degradation is not observed in these specific cases, Putnam et al. investigated an anhydrous deprotection protocol to reduce concern for DHA-based polymers containing additional acid-hydrolyzable bonds.123 They found that a deprotection procedure developed by Hu et al. in which ketal containing compounds are refluxed in an iodine/acetone solution successfully afforded poly(carbonate-esters) of DHA and lactic acid (pLA-pDHA, Figure 12c).123,128 The extent of deprotection to afford the ketone polymer influences polymer solubility. Increased ketone content (i.e., DHA content) decreases solubility in organic solvents like acetone, and therefore, polymers high in DHA content precipitate from solution prior to reaction completion. Despite this challenge, ≥80% deprotection is possible for all of the polymers tested, encompassing a range of 15–100% DHA content.123
Waymouth et al. demonstrated the success of triphenylcar-benium tetrafluoroborate in the deprotection of poly-(carbonate-ester) copolymers of DHA and ε-CL (pDHA-pCL, Figure 12d).121 Similar to pLA-pDHA, polymers high in DHA content precipitated from solution during reaction; however, complete deprotection was still observed and reported. Also, the procedure requires only a small quantity of water (one equivalent to each MeO2DHAC unit) compared to the TFA-based method reported by Putnam et al., thereby offering a second route by which DHA-based polymers can be deprotected with decreased probability for hydrolytic degradation. 93,121
3.2.5. Direct Esterification
While ROP affords a large number of possibilities to synthesize DHA-based polymeric materials, protection of DHA is time-consuming. Ketone protection generally takes several days depending on the desired level of purity of the final product and generates low yields. In 1969, Schrek et al. and Lasslo et al. showed that esters of DHA which retain the ketone functional group can be synthesized using acyl chloride compounds, such as undecanoyl chloride, in the presence of pyridine.84,129,130 The goal of their studies was to develop sustained release mosquito repellent technologies in which DHA would anchor the compound to the skin, then degradation would lead to release of the active repellent molecule.130 The ability to esterify DHA without first protecting the ketone is an attractive feature for the future of DHA-based macromolecules. In 2010, Putnam and Yazdi showed that lipid diesters of DHA synthesized through these methods can be formulated into microparticles and are promising controlled release drug delivery vehicles.131
3.2.6. Cyclic Macromolecules of DHA
Few reports describe the polymerization of the DHA dimer. In 1934, based on a series of X-ray diffraction patterns, Strain and Dore suggested that when the DHA monomer is left in dry form at room temperature for 25–30 days, the molecules would dimerize, and when left for several months, DHA would polymerize further.132 In 1989, Akar and Talinli reported that the DHA dimer polymerizes into poly(spiro-acetals) by mixing DHA dimer in ethanol with heat and an acid catalyst.89 The resulting polymers are insoluble in most organic solvents but slightly soluble in dimethyl sulfoxide (DMSO). Several years later, Alder and Reddy reported a transketalization method to synthesize the poly(spiro-acetals) as shown in Figure 13.133
Figure 13.

Transketalization route to poly(spiro-acetals) of DHA as reported by Alder and Reddy. Reprinted with permission from ref 133. Copyright 1994 Elsevier Ltd.
Putnam et al. reported additional polymers that retain the cyclic nature of the DHA dimer134,135 (Figure 14). In order to prevent dissociation during polymerization, modified forms of the dimer are synthesized, as shown in Figure 14. The ethyl derivative, 2,5-diethoxy-1,4-dioxane-2,5-dimethanol (Figure 14, R=CH2CH3), is used, previously described by Wong et al., as an intermediate in a method for synthesizing DHAP.136 The molecular weight of the resulting polymers ranged from 28 to 48.4 kDa with dispersities of 1.7 to 2.2. The materials exhibited comparable mechanical properties to that of cancellous bone, with compressive yield strengths of 45 ± 5 MPa and Young’s moduli of 0.8 ± 0.01 GPa.134,137,138 The mechanical properties in combination with confirmed NIH-3T3 cell growth on polymer films suggests that polymers generated from the dimeric form of DHA may have applications in tissue engineering.
Figure 14.

Synthetic route to poly(carbonate-acetals) from DHA dimer. (a) R′: triethyl orthoformate, EtOH, p-TsOH, RT, and R″: triisopropyl orthoformate, 2-propanol, p-TsOH, and RT and (b) triphosgene, pyridine, and CH2Cl2. Adapted with permission from ref 134. Copyright 2005 American Chemical Society.
3.3. Applications
3.3.1. Functionalizable Surfaces
Surfaces bearing reactive functional groups are of widespread interest for uses including, but not limited to cell adhesion,139 modifying degradation rates,140 protein immobilization,141 drug attachments, 142,143 and layer-by-layer assembly of nanostructures.144 As previously discussed, DHA reacts with primary amines to form an imine or Schiff base (Figure 9c). Therefore, a number of surface modifications of DHA-based materials are possible.
The Schiff base itself is a powerful tool with applications in drug delivery,143,145,146 bioreactor design,147,148 biosensors,149 protein microarrays,150,151 preparation of nano152/microstructures, 153 etc. The imine possesses several unique properties which enable its widespread use including: (1) pH sensitivity, (2) autofluorescence, (3) formation under mild conditions without added reagents, (4) occurrence in both aqueous and organic solvents, and (5) reversibility (with the option of irreversibility through reductive amination).97 In most biomedical applications, Schiff bases are formed by reactions with aldehydes as opposed to ketones due to the increased reactivity of the carbonyl group. For example, as a tool to prevent infection from medical implants over a prolonged period of time, Meier et al. synthesized polymersomes with outer aldehyde groups that permanently attach to amine-coated silicon surfaces via reductive amination of imine linkages and sustain local release of antibiotics.154 Li et al. synthesized biodegradable and biocompatible microcapsules containing covalently assembled layers of chitosan (amine-containing polysaccharide) and derivatized alginate (oxidized to contain aldehyde groups). The Schiff base linkages between layers resulted in autofluoroescence of the microcapsules and pH dependent permeability, indicating their potential use in drug delivery applications with improved in vivo tracking capabilities. 155 An extensive review of Schiff base forming technologies based on aldehyde-amine reactions, and their applications, is recently covered by Jia and Li.97
Putnam et al. performed several studies to show that the ketone functionality of DHA-based polymers does not lose reactivity upon polymerization of the alpha hydroxyl groups.93 They spin-coated ketone-protected DHA polymer [p-(MeO2DHAC)] onto glass slides and spotted a TFA/water solution in specified regions to generate areas of deprotected (i.e., ketone-containing) polymer. Subsequent incubation with fluorescently tagged poly(lysine) showed immobilization only in regions containing deprotected polymer, indicating that the ketone is essential to the immobilization mechanism (Figure 15a). Similar results are obtained for studies with fluorescently tagged albumin, demonstrating that pDHA is also reactive with proteins. To further verify that Schiff base formation is feasible postpolymerization, Putnam et al. synthesized a small molecular weight analog (I, Figure 15b) of pDHA and studied its reaction with phenylethylamine in the presence of a reducing agent. 13C NMR data showed the expected product from reductive amination of a Schiff base formed between the analog and amine compounds (II, Figure 15b).93 The results of Putnam et al. indicate that pDHA, and copolymers thereof, are promising functional biomaterials. In addition, pDHA is reported to possess high strength, similar to that of cancellous bone, and a Tg above physiological temperature (~60 °C). These properties further support the potential for DHA-based materials in medical devices, drug delivery, tissue engineering, and more.93
Figure 15.

(a) Fluorescent imaging of poly(lysine) surface conjugation onto glass microscope slides coated with A, pDHA; B, p-(MeO2DHAC) and pDHA; and C, p(MeO2DHAC). (b)13C NMR spectra of a small molecular weight analog of pDHA both (I) before and (II) after reaction with phenylethylamine and a reducing agent. Reprinted with permission from ref 93. Copyright 2006 American Chemical Society.
3.3.2. Hydrogels
Hydrogels are cross-linked polymer networks that swell in aqueous solutions and have varied applications in tissue engineering,156–158 wound healing,159–163 and drug delivery.164–167 Hydrogels are prepared using naturally occurring polymers [cellulose,168 hyaluronan,169 alginate,170 etc.), synthetic materials (poly(vinyl alcohol),171 poly(ethylene glycol),172,173 poly(2-hydroxyethyl methacrylate), 174,175 etc.] and genetically engineered protein-based materials created using recombinant DNA technology.176–178
Putnam et al. reported a new class of synthetic hydrogels using DHA-based polymers.122,179 They found that diblock copolymers comprised of pDHA, a water-insoluble polymer, and the highly hydrophilic polymer PEG afford biocompatible hydrogels with tunable properties (MPEG-pDHA, Figure 16a). Studies performed on diblock copolymers with fixed-length MPEG segments (5 kDa) and variable pDHA segments showed that as the pDHA chain increases, hydrogel pore size, swelling, and hydrolytic degradation rates decrease, whereas entanglement density and viscosity increases. Physical cross-linking in MPEG-pDHA hydrogels is hypothesized to occur due to charge interactions arising from the dipole moment of the carbonyl groups on dihydroxyacetone. It is hypothesized that the transient nature of such attractions contribute to the ability to extrude MPEG-pDHA through a 26-gauge needle (Figure 16b).179
Figure 16.

(a) SEM image of lyophilized MPEG-pDHA (5000–3000) hydrogel. (b) MPEG-pDHA (5000–5000) hydrogel extrusion through a 26-gauge needle. (c) Seroma volumes measured using a rat mastectomy model following treatment with various MPEG-pDHA formulations and a saline control. Figures are reprinted from ref 179 with permission. Copyright 2010 National Academy of Sciences, USA.
A unique property of DHA-based hydrogels is the relatively rapid degradation rate compared to other polycarbonates [e.g., poly(trimethylene carbonate), (PTMC)]. In vitro, under conditions mimicking the physiological media, MPEG-DHA hydrogels degrade within a 24 h period.179 The rapid degradation rate of DHA hydrogels, in combination with the ease of extrusion through a needle, is advantageous in the prevention of seromas, accumulation of serous fluid resulting from surgical complications. In an in vivo rat model of lymphandenctomy, the polymers with molecular weights ~3 kDa in pDHA and ~5 kDa in MPEG are highly effective at preventing seroma formation (Figure 16c).179 Three days after treatment, no polymer is observed at the surgical site, indicating that MPEG-pDHA hydrogels are capable of serving a transient distinct function before being eliminated from the body.
The rapid degradation of MPEG-pDHA polymers along with the ability to prevent seroma provides a rationale for investigation of other potential uses, including one as a biodegradable hemostatic agent.180 With use of an in vivo liver resection rat model, application of the MPEG-pDHA reduced the bleeding time to 97 s compared to a saline control (464 s) and Instat, an industry standard (165 s).180 The total blood loss is not statistically different between MPEG-pDHA and Instat trials, indicating that MPEG-pDHA hydrogels are adequate hemostatic agents with the added bonus of a rapid in vivo resorption rate. Histological studies performed 3 weeks postoperation showed a similar inflammatory response of MPEG-pDHA samples to saline controls.180 The combination of biocompatibility, reduced bleeding time, synthetic origin of the material, and rapid degradation of MPEG-pDHA hydrogels suggest that MPEG-pDHA hemostatic agents would lead to minimized risk of infection and long-term inflammation.
The reactive carbonyl functionality present within DHA hydrogels provides opportunities for chemical cross-linking, drug attachments, or surface modifications, thereby opening doors to additional applications. Furthermore, chemical alterations, such as the introduction of small hydrophobic regions or the use of urethane polymer linkages, may lead to enhanced control in hydrogel swelling and degradation, expanding the potential for DHA-based hydrogels in biomedical applications.
3.3.3. Controlled Drug Delivery
Polymeric materials are of significant interest in the field of controlled drug delivery owing to the vast opportunities in synthetic design leading to various tunable features as well as their ability to form macro, micro, or nanostructures. Several review articles cover polymers used for controlled drug delivery that describe their unique advantages and specific applications in detail, and the reader is referred to these articles.181–185 DHA-based polymers contain several desirable characteristics for controlled drug release systems. First, the degradation of DHA-based polymers into biocompatible compounds reduces risks of inflammation and toxicity while also eliminating concerns over invasive post-treatment device removal. Second, through ROP, DHA can be easily copolymerized in a controlled manner with other biocompatible materials such as lactide123 and caprolactone.121 By altering initial monomer, comonomer, initiator, and catalyst ratios, the final product composition can be predicted, creating a reliable parameter by which drug release rates are controlled. Overall, there is a large realm of possibilities for DHA-based polymers with regard to hydrophilicity, rigidity, glass-transition temperatures, melting temperatures, rheological properties, and pH sensitivity, depending on the chosen comonomer and reaction conditions. Furthermore, variations in these properties yield opportunities for different 3D structures. Diblock copolymers of DHA and MPEG afford hydrogels179 and nanoparticles,122 whereas random copolymers of DHA and LA give powders that can be compressed into solid tablets.123 Putnam and Yazdi showed that DHA-based microparticles are also feasible; however, these materials employ a separate synthetic technique, as previously discussed.131 A third attractive feature of DHA-based materials is the reactive ketone functionality. Although this has yet to be specifically explored, it is an extension of the research and opens up possibilities for the attachment of active agents or affinity-ligands.
Putnam et al. showed that random copolymers of DHA and LA, synthesized via ROP with stannous octoate catalysts, are promising candidates for controlled release of protein therapeutics.186 Cylindrical tablets, for subsequent drug release studies, are prepared by mixing the powdered polymers containing approximately 50 to 85% DHA with dry powders of model proteins, bovine serum albumin (BSA) or lysozyme, and compressed. Copolymers containing less than 50% DHA are too tough to process into tablets and therefore were not investigated. Thermal studies revealed that for all LA:DHA ratios, glass transition temperature (53–68 °C) and degradation temperatures (Td, 50 wt %: 230–330 °C) are above physiological temperature, indicating that the tablets will retain structural integrity when subject to in vivo temperatures.123,186
In vitro controlled release experiments showed a first-order release of BSA and lysozyme over 2.5–70 days, depending on the percent drug loading and percent DHA in the polymer backbone.186 Lysozyme activity tests following release indicated that the protein retained significant activity throughout release and was not highly inactivated due to the environment of the delivery device. For any copolymer composition, at least 50% of the protein activity is retained after one month of release. Protein release rates increased with increasing pDHA content.186 This result is not surprising given the results of in vitro degradation studies, which showed that polymer erosion occurs more rapidly with increasing DHA content within the polymer backbone. In addition to tablet erosion studies visualized by scanning electron microscopy, degradation of the polymer backbone can be tracked utilizing the bicinchoninic acid assay, which Putnam et al. discovered to be a valuable tool in quantitatively measuring α-hydroxy ketones, such as DHA, in solution.187 Zhuo et al. report that upon deprotection of the pDHA homopolymer, hydrolytic degradation of the associated carbonate bonds drastically increases.117 The results suggest that percent deprotection of pDHA in DHA-based materials is a potential control parameter in drug release technologies.
Zhuo and co-workers performed release studies on polymers containing EOPDC (Figure 10b), a cyclic-acetal protected form of DHA, using the chemotherapeutic agent Tegafur.125–127 Results from EOPDC:1,4-dioxane-2-one copolymers and EOPDC:caprolactone copolymers show a decreased drug release rate with increasing EOPDC content, resulting in release of less than 4% in 350 h in any formulation.126,127
Diblock copolymers of pDHA and MPEG (Figure 12b) are also proposed for controlled release applications; however, no formal testing is reported. Putnam et al. showed that in addition to hydrogels, MPEG-pDHA polymers can be formulated into nanoparticles that take on a micellar shape with a pDHA core and PEG corona.122 Nanoparticles, prepared by control precipitation (via stirring) from DMSO/polymer solutions in water, ethanol, or dichloromethane, afforded average particle diameters of 45 ± 1 nm, 70 ± 1 nm, and 94 ± 1 nm, respectively, from polymers with molecular weights of 1.8 kDa for pDHA and 4.5 kDa PEG.
3.3.4. Green Thermoplastics
Today, concerns over landfill sizes, depletion of scarce resources, and greenhouse gas production are significant given the increasing world population. These issues spurred the investigation, development, and use of biodegradable plastics from renewable resources, as opposed to traditional petroleum-based materials such as polyethylene or polypropylene. Examples of “green” plastics include those composed of starch, soy protein, cellulose, PLA, and poly(hydroxyalkanoate)s.188–192 Waymouth et al. noticed that the structure of pDHA is similar to Carilon, a polyketone thermoplastic derived from ethylene (E) and carbon monoxide (CO).121 Since DHA can be synthesized from glycerol,193,194 a byproduct in the production of bio-diesel, the authors investigated the potential use of DHA-based polymers in the field of renewable and biodegradable thermoplastic materials.121 Similar to E/CO polymers, pDHA possesses a melting temperature, Tm of 246 °C, close to the thermal degradation temperature, Td = 273 °C, which is difficult for processing due the high Tm and close Tm and Td values.121 As a solution, Waymouth et al. copolymerized DHA with ε-CL, which lowered the Tm of the polymer, with the extent of Tm lowering depending on the percent of caprolactone added to the composition.121 These results indicate that pDHA-CL copolymers are thermoplastic materials with tunable thermal properties.
Guillaume et al., also interested in creating thermoplastic materials from biorenewable resources, synthesized diblock and triblock copolymers of dimethoxy-acetal protected DHA with the well-known polycarbonate PTMC.124 It was found that much like PLA, a popular “green” choice for polymers, segments of protected DHA added rigidity to PTMC polymers which is a control parameter for the generation of well-performing thermoplastic materials. The results from the Guillaume and Waymouth laboratories, show that DHA-based polymers are not limited to biomedical applications. The features that make these polymers appeal to biomedical research, their biocompatibility and biodegradability, also open doors to the possibility of producing materials with improved environmental preservation capabilities.
3.4. Dihydroxyacetone Synthon Summary
Dihydroxyacetone is a natural metabolite used in the synthesis of several polymeric biomaterials including hydrogels, nanoparticles, microparticles, powders, and thermoplastics. The resulting DHA-based macromolecules successfully demonstrated their use in controlled drug delivery, seroma prevention, hemostasis, and tissue engineering applications. ROP is the primary synthetic route to DHA-based polymers, offering facile copolymerization with additional monomers and various control parameters. ROP, however, requires timely protection/deprotection of the active ketone. The use of acyl chloride reactions provides an alternative synthetic route that avoids ketone modification procedures.
While a diverse set of DHA-based materials are known and useful in several applications, a number of opportunities remain. First, the carbonyl group present on DHA-based polymers offers unique capabilities for drug attachments, chemical cross-linking, and surface modifications via Schiff base formation, which are yet to be reported. Second, the α-hydroxy groups present on dihydroxyacetone offer the possibility of polymerization into urethane bonds, which is not extensively studied. Lastly, 2,2-dimethoxy-propane-1,3-diol (MeO2DHA, Figure 10a), the intermediate product in the synthesis of MeO2DHAC, is a promising candidate for synthesis of future DHA-based polymers.
Similarly to other metabolic synthons that have gained widespread attention for use in polymeric biomaterials, such as lactic acid, dihydroxyacetone offers a diverse set of synthetic opportunities that can yield adjustable mechanical and thermal properties for a broad range of applications, both inside and outside the realm of biomedical engineering.
4. GLYCEROL SYNTHONS
Glycerol, like DHA, is a three-carbon polyol and contains two primary and one secondary hydroxyl groups. Oxidation of the secondary hydroxyl to a ketone in glycerol affords DHA, discussed in the previous section. GL (also known as glycerin or glycerine) is generally considered to be nontoxic and has an oral LD50 of 12600 mg/kg in rats.195 GL is widely used in cosmetic and pharmaceutical products as a thickener, plasticizer, emollient, bodying agent, humectant, and lubricant. Its use as a synthon for the preparation of copolymers with other monomers is well-established via thermal condensation polymerization approaches; however, the preparation and subsequent use of cyclic derivatives for more controlled polymerization reactions is only recently described. As discussed below, these newer approaches afford polymers with specific molecular weights and lower dispersities, as well as opportunities to synthesize chiral polymers and copolymers with other monomers.
4.1. General
4.1.1. Glycerol Metabolism
GL is a well-known natural metabolite with many roles, including serving as the backbone unit of triglycerides and phospholipids. For example, GL phospholipids, the most abundant lipids in cell membranes, can possess a variety of different head groups. Thus, GL (and more accurately, GL 3-phosphate) is a key intermediate in the biosynthesis of these important lipids. GL is also a precursor for the biosynthesis of glucose. Catabolism of triacylglycerols to give GL, which is then phosphorylated at the C3 position, enables entry into gluconeogenesis (discussed above) and subsequent glucose production.
4.1.2. Structure and Reactivity
Although GL only possesses two primary and one secondary hydroxyl groups, it is amenable to a large number of chemical transformations (e.g., dehydration, selective oxidation, and hydrogenolysis),196 providing a means to prepare GL-derived synthons for subsequent polymerization. For example, with primary alcohols at the C1 and C3 positions and a protected secondary alcohol at the C2 position, 6-membered cyclic carbonates are obtained upon treatment with triphosgene or ethyl chloroformate. This cyclic structure is the monomer for the synthesis of linear poly(1,3-glycerol carbonates) [P(1,3-GLCs)] described next.
4.2. Linear Poly(1,3-glycerol Carbonates)
In general, aliphatic polycarbonates degrade at faster rates than aromatic polycarbonates, prompting investigation into their potential utility as scaffolds for engineered tissues and drug delivery systems.111 When the aliphatic building block is GL, the added functionalities of biocompatibility and biodegradability become key for subsequent use. A number of protected GL-based cyclic carbonates have been described and reviewed.112 For example, benzyloxy trimethylene carbonate (BTMC), in which the 2-hydroxyl group is protected with a benzyl ether protecting group, is a prominent example in the literature. The resulting 6-membered cyclic carbonate ring is amenable to ROP (facilitated by either aluminum or tin coordination catalysts), and the benzyl protecting group can be removed to expose the alcohol functional group with ~95% completion using hydrogenolysis catalyzed by palladium/carbon to afford P(1,3-GLC) (Figure 17).197,198
Figure 17.

Structures of glycerol, 5-benzyloxy-trimethylene carbonate, poly(1,3-glycerol carbonate), poly(glycerol-co-ε-caprolactone), and alkyl-substituted poly(glycerol-co-ε-caprolactone).
The alcohol functional group of the deprotected poly(5-benzyloxy-trimethylene carbonate) enhances the hydrophilicity as well as the hydrolytic degradation rate of the polymer, opening potential avenues for applications requiring degradability. Modulation of the degradation rate is also possible through substitution of the alcohol with other functional groups and by copolymerization with other hydrophobic monomers. For example, poly(carbonate-ester)s of GL and 6-hydroxyhexanoic acid (through ε-CL) are synthesized by ROP followed by functionalization at the alcohol functional group with both hydrophilic and hydrophobic moieties.199 Additionally, a series of hydrophobic alkane substitutions (C12, C14, C16, and C18) can be conjugated to the poly(glycerol-co-ε-caprolactone) [P(GL-co-ε-CL)] by esterification with DCC (Figure 17).200 The static contact angles of thin films cast from these polymers ranged from ~80° (C12) to ~120° (C18), and films loaded with the anticancer drug, 10-hydroxycamptothecin, released the drug as a first-order process with the C12 side chain and became more zero order as the hydrophobicity of the side chain increased to C18.
4.3. Linear Poly(1,2-glycerol Carbonates)
Recently, linear poly(1,2-glycerol carbonates) [P(1,2-GLCs)] (Figure 18) are reported and represent a new class of aliphatic polycarbonates. These polymers are of interest as potential bioengineered materials due to their biocompatibility and biodegradability, important requisites for in vivo use. These polymers possess free primary hydroxyl pendant groups within their structures that can be further functionalized with active drugs, fluorescent dyes, and bioactive species. Furthermore, they contain glycerol units, which, upon degradation, will be eliminated from the human body via their normal metabolic pathways, as discussed above.
Figure 18.

Structures of linear poly(1,2-glycerol carbonate) and poly(1,3-glycerol carbonate).
While linear [P(1,3-GLC)] (Figure 18) derivatives are more commonly used in drug delivery systems and medical devices,123,179,201,202 P(1,2-GLCs) are less well-explored. Linear P(1,3-GLCs) are usually synthesized via the ROP of functionalized 6-membered ring carbonate monomers, such as 2-BTMC or dimethylacetal DHA carbonate monomers, as described in the preceding section.112,196 However, P(1,2-GLCs) cannot be obtained by the same strategy because 5-membered ring glycerol carbonates are thermodynamically stable, and the polymerization reactions generally do not occur under mild conditions or involve partial decarboxylation and loss of carbon dioxide (CO2), under vigorous conditions, leading to poly-(alkene ether-carbonates) with low content of carbonate units (less than 50%).203 On the other hand, the polycondensation of 1,2-diols with phosgene mainly generates 5-membered cyclic carbonates instead of P(1,2-GLCs).204 Therefore, other potential synthetic routes are required to access these novel polymers.
The ring-opening copolymerization of epoxides and CO2 using various catalysts (e.g., cobalt complexes, phenoxide zinc, and β-diiminate–zinc complexes) represents a route for the synthesis of linear P(1,2-GLCs) (Figure 19).205–212 This elegant strategy affords P(1,2-GLC) derivatives starting from glycidyl ethers,213–216 which upon degradation generate nontoxic 1,2-GL moieties and CO2, and is discussed in the following sections.
Figure 19.

Coupling reaction of epoxides and CO2. Adapted with permission from ref 205. Copyright 2005 American Chemical Society.
In 2013, Grinstaff et al. reported the synthesis of atactic and isotactic linear poly(benzyl-1,2-GLCs) via the ring-opening copolymerization of rac/(R) benzyl glycidyl ethers (BGEs) with CO2 using [SalcyCoIIIX] complexes with high carbonate linkage and polymer/cyclic carbonate selectivities (>99%).216 After a reductive hydrogenation step to remove benzyl groups, P(1,2-GLCs) with pendant primary hydroxyl groups are obtained (Figure 20). These polymers degrade more rapidly than the P(1,3-GLCs) with a half-life (t1/2) of ≈3 days. NMR spectroscopy, GPC, and polarimetry data on the benzyl-protected polymers are consistent with the proposed structures. 1H NMR confirmed that all polymers contained >99% carbonate linkages and a selectivity of P(Bn-1,2-GLCs) over cyclic carbonates between 70 and 98% depending on the axial ligand. The atactic/isotactic nature of the polymers is confirmed by 13C NMR as the atactic polymer exhibited multiple overlapping carbonyl peaks of the same intensity, whereas the isotactic polymer showed only one sharp peak at 154 ppm. The 13C NMR data also confirmed the head-to-tail selectivity of the polymers. GPC analysis showed number-average molecular weights (Mn) lower than the expected theoretical values (20–50 kDa), with bimodal and narrow Đ of 1.1. The specific rotation of the isotactic polymer of 20.3 kDa in THF at 27 °C is −15.2 °C. After deprotection of the benzyl groups, the P(1,2-GLCs) are soluble in DMF and DMSO. On the basis of GPC analysis, the degradation of P(1,2-GLCs) in DMF at 37 °C is faster than P(1,3-GLCs) with a t1/2 ≈ 3 days, which is attributed to the stronger nucleophilic nature of the primary hydroxyl of the 1,2-GL moiety as compared to the secondary hydroxyl group of the 1,3-GL moiety, which attack intramolecularly the carbonate linkage, with the formation of a stable 5-membered glycerol carbonate ring.
Figure 20.

Synthesis of poly(1,2-glycerol carbonates) using the [SalcyCoIIIX]/PPNY complex. Adapted with permission from ref 216. Copyright 2013 American Chemical Society.
In the same year, Frey et al. simultaneously reported the synthesis of protected P(1,2-GLCs) starting from BGEs or ethoxy ethyl glycidyl ethers (EEGEs) and CO2, using a ZnEt2/pyrogallol catalyst.213 The desired P(1,2-GLCs) are obtained after a subsequent deprotection step (Figure 21), with no formation of polyether linkages or cyclic carbonates. Again, 1H and 13C NMR spectroscopy data confirmed the protected polycarbonate structures. GPC showed a monomodal molecular weight distribution, with Mn in the range of 16–25 kDa, and moderate Đ between 1.4 and 1.8. However, some copolymers exhibited bimodal distributions, typical in the copolymerization of epoxides with CO2 using a binary catalyst system. FT-IR spectra showed the expected absorption band of carbonyl groups, which confirmed the introduction of the carbonate linkage within the polymer backbone. FT-IR also confirmed the absence of a cyclic carbonate vibration band, which is usually visible around 1800 cm−1. The polymers did not exhibit a melting point, as determined by DSC, confirming their amorphous nature. The Tg ranged between −22 and −17 °C, depending on the length and structure of the polymer. After deprotection of the polymer, the 1H NMR spectrum of P(1,2-GLC)s showed the complete disappearance of the benzyl and EE signals, and all polymers possessed monomodal distributions with moderate Đ (1.7–2.3) and Mn in the range of 5–18 kDa, as shown by GPC. P(1,2-GLCs) exhibited higher Tg (−4 °C) than those of the protected ones, as a likely consequence of the interactions between the hydroxyl groups. In both DMF and THF solvents, the molecular weight of the polymers decreased in time with the formation of only cyclic carbonate (THF) or along with oligomers (DMF); however, the degradation in THF is faster than in DMF. Similarly, when the polymers are stored in bulk at −28 °C for 8 weeks, a significant amount of cyclic carbonate is observed. In all cases, the degradation of P(1,2-GLCs) is faster than P(1,3-GLCs).
Figure 21.

Synthesis of poly(1,2-glycerol carbonates) using ZnEt2/pyrogallol catalyst.213
The terpolymerization of BGE, propylene oxide (PO), and CO2 is recently reported using two catalysts, the binary system [rac-Salcy-CoIIIDNP]/PPNDNP and the bifunctional system [rac-SalcyCoIIIDNP] bearing a quaternary ammonium salt by Grinstaff et al.217 Both catalysts afforded P(Bn-1,2-GL-co-PCs) (PC = propylene carbonate), with high polymer and carbonate linkage selectivities (>99%), Mn ranging between 5 and 30 kDa, and narrow Đ (1.1–1.2) (Figure 22), as shown by 1H NMR spectroscopy and GPC, respectively. 1H NMR spectrum of the terpolymer confirmed the presence of two separate peaks at δ = 5.0 and 4.99 ppm, attributed to the CH in 1,2-GL and PC units, respectively. On the other hand, the 13C NMR spectrum of the terpolymer indicated that the GL and PC units may be alternating within the polymer structure, due to the splitting of their carbon signals as compared to the single sharp peaks observed with their respective copolymers. Incorporation of the third monomer, PO, should afford polymers with a higher Tg because polypropylene carbonates exhibit high impact and scratch resistance properties with a Tg of 40 °C. On the other hand, P(benzyl-1,2-GLCs) are relatively soft materials with a Tg of 9 °C. Therefore, the terpolymerization of these two monomers with CO2 will give materials with distinct thermal and mechanical properties that differ from their respective copolymers. By DSC and TGA, it was noted that the increase of GLC units within the terpolymer structure decreased the Tg and increased the Tdecomp of the resulting polymer. After deprotection of the benzyl-groups, P(1,2-GL-co-PCs) exhibited lower Tg and Tdecomp than their respective protected ones. Both benzyl-protected (Mn = 32.3 kDa) and deprotected polymers (Mn = 17.3 kDa) exhibited high storage (G′ = 1 and 2 *106 Pa, respectively, at 4 Hz of frequency) and loss (G′′ = 1 and 3.5 *106 Pa, respectively, at 4 Hz of frequency) moduli, which increased with the incorporation of PC units in the polymer chains, as determined from rheological measurements on cast molds of the terpolymers.
Figure 22.

Synthesis of poly(1,2-glycerol-co-propylene carbonates) using [rac-SalcyCoIIIX] catalysts. Adapted with permission from ref 217. Copyright 2013 Wiley Periodicals Inc.
Similarly, Frey et al. and Luinstra et al. independently reported the synthesis of GL-PC terpolymers starting from BGE/glycidyl methyl ether (MGE)/CO2 (Figure 23A)218 and glycidyl nitrobenzyl (NGE)/PO/CO2 (Figure 23B),219 respectively, using a zinc-based catalyst. Mn values of the resultant terpolymers varied between 9–30 kDa and 20–90 kDa for both syntheses, respectively, with Đ ranging between 2.4 and 3.9. High carbonate linkages (>90%) are achieved during synthesis. Upon increasing the quantity of MGE units within the P((Bn-1,2-GL-co-MGE)-C) structure, the Tg increased from −21 °C for 10% of MGE to 1.3 °C for 87% of MGE. In contrast, a different trend is observed for P(NBn-1,2-GL-co-PCs): the Tg increased from 29.3 to 30.9 °C for the NGE/PO ratio up to 4.6 mol % and then decreased to 27.7 °C as the ratio continued to increase. This trend is rationalized as a greater number of NBn-groups increases Tg by acting as bulky side groups, whereas Tg decreases with decreasing molecular weight. After deprotection of the Bn- and NBn-groups, the terpolymers exhibited higher Tg than their respective protected ones, likely due to the interaction of the hydroxyl groups within the polymer structures.
Figure 23.

Synthesis of glycerol-based polycarbonates using Zn catalysts. (A) Adapted from ref 218 with permission of The Royal Society of Chemistry. (B) Adapted with permission from ref 219. Copyright 2014 American Chemical Society.
Both cobalt and zinc catalysts produced copolymers and terpolymers with high carbonate linkage (>90%) and polymer over cyclic carbonate selectivities. Salen cobalt complexes, on the other hand, exhibited greater activity and generated polymers with high molecular weights (Mn = 50 kDa) and narrow dispersity (Đ = 1.1). However, the cobalt metal is toxic and the synthesis of the salen cobalt complexes requires several steps. Importantly, the development of poly(1,2-glycerol carbonates) using the catalytic epoxide/CO2 systems presents a synthetic challenge as compared to poly(1,2-glycerol ether)s because this system is sensitive to trace amounts of water acting as a chain transfer agent, which results in lower molecular weight polymers than the theoretical ones, with bimodal distributions in most cases. Continued efforts in this area will provide new synthetic routes to tailored polymers and opportunities to evaluate these materials for potential clinical applications.
4.4. Applications
The prevention of tumor recurrence after surgical removal of a tumor is of significant clinical importance. For many early-stage cancer patients, surgery is the standard of care because systemically delivered chemotherapy results in limited benefit (due to only a small percentage of the injected drug reaching the target tissue) and significant morbidity. For example, in patients suffering from localized (early stage) nonsmall cell lung cancer, the most effective treatment is surgical resection. Yet, if recurrence occurs the long-term survival is poor with a resultant two-year survival of only 18–24%.220–222 Similarly, for sarcoma patients, the large tumor size, number of tumors, and anatomic complexity of the disease often result in positive microscopic margins after a macroscopically complete resection. The five-year recurrence rates are 50% or greater, with death generally due to sequelae of locoregional relapse.223–225 Recognizing this unmet need, Grinstaff and Colson et al. developed a local drug delivery strategy aimed at improving overall survival in these patients by reducing the rates of locoregional recurrence. The two key requirements for this drug delivery device are that it will (1) locally deliver a high concentration of the chemotherapeutic agent for a duration of more than 50 days and (2) be affixed to the resected tumor site via staples or sutures.
Specifically, Grinstaff et al. copolymerized 5-BTMC and ε-CL using Sn(Oct)2 to give a poly(carbonate-ester) followed by hydrogenolysis to yield a biocompatible copolymer containing pendant secondary hydroxyl groups. Subsequent coupling of various fatty acids of chain lengths C8, C12, C14, C16, and C18 to the secondary hydroxyl afforded polymers with different degrees of hydrophobicity, crystallinity, and film forming capacity.200 Encapsulation of chemotherapeutic agents [e.g., 10-hydroxycamptothecin (10HCPT) or paclitaxel (Pax)] into cast films of the polymer results in drug release rates dependent on the fatty acid chain length with extended, prolonged release of the agent occurring over 7 weeks for the most hydrophobic film, the C18 analog. Repeated exposure of the 10HCPTloaded or paclitaxel-loaded polymer films to fresh lung cancer cells in vitro afforded cell death for over 50 days.201,202 The paclitaxel-loaded poly(glycerol monostearate-co-caprolactone) films prevented locoregional recurrence of tumor in vivo.201 The overall freedom from locoregional recurrence is 83% with the use of the paclitaxel-loaded films, while the freedom from recurrence in mice treated with unloaded films or paclitaxel administered intraperitoneally or locally at the surgical site is significantly lower at 12, 22, and 0%, respectively. Similar results are observed in the sarcoma model studies, where the paclitaxel-loaded film, but not unloaded films, reduced CS-1 viability in vitro for >50 days (Figure 24). In vivo, locoregional recurrence is observed in 2 of 12 (17%) Pax-films, 9 of 13 (69%) unloaded films, and 8 of 9 (89%) Pax IV treated mice versus 7 of 8 (88%) untreated mice (P < 0.01). Moreover, the median overall survival increased to 81 compared to 64, 48, and 56 days, respectively. The increase in in vivo efficacy with the paclitaxel-loaded film is attributed to the higher localized concentrations of paclitaxel in tissues (50- to 300-fold) as compared to paclitaxel delivered via IV, confirmed by quantitative mass spectrometry data.226
Figure 24.

(A) Photograph of a polymer film coated on a collagen matrix. Reprinted from ref 201 with permission of Springer. Copyright 2009 Society of Surgical Oncology. (B) In vivo cytotoxicity data for a paclitaxel loaded film against human sarcoma cells. (C) Paclitaxel tissue levels as a function of time after administration of paclitaxel IV or implantation of the paclitaxel loaded films. (D) Paclitaxel loaded films prevent tumor recurrence in an in vivo model. Adapted from ref 226 with permission of Springer. Copyright 2011 Society of Surgical Oncology.
4.5. Polyglycerol-Based Dendrimers
Among various dendritic structures being investigated for biomedical applications, dendrimers composed of the GL metabolite are promising because they are biocompatible and biodegradable.31 In fact, the first example of a GL dendrimer, poly(1,2-glycerol ether), is reported by Haag.227 Starting from a trimethylolpropane core, allylation of the hydroxyl group using allyl chloride and a base, followed by dihydroxylation of the double bond using OsO4 to give a diol afforded the subsequent generation. Repeating this simple and efficient two-step approach provides globular poly(1,2-glycerol ether) dendrimers in good yield (e.g., G3 dendrimer is 75% overall yield) (Figure 25).
Figure 25.

Synthesis of a polyglycerol dendrimer.227
Polyesters based on GL and SA, LA (vide supra) and AA, are also of significant interest and are being evaluated for drug delivery, tissue engineering, and wound repair.31–37 For example, GL-SA dendrimers (Gn-PGLSA) are prepared via subsequent esterification and hydrogenolysis reactions similar to the divergent procedure to synthesize PGLLA described above (Figure 26).228 Specifically, a benzylidene acetal-protected GLSA monomer is reacted with an unprotected Gn–1-PGLSA–OH dendrimer, followed by hydrogenolysis of the protecting groups. At each sequence, the protected dendrimers are purified by silica gel chromatography to remove unreacted monomers and coupling reagents. Each reaction step can be monitored by NMR and/or FT-IR spectroscopy. Molecular weights of Gn dendrimers ranged from 0.96 kDa for G1-PGLSA–OH to 10.712 kDa for G4-PGLSA–OH as determined by matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) and size exclusion chromatography (SEC) against polystyrene standards, with Đ values equal to less than 1.02. The Tg for all of dendrimers, obtained with modulated differential scanning calorimetry (MDSC), ranged from 38–47 °C and from −20 to −13 °C for protected and deprotected dendrimers, respectively. With the use of this same strategy, GLSA–adipic acid Gn-dendrimers possessing alternating or block succinate/adipate layers229 and glycerol– succinic acid–salicylic acid (GLSASAL) Gn-dendrimers (Figure 26)230 can be synthesized.
Figure 26.

Chemical structures of polyester dendrimers based on metabolically derived synthons. [G], generation; P, poly; GL, glycerol; SA, succinic acid; SAL, salicylic acid; and LA, lactic acid. Top structure, work by Grinstaff et al.228 Bottom structure, work by Chai et al.230
An example of biocompatible polyester dendrimers based on the convergent approach is reported by Grinstaff et al. for the development of dendritic poly(glycerol-succinic acids) (PGLSA) dendrimers.231 The synthesis of Gn-dendrons is achieved using a benzilydene acetal-protected GL and tert-butyldiphenylsilyl protected SA derivatives. Biocompatible and biodegradable G4-PGLSA dendrons, G3-PGLSA dendrimers, and G3–PGLSA-PEG linear–dendritic hybrid macromolecules are synthesized using this strategy, and these structures can be further functionalized with methacrylate groups for the generation of cross-linked hydrogels.
4.6. Polyglycerol-Based Hyperbranched Polymers
Hyperbranched polyglycerols are extensively studied, and the reader is referred to thorough reviews by both Haag232,233 and Frey.234 Hyperbranched polyethers and poly(ether-ester)s based on GL are typically prepared from cyclic AB2 type monomers via a ring-opening multibranching polymerization, which gives relatively well-defined structures with narrow molecular weight distributions. Examples of monomers for polymerization include glycidol,235 glycerol carbonate,236 6-hydroxymethyl-1,4-dioxan-2-one,237 5-hydroxymethyl-1,4-dioxan-2-one,238 and 5-(3-[(2-hydroxyethyl)thio]propoxy)-1,3-dioxan-2-one.239 For the polymerization of glycidol, as an example, 1,1,1-tris(hydroxymethyl)propane is used as the initiator and for decarboxylation which affords a latent hydroxyl group for continued polymerization. An exponential increase in the number of both branching points and end groups is achieved giving the hyperbranched poly(glycerol ether) structure.
Building from these results, both the groups of Zhuo237 and Rokicki238 describe elegant approaches to hyperbranched poly(ether-ester)s composed of GL and GA via a self-condensing ROP of a GL synthon [e.g., a cyclic glycerol-1,2 glycerate (6-hydroxymethyl-1,4-dioxan-2-one) (Figure 27)].237
Figure 27.

Synthesis of a hyperbranched poly(ether-ester)s composed of glycerol and glycolic acid via a self-condensing ring-opening polymerization of a cyclic glycerol-glycolic acid synthon. 1: Sn(Oct)2, 110 °C in the bulk phase. Adapted with permission from ref 237. Copyright 2005 American Chemical Society.
4.7. Applications
Polyester, polyether, and poly(ether-ester) dendrimers, as well as hybrid dendritic-linear ester-ether copolymers possessing GL as one of the building blocks are of significant interest in the biomedical field. The ester linkages present within their structures allow for their in vivo degradation. Furthermore, because they are primarily composed of naturally occurring metabolites (e.g., GL, SA, GA, SAL, and LA) or biocompatible units (e.g., PEG), they will degrade into safe components that will be eliminated from the body without causing (or minimizing) toxicity to the tissues. The following paragraphs discuss some example applications of these biomaterials and describe their performances for a number of clinical unmet needs.
4.7.1. Drug Delivery
Dendrimers are interesting carriers for drug delivery because of their monodispersity, which allows for reproducible pharmacokinetic properties, their well-defined structures, which controls the encapsulation, or the covalent/ionic attachment of the drug and their flexible tailored functionalization for specific targeting of biological sites.
Grinstaff et al. reported the encapsulation of hydrophobic anticancer drugs, 10HCPT and 7-butyl-10-aminocamptothecin (BACPT), within G4.5-PGLSA-CO2Na dendrimers and studied their in vitro cytotoxicity on various human cancer cell lines (Figure 28).240,241 Both anticancer agents exhibit poor water solubility (<50 μM), and different cell line cytotoxicity profiles, and thus are ideal candidates for study. BACPT is more lipophilic and cytotoxic than 10HCPT. The dendrimer, free camptothecin, or dendrimer-encapsulated camptothecin are incubated with human adenocarcinoma (MCF-7), nonsmall cell lung carcinoma (NCI-H460), glioblastoma (SF-268), and colorectal adenocarcinoma (HT-29) cell lines for 3 days and their anticancer activity evaluated using a sulforhodamine B (SRB) protein binding assay. Cell viability significantly decreases in the presence of 10HCPT/dendrimer system as compared to free camptothecin dissolved in DMSO, while no cytotoxicity is observed with the dendrimer alone. Specifically, the half maximal inhibitory concentration (IC50) values for the DMSO dissolved 10HCPT compared to the dendrimer-encapsulated 10HCPT reduced from 113.05 to 32.3 nM for the HT-29-treated cells, 72.0 to 10.1 nM for the MCF-7-treated cells, 32.4 to 16.7 nM for the NCI-H460-treated cells, and 13.1 to 4.6 nM for the SF-268-treated cells. Additional studies of cellular uptake and retention in MCF-7 cell lines demonstrate that the dendrimer-encapsulated 10HCPT is taken up by cells at a greater rate and retained longer within the cell than the free 10HCPT drug, which may explain its enhanced cytotoxicity toward the cell line. A similar cytotoxicity profile is observed for BACPT. Specifically, when comparing the DMSO-dissolved BACPT to the dendrimer-encapsulated BACPT, the IC50 values reduced from 11.9 to 10.0 nM for the HT-29-treated cells, 26.7 to 8.3 nM for the MCF-7-treated cells, 1.2 to 0.6 nM for the NCI-H460-treated cells, and 6.6 to 1.2 nM for the SF-268-treated cells. The dendrimer-encapsulated camptothecin showed enhanced cytotoxicity toward human cancer cell lines for both 10HCPT and BACPT over the DMSO-dissolved drugs, with the largest enhancement observed in MCF-7 cell lines for 10HCPT and in SF-268 cell lines for BACPT. Moreover, the dendritic systems allowed for enhanced solubility and uptake of the drug within the system and an increase in cellular uptake and retention, again resulting in the enhanced cytotoxicity toward cancer cell lines. The same group developed another variation of this system composed of two [G4]-PGLSA–OH dendrons, linked via their focal points to a PEG chain and evaluated it for drug delivery.242 Again, the anticancer agent, 10HCPT, can be encapsulated within the linear–dendritic hybrid system. The dendrimer did not display any cytotoxic effects against the human colon cancer cells (HT-29) cells, whereas a decrease in cell viability is observed for both free and encapsulated 10HCPT. At the highest concentration of encapsulated 10HCPT (20 μM), less than 5% of cells remained viable. These experiments demonstrated that the activity of the anticancer drug is retained after its encapsulation within the dendrimer structures and that polyglycerol-based dendrimers are a suitable carrier for hydrophobic anticancer drugs.
Figure 28.

(A) Chemical structure of [G4.5]-PGLSA-CO2Na dendrimer-encapsulated 10HCPT/BACPT. (B) Cytotoxicty profile of BACT vs encapsulated BACPT in several human cancer cell lines. Reprinted with permission from ref 241. Copyright 2006 American Association for Cancer Research.
4.7.2. Wound Closure
Traumatic and surgical wounds require immediate closure to prevent infection and promote healing. Sutures and staples are the most commonly used closure techniques. However, suturing is a time-consuming method, may require anesthesia, and can cause inflammation, nerve damage, and infection, while staples can generate less precise wound edge alignment. As a consequence, dendritic hydrogel adhesives have emerged as promising materials to supplement or replace the conventional closure techniques in the repair of wounds.162 Grinstaff et al. reported the synthesis of degradable and photo-cross-linkable linear–dendritic hybrid adhesives for corneal tissue closure (Figure 29).33,243 Corneal lacerations and perforations are generally caused by trauma, inflammation, and infection and if not treated quickly and effectively can lead to cataract formation, corneal scarring, glaucoma, and blindness. The Gn-dendron is composed of naturally occurring metabolites, SA and GL, linked by ester groups and coupled via their focal points to a PEG spacer of 3.4 kDa in molecular weight (Figure 29). The GL units on the periphery of the dendrons are further functionalized with methacrylate (MA) groups, which upon photopolymerization, cross-link and form a hydrogel, ([Gn]PGLSA-MA)2-PEG3400, in water.33,243 The hydrogel, once formed, adhered to moist corneal surfaces, and secured the corneal lacerations better than conventional nylon sutures. Moreover, the repair procedure was five times faster than suturing the wound and did not inflict any additional trauma to the wound. In order to evaluate whether the hydrogel sealed the wound, the burst pressure for both the nylon suture and the dendritic-linear hydrogel treated eyes was measured using a pressure transducer probe inserted in the eye through the optic nerve. A 4.1 mm corneal laceration was made in an enucleated ex vivo eye and then sealed with either the hydrogel adhesive, ([Gn]PGLSA-MA)2-PEG3400, or nylon suture. The leaking pressure measured after treatment with the ([G1]PGLSA-MA)2-PEG3400 sealant is 171 ± 44 mmHg, as compared to 90 ± 18 mmHg obtained with sutures (IOP in human eyes is around 15–20 mmHg).33 This linear–dendritic hybrid sealant is also efficient in securing 4.1 mm stellate corneal wounds as well as closing corneal lacerations of enucleated human eyes, produced in a laser-assisted in situ keratomileusis (LASIK) procedure.244–246 Building on the successful ex vivo studies, the authors conducted an in vivo corneal laceration repair study in white leghorn chickens.34,247 All of the animals received a 4 mm full thickness linear wound in the right eye and were then treated with either the biodendrimer sealant or the standard of care, conventional nylon sutures. The dendritic-linear hybrid sealant did not show any toxic effect during the testing period and successfully sealed 100% of the lacerations, by postoperative day 2. Moreover, histological examinations at day 28 showed that the wounds treated with the sealant presented less scarring and possessed a more uniform corneal structure than the ones treated with sutures. Finally, the sealant completely disappeared from the wound site after day 14.34 This study supports the use of dendritic-linear hydrogels as sealants for corneal wound repair, as they secure and seal corneal lacerations better than conventional sutures, do not inflict additional trauma or toxicity to the treated tissue, and are completely removed from the wound site once the wound is healed. Clinically, the nylon sutures would be removed, which highlights another advantage of using a biocompatible and biodegradable adhesive.
Figure 29.
(A) Chemical structure of ([G1]PGLSA-MA)2-PEG3400 for the formation of photo-cross-linked dendritic hydrogel. G: generation, P: poly, GL: glycerol (in blue), SA: succinic acid (in red), MA: methacrylate (in black), PEG: poly(ethylene glycol) (in green). Adapted with permission from ref 34. Copyright 2007 Elsevier. (B) Photograph of a full-thickness corneal laceration in an enucleated ex vivo eye sealed with the dendritic hydrogel adhesive. Reprinted with permission from ref 33. Copyright 2002 Wiley-VCH. (C) Photograph of a corneal laceration in an in vivo chicken eye sealed with the dendritic.
4.7.3. Antibacterial Agents
Polyester dendrimers based on metabolically derived synthons are also investigated for their potential antibacterial properties due to the increasing need for novel antibacterial biomaterials and alternative mechanisms of action, given the rise in resistance among bacteria. Among antibacterial biocides, amphiphilic compounds are known to cause perturbation and disruption of the bacterial cell wall/cytoplasmic membrane.248 Grinstaff et al. thus investigated the antibacterial activity of anionic amphiphilic dendrons A and B composed of SA, GL, and myristic acid, since dendrimers with terminal negative charges are generally not cytotoxic to eukaryotic cell lines (Figure 30).249 Cytotoxicological experiments against a wild-type Gram-positive bacterial strain (Bacillus subtilis AG174) revealed that the dendrimer constituents (SA, GL, and myristic acid) are not toxic to B. subtilis over the concentration range tested (from 1.0 × 10−5 to 1.0 × 10−3 M), whereas SDS and Triton X-100 (positive controls) are toxic with half-maximal effective concentration (EC50) of 1.4 × 10−4 and 8.8 × 10−5 M, respectively, and amphiphilic dendrons A and B exhibit an antibacterial activity with EC50 of 6.0 × 10−5 and 4.1 × 10−5 M, respectively. However, the amplitude of the sigmoid curve for the amphiphilic dendron samples is comparatively compressed, which the authors attributed to a bacteriostatic mechanism of action.249 The eukaryotic cytotoxicity of the dendritic amphiphiles, their constituents, and the positive controls were also conducted against a primary cell line of human umbilical vein endothelial cells (HUVECs). Similar to the results obtained with Gram-positive bacteria, SA, GL, and myristic acid did not show any toxic effect to cells, while both SDS and Triton X-100 are cytotoxic, with EC50 of 5.4 × 10−4 and 1.1 × 10−4 M, respectively. As for the synthetic dendritic amphiphiles, dendron A showed cytotoxicity with EC50 of 1.3 × 10−4 M, whereas dendron B did not, in the concentration range tested. Increasing the concentration of B to its aqueous solubility limit of 2.0 × 10−3 M resulted in a reduction of 50% of the negative untreated control, and thus the EC50 is estimated to be higher than 1.5 × 10−3 M. While SDS, Triton X-100, and dendron A are not ideal antibacterial candidates due to their toxicity toward eukaryotic cells, dendron B exhibits a promising eukaryotic/prokaryotic EC50 ratio (≥36). The difference in eukaryotic cytotoxic effect between the two synthetic dendrons A and B is attributed to the supramolecular assemblies that these materials form in water at a given concentration. Further studies are underway to understand the structure–activity of these antibacterial dendritic materials as well as their mechanism of action.
Figure 30.

Chemical structures of anionic amphiphilic antibacterial dendrons.249
4.7.4. Cartilage Tissue Engineering
Natural and synthetic biopolymers are widely studied as scaffolds for tissue engineering due to their tunable mode of administration, mechanical properties, surface modification/functionalization, biocompatibility, biodegradability, and capacity of drug delivery and cell adhesion.250 Specifically, hydrogels are particularly attractive materials as tissue-engineering scaffolds because they can mimic many roles of extracellular matrices (ECM) found in human tissues, such as diffusion of oxygen and nutrients to cells, release of waste and CO2 from cells, attachment sites for cell adhesion, resemblance of the tissue structure, control of mechanical properties, and regulation of the cells function.251 Grinstaff et al. evaluated the use of GL, SA, and PEG based-linear– dendritic polyester hydrogels as scaffolds for articular cartilage tissue engineering by studying their mechanical and degradation properties as well as their ability to support articular chondrocytes and ECM synthesis in vitro.252 Various concentrations (7.5%–20%) of ([G1]PGLSA-MA)2-PEG3400 dendritic macromers (Figure 31) are photo-cross-linked with a UV or argon-ion laser source (with the appropriate photoiniating system) to afford 3D hydrogel networks for further investigation of their mechanical properties and use as a cell scaffold for chondrocytes. The complex modulus |G*| is slightly dependent on the concentration of the macromers and increased from 1.2 to 1.7 kPa, whereas the loss angle, δ, increased from 6.3 ± 0.5° to 70.6 ± 0.3°, over the concentration range tested, indicating a transition from an elastic to a viscoelastic behavior of the material. Additionally, the equilibrium compressive modulus, E, also increased from 3.7 ± 0.1 to 34 ± 5 kPa, in the concentration range tested, indicating an increasing stiffness of the material with increasing macromer concentration. In order to evaluate the ability of the hydrogel to encapsulate cells and construct cartilaginous ECMs, chondrocytes are mixed with the dendritic macromer at 7.5 and 15% concentrations and cross-linked to form the 3D network. Stained histological sections of the hydrogel-cell constructs at 2, 4, and 12 weeks post encapsulation showed no loss of cell viability or signs of dedifferentiation at either macromer concentration. Additionally, at the lower macromer concentration, chondrocytes produced abundant ECM rich in proteoglycans and type II collagen after 2 weeks similar to native articular cartilage, whereas at the higher macromer concentration, encapsulated cells produced less ECM only in their immediate proximity. After 4 weeks of incubation, the hydrogel-cell constructs, at the lower concentration, started to degrade after having accumulated a significantly protein-rich ECM, while at the higher concentration, no degradation is observed even after 12 weeks. The degree of swelling of the hydrogel is another important criteria when evaluating them for tissue repair because excessive swelling can be detrimental to in vivo applications, especially when these materials are applied in confined areas such as cartilage defect sites. The dendritic hydrogel tested in this study showed a small percentage of swelling after cross-linking at 37 °C and equilibrium in phosphate-buffered saline (PBS) or chondrocytes medium, due to the efficient cross-linking of the material after photopolymerization. The results obtained with the dendritic cross-linked biomaterial demonstrate the ability of these scaffolds to encapsulate cells, support the production of cartilaginous ECMs, and degrade once their function is fulfilled. However, these properties depend significantly on the macromer concentration and the mechanical properties of the hydrogel (i.e., hydrogels at lower concentration exhibit lower mechanical properties than for the native articular cartilages but support the production of ECM and degrade in time, whereas hydrogels at higher concentration possess improved mechanical properties but are not capable of constructing abundant ECMs and degrading). Therefore, it is important to find a compromise between the mechanical, biochemical, diffusion, and degradation properties of the biomaterial in order to successfully apply it for a tissue engineering application like cartilage repair, and dendritic macromolecules are particularly promising as one is able to control and tune the hydrogel properties for a desired application.
Figure 31.

Photo-cross-linking of the [G1]PGLSA-MA)2-PEG3400 dendritic macromers in the presence of chondrocytes affords hydrogel-cell constructs which support the growth of chondrocytes as evidenced by significant Saffarin-O staining for the production of glycosaminoglycans. Adapted with permission from ref 252. Copyright 2006 American Chemical Society.
5. NATURAL AMINO ACID SYNTHONS
Natural L-amino acids are the basic unit of peptides and proteins. When connected in a specific sequence through amide bonds, the polymer folds into secondary and tertiary structures, and given the number of amino acid building blocks (or synthons), the variety of post-translational protein modifications, and the manner by which they fold affords an almost unlimited number of possibilities. Proteins perform diverse biological functions including structural support, metabolic catalysis, signaling, molecular recognition, transportation of biologics, etc. To distinguish from natural short polypeptides or proteins and synthetic polypeptides prepared via solid phase stepwise synthesis, we will refer to “poly(amino acid)s” for polymers synthesized from synthons derived from natural amino acids. Working with synthetic poly(amino acid)s offers a number of advantages including (1) a large pool of monomers derived from natural amino acids (21 proteinogenic amino acids in human), (2) a wide range of polymer properties attainable via judicious choice and combinations of synthons, and (3) the ability to self-assemble into 3D structures. Because of these benefits, poly(amino acid)s are an intriguing target for chemical synthesis, and preparation routes to such structures are actively investigated. Once synthesized and characterized, these polymers are of interest for a number of potential biomedical applications.
5.1. Linear poly(amino acid)s
Synthesizing poly(amino acid)s by polymerization of N-carboxyanhydrides (NCAs) is an established method since the 1940s (Figure 32). After more than a hundred years of development, since its first report by Leuchs in 1906,253 there are a large number of diverse monomers available for NCA polymerization as well as catalysts, including those that afford controlled living polymerization.254–258 Various poly(amino acid)s homopolymers, random copolymers, and block copolymers are reported and characterized. Overall, NCA polymerization possesses the following advantages: (1) a large pool of readily available monomers derived from natural amino acids (21 proteinogenic L-amino acids), (2) facile access to high molecular weight polymer and different polymer structures ranging from homopolymer to random copolymer to block copolymer, and (3) polymerization proceeds with no detectable racemization.259 Importantly, a significant critical review of NCA polymerization chemistry and the types of monomers that can be polymerized is recently reported (2015), and the reader is referred to this article,260 as well as to two other excellent reviews from 2006 and 2013.259,261
Figure 32.
Synthesis of poly(amino acid)s by polymerizing N-carboxyanhydrides (NCAs).
5.2. Poly(amino acid)s Dendrimers and Dendrons
Dendritic structure constitutes an important class of polymer architecture as described earlier. Peptide dendrimers can be divided into two general classifications. First are grafted peptide dendrimers where the branching core is an organic compound and peptides or proteins are attached as surface functional groups. Alternatively, a G1 to G4 dendrimer is decorated with peptides or proteins via coupling to the surface functional groups. Peptide dendrimers of the second type are branching poly(amino acid)s, and these poly(amino acid) dendrimers and dendritic polymers are the focus of this section.
Poly(amino acid) dendrimers are synthesized from monomers such as L-lysine, L-glutamic acid, and L-aspartic acid because these monomers possess three functional groups where two of the functionalities are similar, and the two dissimilar but complementary groups can be linked together to form a covalent bond in the presence of a suitable coupling reagent (e.g., DCC). For example, lysine contains two primary amines and one carboxylic acid, and appropriate protection/deprotection schemes can be used as the monomer to prepare poly(L-lysine) dendrimers. In fact, poly(L-lysine) dendrimers were first synthesized in 1981 by Denkewalter.262 Since then, a variety of different poly(amino acid) compositions, structural features, and polymer architectures are reported and their chemical/physical properties described.263 For example, in 1999 and 2000, Park et al. reported the synthesis of a barbell-like triblock copolymer, poly(L-lysine) dendrimer-block-poly-(ethylene glycol)-block-poly(L-lysine) dendrimer, by the liquid-phase peptide synthesis method.264,265 Poly(ethylene glycol) bis(amine) is coupled to two di-Fmoc-L-lysines (Fmoc = fluorenylmethyloxycarbonyl). Deprotection of the Fmoc-L-lysines using piperidine gives four amines for further coupling-deprotection cycles to give the final triblock barbell-like dendrimer up to G4 (Figure 33).
Figure 33.

Barbell-like poly(L-lysine) dendrimer with a PEG supporter.265
In 2002, Aoyagi et al. reported the synthesis of globular poly(L-lysine) dendrimers up to G6.266 The synthesis begins by coupling the hexamethylenediamine core with Boc-protected lysine using standard coupling chemistry (Boc = tert-butyloxycarbonyl). Then the Boc-protecting groups are removed by acid treatment to give the G1 dendron with four free amine groups. The coupling and deprotection reactions are repeated to give monodisperse lysine dendrimer up to G6.
In 2004, Grinstaff et al. reported the synthesis of G3 and G4 poly(L-lysine) dendrons with cysteines (cys) attached to the periphery.267 The dendrons are prepared using the penta-fluoroester (PFP) strategy since this approach afforded higher amide coupling reaction yields than coupling reactions employing (benzotriazol-1-yloxy)tris(dimethylamino)-phosphonium hexafluorophosphate (BOP), DCC, 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDCI), or oxalyl chloride. The trilysine core is prepared by reacting two equivalents of Z-Lys(Z)OPFP with LysOMe·2HCl (Z = carboxybenzyl, OMe = methoxy, Lys = lysine) in the presence of N,N-diisopropylethylamine (DIPEA) and hydroxybenzotriazole (HOBt). Subsequent removal of the Z group via catalytic hydrogenolysis and reaction with IsoCys(Boc)OPFP afforded protected dendron. Removal of the Boc and iso-protecting groups of cysteine using TFA and 1 N HCl in MeOH, respectively, gave the thiol-terminated dendron of interest. The larger G4 dendron is prepared in a similar manner, except that an additional Lys coupling reaction is performed (Figure 34). Recently, Grinstaff et al. reported a similar poly(L-lysine) dendron with thiol groups on its surface possessing a PEG attached to the carboxylic acid of the lysine core to improve aqueous solubility. All three dendrons form hydrogels in the presence of poly(ethylene glycol disuccinimidyl valerate) or similar activated ester via formation of thioester linkages.268
Figure 34.

Synthesis of poly(L-lysine) dendrimer with cysteine on the surface by Grinstaff et al. Adapted with permission from ref 267. Copyright 2004 American Chemical Society.
Besides conventional solution-phase synthesis discussed above, solid-phase chemistry can also be used for the synthesis of poly(L-lysine) dendrimers. Florence et al. reported a lipidic poly(lysine) dendrimer with lipidic molecules on the surface of the dendrimer.269 Solid phase coupling reactions are achieved using a 3-fold excess of (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) (HBTU) activated Boc protected L-lysine on a MBHA resin (MBHA = 4-methyl benzhydrylamine). Repeated cycles of coupling and deprotection afforded the L-lysine dendrimer up to G6. Attempts to synthesize a complete G7 dendrimer were unsuccessful because of steric congestion at the surface inhibiting the coupling reactions.
Another example of dendritic poly(amino acid)s is that based on glutamic acid and aspartic acid, which possess two carboxylic acids and one amine functional group. The synthetic approach to these dendritic structures is similar to that for the poly(L-lysine) structures, where a series of repetitive conjugation and deprotection steps are performed. Therefore, we will only discuss a couple of examples. Mitchell and co-workers prepared dendrimers based on aspartic acid as well as dendrimers based on glutamic acid (Figure 35) using a divergent approach.270 Also, Ranganathan published the results on glutamic acid-based dendrimers via a convergent approach using a 1,3,5-benzene tricarbonyl, 1,3-benzene dicarbonyl, and admantane as dendrimer cores.271,272
Figure 35.

Synthesis of poly(glutamic acid) dendrimer using a divergent approach. Adapted from ref 270 with permission of The Royal Society of Chemistry.
5.3. Applications
Linear and dendritic poly(amino acid)s are of interest for a number of biomedical applications given their composition from biocompatible, nontoxic building blocks.273 The dendritic poly(amino acid)s possess additional unique characteristics, as discussed above, such as high ratios of surface area to volume, a multitude of functionalizable end groups, and nearly monodisperse polymers of controlled interior and exterior structure. These characteristics lead to advantageous properties such as high solubility, low viscosity, adhesiveness, and ability to encapsulate low molecular weight compounds.
5.3.1. Drug and Gene Delivery
A well-known example of a poly(amino acid) used for a drug delivery application is the paclitaxel poliglumex conjugate (XYOTAX, OPAXIO).274 Paclitaxel poliglumex (PPX), developed by Cell Therapeutics, is a conjugate of paclitaxel and poly(L-glutamic acid) as shown in Figure 36. Use of the poly(L-glutamic acid) ensures aqueous solubility of the formulation even at a 40 wt % loading of drug, eliminating one of the major challenges associated with paclitaxel: its limited solubility in aqueous solution. Due to the enhanced permeability of tumor vasculature, the macromolecular pro-drug, PPX, accumulates in tumor tissues after intravenous administration. In an in vivo small animal tumor model, PPX is more efficacious than paclitaxel alone. This increase in efficacy is a consequence of increased tumor drug concentration and exposure. Results from phase 1 and 2 clinical studies demonstrated positive outcomes with PPX performing similar to paclitaxel with reduced neutropenia and alopecia. PPX currently remains under investigation for treatment of various forms of cancer.
Figure 36.
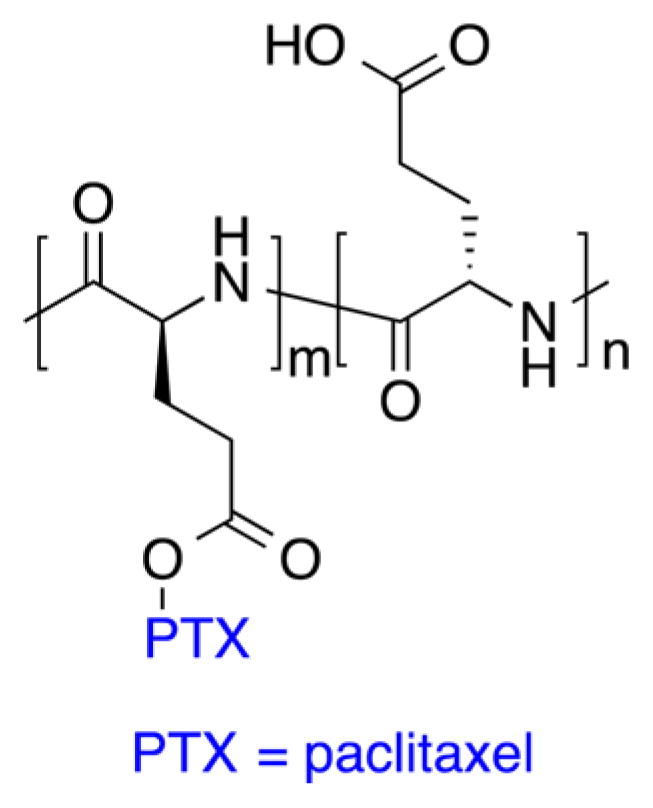
Chemical structure of paclitaxel poliglumex.
The triblock barbell-like and globular poly(L-lysine) dendritic polymers mentioned above are cationic at neutral pH, due to the high surface amine group density and, thus, are of interest for DNA complexation and gene delivery. For example, the barbell-like poly(lysine) dendrimer-block-PEG-block-poly-(lysine) dendrimer forms spherical water-soluble complexes with plasmid DNA between 50 to 150 nm in diameter. The self-assembling ability of G3 and G4 polymers with plasmid DNA is an important factor, and this likely reflects the size and the number of positively charged amines per polymer molecule available for DNA complex formation.265 This dendritic linear hybrid polymer exhibited less cytotoxicity compared to poly(L-lysine), poly(D-lysine), and poly(ethylenimine).
A similar study is described by Aoyagi et al. using monodisperse globular poly(L-lysine) dendrimers, who observed a dendrimer generational dependence on gene transfection. 266 Dendrimers of third generation and larger complexed with plasmid DNA to afford a dendriplex, with increasing generations affording a greater degree of compaction. This result is in agreement with Park et al.’s above results, which show that the generation number is an important factor for DNA complexation. Significant gene transfection and minimal cytotoxicity was observed for several cell lines when using the fifth and sixth generation dendrimers.
5.3.2. Adhesives and Sealants
Marine adhesive proteins (MAPs) are superior adhesives for wet environments and, therefore, are of particular interest for use as surgical adhesives and sealants. However, a potential limitation of using MAPs as a surgical adhesive is that the adhesive proteins and oxidase enzymes afforded toxicity and antigenicity in an animal model.275 With the idea of mimicking natural MAPs, Deming et al. designed and synthesized a mimic of MAPs with a synthetic poly(amino acid) backbone (Figure 37), a copolymer from lysine and 3,4-dihydroxyphenyl-L-alanine (DOPA). These materials have a number of advantages for biomedical applications such as a ready supply of polymer and the use of a simple oxidizing agent for the adhesive.
Figure 37.

(Left) Chemical structure of the adhesive mimicking MAPs. (Right) Evaluation of the adhesive using different adherents. Adapted with permission from ref 275. Copyright 1998 American Chemical Society.
The high density of functional end group for cross-linking and the low intrinsic viscosity of dendritic polymers make them ideal materials for the preparation of hydrogel-based sealants, which require fast cross-linking rates and administration through a small diameter needle. In 2004, Grinstaff et al. reported a dendritic macromer that undergoes fast cross-linking with a PEG based cross-linker to form a sealant for securing cataract incisions.267 Specifically, they designed and synthesized dendritic G3 and G4 poly(lysine) dendrons possessing cysteine residues at the periphery. When the dendron solution is mixed with poly(ethylene glycol) dialdehyde, gelation occurred rapidly with the formation of the thiazolidine cross-linking hydrogel. Using an ex vivo enucleated eye model, the mean leaking pressure for the eyes repaired with the hydrogel sealant (N = 8) was 184 ± 79 mmHg. In contrast, the mean leaking pressure for the current standard of care nontreated (N = 7) and suture (N = 2)-treated eyes were 24 ± 8 and 54 ± 16 mmHg, respectively. As normal intraocular pressure is between 12 and 16 mmHg, the hydrogel sealant efficiently seals the wound.
Another hydrogel sealant system which enables controlled dissolution after application is described by Grinstaff et al. where the chemistry of native chemical ligation (NCL) is used to disassemble the hydrogel sealant.268 Specifically, a poly-(lysine) dendrimer with thiols at its surface is reacted with PEG disuccinimidyl valerate to afford a thioester cross-linked hydrogel. Rapid gel formation occurs, and upon subsequent exposure to a solution of cysteine methyl ether (CME), the hydrogel sealant dissolves (Figure 38). This intricate, yet practical, design of smart materials fulfills the unmet need for a sealant system for trauma scenarios sustained in military injuries or in rural or wilderness settings. Such a hydrogel sealant system would enable immediate management of the wound by adhering to the tissue and stopping the blood flow, and its subsequent controlled dissolution would allow for gradual wound re-exposure during definitive surgical care.
Figure 38.

(a and b) Photographs of hydrogels PEG-LysSH (green) and PEG-LysNH2 (pink) adhered to human skin tissue, under torsion. (c) Dissolution of PEG-LysSH hydrogel in CME solution (0.3 M) in PBS at pH 8.5 at different time intervals (0, 10, 20, and 30 min). Reprinted with permission from ref 268. Copyright 2013 Wiley-VCH.
5.4. Linear Poly(desaminotyrosyl-tyrosine-alkyl Ester Carbonate)s
Kohn et al. pioneered the exploration of pseudopoly(amino acids) as potential biomaterials with particular focus on tyrosine-based polycarbonates as degradable biomaterials.276 Early work focused on characterizing the influence of hydrophobic side chains of desaminotyrosyl-tyrosine alkyl esters on the polycarbonate’s functional characteristics (i.e., rate of degradation, tensile strength, and cell-polymer compatibility). The polycarbonates are synthesized from their respective diol monomers in solution by treatment with phosgene,277 reaching weight-average molecular weights ranging between 120 and 450 kDa with dispersities consistent with step polymerizations ranging from 1.4 to 1.8 (Figure 39). Rates of degradation followed first-order kinetics and aliphatic side chain substitutions ranging from ethyl to octyl did not significantly change the degradation rate or pattern.
Figure 39.

Structures of poly(desaminotyrosyl-tyrosine alkyl ester carbonate) and terpolymer of poly(desaminotyrosyl-tyrosine alkyl ester-co-desaminotyrosyl-tyrosine-co-oligoethylene glycol).276,281
This class of polycarbonates is also one of the first polymer classes examined in a combinatorial fashion through automated synthesis, wherein the synthetic methods can be rapidly optimized278 and the resulting materials efficiently screened for function, such as interactions with growing cells.279 Recently, terpolymers of desaminotyrosyl-tyrosine alkyl ethyl ester, desaminotyrosyl-tyrosine and oligoethylene glycol, are described as structural biomaterials with rapid degradation rates to assist in the implantation of cortical neural probes.280,281 The addition of the pendant carboxylic acid and the hydrophilic ethylene glycol groups facilitates the rapid influx of water, leading to faster rates of polymer degradation with increasing content and the corresponding decrease in electrical impedance upon implantation (~10 min). Titration of the hydrophobic monomer, combined with the carbonate backbone functionality, allows maintenance of an initial structural intrinsic strength of ~4 MPa, which is sufficiently greater than the critical strength for penetration (~1.3 MPa).
6. FUMARATE SYNTHONS
Fumaric acid is a trans-diacid metabolized through the Krebs cycle. In the mid 1990s Mikos et al. reported the first synthesis of poly(propylene fumarate) (PPF) and its use as a biomaterial (Figure 40).282 In the most recent synthesis advances for PPF, the fumaric carboxylic acid functionalities are esterified with the hydroxyl groups of propylene glycol, an FDA-approved pharmaceutical excipient, in a two-step synthesis through a bis(hydroxypropyl) fumarate diester intermediate with zinc chloride as a catalyst.283 The highest molecular weight of the homopolymer is fairly low (on the order of ~4 kDa), and the polymer is a viscous liquid at room temperature. However, PPF is an intriguing material owing to its double bond functionality that allows facile cross-linking to create solid thermoset matrices. This dichotomy of liquid to solid character fosters the investigation of this material’s potential utility in a range of areas such as an injectable scaffold for drug delivery and tissue engineering284 and as a patternable material through laser stereolithography.285
Figure 40.
Chemical structure of poly(propylene fumarate) (PPF).
7. SACCHARIDE SYNTHONS
Carbohydrates are attractive building block synthons for biomaterials given their prevalence and diversity within the human metabolome. Moreover, natural polysaccharides play essential roles in many biological processes,286–288 and natural polysaccharides themselves are investigated for various biomedical applications such as drug delivery, wound repair, and tissue engineering.159,289–292 However, the polyol nature of carbohydrates (large number of the same functional groups and reactivity) limits the control over the resulting polymeric structures by traditional synthetic methods. Indeed, the synthesis of linear polyether or polyester carbohydrates through traditional stepwise synthetic routes requires protection/deprotection steps, which afford low yields and quantities of the final materials or through condensation polymerization reactions give lower molecular weight samples with a broad molecular weight.293 In this section, we describe recently reported synthetic approaches which circumvent the inherent challenges of multiple functional groups and allow the preparation of well-defined polymers containing saccharides to include polyester saccharides, glyco(amidoamine)s, poly-(amidosaccharide)s, sugar-derived polycarbonates, and glycopolymers.
7.1. Polyester Saccharides
We begin with the application of enzymatic catalysis to prepare polyester saccharides, which is an active area of investigation by many groups including Gross and co-workers.294 Lipases are regioselective catalysts for esterification. When Lipase B from Candida Antarctica is immobilized on beads, polyesters can be prepared containing nonreducing sugars (e.g., erythritol, xylitol, ribitol, mannitol, glucitol, and galactitol), 1,8-octanediol, and adipic acid.295 Surprisingly, there is no correlation between carbohydrate chain length and molecular weight, with weight-average molecular weights ranging from ~10 to ~75 kDa, leading to the conclusion that longer chains are derived from chain branching at secondary hydroxyl groups with different reactivity.
The utility of lipase regioselectivity under mild reaction conditions is demonstrated by the capping of organosilicon carboxylic acids and the primary alcohol of α,β-ethyl glucoside, 296 by the polyesterification of sorbitol and glycerol with adipic acid and 1,8-octane diol,297 and by the selective deacetylation of sophorolipids to expand their function group capacity as polymers synthesized by ring-opening metathesis polymerization.298
7.2. Poly(glycoamidoamine)s
In addition to polymers containing cyclic carbohydrate building blocks, Reineke et al. advanced the study of polymers based on the open form, rather than the pyranose ring, of the carbohydrate to form a series of poly(glycoamidoamine)s. The sugars are linked with amine-containing spacers with the primary goal of determining the structure–function relationships associated with the delivery of nucleic acids to mammalian cells. Structures containing D-glucaramide, meso-glactaramide, D-mannaramide, and L-tartaramide are described and have been actively studied since the early 2000s (Figure 41).299–302 The synthesis of these polymers is relatively straightforward and involves reacting the esterified sugar (e.g., esterified D-glucaric acid) with amine-containing comonomers such as diethylenetriamine, triethylenetetramine, or tetraethylenepentamine at room temperature in methanol. The polymers are then isolated and purified via dialysis versus a 1000 MWCO membrane.301
Figure 41.

Examples of poly(glycoamidoamine)s reported by the Reineke group containing (i) D-glucaramide, (ii) meso-glactaramide, (iii) D-mannaramide, and (iv) L-tartaramide.299–302 Adapted with permission from ref 306. Copyright 2010 American Chemical Society.
7.3. Applications
The inclusion of cationic spacers into the glycopolymer provides an opportunity to design and evaluate these polymers for complexing and delivering nucleic acids (DNA and RNA) to cells. The optimal balance between nucleic acid binding affinity, polycation associated cytotoxicity, and overall transfection efficiency is compositionally dependent, as well as cell dependent.303,304 Polymers with similar composition, but containing guanidine groups to enhance polymer/DNA complex stability, are also described with one construct approaching the transfection efficiency of the positive controls (Glycofect and poly-L-arginine).305 One challenge associated with the amidoamine class of polymers is their propensity to undergo autocatalytic hydrolysis.306 In an attempt to enhance polymer stability, increase polymer molecular weight, and promote stronger polymer interactions with DNA through interaction with the minor grooves, glycopolycations possessing the disaccharide, trehalose, are synthesized using the Huisgen cycloaddition, or “click” reaction, and subsequently investigated. 307 These polymers promoted nucleic acid delivery into cells grown in culture, surpassing Jet-PEI, which served as the positive control. Perhaps most importantly, the trehalose residues provided the aqueous solubility necessary to maintain the heterocycle structures in solution, which can be challenging for poly triazoles. Excitingly, these activities have led to one of the poly(galactaramidoamine) being commercialized as a transfection reagent, under the trade name, Glycofect.
7.4. Poly(amidosaccharide)s
As mentioned above, natural polysaccharides are of significant utility; however, their isolation and extensive purification processes, high dispersity, trace contamination by endotoxins, and batch-to-batch variation limit their use as bio-engineered materials. Therefore, water-soluble carbohydrate-based polymers, which retain the stereochemically defined, cyclic backbone of natural polysaccharides, and which can be prepared by controlled and robust synthetic routes are of interest from both fundamental and applied perspectives. Specifically, amide-linked polysaccharides, termed poly-(amidosaccharide)s (PAS)s, are novel polymeric structures, which possess hydrophilic pyranose rings linked through 1- and 2-positions by α-amide-linkages (Figure 42), recently reported by Grinstaff et al.308–312 These synthetic polymers are not found in nature. However, they structurally mimic natural polysaccharides due to the chiral and rigid hydrophilic pyranose backbones within their structures, and at the same time, they can be easily and rapidly synthesized with higher purity, low dispersity, and controlled molecular weight and derivatization. PASs can also be considered as highly functionalized β-polypeptides due to the 1,2-peptide linkages within their structures. β-polypeptides form defined secondary structures and therefore represent an interesting class of synthetic polymers, themselves due to their promising biological applications. The reader is referred to several recent reviews on this topic for additional information.313–315 The following paragraphs discuss the synthesis and characterization of PASs and emphasize their potential biological applications.
Figure 42.

Comparison between a natural polysaccharide (e.g., amylose) and poly(amidosaccharide) (PAS). Adapted with permission from ref 310. Copyright 2012 American Chemical Society.
Hydrophilic α-1,2-linked pyranose-based PASs are synthesized via a controlled anionic ROP of a β-lactam sugar monomer derived from benzyl-protected D-glucal/galactal (D-glc/gal), as depicted in Figure 43.308–312 Enantioselective PASs with defined molecular weights and low dispersities are obtained, with no epimerization occurring at the 1- or 2- position of the pyranose ring. The β-lactam sugar monomer is obtained in moderate yield via the stereoselective cycloaddition of tri-O-benzyl-D-glc/gal and chlorosulfonyl isocyanate (CSI) followed by in situ reduction to remove the sulfonyl group. The sugar monomer is then reacted with acyl chlorides or PFP-activated esters initiators, and the ROP is performed at different initiator loadings (e.g., X = 5, 2, and 1 mol %) to prepare PASs of different degrees of polymerization (DP) (e.g., n = 20, 50 and 100, respectively). Debenzylation of the polymer is successfully performed via the Birch reduction, and the deprotected PASs are further purified by dialysis and lyophilization. By comparing the integration of the initiator’s group signals (e.g., phenyl, carbon chains) to the polymer integration in the 1H NMR spectrum, the DP of polymers is estimated and agrees with the data collected by GPC. By 13C NMR, signals of amides, C1 and C2 carbons, within the polymer backbone are apparent. The FT-IR spectra shows strong amide stretches (amide I and amide II) indicative of the formation of the polymer backbone. By the GPC, the Mw, DP, and Đ of PASs range from 4.6 to 54.9 kDa, 22–186, and 1.1– 1.6, respectively. The circular dichroism (CD) spectra of the PAS recorded in water, at room temperature, shows one minimum at 221 nm and one maximum at 190 nm, similar to those reported for oligo-β-peptides with a left-handed 31 helical conformation.310,312
Figure 43.

7.5. Applications
As PASs are composed of rigid and enantiopure pyranose rings linked together via α-amide linkages and exhibit similarities with natural polysaccharides and β-polypeptides, it is of interest to evaluate these biomimetic materials in biological assays as well as for various biomedical applications.
7.5.1. Interactions with Natural Carbohydrate Receptors
Lectin concanavalin A (ConA) is a carbohydrate-binding protein, known to bind specifically α-D-glucosyl and α-D-mannosyl residues of natural polysaccharides through predominant interactions with C3, C4, and C6 alcohols. Thus, having the same α-configuration linkage and free hydroxyl groups at C3, C4, and C6 positions, α-N-1,2-D-glc PASs (Figure 44) may bind ConA at the same site as natural glucose derivatives. To determine if the Glc-PASs of different molecular weights (Mw ≈ 5, 10, and 40 kDa) form aggregated structures with the tetravalent lectin, Glc-PASs and ConA are mixed together and the turbidity of the sample solution is measured based on the scattering at 405 nm.310 Higher molecular weight PASs (Mw = 10 and 40 kDa) interacted strongly with the natural carbohydrate receptors, whereas lower molecular weight PAS (Mw = 5 kDa) showed lower binding affinity, likely due to the short length of its polymeric backbone (6–7 nm), reducing the number of multivalent interactions with ConA.
Figure 44.

Structures of PASs tested for biological applications.
The asialoglycoprotein (ASGP) receptor, a hepatic lectin, is another example of a receptor that binds natural polysaccharides. Specifically, ASGP binds galactose residues with free hydroxyls at the C3 and C4 positions and internalizes these structures into cells. Therefore, the specific uptake of α-N-1,2-D-gal PASs (Figure 44) in a human hepatocyte line expressing the ASGP receptor is investigated to determine whether these polymers are transported into cells.309 First, PASs (Mw = 7 and 50 kDa) are labeled with a fluorescent rhodamine derivative, incubated with cells for 24 h, and then imaged with a confocal microscope. PAS uptake is confirmed with the fluorescent signal localized within the cytosol of cells. In order to confirm whether the uptake of PASs is mediated by the ASGP receptors, asialofetuin (AF), a competitive binder for the ASGP receptor, is added to PASs during their incubation with cells. However, the addition of AF did not affect the uptake of the polymers, concluding that PASs are not recognized by the ASGP receptors on hepatocytes and are taken into cells via a general pathway of endocytosis.
7.5.2. Protein Stabilizing Agents
The potential use of oxidized PASs, containing carboxylic acid residues, as protein stabilizing agents is next discussed and compared to a widely used stabilizing agent, trehalose.312 Proteins are widely used in biochemical research and biomedical applications, however, due to their potential instability and loss of activity during freeze-drying, stabilizing agents, such as trehalose, are frequently added to proteins before lyophilization. Trehalose is a natural α-linked glucose disaccharide, which stabilizes proteins during lyophilization through water retention near the protein structure via hydrogen bonding.
First, the primary alcohols of the various α-N-1,2-D-glc PASs (Mw = 4, 5, and 9 kDa) are oxidized to carboxylic acids by TEMPO-mediated oxidation to afford the PAS polymers of interest (Figure 44). On the basis of CD analysis, the polymers exhibit a helical structure, with a maximum at 191 nm and a minimum at 219 nm, which is maintained over a wide range of pH (pH = 2–8). Next, the activity of PASs as stabilizing agents against protein denaturation is evaluated using the enzyme lysozyme. A mixture of ox-PASs and lysozyme are subjected to 10 lyophilization cycles, and the activity of the protein is compared to its activity before lyophilization and after treatment with trehalose as the stabilizing agent. Proteins treated with the PAS or trehalose stabilizing agents performed better than the untreated control. Furthermore, ox-PASs exhibited higher performance compared to trehalose. The stabilizing effect of PASs may be due to the electrostatic interactions between the polymers and the protein and the formation of a complex as shown by gel electrophoresis.
7.5.3. Promotion/Inhibition of Biofilm Formation
The control of biofilm formation and subsequent growth or inhibition is of significant interest (e.g., in bioenergy and biomedical applications, respectively). Amphiphilic structures and polysaccharides are both known to influence the bacteria life cycle, and thus, Grinstaff et al. investigated PAS structures possessing one or two fatty acid chains as modulators of bacteria activity.308 Gal- and glc-derived PASs (DP = 10 and 20) functionalized with either one or two palmitamide hydrophobic chains at the tail of the polymer (Figures 44 and 45) are prepared where the hydrophilic headgroup contained either gal-derived homopolymers (Figure 45A) or random gal- and glc-derived copolymers (Figure 45B). Several of the PAS amphiphiles, exhibited stable suspensions in aqueous solutions and are thus candidates for evaluating their activity toward modulating the biofilm formation produced by P. aeruginosa strain PA14. The random copolymers (DP = 10 and 20) with one palmitamide chain (serie-1) inhibited biofilm formation by bacteria, whereas the gal-derived homopolymer (DP = 20) with two palmitamide chains (serie-2) promoted biofilm formation. The observation of biofilm inhibition suggests these types of polymers may have utility by themselves or in combination with other agents for the treatment of biofilm-associated infections. The amphiphilic nature of PASs and their capability to form supramolecular assemblies is essential for the observed bioactivity. The Serie-1 amphiphiles inhibited biofilm formation by P. aeruginosa via a similar mechanism to rhamnolipid surfactants, by lowering the surface tension of water. In contrast, the mechanism of biofilm promotion observed with serie-2 is not yet fully understood and it is hypothesized that the ordered rodlike aggregates formed with these polymers might promote this bioactivity.
Figure 45.

Schematic representation of PAS amphiphiles. Adapted from ref 308 with permission of The Royal Society of Chemistry.
Poly(amidosaccharide)s are novel synthetic carbohydrate-derived polymers linked by α-amide linkages, which exhibit structural similarities to natural polysaccharides. These enantiopure polymers are easily synthesized by ring-opening polymerization of a glucal/galactal-derived β-lactam monomer, in high yields, controlled molecular weights, and low dispersities. Poly(amidosaccharide)s exhibit biological properties similar to those of natural polysaccharides and thus can be considered as biomimetics of such polymers. Furthermore, they possess advantages over the natural version, such as the possibility to tune the chemical structure and molecular weight as well as to functionalize the structure for a given property and application.
7.6. Sugar-Derived Polycarbonates
Sugar-derived polycarbonates are recently reported by Wooley et al. through a controlled organocatalyzed ROP of a glucose-based six-membered bicylic carbonate monomer (Figure 46A).316 Use of the initiator 4-methylbenzyl alcohol (monomer/initiator ratio =15–51) and commercially available TBD (1–2 mol %) organocatalyst afforded amorphous poly(D-glucose carbonate)s with controlled Mn (2.3–14.7 kDa) and narrow Đ (1.11–1.16) values, as well as random head-to-head, head-to-tail, and tail-to-tail regiochemistries. The polymers exhibit high Td of 250–320 °C and a Tg of 106 °C (Mn = 2.3 kDa), as measured by TGA and DSC, respectively. The Tg value is significantly higher than the common aliphatic polycarbonates such as P(1,3-TMC)s which possess a Tg of −18 °C.
Figure 46.

Sugar-derived poly(carbonate)s formed by ring opening polymerization of (A) six-membered and (B) five-membered bicyclic carbonate monomers containing glucopyranoside structures. (A) Adapted with permission from ref 316. Copyright 2013 American Chemical Society. (B) Adapted with permission from ref 317. Copyright 2005 American Chemical Society.
Similarly, Endo et al. reported the anionic ROP of a five-membered cyclic glucopyranoside carbonate, with various initiators such as n-butyllithium (n-BuLi), lithium tert-butoxide (t-BuOLi), potassium tert-butoxide (t-BuOK), and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (Figure 46B).317,318 The polymerization proceeded smoothly at room temperature and afforded sugar-derived polycarbonates in good yields (70– 98%) and without polyether moieties. However, broader Đ values (1.6–2.08) with significant formation of cyclic polymer structures due to a backbiting reaction are observed as compared to the ROP of six-membered bicyclic carbonate monomers.
7.7. Glycopolymers
Polymers bearing pendant carbohydrates (i.e., glycopolymers) constitute another interesting strategy for the development of polysaccharide mimics (Figure 47). Glycopolymers can be obtained in a controlled fashion with variable lengths, architectures, and functionalities from (i) the polymerization of glycomonomers using free-radical polymerization (FRP), living anionic polymerization, ROP, ring-opening metathesis polymerization (ROMP), and controlled/living free-radical polymerization (CLRP) or (ii) the postfunctionalization of polymers with sugar moieties. Glycopolymers are being explored as glycan analogs in drug delivery, vaccine development, biosensing, and signal transduction processes, and there are several excellent reviews on the topic, which summarize their synthesis and biological applications.319–326 Therefore, in this section, we will briefly describe a couple of examples of synthetic glycopolymers, which illustrate their importance for the investigation of cell viability and function.
Figure 47.

Examples of glycopolymers developed by the Kiessling328 and Bertozzi groups.329
The Kiessling group has described a number of glycopolymers prepared via ROMP, as tools to study the molecular mechanisms behind carbohydrate-mediated signal transduction. 321 One example is the evaluation of various lengths of glycopolymers containing a 3,6-disulfogalactose ligand and a fluorescent dye as inhibitors of L-selectin, a leukocyte surface receptor. The authors showed that the glycopolymers bind to L-selectin at the surface of the cell with a receptor-to-ligand binding ratio that increases with increasing valency of the polymer, demonstrating receptor clustering.327 Additional investigations of sulfated neoglycopolymers (Figure 47) showed that the polymers are able to facilitate the down-regulation of L-selectin by triggering its shedding.328
The Bertozzi group described the synthesis of mucinlike glycopolymers with low dispersities (Đ < 1.15) through the reversible addition–fragmentation chain transfer (RAFT) polymerization of (2-oxopropyl)acrylate (Figure 47).329 The polymers were then arrayed on chips functionalized with azide groups, using the microcontact printing (μCP) technique followed by the copper(I)-catalyzed alkyne–azide cycloaddition (CuACC). The α and β forms of the printed glycopolymers were then incubated with Texas Red-Helix pomatia agglutinin (HPA), a lectin that recognizes α-N-acetylgalactosamine (α-GalNAc), and the protein bound selectively to the α-form of the polymer. This example highlights the specific interaction of these printed glycopolymers with glycan-binding proteins, based on their ligand specificity, which make them suitable for microarray applications.
7.8. Saccharide Synthon Summary
In comparison to the progress reported using hydroxy acid, dihydroxyacetone, and glycerol synthons to prepare polymers, the use of saccharide-based synthons is still in its infancy. Importantly, the diversity of structures and compositions present in carbohydrates provides a wealth of opportunities to create interesting and versatile synthons. An additional advantage (and at times one of the complexities) of working with carbohydrates is the multifunctionality present within the structure either in the ring-closed or open configuration giving rise to sites for additional modification or stereoselective presentation of functional groups to a biological target.
8. CONCLUSIONS AND OUTLOOK
Biomaterials are prominent in the surgical, medical device, and pharmaceutical industries, and perform various roles from structural materials to drug depots. The results and findings described in this review highlight the successful use of molecules derived from those found in natural metabolic pathways as synthons to create new polymers and biomaterials with unique functionalities. Some of the early biomaterials synthesized from metabolic synthons were linear polyesters based on lactic acid, glycolic acid, and 6-hydroxyhexanonic acid. The degradable sutures prepared from these polyesters formed the foundation for how molecularly engineered materials can benefit human health and improve patient outcomes. Interest in poly(hydroxy acid)s continues to grow with new compositions and synthetic routes being explored. For example, microbial synthesis of polyesters from nonpetrochemical feedstocks are an attractive alternative to the commercialization of “green” consumable biomaterials. Advances arising from CRISPRi technology23 are promising for further control of microbial synthetic pathways to produce more functional and inexpensive materials. The intersection of synthetic biology and polymer synthesis will yield new discoveries in the future.
Given the structural complexity, multifunctional groups present, and differing (or similar) reactivity between functional groups on certain metabolic synthons, a number of possibilities are available for synthesis of new polymers and for the formation of interesting polymer architectures. Carbohydrate-based synthons glycerol and glucose highlight this finding. Linear poly(1,3-glycerol carbonate)s are synthesized from cyclic 1,3-carbonates of glycerol with the secondary alcohol protected during the polymerization reaction. Alternatively, poly(1,2-glycerol carbonate)s are prepared by the coupling reaction between an epoxide (glycidyl ether) and CO2. Both polymers possess a pendant hydroxyl functional group for further derivitization, but poly(1,2-glycerol carbonate)s degrade more rapidly than poly(1,3-glycerol carbonate)s. Glycerol by itself or when paired with lactic acid or succinic acid can also be used to prepare dendrimers through both divergent (dendrimer synthesis from an initial core outward over several steps) and convergent synthesis (dendrimer synthesized via couplings of a dendron to core). These dendrimeric biomaterials are of interest for drug delivery and wound healing. Finally, glucose can be converted into a bicyclic structure possessing a β-lactam, and this monomer can be polymerized via anionic ring opening polymerization to afford chiral polysaccharide mimics, termed poly(amidosaccharide)s. Poly(amidosaccharide)s modulate biofilm formation and are of interest for biomedical, as well as bioenergy applications.
Polymers from the 3-membered ketose, dihydroxyacetone, represent another important milestone in the synthesis of biomaterials from metabolically derived synthons. Dihydroxyacetone is an example of where multifunctional reactivity of the monomeric building block is unfavorable. Unlike the multifunctional glycerol, dihydroxyacetone contains a highly reactive ketone that undergoes rapid rearrangement in the presence of amines. Various means of protecting/deprotecting, polymerizing, and copolymerizing this molecule to make biomaterials are reported by different groups to prepare well-characterized biomaterials. The monomer structure is surprisingly simple, yet synthetically challenging. It is an example of how metabolic synthons can bring new functionality to biomaterials and, at the same time, present interesting challenges to their realization.
Other examples of metabolic synthons discussed include linear polymers containing fumarate which allows a liquid-to-solid transition following cross-linking of the double bond (promising as scaffolds for tissue engineered bone), polycarbonates from modified tyrosine (that can be designed to coat and provide structural integrity to neuro-sensor leads and rapidly degrade following implantation into the brain), and poly(glycoamidoamine)s (commercialized as Glycofect for nucleic acid transfection in cell culture).
Significant opportunities still exist for the synthesis of new polymers, the development of biomaterials, and their evaluation in in vitro and in vivo models of diseases, conditions, or trauma. The clinical application space is large and the needs are great. Synthetic biomaterials prepared from metabolically derived synthons will play an important role in meeting such currently unmet needs as scaffold-based heart valves, accommodating intraocular hydrogel lens, seroma preventing adhesives, biodegradable lubricants for cartilage surfaces, absorbable bone fillers for large defects, and polymer conduits for nerve regeneration and growth. Moreover, some currently used biomaterials need to be improved. For example, although sutures have been used in the clinic for 40+ years, the number of new advances in suture design and composition have been few over the past decades, with one example being the discovery and report by Uhrich et al. of a polyasprin.330 Uhrich’s results highlight the fact that current biomaterials can be improved via further refinement of physical or mechanical properties or the introduction of a new feature such as drug delivery. These activities will continue to be driven by curiosity and basic science, as well as patient needs and industry interest.
Looking into the future, the metabolome is replete with complex and fascinating structures that, when harnessed, form well-defined and functional materials. As investigators continue to explore new chemistries, catalysts, and synthetic means of production, new and unique polymers and biomaterials will continue to address unmet needs in medicine. The performance benefits of these new materials in clinical applications are clear and are based on their composition and structure, as well as their physical and rheological properties. Our motivation for writing this review stems from our conviction that by molecularly engineering specific functions into new biomaterials (that, upon implantation, function as designed, degrade accordingly, and then are eliminated from the body via their natural metabolic pathways), we will meet the performance requirements of the next generation of biomaterials. The use of metabolically derived synthons is central to our approach.
Acknowledgments
The authors would like to thank the Cornell Center for Materials Research, the Cornell Center for Advanced Technology, Boston University, the Coulter Foundation, the Pew Foundation, the National Science Foundation, and National Institutes of Health (R01, R21, R56, and T32 mechanisms) for support of our research and educational programs.
Biographies
Nicole G. Ricapito received her B.S. degree in Chemical Engineering from Rensselaer Polytechnic Institute in 2010 where she performed undergraduate research under the guidance of Dr. Pankaj Karande. She is currently a Ph.D. candidate in the Department of Chemical and Biomolecular Engineering at Cornell University working under the direction of Dr. David Putnam. Her thesis research focuses on the design, synthesis, characterization, and evaluation of dihydroxyacetone- based polymers for use in biomedical applications.
Cynthia Ghobril obtained her engineering diploma in 2005 from the Ecole Européenne de Chimie, Polymères et Matériaux of Strasbourg, France. The same year she joined the group of Dr. Charles Mioskowski at the Université de Strasbourg for her graduate studies. Her research focused on the total synthesis of Nigellamine A, an active compound extracted from the plant Nigella Sativa. After obtaining her Ph.D. in November 2009, she began her postdoctoral fellowship under the direction of Dr. Delphine Felder-Flesch at the Institut de Physique et Chimie des Matériaux de Strasbourg, where she mainly worked on the development of dendritic iron oxide nanoparticles for multimodal imaging. In 2011, she joined the group of Professor Mark Grinstaff at Boston University as a postdoctoral fellow, and her research focuses on the design and development of new dendritic polymers and biomaterials.
Mr. Heng Zhang is a Ph.D. candidate in the Department of Chemistry at Boston University. He received his B.S. in Pharmaceutics from Nanjing University of Technology in 2009 and M.S. in Pharmacy from University of Iowa in 2011. Since joining Professor Mark Grinstaff’s group, his research interests have focused on the design and synthesis of novel biocompatible and biodegradable polymeric materials based on glycerol for biomedical applications, as well as organometallic chemistry and polymerization methods.
Mark W. Grinstaff is the Distinguished Professor of Translational Research and Professor of Biomedical Engineering, Chemistry, and Materials Science and Engineering, and Medicine at Boston University. He is also the Director of the NIH T32 Program in Translational Research in Biomaterials. Mark’s awards include the ACS Nobel Laureate Signature Award, NSF Career Award, Pew Scholar in the Biomedical Sciences, Camille Dreyfus Teacher-Scholar, Alfred P. Sloan Research Fellowship, the Edward M. Kennedy Award for Health Care Innovation, a Fellow of the American Academy of Nanomedicine, a fellow of AIMBE, and a Charter Fellow of the National Academy of Inventors. He has published more than 200 peer-reviewed manuscripts. He is a cofounder of four companies that are commercializing his ideas, and he has three products being sold and used in the clinic. His current research activities involve the synthesis of new macromolecules and amphiphiles, self-assembly chemistry, drug delivery, wound repair, and contrast agents for medical imaging (http://people.bu.edu/mgrin/).
David Putnam is an Associate Professor in both the Meinig School of Biomedical Engineering and the School of Chemical and Biomolecular Engineering at Cornell University. His graduate training was under Professor Jindrich Kopecek at the University of Utah, and his postdoctoral training was under Professor Robert Langer at MIT as an NIH Fellow. He was a Scientific co-Founder of TransForm Pharmaceuticals, Inc, which was acquired by Johnson & Johnson in March, 2005. In 2008–2009, he was an Entrepreneur-in-Residence at PureTech Ventures in Boston, MA, where he focused on emerging technologies in the field of drug delivery. He currently serves as CEO of Articulate Biomedical, LLC, a start-up biomaterials company located in Ithaca, NY. He is a member of several Editorial Advisory Boards including Advanced Drug Delivery Reviews, Pharmaceutical Research, Journal of Controlled Release, Biomacromolecules, ACS Biomaterials Science & Engineering, and the Journal of Applied Polymer Science. His funding sources include NIH, NSF, Found Animals Foundation, Total, Inc., and New York State. He is a Fellow of AIMBE and the Coulter Foundation.
Footnotes
Notes
The authors declare the following competing financial interest(s): M.W.G. is the scientific co-founder of HyperBranch Medical Technology, AcuityBio, and Affinergy which are or have developed new biomaterials for clinical use, and thus, he has a conflict of interest. D.P. in the founder of Articulate Biomedical, LLC, which has biomaterials in pre-clinical trials and, thus, has a potential conflict of interest.
References
- 1.Frazza EJ, Schmitt EE. A New Absorbable Suture. J Biomed Mater Res. 1971;5:43–58. doi: 10.1002/jbm.820050207. [DOI] [PubMed] [Google Scholar]
- 2.Schmitt EE, Albert R. Surgical Sutures. 3297033. US Patent. 1967
- 3.Greenberg JA, Clark RM. Advances in Suture Material for Obstetric and Gynecologic Surgery. Rev Obstet Gynecol. 2009;2:146–158. [PMC free article] [PubMed] [Google Scholar]
- 4.Weldon CB, Tsui JH, Shankarappa SA, Nguyen VT, Ma M, Anderson DG, Kohane DS. Electrospun Drug-Eluting Sutures for Local Anesthesia. J Controlled Release. 2012;161:903–909. doi: 10.1016/j.jconrel.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright JC, Hoffman AS. Historical Overview of Long Acting Injections and Implants. In: Wright JC, Burgess DJ, editors. Long Acting Injections and Implants. Advances in Delivery Science and Technology. Springer; Dordrecht, Heidelberg, London, NY: 2012. pp. 11–24. [Google Scholar]
- 6.Lo CT, van Tassel PR, Saltzman WM. Biodegradable Poly(lactide-Co-Glycolide) Nanoparticle Assembly for Continuous Release of Bioactive Agents from Medical Devices. Biomaterials. 2010;31:3631–3642. doi: 10.1016/j.biomaterials.2010.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dash TK, Konkimalla VB. Poly-ε-Caprolactone Based Formulations for Drug Delivery and Tissue Engineering: A Review. J Controlled Release. 2012;158:15–33. doi: 10.1016/j.jconrel.2011.09.064. [DOI] [PubMed] [Google Scholar]
- 8.Makadia HK, Siegel SJ. Poly Lactic-Co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel, Switz) 2011;3:1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gollwitzer H, Thomas P, Diehl P, Steinhauser E, Summer B, Barnstorf S, Gerdesmeyer L, Mittelmeier W, Stemberger A. Biomechanical and Allergological Characteristics of a Biodegradable poly(D, L-Lactic Acid) Coating for Orthopaedic Implants. J Orthop Res. 2005;23:802–809. doi: 10.1016/j.orthres.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Mano JF, Sousa RA, Boesel LF, Neves NM, Reis RL. Bioinert, Biodegradable and Injectable Polymeric Matrix Composites for Hard Tissue Replacement: State of the Art and Recent Developments. Compos Sci Technol. 2004;64:789–817. [Google Scholar]
- 11.Pratt C, Voet D, Voet J. Fundamentals of Biochemistry: Life at the Molecular Level. 3. John Wiley & Sons; Hoboken, NJ: 2008. [Google Scholar]
- 12.Shinno K, Miyamoto M, Kimura Y, Hirai Y, Yoshitome H. Solid-State Postpolymerization of L-Lactide Promoted by Crystallization of Product Polymer: An Effective Method for Reduction of Remaining Monomer. Macromolecules. 1997;30:6438–6444. [Google Scholar]
- 13.Moon SIL, Lee CW, Miyamoto M, Kimura Y. Melt Polycondensation of L-Lactic Acid with Sn(II) Catalysts Activated by Various Proton Acids: A Direct Manufacturing Route to High Molecular Weight Poly(L-Lactic Acid) J Polym Sci, Part A: Polym Chem. 2000;38:1673–1679. [Google Scholar]
- 14.Ajioka M, Enomoto K, Suzuki K, Yamaguchi A. The Basic Properties of Poly(lactic Acid) Produced by the Direct Condensation Polymerisation of Lactic Acid. J Environ Polym Degrad. 1995;3:225– 234. [Google Scholar]
- 15.Gupta AP, Kumar V. New Emerging Trends in Synthetic Biodegradable Polymers - Polylactide: A Critique. Eur Polym J. 2007;43:4053–4074. [Google Scholar]
- 16.Groot W, van Krieken J, Sliekersl O, de Vos S. Production and Purification of Lactic Acid and Lactide. In: Auras R, Lim L-T, Selke SEM, Tsuji H, editors. Poly(Lactic Acid): Synthesis, Structures, Properties, Processing, and Applications. John Wiley & Sons; Hoboken, NJ, USA: 2010. pp. 1–18. [Google Scholar]
- 17.Anderson AJ, Dawes EA. Occurrence, Metabolism, Metabolic Role, and Industrial Uses of Bacterial Polyhydroxyalkanoates. Microbiol Rev. 1990;54:450–472. doi: 10.1128/mr.54.4.450-472.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snell KD, Singh V, Brumbley SM. Production of Novel Biopolymers in Plants: Recent Technological Advances and Future Prospects. Curr Opin Biotechnol. 2015;32:68–75. doi: 10.1016/j.copbio.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 19.Bugnicourt E, Cinelli P, Lazzeri A, Alvarez V. Polyhydroxyalkanoate (PHA): Review of Synthesis, Characteristics, Processing and Potential Applications in Packaging. eXPRESS Polym Lett. 2014;8:791–808. [Google Scholar]
- 20.Misra SK, Valappil SP, Roy I, Boccaccini AR. Polyhydroxyalkanoate (PHA)/Inorganic Phase Composites for Tissue Engineering Applications. Biomacromolecules. 2006;7:2249–2258. doi: 10.1021/bm060317c. [DOI] [PubMed] [Google Scholar]
- 21.Masood F, Chen P, Yasin T, Hasan F, Ahmad B, Hameed A. Synthesis of Poly-(3-Hydroxybutyrate-Co-12 Mol % 3-Hydroxyvalerate) by Bacillus Cereus FB11: Its Characterization and Application as a Drug Carrier. J Mater Sci: Mater Med. 2013;24:1927–1937. doi: 10.1007/s10856-013-4946-x. [DOI] [PubMed] [Google Scholar]
- 22.Wu Q, Wang Y, Chen GQ. Medical Application of Microbial Biopolyesters Polyhydroxyalkanoates. Artif Cells, Blood Substitutes, Imobilization Biotechnol. 2009;37:1–12. doi: 10.1080/10731190802664429. [DOI] [PubMed] [Google Scholar]
- 23.Lv L, Ren Y, Chen J, Wu Q, Chen G. Application of CRISPRi for Prokaryotic Metabolic Engineering Involving Multiple Genes, A Case Study: Controllable P (3HB-Co-4HB) Biosynthesis. Metab Eng. 2015;29:160–168. doi: 10.1016/j.ymben.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 24.Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-Jebria A, Eskew M, Lou, Mintzes J, Deaver D, Lotan N, Langer R. Large Porous Particles for Pulmonary Drug Delivery. Science. 1997;276:1868–1872. doi: 10.1126/science.276.5320.1868. [DOI] [PubMed] [Google Scholar]
- 25.Kwon DY, Kim JI, Kim DY, Kang HJ, Lee B, Lee KW, Kim MS. Biodegradable Stent. J Biomed Sci Eng. 2012;05:208–216. [Google Scholar]
- 26.Esfand R, Tomalia DA. Poly(amidoamine) (PAMAM) Dendrimers: From Biomimicry to Drug Delivery and Biomedical Applications. Drug Discovery Today. 2001;6:427–436. doi: 10.1016/s1359-6446(01)01757-3. [DOI] [PubMed] [Google Scholar]
- 27.Grayson SM, Fréchet JMJ. Convergent Dendrons and Dendrimers: From Synthesis to Applications. Chem Rev. 2001;101:3819–3867. doi: 10.1021/cr990116h. [DOI] [PubMed] [Google Scholar]
- 28.Majoral JP, Caminade AM. Dendrimers Containing Heteroatoms (Si, P, B, Ge, or Bi) Chem Rev. 1999;99:845–880. doi: 10.1021/cr970414j. [DOI] [PubMed] [Google Scholar]
- 29.Mintzer MA, Grinstaff MW. Biomedical Applications of Dendrimers: A Tutorial. Chem Soc Rev. 2011;40:173–190. doi: 10.1039/b901839p. [DOI] [PubMed] [Google Scholar]
- 30.Newkome GR, He E, Moorefield CN. Supra-supermolecules with Novel Properties: Metallodendrimers. Chem Rev. 1999;99:1689–1746. doi: 10.1021/cr9800659. [DOI] [PubMed] [Google Scholar]
- 31.Medina SH, El-Sayed MEH. Dendrimers as Carriers for Delivery of Chemotherapeutic Agents. Chem Rev. 2009;109:3141– 3157. doi: 10.1021/cr900174j. [DOI] [PubMed] [Google Scholar]
- 32.Cheng Y, Zhao L, Li Y, Xu T. Design of Biocompatible Dendrimers for Cancer Diagnosis and Therapy: Current Status and Future Perspectives. Chem Soc Rev. 2011;40:2673–2703. doi: 10.1039/c0cs00097c. [DOI] [PubMed] [Google Scholar]
- 33.Grinstaff MW. Biodendrimers: New Polymeric Biomaterials for Tissue Engineering. Chem –Eur J. 2002;8:2838–2846. [PubMed] [Google Scholar]
- 34.Grinstaff MW. Designing Hydrogel Adhesives for Corneal Wound Repair. Biomaterials. 2007;28:5205–5214. doi: 10.1016/j.biomaterials.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grinstaff MW. Dendritic Macromers for Hydrogel Formation: Tailored Materials for Ophthalmic, Orthopedic, and Biotech Applications. J Polym Sci, Part A: Polym Chem. 2008;46:383–400. [Google Scholar]
- 36.Twibanire JK, Grindley TB. Polyester Dendrimers: Smart Carriers for Drug Delivery. Polymers (Basel, Switz) 2014;6:179–213. [Google Scholar]
- 37.Wolinsky JB, Grinstaff MW. Therapeutic and Diagnostic Applications of Dendrimers for Cancer Treatment. Adv Drug Delivery Rev. 2008;60:1037–1055. doi: 10.1016/j.addr.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 38.Carnahan MA, Grinstaff MW. Synthesis and Characterization of Polyether-Ester Dendrimers from Glycerol and Lactic Acid. J Am Chem Soc. 2001;123:2905–2906. doi: 10.1021/ja005726+. [DOI] [PubMed] [Google Scholar]
- 39.Namazi H, Adeli M. Novel Linear–Globular Thermoreversible Hydrogel ABA Type Copolymers from Dendritic Citric Acid as the A Blocks and Poly(ethyleneglycol) as the B Block. Eur Polym J. 2003;39:1491–1500. [Google Scholar]
- 40.Namazi H, Adeli M. Dendrimers of Citric Acid and Poly(ethylene Glycol) as the New Drug-Delivery Agents. Biomaterials. 2005;26:1175–1183. doi: 10.1016/j.biomaterials.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 41.Haddleton DM, Sahota HS, Taylor PC, Yeates SG. Synthesis of Polyester Dendrimers. J Chem Soc, Perkin Trans. 1996;1:649–656. [Google Scholar]
- 42.Hawker CJ, Frechet J, MJ Unusual Macromolecular Architectures: The Convergent Growth Approach to Dendritic Polyesters and Novel Block Copolymers. J Am Chem Soc. 1992;114:8405–8413. [Google Scholar]
- 43.Hawker CJ, Fréchet JMJ. Monodispersed Dendritic Polyesters with Removable Chain Ends: A Versatile Approach to Globular Macromolecules with Chemically Reversible Polarities. J Chem Soc, Perkin Trans. 1992;1:2459–2469. [Google Scholar]
- 44.Twibanire JK, Grindley TB. Polyester Dendrimers. Polymers (Basel Switz) 2012;4:794–879. [Google Scholar]
- 45.Do JY, Ju JJ. Polyester Dendrimers Carrying NLO Chromophores: Synthesis and Optical Characterization. Macromol Chem Phys. 2005;206:1326–1331. [Google Scholar]
- 46.Do JY, Park SK, Ju JJ, Park S, Lee MH. Electro-Optic Materials: Hyperbranched Chromophores Attached Linear Polyimide and Dendritic Polyesters. Polym Adv Technol. 2005;16:221–226. [Google Scholar]
- 47.Seebach D, Herrmann GF, Lengweiler UD, Bachmann BM, Amrein W. Synthesis and Enzymatic Degradation of Dendrimers from (R)-3-Hydroxybutanoic Acid and Trimesic Acid. Angew Chem, Int Ed Engl. 1996;35:2795–2797. [Google Scholar]
- 48.Gao C, Yan D. Hyperbranched Polymers: From Synthesis to Applications. Prog Polym Sci. 2004;29:183–275. [Google Scholar]
- 49.Jin H, Huang W, Zhu X, Zhou Y, Yan D. Biocompatible or Biodegradable Hyperbranched Polymers: From Self-Assembly to Cytomimetic Applications. Chem Soc Rev. 2012;41:5986–5997. doi: 10.1039/c2cs35130g. [DOI] [PubMed] [Google Scholar]
- 50.Wang D, Zhao T, Zhu X, Yan D, Wang W. Bioapplications of Hyperbranched Polymers. Chem Soc Rev. 2015;44:4023–4071. doi: 10.1039/c4cs00229f. [DOI] [PubMed] [Google Scholar]
- 51.Zhu Q, Qiu F, Zhu B, Zhu X. Hyperbranched Polymers for Bioimaging. RSC Adv. 2013;3:2071–2083. [Google Scholar]
- 52.Žagar E, Žigon M. Aliphatic Hyperbranched Polyesters Based on 2,2-Bis(methylol)propionic Acid-Determination of Structure, Solution and Bulk Properties. Prog Polym Sci. 2011;36:53–88. [Google Scholar]
- 53.Zhang X. Hyperbranched Aromatic Polyesters: From Synthesis to Applications. Prog Org Coat. 2010;69:295–309. [Google Scholar]
- 54.Zhang X. Modifications and Applications of Hyperbranched Aliphatic Polyesters Based on Dimethylolpropionic Acid. Polym Int. 2011;60:153–166. [Google Scholar]
- 55.Reul R, Nguyen J, Kissel T. Amine-Modified Hyperbranched Polyesters as Non-Toxic, Biodegradable Gene Delivery Systems. Biomaterials. 2009;30:5815–5824. doi: 10.1016/j.biomaterials.2009.06.057. [DOI] [PubMed] [Google Scholar]
- 56.Flory PJ. Molecular Size Distribution in Three Dimensional Polymers. VI. Branched Polymers Containing A—R—Bf-1 Type Units. J Am Chem Soc. 1952;74:2718–2723. [Google Scholar]
- 57.Frechet JMJ, Hawker CJ. Hyperbranched Polyphenylene and Hyperbranched Polyesters: New Soluble, Three-Dimensional, Reactive Polymers. React Funct Polym. 1995;26:127–136. [Google Scholar]
- 58.Hawker CJ, Lee R, Fréchet JMJ. One-Step Synthesis of Hyperbranched Dendritic Polyesters. J Am Chem Soc. 1991;113:4583–4588. [Google Scholar]
- 59.Wooley KL, Hawker CJ, Lee R, Fréchet JMJ. One-Step Synthesis of Hyperbranched Polyesters. Molecular Weight Control and Chain End Functionalization. Polym J. 1994;26:187–197. [Google Scholar]
- 60.Turner SR, Voit BI, Mourey TH. All-Aromatic Hyperbranched Polyesters with Phenol and Acetate End Groups: Synthesis and Characterization. Macromolecules. 1993;26:4617–4623. [Google Scholar]
- 61.Kricheldorft HR, Stoeber O, Luebbers D. New Polymer Syntheses. 78. Star-Shaped and Hyperbranched Polyesters by Polycondensation of Trimethylsilyl 3, 5-Diacetoxybenzoate. Macromolecules. 1995;28:2118–2123. [Google Scholar]
- 62.Hawker CJ, Chu F, Pomery PJ, Hill DJT. Hyperbranched Poly(ethylene Glycol)s: A New Class of Ion-Conducting Materials. Macromolecules. 1996;29:3831–3838. [Google Scholar]
- 63.Kumar A, Ramakrishnan S. Structural Variants of Hyperbranched Polyesters. Macromolecules. 1996;29:2524–2530. [Google Scholar]
- 64.Rabinowitch IM. Observations on the Use of Dihydroxyacetone in the Treatment of Diabetes Mellitus. Can Med Assoc J. 1925;15:374–381. [PMC free article] [PubMed] [Google Scholar]
- 65.Ringer AI, Frankel EM. The Chemistry of Gluconeogenesis: IX. The Formation of Glucose in the Diabetic Organism. J Biol Chem. 1914;18:233–236. [Google Scholar]
- 66.Macdermot H. Dioxyacetone. Can Med Assoc J. 1925;15:412. [PMC free article] [PubMed] [Google Scholar]
- 67.Rabinowitch IM. Blood Sugar Time Curves Following the Ingestion of Dihydroxyacetone. J Biol Chem. 1925;65:55–58. [Google Scholar]
- 68.Rabinowitch IM. A Case of Diabetic Coma Treated with Dioxyacetone, with Recovery. Can Med Assoc J. 1925;15:520–522. [PMC free article] [PubMed] [Google Scholar]
- 69.Goldman L, Barkoff J, Blaney D, Nakai T, Suskind R. Investigative Studies with the Skin Coloring Agents Dihydroxyacetone and Glyoxal. J Invest Dermatol. 1960;35:161–164. doi: 10.1038/jid.1960.100. [DOI] [PubMed] [Google Scholar]
- 70.Maillard LC. Action des acides amines sur les sucres: formation des melanoidines par voie methodique (English translation: Action of amino acids on sugars. Formation of melanoidins in a methodical way) Compt Rend Hebd Seances Acad Sci. 1912;154:66– 68. [Google Scholar]
- 71.Hodge JE. Chemistry of Browning Reactions in Model Systems. J Agric Food Chem. 1953;1:928–943. [Google Scholar]
- 72.Wittgenstein E, Berry HK. Staining of Skin with Dihydroxyacetone. Science. 1960;132:894–895. doi: 10.1126/science.132.3431.894. [DOI] [PubMed] [Google Scholar]
- 73.Wittgenstein EVA, Berry HK. Reaction of Dihydroxyacetone (DHA) with Human Skin Callus and Amino Compounds. J Invest Dermatol. 1961;36:283–286. doi: 10.1038/jid.1961.46. [DOI] [PubMed] [Google Scholar]
- 74.Fu JM, Dusza SW, Halpern AC. Sunless Tanning. J Am Acad Dermatol. 2004;50:706–713. doi: 10.1016/j.jaad.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 75.Petersen AB, Wulf HC, Gniadecki R, Gajkowska B. Dihydroxyacetone, the Active Browning Ingredient in Sunless Tanning Lotions, Induces DNA Damage, Cell-Cycle Block and Apoptosis in Cultured HaCaT Keratinocytes. Mutat Res, Genet Toxicol Environ Mutagen. 2004;560:173–186. doi: 10.1016/j.mrgentox.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 76.Jung K, Seifert M, Herrling T, Fuchs J. UV-Generated Free Radicals (FR) in Skin: Their Prevention by Sunscreens and Their Induction by Self-Tanning Agents. Spectrochim Acta, Part A. 2008;69:1423–1428. doi: 10.1016/j.saa.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 77.Pagoto S. Sunless Tanning. In: Heckman CJ, Manne SL, editors. Shedding Light on Indoor Tanning. Springer; Dordrecht: 2012. pp. 165–178. [Google Scholar]
- 78.O’Leary; RE, Diehl J, Levins PC. Update on Tanning: More Risks Fewer Benefits. J Am Acad Dermatol. 2014;70:562–568. doi: 10.1016/j.jaad.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 79.Sanner T. Opinion on Dihydroxyacetone; Scientific Committee on Consumer Safety; December 14, 2010; pp. 1–35. SCCS/1347/10. [Google Scholar]
- 80.Taylor C, Kwangsukstith C, Wimberly J, Kollias N, Anderson RR. Turbo-PUVA: Dihydroxyacetone-Enhanced Photochemotherapy for Psoriasis. Arch Dermatol. 1999;135:540–544. doi: 10.1001/archderm.135.5.540. [DOI] [PubMed] [Google Scholar]
- 81.Lentini PJ. Compositions Useful in the Phototherapeutic Treatment of Proliferative Skin Disorders. 5,885,557. US Patent. 1999
- 82.Rajatanavin N, Suwanachote S, Kulkollakarn S. Dihydroxyacetone: A Safe Camouflaging Option in Vitiligo. Int J Dermatol. 2008;47:402–406. doi: 10.1111/j.1365-4632.2008.03356.x. [DOI] [PubMed] [Google Scholar]
- 83.Fesq H, Brockow K, Strom K, Mempel M, Ring J, Abeck D. Dihydroxyacetone in a New Formulation - A Powerful Therapeutic Option in Vitiligo. Dermatology. 2001;203:241–243. doi: 10.1159/000051757. [DOI] [PubMed] [Google Scholar]
- 84.Quintana RP, Garson LR, Lasslo A, Sanders SI, Buckner JH, Couck HK, Gilbert IH, Weidhaas DE, Schreck CE. Mosquito-Repellent Dihydroxyacetone Monoesters. J Econ Entomol. 1970;63:1128–1131. doi: 10.1093/jee/63.4.1128. [DOI] [PubMed] [Google Scholar]
- 85.Stanko RT, Robertson RJ, Galbreath RW, Reilly JJ, Greenawalt KD, Goss FL. Enhanced Leg Exercise Endurance with a High-Carbohydrate Diet and Dihydroxyacetone and Pyruvate. J Appl Physiol. 1990;69:1651–1656. doi: 10.1152/jappl.1990.69.5.1651. [DOI] [PubMed] [Google Scholar]
- 86.Niknahad H, Ghelichkhani E. Antagonism of Cyanide Poisoning by Dihydroxyacetone. Toxicol Lett. 2002;132:95–100. doi: 10.1016/s0378-4274(02)00016-4. [DOI] [PubMed] [Google Scholar]
- 87.Turner BB, Hulpieu HR. A Study of the Antidotal Action of Sodium Thiosulphate and Dihydroxyacetone in Cyanide Poisoning, and the Alleged Anti-Dotal Action of Glucose. J Pharmacol Exp Ther. 1933;48:445–469. [Google Scholar]
- 88.Erni B, Siebold C, Christen S, Srinivas A, Oberholzer A, Baumann U. Small Substrate, Big Surprise: Fold, Function and Phylogeny of Dihydroxyacetone Kinases. Cell Mol Life Sci. 2006;63:890–900. doi: 10.1007/s00018-005-5518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Akar A, Talinli N. Synthesis of a Polyspiroacetal. Makromol Chem Rapid Commun. 1989;10:127–130. [Google Scholar]
- 90.Davis L. The Structure of Dihydroxyacetone in Solution. Bioorg Chem. 1973;2:197–201. [Google Scholar]
- 91.Slepokura K, Lis T. Crystal Structures of Dihydroxyacetone and Its Derivatives. Carbohydr Res. 2004;339:1995–2007. doi: 10.1016/j.carres.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 92.Reynolds SJ, Yates DW, Pogson CI. Dihydroxyacetone Phosphate. Its Structure and Reactivity with Alpha-Glycerophosphate Dehydrogenase, Aldolase and Triose Phosphate Isomerase and Some Possible Metabolic Implications. Biochem J. 1971;122:285–297. doi: 10.1042/bj1220285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zelikin AN, Zawaneh PN, Putnam D. A Functionalizable Biomaterial Based on Dihydroxyacetone, an Intermediate of Glucose Metabolism. Biomacromolecules. 2006;7:3239–3244. doi: 10.1021/bm060544e. [DOI] [PubMed] [Google Scholar]
- 94.Grazi E, Rowley PT, Cheng T, Tchola O, Horecker BL. The Mechanism of Action of Aldolases III. Schiff Base Formation with Lysine. Biochem Biophys Res Commun. 1962;9:38–43. doi: 10.1016/0006-291x(62)90083-9. [DOI] [PubMed] [Google Scholar]
- 95.Jia J, Schorken U, Lindqvist Y, Sprenger GA, Schneider G. Crystal Structure of the Reduced Schiff-Base Intermediate Complex of Transaldolase B from Escherichia Coli: Mechanistic Implications for Class I Aldolases. Protein Sci. 1997;6:119–124. doi: 10.1002/pro.5560060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carnali JO, Madison SA, Shah P, Qiu Q. Structure/Property Relationship for Ethylenediamine Derivatives as Aids in Sunless Tanning. Ind Eng Chem Res. 2012;51:15573–15581. [Google Scholar]
- 97.Jia Y, Li J. Molecular Assembly of Schiff Base Interactions: Construction and Application. Chem Rev. 2015;115:1597–1621. doi: 10.1021/cr400559g. [DOI] [PubMed] [Google Scholar]
- 98.Nguyen BC, Kochevar IE. Influence of Hydration on Dihydroxyacetone-Induced Pigmentation of Stratum Corneum. J Invest Dermatol. 2003;120:655–661. doi: 10.1046/j.1523-1747.2003.12089.x. [DOI] [PubMed] [Google Scholar]
- 99.Johnson JA, Fusaro RM. Alteration of Skin Surface Protein with Dihydroxyacetone: A Useful Application of the Maillard Browning Reaction. In: Labuza TP, Reineccius GA, Monnier VM, O’Brien J, Baynes JW, editors. Maillard Reactions in Chemistry, Food, and Health. The Royal Society of Chemistry; Cambridge: 1994. pp. 114–119. [Google Scholar]
- 100.van Boekel MAJS. Kinetic Aspects of the Maillard Reaction: A Critical Review. Nahrung. 2001;45:150–159. doi: 10.1002/1521-3803(20010601)45:3<150::AID-FOOD150>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 101.Martins SIFS, Jongen WMF, van Boekel MAJS. A Review of Maillard Reaction in Food and Implications to Kinetic Modelling. Trends Food Sci Technol. 2000;11:364–373. [Google Scholar]
- 102.Nuyken O, Pask S. Ring-Opening Polymerization—An Introductory Review. Polymers (Basel, Switz) 2013;5:361–403. [Google Scholar]
- 103.Lee ALZ, Ng VWL, Gao S, Hedrick JL, Yang YY. Injectable Hydrogels from Triblock Copolymers of Vitamin E-Functionalized Polycarbonate and Poly(ethylene Glycol) for Subcutaneous Delivery of Antibodies for Cancer Therapy. Adv Funct Mater. 2014;24:1538–1550. [Google Scholar]
- 104.Stevens DM, Rahalkar A, Spears B, Gilmore K, Douglas E, Muthukumar M, Harth E. Semibranched Polyglycidols as “fillers” in Polycarbonate Hydrogels to Tune Hydrophobic Drug Release. Polym Chem. 2015;6:1096–1102. [Google Scholar]
- 105.Lee ALZ, Ng VWL, Gao S, Hedrick JL, Yang YY. Injectable Biodegradable Hydrogels from Vitamin D-Functionalized Polycarbonates for the Delivery of Avastin with Enhanced Therapeutic Efficiency against Metastatic Colorectal Cancer. Biomacromolecules. 2015;16:465–475. doi: 10.1021/bm5015206. [DOI] [PubMed] [Google Scholar]
- 106.Jiang T, Li YM, Lv Y, Cheng YJ, He F, Zhuo RX. Amphiphilic Polycarbonate Conjugates of Doxorubicin with pH-Sensitive Hydrazone Linker for Controlled Release. Colloids Surf, B. 2013;111:542–548. doi: 10.1016/j.colsurfb.2013.06.054. [DOI] [PubMed] [Google Scholar]
- 107.Ke X, Coady DJ, Yang C, Engler AC, Hedrick JL, Yang YY. pH-Sensitive Polycarbonate Micelles for Enhanced Intracellular Release of Anticancer Drugs: A Strategy to Circumvent Multidrug Resistance. Polym Chem. 2014;5:2621–2628. [Google Scholar]
- 108.Magno MHR, Kim J, Srinivasan A, McBride S, Bolikal D, Darr A, Hollinger JO, Kohn J. Synthesis, Degradation and Biocompatibility of Tyrosine-Derived Polycarbonate Scaffolds. J Mater Chem. 2010;20:8885–8893. [Google Scholar]
- 109.Kim J, Magno MHR, Waters H, Doll BA, McBride S, Alvarez P, Darr A, Vasanji A, Kohn J, Hollinger JO. Bone Regeneration in a Rabbit Critical-Sized Calvarial Model Using Tyrosine-Derived Polycarbonate Scaffolds. Tissue Eng, Part A. 2012;18:1132–1139. doi: 10.1089/ten.TEA.2011.0582. [DOI] [PubMed] [Google Scholar]
- 110.Xu J, Filion TM, Prifti F, Song J. Cytocompatible Poly(ethylene Glycol)-Co-Polycarbonate Hydrogels Crosslinked by Copper-Free, Strain-Promoted “Click” Chemistry. Chem - Asian J. 2011;6:2730–2737. doi: 10.1002/asia.201100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu J, Feng E, Song J. Renaissance of Aliphatic Polycarbonates: New Techniques and Biomedical Applications. J Appl Polym Sci. 2014;131:39822. doi: 10.1002/app.39822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feng J, Zhuo RX, Zhang XZ. Construction of Functional Aliphatic Polycarbonates for Biomedical Applications. Prog Polym Sci. 2012;37:211–236. [Google Scholar]
- 113.Wang LS, Jiang XS, Wang H, Cheng SX, Zhuo RX. Preparation and Cytotoxicity of Novel Aliphatic Polycarbonate Synthesized from Dihydroxyacetone. Chin Chem Lett. 2005;16:572–574. [Google Scholar]
- 114.Wang LS, Cheng SX, Zhuo RX. Novel Biodegradable Aliphatic Polycarbonate Based on Ketal Protected Dihydroxyacetone. Macromol Rapid Commun. 2004;25:959–963. [Google Scholar]
- 115.Cesarotti E, Antognazza P, Pallavicini M, Villa L. Synthesis and Ruthenium-Catalyzed Enantioselective Hydrogenation of 3-OSubstituted 1,3-Dihydroxypropan-2-Ones. Helv Chim Acta. 1993;76:2344–2349. [Google Scholar]
- 116.Ferroni EL, DiTella V, Ghanayem N, Jeske R, Jodlowski C, O’Connell M, Styrsky J, Svoboda R, Venkataraman A, Winkler BM. A Three-Step Preparation of Dihydroxyacetone Phosphate Dimethyl Acetal. J Org Chem. 1999;64:4943–4945. doi: 10.1021/jo9823902. [DOI] [PubMed] [Google Scholar]
- 117.Wang LS, Cheng SX, Zhuo RX. Synthesis and Hydrolytic Degradation of Aliphatic Polycarbonate Based on Dihydroxyacetone. Polym Sci Ser B. 2013;55:604–610. [Google Scholar]
- 118.Pearson DM, Conley NR, Waymouth RM. Palladium-Catalyzed Carbonylation of Diols to Cyclic Carbonates. Adv Synth Catal. 2011;353:3007–3013. [Google Scholar]
- 119.Helou M, Brusson JM, Carpentier JF, Guillaume SM. Functionalized Polycarbonates from Dihydroxyacetone: Insights into the Immortal Ring-Opening Polymerization of 2,2-Dimethoxytrimethylene Carbonate. Polym Chem. 2011;2:2789–2795. [Google Scholar]
- 120.Helou M, Miserque O, Brusson JM, Carpentier JF, Guillaume SM. Organocatalysts for the Controlled “Immortal” Ring-Opening Polymerization of Six-Membered-Ring Cyclic Carbonates: A Metal-Free, Green Process. Chem - Eur J. 2010;16:13805–13813. doi: 10.1002/chem.201001111. [DOI] [PubMed] [Google Scholar]
- 121.Simon J, Olsson JV, Kim H, Tenney IF, Waymouth RM. Semicrystalline Dihydroxyacetone Copolymers Derived from Glycerol. Macromolecules. 2012;45:9275–9281. [Google Scholar]
- 122.Zawaneh PN, Doody AM, Zelikin AN, Putnam D. Diblock Copolymers Based on Dihydroxyacetone and Ethylene Glycol: Synthesis, Characterization, and Nanoparticle Formulation. Biomacromolecules. 2006;7:3245–3251. doi: 10.1021/bm0605457. [DOI] [PubMed] [Google Scholar]
- 123.Weiser JR, Zawaneh PN, Putnam D. Poly(carbonate-Ester)s of Dihydroxyacetone and Lactic Acid as Potential Biomaterials. Biomacromolecules. 2011;12:977–986. doi: 10.1021/bm101342p. [DOI] [PubMed] [Google Scholar]
- 124.Guerin W, Helou M, Slawinski M, Brusson JM, Carpentier JF, Guillaume SM. Macromolecular Engineering via Ring-Opening Polymerization (3): Trimethylene Carbonate Block Copolymers Derived from Glycerol. Polym Chem. 2014;5:1229– 1240. [Google Scholar]
- 125.Wang L, Cheng S, Zhuo R. Syntheses and Properties of Novel Copolymers of Polylactide and Aliphatic Polycarbonate Based on Ketal-Protected Dihydroxyacetone. Polym-Plast Technol Eng. 2013;52:1063–1067. [Google Scholar]
- 126.Wang L, Cheng S, Zhuo R. Syntheses and Properties of Novel Copolymers of Polycaprolactone and Aliphatic Polycarbonate Based on Ketal-Protected Dihydroxyacetone. Polym Bull. 2014;71:47–56. [Google Scholar]
- 127.Wang L, Cheng S, Zhuo R. Syntheses and Properties of Novel Copolymers of Poly (1,4-Dioxane-2-One) and Aliphatic Polycarbonate Based on Ketal-Protected Dihydroxyacetone. Macromol Chem Phys. 2013;214:458–463. [Google Scholar]
- 128.Sun J, Dong Y, Cao L, Wang X, Wang S, Hu Y. Highly Efficient Chemoselective Deprotection of O,O-Acetals and O,O-Ketals Catalyzed by Molecular Iodine in Acetone. J Org Chem. 2004;69:8932–8934. doi: 10.1021/jo0486239. [DOI] [PubMed] [Google Scholar]
- 129.Garson LR, Quintana RP, Lasslo A. Monomer and Dimer Formation in Esters of Dihydroxyacetone. Can J Chem. 1969;47:1249–1251. [Google Scholar]
- 130.Quintana RP, Garson LR, Lasslo A. Monomolecular Films of Compounds with Potential Dermophilic and Prophylactic Properties. Esters of Dihydroxyacetone. Can J Chem. 1969;47:853– 856. [Google Scholar]
- 131.Putnam D, Yazdi S. Biodegradable Compositions and Materials. WO2008101173. US Patent. 2008
- 132.Strain HH, Dore WH. Polymerization of Dihydroxyacetone. J Am Chem Soc. 1934;56:2649–2650. [Google Scholar]
- 133.Alder RW, Reddy BSR. Attempted Equilibration of an Insoluble Spiran Polymer with Monomers and Oligomers through Reversible Chemical Reactions: Transketalization Route to Spiropolymers from 1,4-Cyclohexanedione and Pentaerythritol. Polymer. 1994;35:5765–5772. [Google Scholar]
- 134.Zelikin AN, Putnam D. Poly (carbonate-Acetal)s from the Dimer Form of Dihydroxyacetone. Macromolecules. 2005;38:5532– 5537. [Google Scholar]
- 135.Putnam D, Zelikin A. Dihydroxyacetone-Based Polymers. US7659420. US Patent. 2010
- 136.Jung S-H, Jeong J-H, Miller P, Wong C-H. An Efficient Multigram-Scale Preparation of Dihydroxyacetone Phosphate. J Org Chem. 1994;59:7182–7184. [Google Scholar]
- 137.Linde F, Hvid I, Madsen F. The Effect of Specimen Geometry on the Mechanical Behaviour of Trabecular Bone Specimens. J Biomech. 1992;25:359–368. doi: 10.1016/0021-9290(92)90255-y. [DOI] [PubMed] [Google Scholar]
- 138.Goldstein SA. The Mechanical Properties of Trabecular Bone: Dependence on Anatomic Location and Function. J Biomech. 1987;20:1055–1061. doi: 10.1016/0021-9290(87)90023-6. [DOI] [PubMed] [Google Scholar]
- 139.Arima Y, Iwata H. Effects of Surface Functional Groups on Protein Adsorption and Subsequent Cell Adhesion Using Self-Assembled Monolayers. J Mater Chem. 2007;17:4079–4087. doi: 10.1016/j.biomaterials.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 140.McCoy CP, Morrow RJ, Edwards CR, Jones DS, Gorman SP. Neighboring Group-Controlled Hydrolysis: Towards “Designer” Drug Release Biomaterials. Bioconjugate Chem. 2007;18:209–215. doi: 10.1021/bc060238y. [DOI] [PubMed] [Google Scholar]
- 141.Vaisocherová H, Yang W, Zhang Z, Cao Z, Cheng G, Piliarik M, Homola J, Jiang S. Ultralow Fouling and Functionalizable Surface Chemistry Based on a Zwitterionic Polymer Enabling Sensitive and Specific Protein Detection in Undiluted Blood Plasma. Anal Chem. 2008;80:7894–7901. doi: 10.1021/ac8015888. [DOI] [PubMed] [Google Scholar]
- 142.Zhang XQ, Xu X, Lam R, Giljohann D, Ho D, Mirkin CA. A Strategy for Increasing Drug Solubility and Efficacy through Covalent Attachment to Polyvalent DNA-Nanoparticle Conjugates. ACS Nano. 2011;5:6962–6970. doi: 10.1021/nn201446c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Saito H, Hoffman ASH, Ogawa HI. Delivery of Doxorubicin from Biodegradable PEG Hydrogels Having Schiff Base Linkages. J Bioact Compat Polym. 2007;22:589–601. [Google Scholar]
- 144.Cui W, Cui Y, Zhu P, Zhao J, Su Y, Yang Y, Li J. An Anticoagulant Activity System Using Nanoengineered Autofluorescent Heparin Nanotubes. Chem - Asian J. 2012;7:127–132. doi: 10.1002/asia.201100425. [DOI] [PubMed] [Google Scholar]
- 145.Ren K, Ji J, Shen J. Tunable DNA Release from Cross-Linked Ultrathin DNA/PLL Multilayered Films. Bioconjugate Chem. 2006;17:77–83. doi: 10.1021/bc050264g. [DOI] [PubMed] [Google Scholar]
- 146.Wu X, Yu G, Luo C, Maeda A, Zhang N, Sun D, Zhou Z, Puntel A, Palczewski K, Zheng-Rong L. Synthesis and Evaluation of a Nanoglobular Dendrimer 5- Aminosalicylic Acid Conjugate with a Hydrolyzable Schiff Base Spacer for Treating Retinal Degeneration. ACS Nano. 2014;8:153–161. doi: 10.1021/nn4054107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Girelli AM, Mattei E, Messina A. Immobilized Tyrosinase Reactor for on-Line HPLC Application. Development and Characterization. Sens Actuators, B. 2007;121:515–521. [Google Scholar]
- 148.Bao H, Liu S, Zhang L, Chen G. Efficient Sample Proteolysis Based on a Microchip Containing a Glass Fiber Core with Immobilized Trypsin. Microchim Acta. 2012;179:291–297. [Google Scholar]
- 149.Marques ME, Mansur AAP, Mansur HS. Chemical Functionalization of Surfaces for Building Three-Dimensional Engineered Biosensors. Appl Surf Sci. 2013;275:347–360. [Google Scholar]
- 150.Macbeath G, Schreiber SL. Printing Proteins as Microarrays for High-Throughput Function Determination. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 151.Avseenko NV, Morozova TY, Ataullakhanov FI, Morozov VN. Immobilization of Proteins in Immunochemical Microarrays Fabricated by Electrospray Deposition. Anal Chem. 2001;73:6047–6052. doi: 10.1021/ac010460q. [DOI] [PubMed] [Google Scholar]
- 152.Tian Y, He Q, Cui Y, Li J. Fabrication of Protein Nanotubes Based on Layer-by-Layer Assembly. Biomacromolecules. 2006;7:2539–2542. doi: 10.1021/bm060412l. [DOI] [PubMed] [Google Scholar]
- 153.Duan L, He Q, Yan X, Cui Y, Wang K, Li J. Hemoglobin Protein Hollow Shells Fabricated through Covalent Layer-by-Layer Technique. Biochem Biophys Res Commun. 2007;354:357–362. doi: 10.1016/j.bbrc.2006.12.223. [DOI] [PubMed] [Google Scholar]
- 154.Langowska K, Kowal J, Palivan CG, Meier W. A General Strategy for Creating Self-Defending Surfaces for Controlled Drug Production for Long Periods of Time. J Mater Chem B. 2014;2:4684–4693. doi: 10.1039/c4tb00277f. [DOI] [PubMed] [Google Scholar]
- 155.Jia Y, Fei J, Cui Y, Yang Y, Gao L, Li J. pH-Responsive Polysaccharide Microcapsules through Covalent Bonding Assembly. Chem Commun. 2011;47:1175–1177. doi: 10.1039/c0cc03578e. [DOI] [PubMed] [Google Scholar]
- 156.Brandl F, Sommer F, Goepferich A. Rational Design of Hydrogels for Tissue Engineering: Impact of Physical Factors on Cell Behavior. Biomaterials. 2007;28:134–146. doi: 10.1016/j.biomaterials.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 157.Nicodemus GD, Bryant SJ. Cell Encapsulation in Biodegradable Hydrogels for Tissue Engineering Applications. Tissue Eng, Part B. 2008;14:149–165. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Billiet T, Vandenhaute M, Schelfhout J, Van Vlierberghe S, Dubruel P. A Review of Trends and Limitations in Hydrogel-Rapid Prototyping for Tissue Engineering. Biomaterials. 2012;33:6020– 6041. doi: 10.1016/j.biomaterials.2012.04.050. [DOI] [PubMed] [Google Scholar]
- 159.Lee KY, Mooney DJ. Alginate: Properties and Biomedical Applications. Prog Polym Sci. 2012;37:106–126. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kokabi M, Sirousazar M, Hassan ZM. PVA-Clay Nanocomposite Hydrogels for Wound Dressing. Eur Polym J. 2007;43:773–781. [Google Scholar]
- 161.Kirker KR, Luo Y, Nielson JH, Shelby J, Prestwich GD. Glycosaminoglycan Hydrogel Films as Bio-Interactive Dressings for Wound Healing. Biomaterials. 2002;23:3661–3671. doi: 10.1016/s0142-9612(02)00100-x. [DOI] [PubMed] [Google Scholar]
- 162.Ghobril C, Grinstaff MW. The Chemistry and Engineering of Polymeric Hydrogel Adhesives for Wound Closure: A Tutorial. Chem Soc Rev. 2015;44:1820–1835. doi: 10.1039/c4cs00332b. [DOI] [PubMed] [Google Scholar]
- 163.Oelker AM, Grinstaff MW. Ophthalmic Adhesives: A Materials Chemistry Perspective. J Mater Chem. 2008;18:2521–2536. [Google Scholar]
- 164.Schweizer D, Schönhammer K, Jahn M, Göpferich A. Protein-Polyanion Interactions for the Controlled Release of Monoclonal Antibodies. Biomacromolecules. 2013;14:75–83. doi: 10.1021/bm301352x. [DOI] [PubMed] [Google Scholar]
- 165.Teles H, Vermonden T, Eggink G, Hennink WE, de Wolf FA. Hydrogels of Collagen-Inspired Telechelic Triblock Copolymers for the Sustained Release of Proteins. J Controlled Release. 2010;147:298–303. doi: 10.1016/j.jconrel.2010.07.098. [DOI] [PubMed] [Google Scholar]
- 166.Oh EJ, Park K, Kim KS, Kim J, Yang JA, Kong JH, Lee MY, Hoffman AS, Hahn SK. Target Specific and Long-Acting Delivery of Protein, Peptide, and Nucleotide Therapeutics Using Hyaluronic Acid Derivatives. J Controlled Release. 2010;141:2–12. doi: 10.1016/j.jconrel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 167.Chen LG, Liu ZL, Zhuo RX. Synthesis and Properties of Degradable Hydrogels of Konjac Glucomannan Grafted Acrylic Acid for Colon-Specific Drug Delivery. Polymer. 2005;46:6274–6281. [Google Scholar]
- 168.Chang C, Zhang L. Cellulose-Based Hydrogels: Present Status and Application Prospects. Carbohydr Polym. 2011;84:40–53. [Google Scholar]
- 169.Burdick JA, Prestwich GD. Hyaluronic Acid Hydrogels for Biomedical Applications. Adv Mater. 2011;23:41–56. doi: 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Augst AD, Kong HJ, Mooney DJ. Alginate Hydrogels as Biomaterials. Macromol Biosci. 2006;6:623–633. doi: 10.1002/mabi.200600069. [DOI] [PubMed] [Google Scholar]
- 171.Baker MI, Walsh SP, Schwartz Z, Boyan BD. A Review of Polyvinyl Alcohol and Its Uses in Cartilage and Orthopedic Applications. J Biomed Mater Res, Part B. 2012;100:1451–1457. doi: 10.1002/jbm.b.32694. [DOI] [PubMed] [Google Scholar]
- 172.Krsko P, Libera M. Biointeractive Hydrogels. Mater Today. 2005;8:36–44. [Google Scholar]
- 173.Zhu J. Bioactive Modification of Poly(ethylene Glycol) Hydrogels for Tissue Engineering. Biomaterials. 2010;31:4639–4656. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lu S, Anseth KS. Photopolymerization of Multilaminated poly(HEMA) Hydrogels for Controlled Release. J Controlled Release. 1999;57:291–300. doi: 10.1016/s0168-3659(98)00125-4. [DOI] [PubMed] [Google Scholar]
- 175.Sun L, Li D, Hemraz UD, Fenniri H, Webster TJ. Self-Assembled Rosette Nanotubes and poly(2-Hydroxyethyl Methacrylate) Hydrogels Promote Skin Cell Functions. J Biomed Mater Res, Part A. 2014;102:3446–3451. doi: 10.1002/jbm.a.35008. [DOI] [PubMed] [Google Scholar]
- 176.Lao UL, Sun M, Matsumoto M, Mulchandani A, Chen W. Genetic Engineering of Self-Assembled Protein Hydrogel Based on Elastin-like Sequences with Metal Binding Functionality. Biomacromolecules. 2007;8:3736–3739. doi: 10.1021/bm700662n. [DOI] [PubMed] [Google Scholar]
- 177.Xu C, Breedveld V, Kopeček J. Reversible Hydrogels from Self-Assembling Genetically Engineered Protein Block Copolymers. Biomacromolecules. 2005;6:1739–1749. doi: 10.1021/bm050017f. [DOI] [PubMed] [Google Scholar]
- 178.Dandu R, Von Cresce A, Briber R, Dowell P, Cappello J, Ghandehari H. Silk–elastinlike Protein Polymer Hydrogels: Influence of Monomer Sequence on Physicochemical Properties. Polymer. 2009;50:366–374. [Google Scholar]
- 179.Zawaneh PN, Singh SP, Padera RF, Henderson PW, Spector JA, Putnam D. Design of an Injectable Synthetic and Biodegradable Surgical Biomaterial. Proc Natl Acad Sci U S A. 2010;107:11014–11019. doi: 10.1073/pnas.0811529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Henderson PW, Kadouch DJM, Singh SP, Zawaneh PN, Weiser J, Yazdi S, Weinstein A, Krotscheck U, Wechsler B, Putnam D, Spector Ja. A Rapidly Resorbable Hemostatic Biomaterial Based on Dihydroxyacetone. J Biomed Mater Res, Part A. 2009;93:776–782. doi: 10.1002/jbm.a.32586. [DOI] [PubMed] [Google Scholar]
- 181.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric Systems for Controlled Drug Release. Chem Rev. 1999;99:3181–3198. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 182.Bajpai AK, Shukla SK, Bhanu S, Kankane S. Responsive Polymers in Controlled Drug Delivery. Prog Polym Sci. 2008;33:1088–1118. [Google Scholar]
- 183.Kumar MNVR, Kumar N. Polymeric Controlled Drug-Delivery Systems: Perspective Issues and Opportunities. Drug Dev Ind Pharm. 2001;27:1–30. doi: 10.1081/ddc-100000124. [DOI] [PubMed] [Google Scholar]
- 184.James HP, John R, Alex A, Anoop KR. Smart Polymers for the Controlled Delivery of Drugs – a Concise Overview. Acta Pharm Sin B. 2014;4:120–127. doi: 10.1016/j.apsb.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kost J, Langer R. Responsive Polymeric Delivery Systems. Adv Drug Delivery Rev. 1991;6:19–50. doi: 10.1016/s0169-409x(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 186.Weiser JR, Yueh A, Putnam D. Protein Release from Dihydroxyacetone-Based Poly(carbonate Ester) Matrices. Acta Biomater. 2013;9:8245–8253. doi: 10.1016/j.actbio.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 187.Weiser JR, Ricapito NG, Yueh A, Weiser EL, Putnam D. A Mechanistic Analysis of the Quantitation of A-Hydroxy Ketones by the Bicinchoninic Acid Assay. Anal Biochem. 2012;430:116–122. doi: 10.1016/j.ab.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 188.Dorgan JR, Lehermeier HJ, Palade LI, Cicero J. Polylactides: Properties and Prospects of an Environmentally Benign Plastic from Renewable Resources. Macromol Symp. 2001;175:55–66. [Google Scholar]
- 189.Kumar R, Choudhary V, Mishra S, Varma IK, Mattiason B. Adhesive and Plastics Based on Soy Protein Products. Ind Crops Prod. 2002;16:155–172. [Google Scholar]
- 190.Mülhaupt R. Green Polymer Chemistry and Bio-Based Plastics: Dreams and Reality. Macromol Chem Phys. 2013;214:159–174. [Google Scholar]
- 191.Mohanty AK, Misra M, Drzal LT. Sustainable Bio-Composites from Renewable Resources: Opportunities and Challenges in the Green Materials World. J Polym Environ. 2002;10:19–26. [Google Scholar]
- 192.Sudesh K, Iwata T. Sustainability of Biobased and Biodegradable Plastics. Clean: Soil, Air, Water. 2008;36:433–442. [Google Scholar]
- 193.Painter RM, Pearson DM, Waymouth RM. Selective Catalytic Oxidation of Glycerol to Dihydroxyacetone. Angew Chem, Int Ed. 2010;49:9456–9459. doi: 10.1002/anie.201004063. [DOI] [PubMed] [Google Scholar]
- 194.Kwon Y, Birdja Y, Spanos I, Rodriguez P, Koper MTM. Highly Selective Electro-Oxidation of Glycerol to Dihydroxyacetone on Platinum in the Presence of Bismuth. ACS Catal. 2012;2:759–764. [Google Scholar]
- 195.XJ Autolysis E. Coli Strains Glycerol Stocks. Zymo Research Coporation; Irvine, CA: CAS # 56-81-5. revised 2013. [Google Scholar]
- 196.Zhang H, Grinstaff MW. Recent Advances in Glycerol Polymers: Chemistry and Biomedical Applications. Macromol Rapid Commun. 2014;35:1906–1924. doi: 10.1002/marc.201400389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang XL, Zhuo RX, Liu LJ, He F, Liu G. Synthesis and Characterization of Novel Aliphatic Polycarbonates. J Polym Sci, Part A: Polym Chem. 2002;40:70–75. [Google Scholar]
- 198.Ray WC, Grinstaff MW. Polycarbonate and Poly (carbonate-Ester)s Synthesized From Biocompatible Building Blocks of Glycerol and Lactic Acid. Macromolecules. 2003;36:3557–3562. [Google Scholar]
- 199.Wolinsky JB, Ray WC, III, Colson YL, Grinstaff MW. Poly(carbonate Ester)s Based on Units of 6-Hydroxyhexanoic Acid and Glycerol. Macromolecules. 2007;40:7065–7068. [Google Scholar]
- 200.Wolinsky JB, Yohe ST, Colson YL, Grinstaff MW. Functionalized Hydrophobic Poly (glycerol-Co-E-Caprolactone) Depots for Controlled Drug Release. Biomacromolecules. 2012;13:406–411. doi: 10.1021/bm201443m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Liu R, Wolinsky JB, Walpole J, Southard E, Chirieac LR, Grinstaff MW, Colson YL. Prevention of Local Tumor Recurrence Following Surgery Using Low-Dose Chemotherapeutic Polymer Films. Annals of Surgical Oncology. 2010;17:1203–1213. doi: 10.1245/s10434-009-0856-z. [DOI] [PubMed] [Google Scholar]
- 202.Wolinsky JB, Liu R, Walpole J, Chirieac LR, Colson YL, Grinstaff MW. Prevention of In Vivo Lung Tumor Growth by Prolonged Local Delivery of Hydroxycamptothecin Using Poly(ester-Carbonate)-Collagen Composites. J Controlled Release. 2010;144:280–287. doi: 10.1016/j.jconrel.2010.02.022. [DOI] [PubMed] [Google Scholar]
- 203.Rokicki G. Aliphatic Cyclic Carbonates and Spiroorthocarbonates as Monomers. Prog Polym Sci. 2000;25:259–342. [Google Scholar]
- 204.Zhou CH, Beltramini JN, Fan YX, Lu GQ. Chemoselective Catalytic Conversion of Glycerol as a Biorenewable Source to Valuable Commodity Chemicals. Chem Soc Rev. 2008;37:527–549. doi: 10.1039/b707343g. [DOI] [PubMed] [Google Scholar]
- 205.Cohen CT, Chu T, Coates GW. Cobalt Catalysts for the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide: Combining High Activity and Selectivity. J Am Chem Soc. 2005;127:10869–10878. doi: 10.1021/ja051744l. [DOI] [PubMed] [Google Scholar]
- 206.Darensbourg DJ. Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2. Chem Rev. 2007;107:2388–2410. doi: 10.1021/cr068363q. [DOI] [PubMed] [Google Scholar]
- 207.Kim JG, Coates GW. Synthesis and Polymerization of Norbornenyl-Terminated Multiblock Poly(cyclohexene Carbonate)s: A Consecutive Ring-Opening Polymerization Route to Multi-segmented Graft Polycarbonates. Macromolecules. 2012;45:7878–7883. [Google Scholar]
- 208.Kim JG, Cowman CD, LaPointe AM, Wiesner U, Coates GW. Tailored Living Block Copolymerization: Multi-block Poly(cyclohexene Carbonate)s with Sequence Control. Macromolecules. 2011;44:1110–1113. [Google Scholar]
- 209.Lu XB, Darensbourg DJ. Cobalt Catalysts for the Coupling of CO2 and Epoxides to Provide Polycarbonates and Cyclic Carbonates. Chem Soc Rev. 2012;41:1462–1484. doi: 10.1039/c1cs15142h. [DOI] [PubMed] [Google Scholar]
- 210.Lu XB, Wang Y. Highly Active, Binary Catalyst Systems for the Alternating Copolymerization of CO2 and Epoxides Under Mild Conditions. Angew Chem, Int Ed. 2004;43:3574–3577. doi: 10.1002/anie.200453998. [DOI] [PubMed] [Google Scholar]
- 211.Nakano K, Kamada T, Nozaki K. Selective Formation of Polycarbonate over Cyclic Carbonate: Copolymerization of Epoxides with Carbon Dioxide Catalyzed by a Cobalt(III) Complex with a Piperidinium End-Capping Arm. Angew Chem, Int Ed. 2006;45:7274–7277. doi: 10.1002/anie.200603132. [DOI] [PubMed] [Google Scholar]
- 212.Wu GP, Wei SH, Ren WM, Lu XB, Xu TQ, Darensbourg DJ. Perfectly Alternating Copolymerization of CO2 and Epichlorohydrin Using Cobalt(III)-Based Catalyst Systems. J Am Chem Soc. 2011;133:15191–15199. doi: 10.1021/ja206425j. [DOI] [PubMed] [Google Scholar]
- 213.Geschwind J, Frey H. Poly(1,2-Glycerol Carbonate): A Fundamental Polymer Structure Synthesized From CO2 and Glycidyl Ethers. Macromolecules. 2013;46:3280–3287. [Google Scholar]
- 214.Lukaszczyk J, Jaszcz K, Kuran W, Listos T. Synthesis and Modification of Functional Polycarbonates with Pendant Allyl Groups. Macromol Biosci. 2001;1:282–289. [Google Scholar]
- 215.Lukaszczyk J, Jaszcz K, Kuran W, Listos T. Synthesis of Functional Polycarbonates by Copolymerization of Carbon Dioxide with Allyl Glycidyl Ether. Macromol Rapid Commun. 2000;21:754–757. [Google Scholar]
- 216.Zhang H, Grinstaff MW. Synthesis of Atactic and Isotactic Poly(1,2-Glycerol Carbonate)s: Degradable Polymers for Biomedical and Pharmaceutical Applications. J Am Chem Soc. 2013;135:6806–6809. doi: 10.1021/ja402558m. [DOI] [PubMed] [Google Scholar]
- 217.Zhang H, Grinstaff MW. Terpolymerization of Benzyl Glycidyl Ether, Propylene Oxide, and CO2 Using Binary and Bifunctional [rac-SalcyCoIIIX] Complexes and the Thermal and Mechanical Properties of the Resultant Poly(benzyl 1,2-Glycerol-co-Propylene Carbonate)s and poly(1,2-Glycerol-co-propylene carbonate)s. J Appl Polym Sci. 2014;131:39893. [Google Scholar]
- 218.Hilf J, Phillips A, Frey H. Poly(carbonate) Copolymers with a Tailored Number of Hydroxyl Groups from Glycidyl Ethers and CO2. Polym Chem. 2014;5:814. [Google Scholar]
- 219.Wu X, Zhao H, Nörnberg B, Theato P, Luinstra GA. Synthesis and Characterization of Hydroxyl-Functionalized Poly-(propylene Carbonate) Macromolecules. 2014;47:492–497. [Google Scholar]
- 220.Harpole DH, Herndon JE, Young WG, Wolfe WG, Sabiston DC. Stage I Nonsmall Cell Lung Cancer. A Multivariate Analysis of Treatment Methods and Patterns of Recurrence. Cancer. 1995;76:787–796. doi: 10.1002/1097-0142(19950901)76:5<787::aid-cncr2820760512>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 221.Martini N, Burt ME, Bains MS, McCormack PM, Rusch VW, Ginsberg RJ. Survival After Resection of Stage II Non-Small Cell Lung Cancer. Ann Thorac Surg. 1992;54:460–466. doi: 10.1016/0003-4975(92)90435-7. [DOI] [PubMed] [Google Scholar]
- 222.Sugimura H, Nichols FC, Yang P, Allen MS, Cassivi SD, Deschamps C, Williams BA, Pairolero PC. Survival After Recurrent Nonsmall-Cell Lung Cancer After Complete Pulmonary Resection. Ann Thorac Surg. 2007;83:409–418. doi: 10.1016/j.athoracsur.2006.08.046. [DOI] [PubMed] [Google Scholar]
- 223.Bonvalot S, Rivoire M, Castaing M, Stoeckle E, Le Cesne A, Blay JY, Laplanche A. Primary Retroperitoneal Sarcomas: A Multivariate Analysis of Surgical Factors Associated with Local Control. J Clin Oncol. 2008;27:31–37. doi: 10.1200/JCO.2008.18.0802. [DOI] [PubMed] [Google Scholar]
- 224.Karakousis CP, Velez AF, Gerstenbluth R, Driscoll DL. Resectability and Survival in Retroperitoneal Sarcomas. Ann Surg Oncol. 1996;3:150–158. doi: 10.1007/BF02305794. [DOI] [PubMed] [Google Scholar]
- 225.Lehnert T, Cardona S, Hinz U, Willeke F, Mechtersheimer G, Treiber M, Herfarth C, Buechler MW, Schwarzbach MHM. Primary and Locally Recurrent Retroperitoneal Soft-Tissue Sarcoma: Local Control and Survival. Eur J Surg Oncol. 2009;35:986–993. doi: 10.1016/j.ejso.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 226.Liu R, Wolinsky JB, Catalano PJ, Chirieac LR, Wagner AJ, Grinstaff MW, Colson YL, Raut CP. Paclitaxel-Eluting Polymer Film Reduces Locoregional Recurrence and Improves Survival in a Recurrent Sarcoma Model: A Novel Investigational Therapy. Annals of Surgical Oncology. 2012;19:199–206. doi: 10.1245/s10434-011-1871-4. [DOI] [PubMed] [Google Scholar]
- 227.Haag R, Sunder A, Stumbé JF. An Approach to Gylcerol Dendrimers and Pseudo-Dendritic Polyglycerols. J Am Chem Soc. 2000;122:2954–2955. [Google Scholar]
- 228.Carnahan MA, Grinstaff MW. Synthesis and Characterization of Poly(glycerol–succinic Acid) Dendrimers. Macromolecules. 2001;34:7648–7655. [Google Scholar]
- 229.Carnahan MA, Grinstaff MW. Synthesis of Generational Polyester Dendrimers Derived From Glycerol and Succinic or Adipic Acid. Macromolecules. 2006;39:609–616. [Google Scholar]
- 230.Tang S, June SM, Howell BA, Chai M. Synthesis of Salicylate Dendritic Prodrugs. Tetrahedron Lett. 2006;47:7671–7675. [Google Scholar]
- 231.Luman NR, Smeds KA, Grinstaff MW. The Convergent Synthesis of Poly(glycerol-Succinic Acid) Dendritic Macromolecules. Chem - Eur J. 2003;9:5618–5626. doi: 10.1002/chem.200305172. [DOI] [PubMed] [Google Scholar]
- 232.Calderón M, Quadir MA, Sharma SK, Haag R. Dendritic Polyglycerols for Biomedical Applications. Adv Mater. 2010;22:190–218. doi: 10.1002/adma.200902144. [DOI] [PubMed] [Google Scholar]
- 233.Sisson AL, Haag R. Polyglycerol Nanogels: Highly Functional Scaffolds for Biomedical Applications. Soft Matter. 2010;6:4968–4975. [Google Scholar]
- 234.Wilms D, Stiriba SE, Frey H. Hyperbranched Polyglycerols: From the Controlled Synthesis of Biocompatible Polyether Polyols to Multipurpose Applications. Acc Chem Res. 2010;43:129–141. doi: 10.1021/ar900158p. [DOI] [PubMed] [Google Scholar]
- 235.Sunder A, Hanselmann R, Frey H, Mülhaupt R. Controlled Synthesis of Hyperbranched Polyglycerols by Ring-Opening Multibranching Polymerization. Macromolecules. 1999;32:4240–4246. [Google Scholar]
- 236.Rokicki G, Rakoczy P, Parzuchowski P, Sobiecki M. Hyperbranched Aliphatic Polyethers Obtained from Environmentally Benign Monomer: Glycerol Carbonate. Green Chem. 2005;7:529–539. [Google Scholar]
- 237.Yu X, Feng J, Zhuo R. Preparation of Hyperbranched Aliphatic Polyester Derived From Functionalized 1,4-Dioxan-2-One. Macromolecules. 2005;38:6244–6247. [Google Scholar]
- 238.Parzuchowski PG, Grabowska M, Tryznowski M, Rokicki G. Synthesis of Glycerol Based Hyperbranched Polyesters with Primary Hydroxyl Groups. Macromolecules. 2006;39:7181–7186. [Google Scholar]
- 239.Parzuchowski PG, Jaroch M, Tryznowski M, Rokicki G. Synthesis of New Glycerol-Based Hyperbranched Polycarbonates. Macromolecules. 2008;41:3859–3865. [Google Scholar]
- 240.Morgan MT, Carnahan MA, Immoos CE, Ribeiro AA, Finkelstein S, Lee SJ, Grinstaff MW. Dendritic Molecular Capsules for Hydrophobic Compounds. J Am Chem Soc. 2003;125:15485–15489. doi: 10.1021/ja0347383. [DOI] [PubMed] [Google Scholar]
- 241.Morgan MT, Nakanishi Y, Kroll DJ, Griset AP, Carnahan MA, Wathier M, Oberlies NH, Manikumar G, Wani MC, Grinstaff MW. Dendrimer-Encapsulated Camptothecins: Increased Solubility, Cellular Uptake, and Cellular Retention Affords Enhanced Anticancer Activity In Vitro. Cancer Res. 2006;66:11913–11921. doi: 10.1158/0008-5472.CAN-06-2066. [DOI] [PubMed] [Google Scholar]
- 242.Morgan MT, Carnahan MA, Finkelstein S, Prata CAH, Degoricija L, Lee SJ, Grinstaff MW. Dendritic Supramolecular Assemblies for Drug Delivery. Chem Commun. 2005;34:4309–4311. doi: 10.1039/b502411k. [DOI] [PubMed] [Google Scholar]
- 243.Carnahan MA, Middleton C, Kim J, Kim T, Grinstaff MW. Hybrid Dendritic–Linear Polyester–Ethers for in Situ Photopolymerization. J Am Chem Soc. 2002;124:5291–5293. doi: 10.1021/ja025576y. [DOI] [PubMed] [Google Scholar]
- 244.Degoricija L, Johnson CS, Wathier M, Kim T, Grinstaff MW. Photo Cross-Linkable Biodendrimers as Ophthalmic Adhesives for Central Lacerations and Penetrating Keratoplasties. Invest Ophthalmol Visual Sci. 2007;48:2037–2042. doi: 10.1167/iovs.06-0957. [DOI] [PubMed] [Google Scholar]
- 245.Kang PC, Carnahan MA, Wathier M, Grinstaff MW, Kim T. Novel Tissue Adhesives to Secure Laser in Situ Keratomileusis Flaps. J Cataract Refractive Surg. 2005;31:1208–1212. doi: 10.1016/j.jcrs.2004.10.067. [DOI] [PubMed] [Google Scholar]
- 246.Velazquez AJ, Carnahan MA, Kristinsson J, Stinnett S, Grinstaff MW. New Dendritic Adhesives for Sutureless Ophthalmic Surgical Procedures: In Vitro Studies of Corneal Laceration Repair. Arch Ophthalmol. 2004;122:867–870. doi: 10.1001/archopht.122.6.867. [DOI] [PubMed] [Google Scholar]
- 247.Luman NR, Kim T, Grinstaff MW. Dendritic Polymers Composed of Glycerol and Succinic Acid: Synthetic Methodologies and Medical Applications. Pure Appl Chem. 2004;76:1375–1385. [Google Scholar]
- 248.Denyer SP. Mechanisms of Action of Antibacterial Biocides. Int Biodeterior Biodegrad. 1995;36:227–245. [Google Scholar]
- 249.Meyers SR, Juhn FS, Griset AP, Luman NR, Grinstaff MW. Anionic Amphiphilic Dendrimers as Antibacterial Agents. J Am Chem Soc. 2008;130:14444–14445. doi: 10.1021/ja806912a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Kohane DS, Langer R. Polymeric Biomaterials in Tissue Engineering. Pediatr Res. 2008;63:487–491. doi: 10.1203/01.pdr.0000305937.26105.e7. [DOI] [PubMed] [Google Scholar]
- 251.Lee KY, Mooney DJ. Hydrogels for Tissue Engineering. Chem Rev. 2001;101:1869–1880. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 252.Söntjens SHM, Nettles DL, Carnahan MA, Setton LA, Grinstaff MW. Biodendrimer-Based Hydrogel Scaffolds for Cartilage Tissue Repair. Biomacromolecules. 2006;7:310–316. doi: 10.1021/bm050663e. [DOI] [PubMed] [Google Scholar]
- 253.Leuchs H. Ueber Die Glycin-Carbonsäure. Ber Dtsch Chem Ges. 1906;39:857–861. [Google Scholar]
- 254.Fang X, Wang CJ. Recent Advances in Asymmetric Organocatalysis Mediated by Bifunctional Amine–Thioureas Bearing Multiple Hydrogen-Bonding Donors. Chem Commun. 2015;51:1185–1197. doi: 10.1039/c4cc07909d. [DOI] [PubMed] [Google Scholar]
- 255.Deming TJ. Transition Metal-Amine Initiators for Preparation of Well-Defined Poly(gamma-Benzyl L-Glutamate) J Am Chem Soc. 1997;119:2759–2760. [Google Scholar]
- 256.Deming TJ. Cobalt and Iron Initiators for the Controlled Polymerization of Alpha-Amino Acid-N-Carboxyanhydrides. Macromolecules. 1999;32:4500–4502. [Google Scholar]
- 257.Lu H, Cheng J. Hexamethyldisilazane-Mediated Controlled Polymerization of Alpha-Amino Acid N-Carboxyanhydrides. J Am Chem Soc. 2007;129:14114–14115. doi: 10.1021/ja074961q. [DOI] [PubMed] [Google Scholar]
- 258.Peng YL, Lai SL, Lin CC. Preparation of Polypeptide via Living Polymerization of Z-Lys-NCA Initiated by Platinum Complexes. Macromolecules. 2008;41:3455–3459. [Google Scholar]
- 259.Deming TJ. Synthesis and Self-Assembly of Well-Defined Block Copolypeptides via Controlled NCA Polymerization. In: Percec V, editor. Hierarchical Macromolecular Structures: 60 Years After the Staudinger Nobel Prize I. Advances in Polymer Science. Vol. 262. Springer; Cham: 2013. pp. 1–38. [Google Scholar]
- 260.Deming TJ. Synthesis of Side-Chain Modified Polypeptides. Chem Rev. 2015 doi: 10.1021/acs.chemrev.5b00292. accepted. [DOI] [PubMed] [Google Scholar]
- 261.Kricheldorf HR. Polypeptides and 100 Years of Chemistry of Alpha-Amino Acid N-Carboxyanhydrides. Angew Chem, Int Ed. 2006;45:5752–5784. doi: 10.1002/anie.200600693. [DOI] [PubMed] [Google Scholar]
- 262.Denkewalter RG, Kolc J, Lukasavage WJ. Macromolecular Highly Branched Homogeneous Compound Based on Lysine Units. 4289872. US Patent. 1981
- 263.Scholl M, Kadlecova Z, Klok HA. Dendritic and Hyperbranched Polyamides. Prog Polym Sci. 2009;34:24–61. [Google Scholar]
- 264.Choi JS, Lee EJ, Choi YH, Jeong YJ, Park JS. Poly(ethylene Glycol)-Block-poly(L-Lysine) Dendrimer: Novel Linear Polymer/Dendrimer Block Copolymer Forming a Spherical Water-Soluble Polyionic Complex with DNA. Bioconjugate Chem. 1999;10:62–65. doi: 10.1021/bc9800668. [DOI] [PubMed] [Google Scholar]
- 265.Choi JS, Joo DK, Kim CH, Kim K, Park JS. Synthesis of a Barbell-like Triblock Copolymer, Poly(L-Lysine) Dendrimer-Block-Poly(ethylene Glycol)-Block-Poly(L-Lysine) Dendrimer, and Its Self-Assembly with Plasmid DNA. J Am Chem Soc. 2000;122:474–480. [Google Scholar]
- 266.Ohsaki M, Okuda T, Wada A, Hirayama T, Niidome T, Aoyagi H. In Vitro Gene Transfection Using Dendritic Poly(LLysine) Bioconjugate Chem. 2002;13:510–517. doi: 10.1021/bc015525a. [DOI] [PubMed] [Google Scholar]
- 267.Wathier M, Jung PJ, Carnahan MA, Kim T, Grinstaff MW. Dendritic Macromers as in Situ Polymerizing Biomaterials for Securing Cataract Incisions. J Am Chem Soc. 2004;126:12744–12745. doi: 10.1021/ja045870l. [DOI] [PubMed] [Google Scholar]
- 268.Ghobril C, Charoen K, Rodriguez EK, Nazarian A, Grinstaff MW. A Dendritic Thioester Hydrogel Based on Thiol-Thioester Exchange as a Dissolvable Sealant System for Wound Closure. Angew Chem, Int Ed. 2013;52:14070–14074. doi: 10.1002/anie.201308007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Sakthivel T, Toth I, Florence AT. Synthesis and Physicochemical Properties of Lipophilic Polyamide Dendrimers. Pharm Res. 1998;15:776–782. doi: 10.1023/a:1011935406664. [DOI] [PubMed] [Google Scholar]
- 270.Twyman LJ, Beezer AE, Esfand R, Mathews BT, Mitchell JC. The Synthesis of Chiral Dendritic Molecules Based on Amino Acid Repeat Units. J Chem Res Synop. 1998;12:758–759. [Google Scholar]
- 271.Ranganathan D, Kurur S. Synthesis of Totally Chiral, Multiple Armed, Poly Glu and Poly Asp Scaffoldings on Bifunctional Adamantane Core. Tetrahedron Lett. 1997;38:1265–1268. [Google Scholar]
- 272.Ranganathan D, Kurur S, Gilardi R, Karle IL. Design and Synthesis of AB3-Type (A = 1,3,5-Benzenetricarbonyl Unit; B= Glu diOMe or Glu7 Octa OMe) Peptide Dendrimers: Crystal Structure of the First Generation. Biopolymers. 2000;54:289–295. doi: 10.1002/1097-0282(20001005)54:4<289::AID-BIP60>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 273.Deming TJ. Synthetic Polypeptides for Biomedical Applications. Prog Polym Sci. 2007;32:858–875. [Google Scholar]
- 274.Singer JW. Paclitaxel Poliglumex (XYOTAX (TM), CT-2103): A Macromolecular Taxane. J Controlled Release. 2005;109:120–126. doi: 10.1016/j.jconrel.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 275.Yu M, Deming TJ. Synthetic Polypeptide Mimics of Marine Adhesives. Macromolecules. 1998;31:4739–4745. doi: 10.1021/ma980268z. [DOI] [PubMed] [Google Scholar]
- 276.Ertel SI, Kohn J. Evaluation of a Series of Tyrosine-Derived Polycarbonates as Degradable Biomaterials. J Biomed Mater Res. 1994;28:919–930. doi: 10.1002/jbm.820280811. [DOI] [PubMed] [Google Scholar]
- 277.Pulapura S, Kohn J. Tyrosine-Derived Polycarbonates: Backbone-Modified “Pseudo”-Poly(Amino Acids) Designed for Biomedical Applications. Biopolymers. 1992;32:411–417. doi: 10.1002/bip.360320418. [DOI] [PubMed] [Google Scholar]
- 278.Rojas R, Harris NK, Piotrowska K, Kohn J. Evaluation of Automated Synthesis for Chain and Step-Growth Polymerizations: Can Robots Replace the Chemists? J Polym Sci, Part A: Polym Chem. 2009;47:49–58. [Google Scholar]
- 279.Treiser MD, Liu E, Dubin RA, Sung HJ, Kohn J, Moghe PV. Profiling Cell-Biomaterial Interactions Via Cell-Based Fluororeporter Imaging. BioTechniques. 2007;43:361–368. doi: 10.2144/000112533. [DOI] [PubMed] [Google Scholar]
- 280.Lewitus D, Smith KL, Shain W, Kohn J. Ultrafast Resorbing Polymers for Use as Carriers for Cortical Neural Probes. Acta Biomater. 2011;7:2483–2491. doi: 10.1016/j.actbio.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 281.Lo M-c, Wang S, Singh S, Damodaran VB, Kaplan HM, Kohn J, Shreiber DI, Zahn JD. Coating Flexible Probes With An Ultra Fast Degrading Polymer to Aid in Tissue Insertion. Biomed Microdevices. 2015;17:34. doi: 10.1007/s10544-015-9927-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Yaszemski MJ, Payne RG, Hayes WC, Langer R, Mikos AG. In Vitro Degradation of a Poly(propylene Fumarate)-Based Composite Material. Biomaterials. 1996;17:2127–2130. doi: 10.1016/0142-9612(96)00008-7. [DOI] [PubMed] [Google Scholar]
- 283.Kasper FK, Tanahashi K, Fisher JP, Mikos AG. Synthesis of Poly(Propylene Fumarate) Nat Protoc. 2009;4:518–525. doi: 10.1038/nprot.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Kretlow JD, Klouda L, Mikos AG. Injectable Matrices and Scaffolds for Drug Delivery in Tissue Engineering. Adv Drug Delivery Rev. 2007;59:263–273. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 285.Lee KW, Wang S, Fox BC, Ritman EL, Yaszemski MJ, Lu L. Poly(propylene Fumarate) Bone Tissue Engineering Scaffold Fabrication Using Stereolithography: Effects of Resin Formulations and Laser Parameters. Biomacromolecules. 2007;8:1077–1084. doi: 10.1021/bm060834v. [DOI] [PubMed] [Google Scholar]
- 286.Dwek RA. Glycobiology: Toward Understanding the Function of Sugars. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 287.Ernst B, Magnani JL. From Carbohydrate Leads to Glycomimetic Drugs. Nat Rev Drug Discovery. 2009;8:661–677. doi: 10.1038/nrd2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Stern R, Jedrzejas MJ. Carbohydrate Polymers at the Center of Life’s Origins: The Importance of Molecular Processivity. Chem Rev. 2008;108:5061–5085. doi: 10.1021/cr078240l. [DOI] [PubMed] [Google Scholar]
- 289.Bouten PJM, Zonjee M, Bender J, Yauw STK, van Goor H, van Hest JCM, Hoogenboom R. The Chemistry of Tissue Adhesive Materials. Prog Polym Sci. 2014;39:1375–1405. [Google Scholar]
- 290.Kumar MNVR, Muzzarelli RAA, Muzzarelli C, Sashiwa H, Domb AJ. Chitosan Chemistry and Pharmaceutical Perspectives. Chem Rev. 2004;104:6017–6084. doi: 10.1021/cr030441b. [DOI] [PubMed] [Google Scholar]
- 291.Lapčík J, Lapčík L, De Smedt S, Demeester J, Chabreček P. Hyaluronan: Preparation, Structure, Properties, and Applications. Chem Rev. 1998;98:2663–2684. doi: 10.1021/cr941199z. [DOI] [PubMed] [Google Scholar]
- 292.Rinaudo M. Main Properties and Current Applications of Some Polysaccharides as Biomaterials. Polym Int. 2008;57:397–430. [Google Scholar]
- 293.Kumar R, Gao W, Gross RA. Functionalized Polylactides: Preparation and Characterization of [L]-Lactide-Co-Pentofuranose. Macromolecules. 2002;35:6835–6844. [Google Scholar]
- 294.Gross RA, Kumar A, Kalra B. Polymer Synthesis by In Vitro Enzyme Catalysis. Chem Rev. 2001;101:2097–2124. doi: 10.1021/cr0002590. [DOI] [PubMed] [Google Scholar]
- 295.Hu J, Gao W, Kulshrestha A, Gross RA. Sweet Polyesters”: Lipase-Catalyzed Condensation-Polymerizations of Alditols. Macromolecules. 2006;39:6789–6792. [Google Scholar]
- 296.Sahoo B, Brandstadt KF, Lane TH, Gross RA. Sweet Silicones”: Biocatalytic Reactions to Form Organosilicon Carbohydrate Macromers. Org Lett. 2005;7:3857–3860. doi: 10.1021/ol050942e. [DOI] [PubMed] [Google Scholar]
- 297.Kumar A, Kulshrestha AS, Gao W, Gross RA. Versatile Route to Polyol Polyesters by Lipase Catalysis. Macromolecules. 2003;36:8219–8221. [Google Scholar]
- 298.Peng Y, Munoz-Pinto DJ, Chen M, Decatur J, Hahn M, Gross RA. Poly(sophorolipid) Structural Variation: Effects on Biomaterial Physical and Biological Properties. Biomacromolecules. 2014;15:4214–4227. doi: 10.1021/bm501255j. [DOI] [PubMed] [Google Scholar]
- 299.Reineke T, Grinstaff MW. Designer Materials for Nucleic Acid Delivery. MRS Bull. 2005;30:635–639. [Google Scholar]
- 300.Liu Y, Reineke TM. Hydroxyl Stereochemistry and Amine Number within Poly(glycoamidoamine)s Affect Intracellular DNA Delivery. J Am Chem Soc. 2005;127:3004–3015. doi: 10.1021/ja0436446. [DOI] [PubMed] [Google Scholar]
- 301.Liu Y, Wenning L, Lynch M, Reineke TM. New Poly(D-Glucaramidoamine) s Induce DNA Nanoparticle Formation and Efficient Gene Delivery into Mammalian Cells. J Am Chem Soc. 2004;126:7422–7423. doi: 10.1021/ja049831l. [DOI] [PubMed] [Google Scholar]
- 302.Reineke TM, Davis ME. Structural Effects of Carbohydrate-Containing Polycations on Gene Delivery. 2. Charge Center Type. Bioconjugate Chem. 2003;14:255–261. doi: 10.1021/bc025593c. [DOI] [PubMed] [Google Scholar]
- 303.Xue L, Ingle NP, Reineke TM. Highlighting the Role of Polymer Length, Carbohydrate Size, and Nucleic Acid Type in Potency of Glycopolycation Agents for pDNA and siRNA Delivery. Biomacromolecules. 2013;14:3903–3915. doi: 10.1021/bm401026n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Ingle NP, Malone B, Reineke TM. Poly-(glycoamidoamine)s: A Broad Class of Carbohydrate-Containing Polycations for Nucleic Acid Delivery. Trends Biotechnol. 2011;29:443–453. doi: 10.1016/j.tibtech.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 305.Taori VP, Lu H, Reineke TM. Structure-Activity Examination of Poly(glycoamidoguanidine)s: Glycopolycations Containing Guanidine Units for Nucleic Acid Delivery. Biomacromolecules. 2011;12:2055–2063. doi: 10.1021/bm101537f. [DOI] [PubMed] [Google Scholar]
- 306.Liu Y, Reineke TM. Degradation of Poly(glycoamidoamine) DNA Delivery Vehicles: Polyamide Hydrolysis at Physiological Conditions Promotes DNA Release. Biomacromolecules. 2010;11:316–325. doi: 10.1021/bm9008233. [DOI] [PubMed] [Google Scholar]
- 307.Srinivasachari S, Liu Y, Zhang G, Prevette L, Reineke TM. Trehalose Click Polymers Inhibit Nanoparticle Aggregation and Promote pDNA Delivery in Serum. J Am Chem Soc. 2006;128:8176–8184. doi: 10.1021/ja0585580. [DOI] [PubMed] [Google Scholar]
- 308.Dane EL, Ballok AE, O’Toole GA, Grinstaff MW. Synthesis of Bioinspired Carbohydrate Amphiphiles That Promote and Inhibit Biofilms. Chem Sci. 2014;5:551–557. doi: 10.1039/C3SC52777H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Dane EL, Chin SL, Grinstaff MW. Synthetic Enantiopure Carbohydrate Polymers That Are Highly Soluble in Water and Noncytotoxic. ACS Macro Lett. 2013;2:887–890. doi: 10.1021/mz400394r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Dane EL, Grinstaff MW. Poly-Amido-Saccharides: Synthesis via Anionic Polymerization of a B-Lactam Sugar Monomer. J Am Chem Soc. 2012;134:16255–16264. doi: 10.1021/ja305900r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Ghobril C, Heinrich B, Dane EL, Grinstaff MW. Synthesis of Hydrophobic Carbohydrate Polymers and Their Formation of Thermotropic Liquid Crystalline Phases. ACS Macro Lett. 2014;3:359–363. doi: 10.1021/mz5000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Stidham SE, Chin SL, Dane EL, Grinstaff MW. Carboxylated Glucuronic Poly-Amido-Saccharides as Protein Stabilizing Agents. J Am Chem Soc. 2014;136:9544–9547. doi: 10.1021/ja5036804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Abele S, Guichard G, Seebach D. (S)-β3-Homolysine- (S)-β3-Homoserine-Containing B-Peptides: CD Spectra in Aqueous Solution. Helv Chim Acta. 1998;81:2141–2156. [Google Scholar]
- 314.Cheng J, Deming TJ. Synthesis and Conformational Analysis of Optically Active Poly(β-Peptides) Macromolecules. 2001;34:5169–5174. [Google Scholar]
- 315.Seebach D, Overhand M, Kühnle FNM, Martinoni B, Oberer L, Hommel U, Widmer H. B-Peptides: Synthesis by Arndt-Eistert Homologation with Concomitant Peptide Coupling. Structure Determination by NMR and CD Spectroscopy and by X-Ray Crystallography. Helical Secondary Structure of a B-Hexapeptide in Solution and Its Stability Towards Pe. Helv Chim Acta. 1996;79:913–941. [Google Scholar]
- 316.Mikami K, Lonnecker AT, Gustafson TP, Zinnel NF, Pai PJ, Russell DH, Wooley KL. Polycarbonates Derived from Glucose via an Organocatalytic Approach. J Am Chem Soc. 2013;135:6826–6829. doi: 10.1021/ja402319m. [DOI] [PubMed] [Google Scholar]
- 317.Haba O, Tomizuka H, Endo T. Anionic Ring-Opening Polymerization of Methyl 4,6-O-Benzylidene-2,3-O-Carbonyl-Alpha-D-Glucopyranoside: A First Example of Anionic Ring-Opening Polymerization of Five-Membered Cyclic Carbonate without Elimination of CO2. Macromolecules. 2005;38:3562–3563. [Google Scholar]
- 318.Azechi M, Matsumoto K, Endo T. Anionic Ring-Opening Polymerization of a Five-Membered Cyclic Carbonate Having a Glucopyranoside Structure. J Polym Sci, Part A: Polym Chem. 2013;51:1651–1655. [Google Scholar]
- 319.Miura Y, Hoshino Y, Seto H. Glycopolymer Nanobiotechnology. Chem Rev. 2015 doi: 10.1021/acs.chemrev.5b00247. [DOI] [PubMed] [Google Scholar]
- 320.Appelhans D, Klajnert-Maculewicz B, Janaszewska A, Lazniewska J, Voit B. Dendritic Glycopolymers Based on Dendritic Polyamine Scaffolds: View on Their Synthetic Approaches, Characteristics and Potential for Biomedical Applications. Chem Soc Rev. 2015;44:3968–3996. doi: 10.1039/c4cs00339j. [DOI] [PubMed] [Google Scholar]
- 321.Kiessling LL, Grim JC. Glycopolymer Probes of Signal Transduction. Chem Soc Rev. 2013;42:4476–4491. doi: 10.1039/c3cs60097a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Vázquez-Dorbatt V, Lee J, Lin EW, Maynard HD. Synthesis of Glycopolymers by Controlled Radical Polymerization Techniques and Their Applications. ChemBioChem. 2012;13:2478–2487. doi: 10.1002/cbic.201200480. [DOI] [PubMed] [Google Scholar]
- 323.Spain SG, Cameron NR. A Spoonful of Sugar: The Application of Glycopolymers in Therapeutics. Polym Chem. 2011;2:60–68. [Google Scholar]
- 324.Ting SRS, Chen G, Stenzel MH. Synthesis of Glycopolymers and Their Multivalent Recognitions with Lectins. Polym Chem. 2010;1:1392–1412. [Google Scholar]
- 325.Miura Y. Synthesis and Biological Application of Glycopolymers. J Polym Sci, Part A: Polym Chem. 2007;45:5031–5036. [Google Scholar]
- 326.Ladmiral V, Melia E, Haddleton DM. Synthetic Glycopolymers: An Overview. Eur Polym J. 2004;40:431–449. [Google Scholar]
- 327.Owen RM, Gestwicki JE, Young T, Kiessling LL. Synthesis and Applications of End-Labeled Neoglycopolymers. Org Lett. 2002;4:2293–2296. doi: 10.1021/ol0259239. [DOI] [PubMed] [Google Scholar]
- 328.Mowery P, Yang ZQ, Gordon EJ, Dwir O, Spencer AG, Alon R, Kiessling LL. Synthetic Glycoprotein Mimics Inhibit L-Selectin-Mediated Rolling and Promote L-Selectin Shedding. Chem Biol. 2004;11:725–732. doi: 10.1016/j.chembiol.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 329.Godula K, Rabuka D, Nam KT, Bertozzi CR. Synthesis and Microcontact Printing of Dual End-Functionalized Mucin-Like Glycopolymers for Microarray Applications. Angew Chem, Int Ed. 2009;48:4973–4976. doi: 10.1002/anie.200805756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Erdmann L, Uhrich KE. Synthesis and Degradation Characteristics of Salicylic Acid-Derived Poly(anhydride-Esters) Biomaterials. 2000;21:1941–1946. doi: 10.1016/s0142-9612(00)00073-9. [DOI] [PubMed] [Google Scholar]